

Abstract
Background and Purpose
The actions of hydrogen sulfide in human physiology have been extensively studied and, although it is an essential mediator of many biological functions, the underlying molecular mechanisms of its actions are ill-defined. To elucidate the roles of sulfide in inflammation, we have investigated its interactions with human myeloperoxidase (MPO), a major contributor to inflammatory oxidative stress.
Experimental Approach
The interactions of sulfide and MPO were investigated using electron paramagnetic resonance, electronic circular dichroism, UV-vis and stopped-flow spectroscopies.
Key Results
We found favourable reactions between sulfide and the native-ferric enzyme as well as the MPO redox intermediates, ferrous MPO, compound I and compound II. Sulfide was a potent reversible inhibitor of MPO enzymic activity with an IC50 of 1 µM. In addition, the measured second-order rate constants for the reactions of sulfide with compound I [k = (1.1 ± 0.06) × 106 M−1 s−1] and compound II [k = (2.0 ± 0.03) × 105 M−1 s−1] suggest that sulfide is a potential substrate for MPO in vivo.
Conclusion and Implications
Endogenous levels of sulfide are likely to inhibit the activity of circulating and endothelium-bound MPO. The fully reversible inhibition suggests a mediatory role of sulfide on the oxidant-producing function of the enzyme. Furthermore, the efficient HOCl oxidation of sulfide to give polysulfides (recently recognized as important components of sulfide biology) together with MPO-catalysed sulfide oxidation and the lack of interaction between MPO and sulfide oxidation products, predict a modulatory role of MPO in sulfide signalling.
Linked Articles
This article is part of a themed section on Pharmacology of the Gasotransmitters. To view the other articles in this section visit http://dx.doi.org/10.1111/bph.2015.172.issue-6
Introduction
Hydrogen sulfide (and its protonation isomers; commonly called sulfide hereafter) is now a widely recognized low MW signalling molecule and mediator of a variety of human physiological functions (Szabo, 2007; Wang, 2012; Kimura, 2014). Endogenous sulfide is generated by three enzymes: cystathionine-γ-lyase, cystathionine-β-synthase (Singh and Banerjee, 2011) and 3-mercaptopyruvate sulfurtransferase (Shibuya et al., 2009; see also Alexander et al., 2013). In addition, motivated by the reported beneficial physiological effects of sulfide, extensive efforts are now devoted to assess its therapeutic potential, via administration of sulfide-releasing molecules (see Szabo, 2007; Whiteman and Winyard, 2011; Rivers et al., 2012; Wallace et al., 2012).
Of particular relevance to this work, sulfide is gaining increasing attention as a mediator of inflammation (Hegde and Bhatia, 2011; Whiteman and Winyard, 2011; Wallace et al., 2012). However, both pro- and anti-inflammatory properties of sulfide have been reported, in a variety of organs, both in acute and chronic inflammation. The ever increasing controversy around the physiological properties of sulfide is not unique to its role in inflammation as even its in vivo concentrations are highly debated (see Nagy et al., (2014)], which is in part likely to be related to the lack of understanding of the molecular mechanisms of its actions. Some of the biological functions of sulfide are associated with mediation of reactive oxygen species (ROS) production/scavenging and molecular composition. These properties were also suggested to be important in cardiovascular events (Bir and Kevil, 2013; Mani et al., 2013) including protection during reperfusion injury (Elrod et al., 2007; Calvert et al., 2009; King and Lefer, 2011). As we proposed earlier, because of the relatively low biological levels of sulfide compared with other ROS scavengers, direct oxidant capturing is unlikely to play a major role in sulfide-related antioxidant events (Nagy and Winterbourn, 2010). However, sulfide was shown to increase GSH production (Kimura and Kimura, 2004) and mediate enzymic functions [including pro- and antioxidant enzymes and the regulators of their expressions (e.g. Calvert et al., 2009; Sen et al., 2012; Koike et al., 2013; Yang et al., 2013)] via different chemical reactions. Apart from the best studied sulfhydration process of protein thiol residues (Paul and Snyder, 2012), coordination chemical interactions of sulfide with haem proteins and its reducing potential is also gaining ground (Pietri et al., 2011; Miljkovic et al., 2013; Nagy et al., 2014). In fact, these reactions were already investigated in the early 1970s, before the discovery of most of the physiological actions of sulfide, and were focused on toxic effects such as sulfhaemoglobaemia or inhibition of respiration through cytochrome c inactivation (see Pietri et al., 2011).
Myeloperoxidase (MPO) is a haem protein that has been associated with many inflammatory events and cardiovascular diseases (Nussbaum et al., 2013). It is produced in large quantities by leukocytes (such as neutrophils), which are major players in these conditions. The primary function of MPO is the production of reactive oxidants such as hypochlorous acid (HOCl) or hypothiocyanite (OSCN–) (Klebanoff, 1968; van Dalen et al., 1997) that are essential in the clearance of invading pathogens (Klebanoff et al., 2013; Winterbourn and Kettle, 2013). However, uncontrolled production of these oxidants and their reactions with healthy tissue is one of the proposed major contributors of MPO's pathophysiological functions (Klebanoff, 2005; Nussbaum et al., 2013). The detrimental properties are mostly associated with extracellular (circulating or endothelium-bound) MPO. Of particular relevance, where sulfide is proposed to have a protective role, for example during reperfusion injury (Elrod et al., 2007), inhibition of leukocyte adherence (Zanardo et al., 2006), in rheumatoid arthritis (Whiteman et al., 2010), neurodegeneration (Gong et al., 2011) or in atherosclerosis (Mani et al., 2013), active MPO was proposed to act as a protagonist (such as reperfusion injury, Nicholls and Hazen, 2005; leukocyte adherence, Johansson et al., 1997; Wang et al., 2006; rheumatoid arthritis, Stamp et al., 2012; neurodegeneration, Yap et al., 2007 and in atherosclerosis, Heinecke, 1997; Nicholls and Hazen, 2005).
The aim of this study was to investigate the interactions of human MPO with sulfide to assess whether a direct reaction could potentially generate a link between the mediatory roles of MPO and sulfide in the above mentioned physiological events. The enzymic activity of MPO consists of two interrelated cycles via a series of one- and two-electron redox reactions often called the peroxidase and halogenation activities (see Scheme 1). A common first step in both cycles is the reaction of hydrogen peroxide with the ferric enzyme form to give compound I, which contains an oxoiron (IV) haem centre, that is Fe(IV) = O, and a porphyrin π-cation radical. Compound I is reduced by halogenides or SCN− to give the corresponding hypo(pseudo)halous acids and the native-ferric enzyme form in a two-electron redox reaction, which closes the halogenation cycle. Another pathway involves the one-electron reduction of compound I to compound II [an oxoiron(IV) haem]. Typically, compound II is reduced back to the ferric peroxidase in an additional one-electron reduction step to complete the peroxidase cycle.
Figure 10. Scheme.
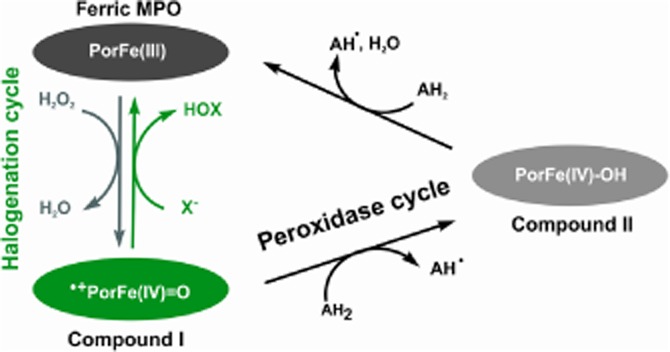
The peroxidase and halogenation cycles of MPO.
We have investigated the interactions of ferric and ferrous MPO, compound I and compound II with sulfide using electron paramagnetic resonance (EPR), electronic circular dichroism (ECD), UV-vis and stopped-flow spectroscopies. We demonstrate favourable interactions of sulfide with all enzyme forms, and our results predicted an efficient and reversible inhibition by sulfide of the catalytic activity of MPO in vivo. In addition, sulfide is likely to be a substrate for compound I and compound II, which together with the previously proposed efficient oxidation of sulfide by the MPO-derived oxidant HOCl, (Nagy and Winterbourn, 2010) predicts a role for MPO in mediating the biological actions of sulfide.
Methods
UV-vis measurements
The recovery of MPO activity after several sulfide oxidation cycles was measured on a Zeiss Specord S10 spectrophotometer (Carl Zeiss Jena GmbH, Germany) and the steady-state enzyme kinetic data were collected on a Bio-Tek Powerwave XS spectrophotometer (Winooski, VT, USA) using quartz cuvettes (1 cm path length).
Steady-state kinetics measurements
The effect of sulfide on the peroxidase activity of MPO was studied by MPO-mediated oxidation of 3,3',5,5'-tetramethylbenzidine (TMB) (Marquez and Dunford, 1997) with slight modifications. Briefly, 6 nM MPO were incubated at 25°C and pH = 5.4 in 100 mM phosphate buffer containing 8% DMF and 1 mM TMB. Reactions were started by adding the indicated amount of H2O2. TMB oxidation was followed spectrophotometrically at 652 nm at various concentrations of sulfide, H2O2 or polysulfides as indicated. The reactions were repeated at least three times for all measurements.
Stopped-flow spectrophotometry
The stopped-flow apparatus (model SX-18MV) equipped for both conventional and sequential stopped-flow measurements was from Applied Photophysics (Surrey, UK). For the 20 µL optical observation cell with 10 mm light path length, the dead time of the instrument is 1.2 ms. All measurements were performed at 25°C and pH = 7.4 in 100 mM phosphate buffer, using a diode-array detector (Applied Photophysics PD.1). At least three determinations were performed for each sample.
Sulfide binding to ferric MPO [followed by reduction of the Fe(III) centre] was measured in the conventional stopped-flow mode. In a typical sulfide binding experiment, one syringe contained 2 µM MPO (200 mM phosphate buffer, pH 7.4), and the second syringe contained at least a 10-fold excess of sulfide.
Ferrous MPO was prepared according to Jantschko et al., (2004). All reaction steps were performed in a glove box (Meca-Plex, Neugebauer, Austria) with a positive pressure of nitrogen (25 mbar, oxygen <3 p.p.m.). Phosphate buffer (100 mM), sulfide and protein solutions (4 µM) were prepared anaerobically by flushing with nitrogen gas (oxygen <3 p.p.m.) for at least 15 min. MPO reduction was achieved by adding 27 µM degassed sodium dithionite solution to 4.5 µM MPO. Although experiments were carefully conducted, very small concentrations of oxygen slowly oxidized ferrous MPO to ferric MPO and the excess of dithionite was consumed by keeping MPO in its ferrous state. Samples were transported to the stopped-flow instrument in gas-tight syringes, where the reaction of sulfide with ferrous MPO was studied using the conventional mixing mode. The reaction was started when dithionite was fully consumed (photometrically tracked by the drop of absorbance at 315 nm) and MPO was still in the ferrous oxidation state.
Because of the inherent instability of compound I, the sequential stopped-flow (multi-mixing) technique was used to measure the rates of the reaction of compound I with sulfide. Typically, MPO (8 µM) was premixed with 80 µM H2O2 and incubated in the ageing loop for 20 ms before the subsequent mixing with sulfide solutions in the second mixing cycle.
Reactivity of compound II with sulfide was also investigated via direct mixing with preformed compound II. In a typical experiment, 8 µM MPO was premixed with 10-fold excess of H2O2 and equimolar homovanillic acid. After a delay time of 40 s, compound II was allowed to react with varying concentrations of sulfide in the second mixing cycle.
Evaluation of kinetic data and kinetic simulation
Polychromatic data were analysed with Pro-Kineticist from Applied Photophysics. The programme simultaneously fits the kinetic traces at all wavelengths to the proposed reaction mechanism and simulates the spectra of all reactant, product and intermediate species as well as their time-dependent distribution on the reaction coordinate.
EPR spectroscopy
EPR measurements were carried out at 10 K on a Bruker EMX continuous wave (cw) spectrometer (Bruker, Germany), operating at X-band (9 GHz) frequencies, equipped with a high-sensitivity resonator and a helium cryostat from Oxford Instruments (ESR900) (UK). EPR spectra were recorded under non-saturating conditions using 2 mW microwave power, 100 kHz modulation frequency, 1 mT modulation amplitude, 20 ms conversion time, 20 ms time constant and 4096 points.
Samples for EPR measurements were prepared at pH = 7.4 in 100 mM phosphate buffer. Addition of sulfide and/or H2O2 to MPO solutions was followed by adding 5% glycerol as a cryoprotectant. One hundred microlitres of these reaction solutions were immediately transferred into a quartz EPR tube (4 mm OD) and frozen in liquid nitrogen. Before measurement, the tube was degassed with argon while the sample was kept frozen on dry ice.
Simulations of high-spin and low-spin Fe(III) forms were carried out using the software EasySpin (Stoll and Schweiger, 2006) and consist of a weighted sum of simulations of the individual high-spin and low-spin compounds. The rhombicity was obtained from gxeff and gyeff (Peisach et al., 1971) and the relative intensities were calculated on the basis of the simulations.
Animals
All animal care and experimental procedures complied with the guidelines of the Canadian Council on Animal Care and were approved by the McMaster University Animal Care Committee. All studies involving animals are reported in accordance with the ARRIVE guidelines for reporting experiments involving animals (Kilkenny et al., 2010; McGrath et al., 2010). A total of 25 animals were used in the experiments described here. Male Wistar rats (200–225g) were obtained from Charles River (Montreal, Canada)
Collection of rat neutrophils
Twenty millilitres of a 1% (w/v) oyster glycogen solution in pH 7.4 PBS (pre-warmed to 37°C) was injected into the peritoneal cavity of rats under isoflurane anaesthesia. Three hours later, the rats were given a lethal overdose of pentobarbital. Twenty millilitres of heparinized (10 U mL−1) PBS (37°C) was injected into the peritoneum. The abdomen was then gently massaged for 1 min. A laparotomy was performed and the peritoneal fluid carefully aspirated using a transfer pipette. The fluid was centrifuged and the pellet was washed three times with cold PBS to remove erythrocytes. The neutrophils were re-suspended in PBS, counted using a haemocytometer, and PBS was added to adjust the concentration to 1 × 107 cells per mL.
Preparation of homogenates of rat colon
Samples of inflamed colon from male Wistar rats (200–225 g) were obtained 3 days after intracolonic treatment with dinitrobenzene sulfonic acid (to induce colitis), as described previously (Wallace et al., 2012). These samples of colon (mean weight 150 mg) were homogenized (Polytron homogeniser for 1 min, on ice) in PBS (pH 7.4; 50 mg/mL−1). Aliquots (0.9 mL) of the homogenized tissue were incubated with 0.1 mL of sulfide solution or vehicle for 7 min. MPO activity was determined, as in the neutrophil lysates (Bradley et al., 1982).
Because considerable sequestering and degradation of sulfide is expected upon addition to biological samples (Flannigan et al., 2013; Nagy et al., 2014), at the end of the period of exposure, a sample was drawn to confirm the levels of ‘acid labile’ sulfide, using the methylene blue (MB) assay in both systems (see Table 2; Stipanuk and Beck, 1982).
Table 2.
Added and measured sulfide concentrations for the rat neutrophil and colon homogenate experiments
| Neutrophils | |
|---|---|
| Added sulfide (µM) | Measured sulfide (µM) |
| 0 | 57 ± 8 |
| 1 | 52 ± 2 |
| 3 | 55 ± 9 |
| 6 | 54 ± 6 |
| 25 | 61 ± 1 |
| 100 | 121 ± 5 |
| 300 | 335 ± 7 |
| 500 | 449 ± 19 |
| Colon homogenate | |
|---|---|
| Added sulfide (µM) | Measured sulfide (µM) |
| 0 | 64 ± 4 |
| 1 | 60 ± 6 |
| 3 | 61 ± 7 |
| 6 | 54 ± 5 |
| 25 | 61 ± 1 |
| 100 | 101 ± 15 |
| 300 | 332 ± 25 |
| 500 | 455 ± 23 |
Added sulfide represents the amount of sulfide that was added to the cells or homogenate extracts. Measured sulfide concentrations were obtained directly from the supernatants after centrifugation by the MB method. Note that the actual sulfide concentrations in the assay solutions are likely to be in the low µM range. We estimate it to be ∼1–2 µM even at 500 µM added sulfide, based on a rough calculation by subtracting the endogenous ‘acid labile’ sulfide levels (measured in the absence of added sulfide) and correcting for the dilution factors (10-fold dilution in HTAB followed by a 30-fold dilution in the assay buffer).
Effects of sulfide on MPO activity in rat neutrophils and colonic tissue homogenates
Aliquots (0.1mL) of the isolated intact neutrophils were exposed to sulfide or vehicle (1µL) for 7 min. They were then added to 0.9 mL of PBS (pH 7.4) containing hexadecyltrimethylammonium bromide (HTAB; 0.5%) and then immediately homogenized with a Polytron disrupter on ice. After centrifugation for 1 min at 9,000× g, MPO activity in the supernatant was measured spectrophotometrically by the dianisidine-H2O2 assay, as described previously (Bradley et al., 1982).
Isolation of neutrophils from human blood
Peripheral venous blood was obtained from healthy adult humans with informed consent. The procedure was approved by the National Ethics Committee under file number BPR-021/00084-2/2014. Neutrophils were isolated by density gradient centrifugation over Ficoll Hypaque, followed by separation from the erythrocytes by dextran sedimentation and hypotonic lysis, as described by Boyum (1968).
The isolated neutrophils were resuspended in 100 mM phosphate buffer (pH = 6.0) containing 0.5% HTAB, at a concentration of 5 × 105 cells per mL and homogenized with a B. Braun Potter S homogenizer to solubilize MPO.
Effects of sulfide on MPO activity in human neutrophil homogenates
MPO peroxidase activity in homogenized neutrophil samples was measured with the TMB assay (see the Steady-state kinetic measurements section) at 25°C with slight modifications. Briefly, the neutrophil homogenate was diluted 50-fold with the assay buffer (100 mM phosphate buffer at pH = 5.4 containing 8% DMF and 1 mM TMB). Reactions were started by the addition of 25 µM H2O2 and TMB oxidation was followed spectrophotometrically at 652 nm in the presence of various concentrations of sulfide (0.5–10 µM).
Data analysis
Results are shown as means ± SEM, unless otherwise stated, from a minimum of 3 assays. Statistical analyses were performed by a one-way analysis of variance, followed by Dunnett's multiple comparison test (P < 0.05 was considered significant) using GraphPad software (Prism, La Jolla, California). Data in Figures represent means ± SEM of 3-6 independent experiments. Representative stopped-flow data, e.g. time dependent spectra, kinetic traces (2000 data points) as well as kobs values for each substrate concentration, were determined at least three times. The second-order rate constants were calculated from the slopes of the linear fits of the kobs-substrate concentration plots using the mean kobs values.
Materials
All reagents and enzymes were purchased from Sigma-Aldrich unless otherwise indicated. Sulfide stock solutions were prepared daily from Na2S × 9H2O in double distilled water. To avoid volatilization, oxidation or interference of photo-induced reactions, stock solutions were kept on ice in the dark using tightly closed plastic bottles. Working solutions were prepared from the stock solution by dilution with phosphate buffer (pH = 7.4) containing 100 µM diethylenetriaminepenta-acetic acid (DTPA; to chelate trace metal ion contaminations) just before the measurements. The concentration of sulfide solutions were determined by measuring their absorbance at 230 nm (ε230 = 7700 M cm−1), and after addition of 5,5'-dithiobis-(2-nitrobenzoic acid) at 412 nm (ε412 = 14100 M cm−1) (Nashef et al., 1977). Highly purified dimeric leukocyte myeloperoxidase (MPO) with a purity index (A428/A280) of at least 0.85 was purchased as lyophilized powder from Planta Natural Products (http://www.planta.at). The concentration of MPO, H2O2 (at pH = 7.4) and hypochlorite (at pH = 12) were determined spectrophotometrically using extinction coefficients of ε428 = 91 000 M cm−1 per haem, ε240 = 43.6 M cm−1 and ε292 = 350 M cm−1 respectively. All other chemicals, buffers and solvents were ACS reagent grade or better.
Results
Interactions of ferric and ferrous MPO with sulfide
To investigate possible reactions of ferric MPO with sulfide we used UV-vis, EPR and ECD spectroscopies for detection of changes at the haem centre of MPO. We observed by UV-vis spectroscopy a sulfide concentration-dependent hypochromic and bathochromic shift of the Soret band from 429 to 432 nm, with a concomitant appearance of a new peak at 625 nm (Figure 1A). The kinetics of this transition is biphasic (see inset to Figure 1A) with both reactions being dependent on the sulfide concentration (with k1 ∼ 3 × 103 M−1 s−1 and k2 25 M−1 s−1 for the faster and slower phases respectively; see Scheme 1). Similar kinetic traces were observed under anaerobic conditions as well as in the presence of 5 mM 5,5-dimethyl-1-pyrroline-N-oxide (DMPO), suggesting that neither O2 nor thiyl radicals are involved in the observed transitions (data not shown).
Figure 1.
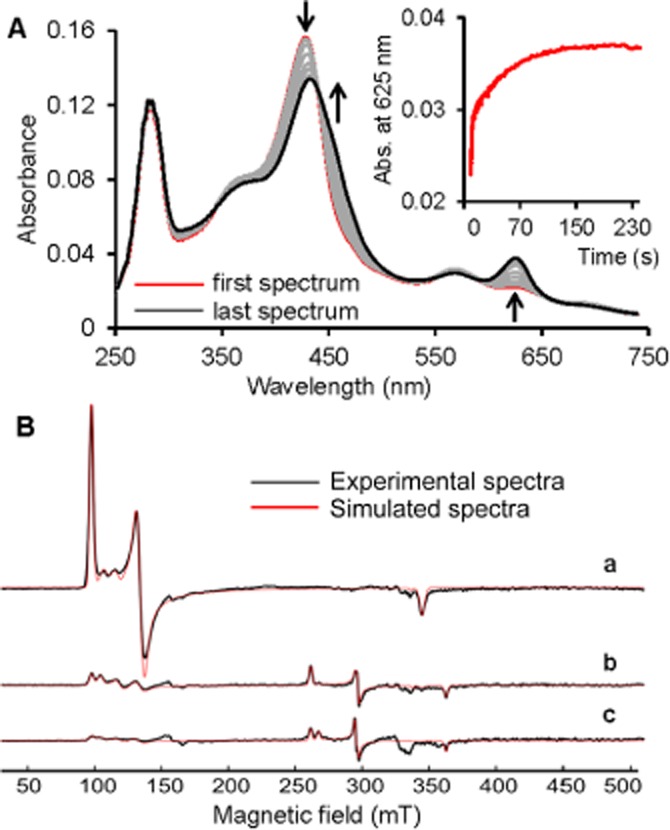
Sulfide binding to ferric MPO observed by stopped-flow and EPR spectroscopies. (A) Spectral changes upon addition of 125 µM sulfide to 2 µM ferric MPO in a conventional stopped-flow experiment. The first spectrum was recorded 0.4 s after mixing, with subsequent spectra taken at 4.35, 10.0, 15.0, 39.7, 66.5, 100.7 and 250 s. Inset shows a typical biphasic time trace at 625 nm. The reaction was carried out in 100 mM phosphate buffer at pH 7.4, and 25°C. (B) Experimental cw EPR spectra (black line) and simulations (red line) of 100 µM MPO with (a) 0 µM (b) 440 µM and (c) 2250 µM sulfide, at pH 7.4. Spectra were recorded at 10 K.
In line with the stopped-flow data, ECD and UV-vis spectroscopies in the visible region revealed a sulfide-dependent loss of Soret ellipticity and absorbance (respectively) without clear isosbestic points when ferric MPO was titrated with increasing amounts of sulfide (Supporting Information Figure S1 A, B). Based on previous data on ferric–haem–sulfide interactions (Nicholls, 1961; English et al., 1984; Pietri et al., 2009; Pietri et al., 2011; Miljkovic et al., 2013), we propose that sulfide binding to ferric MPO forms a low-spin MPOFe(III)–hydrosulfido complex (k1 in Scheme 1) that is subsequently slowly reduced to the MPOFe(II)–H2S complex (k2). Upon plotting the decrease of absorbance at 429 nm versus sulfide concentration (<60 µM) an approximate hyperbolic curve was obtained that allowed the estimation of KD for the MPOFe(III)–hydrosulfido complex to be <12 µM (Supporting Information Figure S1C). Note that the equilibrium constants of consecutive equilibrium processes are dependent on each other; therefore, a more comprehensive study is required to obtain the exact thermodynamic and kinetic constants.
The cw EPR spectrum of MPO is characterized by a rhombically distorted high-spin Fe(III) signal and some smaller contributions of high-spin ferric compounds with less rhombicity [Figure 1B(a) and Table 1]. Upon addition of sulfide (fourfold excess), the intensity of the high-spin area around g = 6 decreased by ∼90% and only ∼17% of the loss of the high-spin signal were detected as a low-spin Fe(III) complex [Figure 1B(b), Table 1]. The g-values of the Fe(III) low-spin signals (Table 1) are similar to those reported for the low-spin sulfide–haem complexes of cytochrome c oxidase (Wever et al., 1975) or Lucina haemoglobin (Kraus et al., 1990). A higher concentration of sulfide [20-fold excess; Figure 1B(c)] further decreased the intensity in the high-spin area and only slightly increased the low-spin Fe(III) intensity. Consequently, at relatively high sulfide concentrations, the formation of the detected low-spin complex is most likely followed by reduction of the Fe(III) centre to give a diamagnetic, EPR-silent species.
Table 1.
EPR simulation parameters from individual high-spin and low-spin forms of MPO with sulfide and H2O2
| HS compoundsa | gxeff | gyeff | gzeff | E/D | R (%) | I (%) | |
|---|---|---|---|---|---|---|---|
| Ferric MPO | HS1 | 5.000 | 6.900 | 1.945 | 0.040 | 12 | 96 |
| HS2 | 5.700 | 6.300 | 1.995 | 0.013 | 4 | 4 | |
| Ferric MPO + 440 µM sulfide | HS1 | 5.000 | 6.900 | 1.945 | 0.040 | 12 | 27 |
| HS2 | 5.605 | 6.443 | 1.995 | 0.018 | 5 | 16 | |
| LS1 | 2.575 | 2.272 | 1.850 | 13 | |||
| LS2 | 2.564 | 2.261 | 1.850 | 38 | |||
| LS3 | 2.508 | 2.245 | 1.850 | 6 | |||
| Ferric MPO + 2.25 mM sulfide | HS1 | 5.000 | 6.840 | 1.945 | 0.038 | 12 | 16 |
| HS2 | 5.605 | 6.443 | 1.995 | 0.018 | 5 | 7 | |
| LS1 | 2.567 | 2.274 | 1.850 | 44 | |||
| LS2 | 2.512 | 2.262 | 1.875 | 33 | |||
| Ferric MPO+ | HS1 | 5.020 | 6.860 | 1.945 | 0.039 | 12 | 97 |
| 100 µM H2O2 | HS2 | 5.700 | 6.300 | 1.995 | 0.012 | 4 | 3 |
| Ferric MPO+ | HS1 | 5.020 | 6.860 | 1.945 | 0.040 | 12 | 87 |
| 10 mM H2O2 | HS2 | 5.150 | 6.650 | 1.945 | 0.030 | 9 | 12 |
| HS3 | 5.800 | 6.280 | 1.995 | 0.009 | 3 | 1 | |
| Ferric MPO+ | HS1 | 5.020 | 6.860 | 1.945 | 0.039 | 12 | 46 |
| 440 µM sulfide+ | HS2 | 5.480 | 6.540 | 1.995 | 0.022 | 7 | 13 |
| 100 µM H2O2 | HS3 | 5.884 | 5.884 | 1.997 | 0 | 0 | 1 |
| LS1 | 2.567 | 2.268 | 1.849 | 20 | |||
| LS2 | 2.567 | 2.260 | 1.849 | 16 | |||
| LS3 | 2.505 | 2.245 | 1.849 | 4 | |||
| Ferric MPO+ | HS1 | 5.000 | 6.870 | 1.945 | 0.040 | 12 | 89 |
| 550 µM sulfide+ | HS2 | 5.260 | 6.580 | 1.975 | 0.028 | 8 | 8 |
| 10 mM H2O2 | HS3 | 5.780 | 6.230 | 1.995 | 0.009 | 3 | 3 |
Minimum number of high-spin and low-spin compounds used for simulation.
E/D, rhombic to axial contribution; HS, high spin; I, relative intensity; LS, low spin; R, rhombicity.
The above stopped-flow, UV-vis and EPR data suggest that sulfide coordination to ferric MPO is followed by the formation of a MPOFe(II)-H2S complex at micromolar concentrations of sulfide. Therefore, we investigated the spectral features of MPOFe(II)-H2S independently via the reaction of ferrous MPO with sulfide (Figure 2). Ferrous MPO is characterized by a Soret maximum at 474 nm and an additional prominent band at 638 nm with a shoulder around 590 nm (Jantschko et al., 2004). Upon addition of sulfide, the formation of a new species was observed with absorbance maxima at 432 nm (shoulder at 473 nm) and 625 nm (green spectrum in Figure 2C). These spectral features closely resemble those of the end products when ferric MPO was reacted with sulfide (red spectrum in Figure 2C), suggesting that the MPOFe(II)–H2S complex is the main product in both reactions of ferric and ferrous MPO with sulfide. A closer inspection of the time traces in Figure 2B indicated that the kinetics of the reaction of ferrous MPO with sulfide was not clearly monophasic, allowing only a rough estimation for the second-order rate constant of k7 ∼2 × 104 M−1 s−1 (see Scheme 1) from the faster phase.
Figure 2.
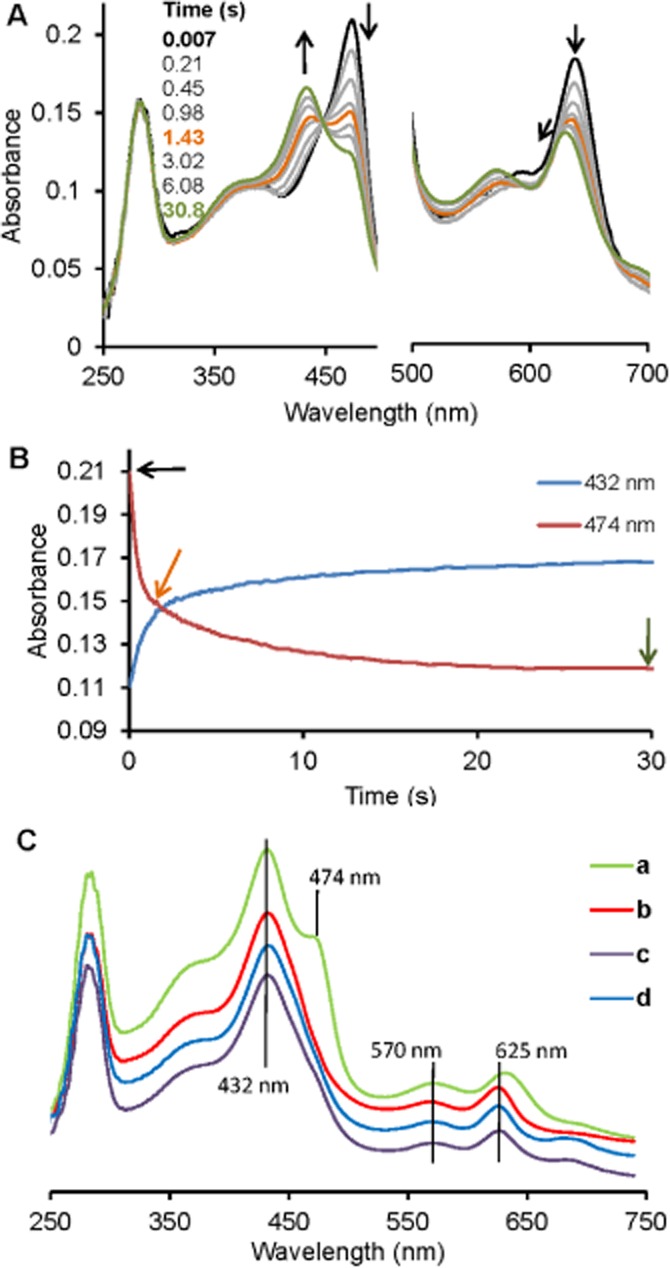
Kinetics of sulfide binding to ferrous MPO monitored by stopped-flow spectroscopy. (A) Spectral changes upon the reaction of 2 µM ferrous MPO with 125 µM sulfide at 25°C in 50 mM phosphate buffer at pH = 7.4 in a conventional stopped-flow experiment. The first spectrum was recorded 7 ms after mixing, with subsequent spectra taken at the indicated time points. The bold black spectrum corresponds to ferrous MPO and arrows indicate the direction of the spectral transitions. The black spectrum was taken at 7 ms, the orange at 1.43 s and the green at 30.8 s. (B) Time traces at 474 and 432 nm, correspond to spectral transitions at (A) with arrows indicating time points where the highlighted spectra in (A) were recorded: (C) Comparison of the spectra of reaction products obtained from the reactions of sulfide with ferrous MPO (a), ferric MPO (b), compound I (c) or compound II (d) at 30.8 s (a), 259 s (b), 1.9 s (c) and 1 s (d) time points.
Sulfide is a potent inhibitor of MPO peroxidase activity
The favourable interaction between native MPO and sulfide predicted a modulatory effect of sulfide on the enzymic activity. Indeed, sulfide was found to be a potent inhibitor of MPO peroxidase activity (Figure 3A) with an IC50 value of ∼1 µM in the 5–60 µM peroxide concentration range (Figure 3B). Because of the promiscuous reactivity of sulfide (Nagy et al., 2014), it is important to confirm that the observed inhibitory effect was on the enzymic turnover and not a result of scavenging TMB oxidation products. When the enzymic reaction was followed until the complete depletion of the added 30 µM peroxide, 1 µM sulfide exhibited its inhibitory effect throughout the reaction, with a similar total final absorbance change (Figure 3C). The facts that (i) the charge-transfer TMB diamine–diimine complex, which is the reporter blue product species in the assay (Josephy et al., 1982), was found to be relatively stable in the presence of sulfide, and that (ii) it is unlikely that 1 µM sulfide could act as an efficient scavenger of as much as 30 µM TMB•+ cation radical [which is an intermediate species in the assay (Josephy et al., 1982)] suggest a direct inhibitory effect on MPO.
Figure 3.
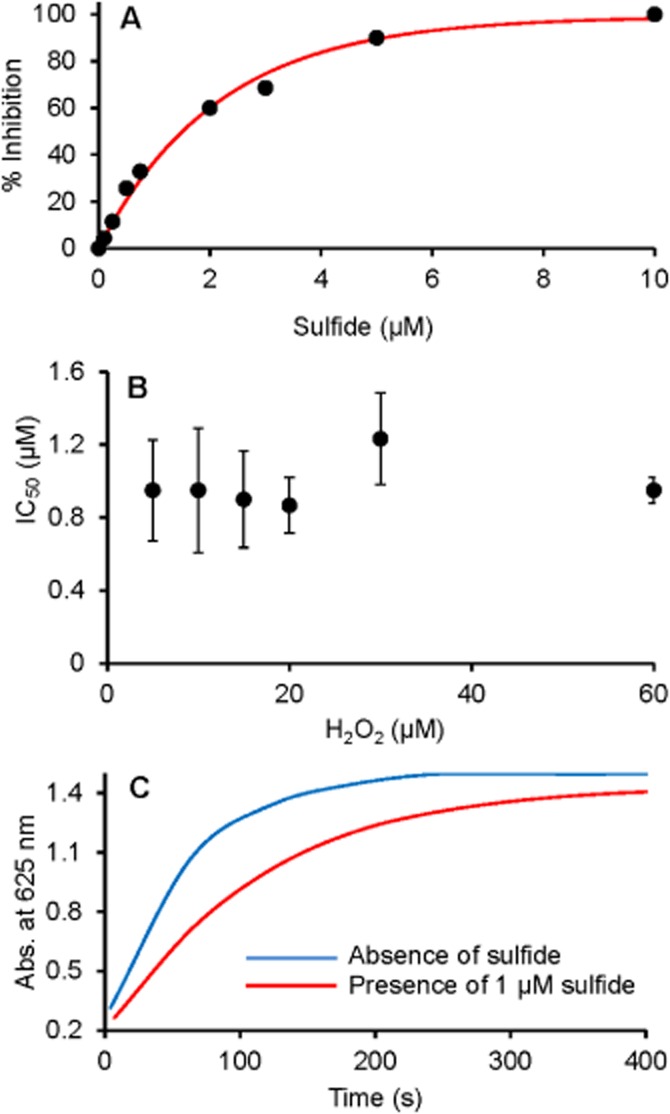
Sulfide concentration-dependent inhibition of MPO peroxidase activity. (A) Representative curve for percentage of inhibition of MPO peroxidase activity at 30 µM H2O2 as a function of added sulfide concentration. (B) Effect of H2O2 concentration on the sulfide-mediated 50% inhibition on MPO peroxidase activity. Data shown are means ± SD of three independent experiments. (C) Representative absorbance changes recorded at 652 nm for the peroxidase activity of MPO in the absence (blue line) and presence of 1 µM sulfide (red line). MPO peroxidase activity was measured by the TMB assay at 6 nM MPO.
A previous study reported less potent MPO peroxidase activity inhibition by sulfide using the guaiacol assay (Laggner et al., 2007). However, in our hands, sulfide inhibition in this assay was just as potent as in the TMB assay (Supporting Information Figure S2A). Laggner et al. incubated MPO with sulfide for 60 min before starting the reactions. As we have shown previously, a substantial loss of sulfide is expected from solutions under these conditions (Nagy et al., 2014), which could contribute to their observed larger IC50 value. In addition, we observed an induction period in the enzyme kinetic traces (Supporting Information Figure S2B), which is an indication that sulfide interferes with the guaiacol assay. However, when the reaction was followed using a H2O2 selective electrode, 1 µM sulfide inhibited the rate of H2O2 consumption by ∼ 50% (Supporting Information Figure S2C), which confirms potent sulfide inhibition of the enzymic activity.
We have shown recently that trace amounts of polysulfide contaminations in sulfide reagent solutions can be responsible for some of the biological effects that have previously been attributed to sulfide (Greiner et al., 2013), and the biological role of polysulfides is gaining increasing attention (Nagy and Winterbourn, 2010; Greiner et al., 2013; Kimura, 2013; Kimura et al., 2013). Therefore, we investigated polysulfide- versus sulfide-mediated inhibition of MPO peroxidase activity. Sulfide reacts with HOCl in a fast reaction to give stoichiometric amounts of polysulfide species (Nagy and Winterbourn, 2010). We measured the relative inhibitory effects of sulfide solutions, treated with or without HOCl, before mixing with MPO. Figure 4 shows that untreated sulfide reagent acts as a potent inhibitor whereas HOCl-treated sulfide lost almost all its inhibitory potential. This observation indicates that inhibition is due to sulfide and that polysulfides have no measurable effect on enzyme catalysis.
Figure 4.

Polysulfide versus sulfide-mediated inhibition of MPO peroxidase activity. Representative absorbance changes recorded at 652 nm for the peroxidase activity of MPO in the absence (blue line) and presence of 2 µM sulfide (green line) or polysulfides (red line). The Figure illustrates potent inhibition by sulfide and an almost complete loss of the inhibitory potential when the sulfide solutions were pretreated with 0.77 µM equivalent of HOCl to consume 75% of the sulfide to give polysulfide species (Nagy and Winterbourn, 2010). MPO peroxidase activity was measured by the TMB assay at 6 nM MPO.
Reaction of sulfide with MPO compound I and II
In order to get a deeper insight into the mechanism of MPO inhibition by sulfide, we used sequential stopped-flow spectrophotometry to investigate the reactions of sulfide with MPO compounds I and II.
Compound I is characterized by a 50% decrease of absorbance in the Soret region. At least a 10-fold excess of H2O2 is needed to get the full hypochromicity at 430 nm. Figure 5A shows the spectral changes of the reaction of 2 µM compound I with 5 µM sulfide. At low sulfide concentrations, compound I is transformed to a compound II-like spectrum with a shift of the Soret maximum to 454 nm and an increase of the absorbance at 625 nm. This transition occurred without clear isosbestic points (see Figure 5A). A typical biphasic kinetic time trace at 456 nm with a double-exponential fit for the reaction of compound I with 5 µM sulfide is shown in Figure 5B. Both obtained pseudo first-order rate constants exhibited a linear dependency on the sulfide concentration (Figures 5C). The apparent second-order rate constants were calculated from the slopes of the linear fits to be (1.1 ± 0.06) × 106 M−1 s−1 for the initial compound II formation (k4 on Scheme 1) and (2.4 ± 0.03) × 105 M−1 s−1 for the second phase respectively. The latter could correspond to a subsequent reaction of compound II with sulfide. Spectral changes depicted in Figure 5A and D indicate that compound II reacts to give a mixture of, most likely, MPOFe(III)–hydrosulfido and MPOFe(II)–H2S complexes. The final spectra are similar to the ones that were obtained when ferric MPO was mixed with sulfide in the absence of peroxide (compare with Figures 1A and 2C).
Figure 5.

Reduction of compound I by sulfide. (A) Spectral changes upon addition of 5 µM sulfide to 2 µM compound I in a sequential-mixing stopped-flow experiment. Compound I was formed by mixing 4 µM MPO with 40 µM H2O2 using a delay time of 20 ms. First spectrum was recorded 9 ms after the second mixing cycle and the subsequent spectra as indicated in the figure. Arrows show the direction of the spectral transitions. (B) Typical biphasic kinetic trace (black line) with a double-exponential fit (red line) showing the reaction of compound I with 5 µM sulfide followed at 456 nm. (C) Dependence of the obtained pseudo first-order rate constants, obtained from the first (green dots) and second (purple dots) phases of the double-exponential kinetic traces upon compound I reduction, on the sulfide concentration. (D) Spectral changes after mixing 2 µM compound I with 20 µM sulfide in a sequential-mixing stopped-flow experiment. Reaction conditions: 100 mM phosphate buffer, pH 7.4, and 25°C.
The kinetics of the reaction of compound II with sulfide was investigated separately using preformed compound II (see Methods). Consistent with the compound I measurements, at low sulfide concentrations, the spectral changes indicate the conversion of compound II to the corresponding mixture of MPOFe(III)–hydrosulfido and MPOFe(II)–H2S complexes (Figure 6A). The lack of isosbestic points is indicative of a multi-step process. The pseudo first-order rate constants for the reaction of compound II with sulfide (that were obtained from the first, faster phase of the kinetic traces with double-exponential fits, see Figure 6B) exhibited linear dependency on sulfide concentration (Figure 6C). The evaluated apparent second-order rate constant (at pH = 7.4) from the slope of the linear fit [(2.0 ± 0.03) × 105 M−1 s−1] is consistent with that of the second phase obtained from the compound I experiments [(2.4 ± 0.03) × 105 M−1 s−1]. Furthermore, this rate constant is similar to the reported second-order rate constant for the reaction of lactoperoxidase (LPO) compound II with sulfide (1.5 × 105 M−1 s−1) (Nakamura et al., 1984).
Figure 6.

Reduction of compound II by sulfide. (A) Spectral changes upon addition of 20 µM sulfide to 2 µM compound II in a sequential-mixing stopped-flow experiment. Compound II was formed by mixing 4 µM MPO with 40 µM H2O2 and 4 µM homovanillic acid using a delay time of 40 s. First spectrum was recorded 9 ms after the second mixing cycle and the subsequent spectra as indicated in the figure. Arrows shows the direction of the spectral transitions. (B) Typical biphasic kinetic trace (black line) with a double-exponential fit (red line) showing the reaction of MPO compound I with 20 µM sulfide followed at 456 nm. (C) Sulfide concentration dependence of the obtained pseudo first-order rate constants for compound II reduction by sulfide. (D) Spectral changes after mixing 2 µM compound II with 200 µM sulfide in a sequential-mixing stopped-flow experiment. Reaction conditions: 100 mM phosphate buffer, pH 7.4 and 25°C.
Upon reaction of compound II with higher sulfide concentrations, the spectral changes in the first phase of the reaction up to 200 ms, indicated the formation of a ferrous MPO-like intermediate species with a pronounced shoulder at ∼473 nm and a shift of the peak maximum of the visible band from 625 to 635 nm (Figure 6D). This observation is suggestive of a two-electron reduction of compound II during the first rapid phase in the reaction of compound II with sulfide (i.e. k6 in Scheme 1). In the second phase of the reaction, from 200 ms up to 20 s, ferrous MPO is converted to the corresponding MPOFe(II)–H2S complex (k7 in Scheme 1). Furthermore, kinetic simulations of the polychromatic data confirm a direct spectral transition from compound II to ferrous MPO followed by the formation of the MPOFe(II)–H2S complex (in both systems, where compound I or compound II reacted with sulfide) at all sulfide concentrations (see Supporting Information Figure S3). However, it cannot be excluded that a small portion of compound II is directly reduced by sulfide to ferric MPO in a one-electron reaction (k5 in Scheme 1). [Note that compound II of MPO was shown to slowly oxidize aliphatic and aromatic thiols (Burner et al., 1999)].
Formation of the MPOFe(II)–H2S complex was found to be independent of the sulfide concentration, but slowed down substantially in oxygen-depleted solutions (Supporting Information Figure S4 and Figure 7, respectively). This might suggest that O2 mediated the oxidation of the MPOFe(II)–H2S complex to the MPOFe(III)–HS− complex (Scheme 1).
Figure 7.
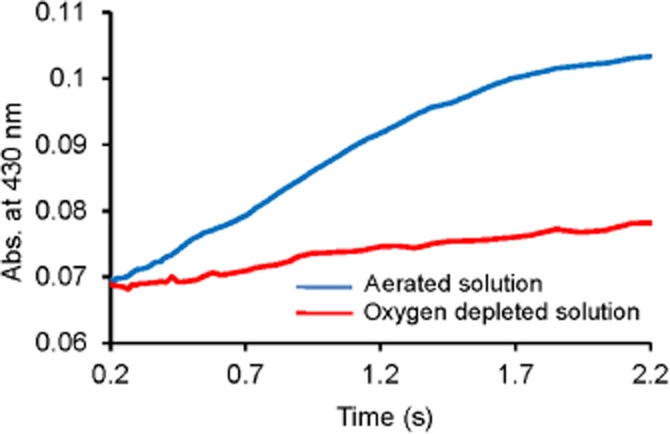
Oxygen-mediated rate-determining reformation of the MPO–sulfide complex mixture. Representative kinetic traces for the reformation of the MPO–sulfide complexes [which we propose is a mixture of MPOFe(III)-hydrosulfido and MPOFe(II)–H2S complexes] recorded at 430 nm in aerated (blue line) and oxygen-depleted (red line) solutions. Conditions: 2 µM compound I with 100 µM sulfide in 100 mM phosphate buffer, pH 7.4 and 25°C. For the anaerobic measurements, nitrogen was bubbled through the solutions for at least 2 h before use.
Sulfide inhibition of MPO is reversible
Some of the potent MPO inhibitors act via irreversible damage to the haem prosthetic group through reactive radical intermediates (see Kettle et al., 1997; Tiden et al., 2011) and some via reversible equilibrium processes (Soubhye et al., 2010; 2013; Aldib et al., 2012; Forbes et al., 2012). Therefore, we have investigated the reversibility of sulfide-mediated inhibition on MPO peroxidase activity. When a mixture of 2 µM MPO and 20 µM sulfide (which is far above the measured IC50) was diluted (333-fold) to reach the enzyme concentration (6 nM) that is used in the peroxidase activity assay, no inhibition was observed (Figure 8A). This dilution results in a 60 nM final sulfide concentration, which is expected to have negligible inhibitory effect under normal assay conditions (i.e. in the absence of preincubation at higher reactant concentrations). Hence, the sulfide that is bound to MPO in the concentrated reaction mixture is released upon dilution. In addition, when the experiment was repeated with 200 µM and 2 mM sulfide pretreatment, the observed inhibitory effects after dilution were proportional to the ones that were observed for the corresponding sulfide levels (600 nM and 6 µM, respectively) in the absence of preincubation at higher reactant concentrations (Figure 8A). These observations confirm that the reaction sequence of ferric MPO → MPOFe(III)–hydrosulfido complex → ferrous–MPOFe(II)–H2S complex is indeed reversible (see Scheme 1). Furthermore, longer incubation of the diluted reaction mixtures resulted in a slow, time-dependent loss of the inhibitory potential of sulfide (Figure 8B). This recovery of MPO peroxidase activity is consistent with the previously observed loss of sulfide (via volatilization of H2S and/or oxidation processes) under these conditions (Nagy et al., 2014).
Figure 8.
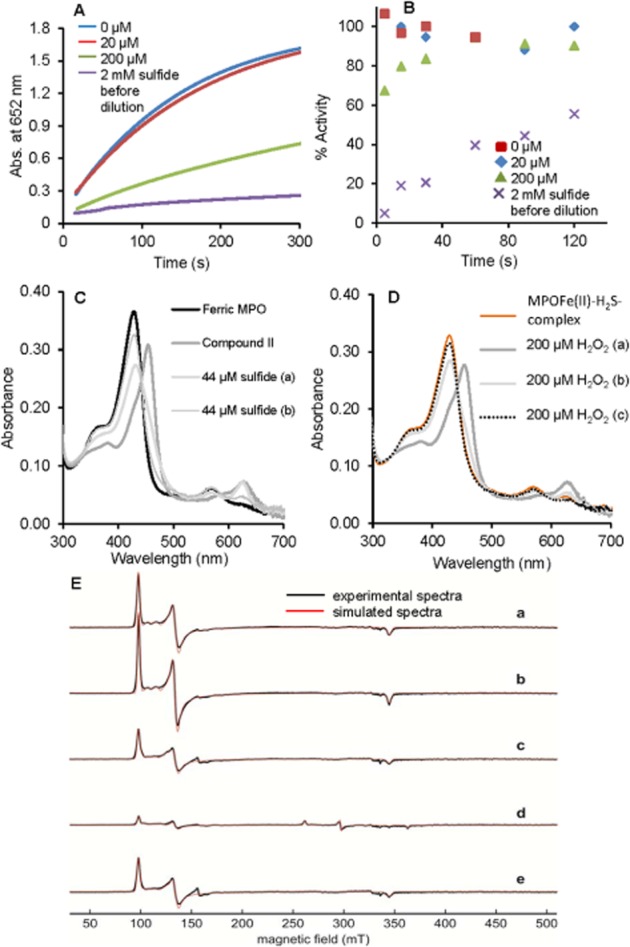
Reversible sulfide inhibition of MPO peroxidase activity. (A) The mixtures of 2 µM MPO with 0 µM (blue), 20 µM (red line), 200 µM (green line) or 2000 µM (purple line) sulfide were diluted 333-fold to a final enzyme concentration of 6 nM (at 25°C and pH 7.4 in 100 mM phosphate buffer). The peroxidase activity of the differently treated MPO solutions were measured as described ∼2 min after dilution. (B) Time-dependent change of the peroxidase activity in the diluted (333-fold as in A) reaction mixtures for 0 µM, 20 µM, 200 µM or 2000 µM sulfide pretreatment. (C) Reaction of MPO compound II with sulfide. Compound II was formed by reaction of 4 µM ferric MPO in 100 mM phosphate buffer pH 7.4 with 70-fold excess of H2O2. Excess of H2O2 was removed after 35 s by adding 10 µg mL−1 bovine liver catalase. Compound II was mixed with 44 µM sulfide and consecutive spectra were obtained at 30 s (trace a) and 10 min (trace b). (D) To the resulting MPO–sulfide complexes that were generated in the reaction of compound II with sulfide (in the experiment shown in C) 200 µM H2O2 was added and subsequent spectra were recorded after 30 s (trace a), 2 min (trace b) and 10 min (trace c). (E) Experimental cw EPR spectra and simulations of 100 µM MPO (a), or 100 µM MPO with (b) 100 µM H2O2, (c) 10 mM H2O2, (d) 100 µM H2O2 and 440 µM sulfide, or (e) 10 mM H2O2 and 550 µM sulfide, at pH 7.4. The spectra were recorded at 10 K.
Irreversible inactivation of MPO can also occur during the catalytic cycle because of the potential formation of damaging radical intermediates. Although, in our peroxidase assay, the reactions of compounds I and II with sulfide are kinetically not competent (because of the large excess of TMB), it is important to establish the effects of sulfide-mediated enzymic turnover on MPO activity, because these reactions could be relevant under physiological conditions (see Discussion). The observed one-electron oxidation of sulfide by Compound I predicts the possible formation of radical intermediates, including the reactive thiyl radical, which could also react with different enzyme forms. Our experiments indicated no formation of new absorbing products, which is suggestive of no irreversible modifications at the active site. Indeed, addition of a second aliquot of peroxide to the re-formed MPOFe(III)–hydrosulfido complex (after catalytic turnover in the presence of sulfide, Figure 8C) resulted in an immediate formation of compound II (Figure 8D), showing that MPO was still fully functional. In addition, we used EPR spectroscopy to investigate the enzyme forms after catalytic turnover in the presence of sulfide. Figure 8E shows the cw EPR spectra of MPO with two different concentrations of H2O2 in the presence and absence of sulfide. Equimolar addition of H2O2 to MPO alone had negligible effect on the EPR spectral characteristics of the enzyme. Even a 100-fold excess of H2O2 resulted in no substantial qualitative changes in the EPR signals, but a ∼40% decrease in the intensity of the high-spin area with a concomitant formation of a small free-radical peak (typical for a protein radical) in the g = 2 region. This loss of intensity could be explained by formation of the EPR silent compounds II and III forms during the redox-cycle of MPO with H2O2. However, when MPO was cycled at an excess of sulfide over H2O2 the intensity of high-spin Fe(III) dropped drastically with the formation of similar low-spin Fe(III) complexes that were detected in the system when MPO and sulfide were reacted in the absence of H2O2. On the other hand, the addition of excess H2O2 (10 mM) over sulfide (550 µM) recovered ∼70% of the high-spin ferric MPO signal (which corresponds to ∼100% recovery when compared with the ferric MPO signal in the presence of 10 mM H2O2 and no sulfide) with no detectable residual low-spin Fe(III) complex (see Table 1). This recovery after several catalytic cycles can be explained by the full consumption of sulfide during the catalytic turnover (Scheme 1) and confirms that sulfide acts as a ligand as well as an electron donor. Furthermore, polysulfides (and other potential MPO-catalysed sulfide oxidation products) do not seem to coordinate to the active site of the enzyme.
Sulfide inhibition of MPO in homogenates of rat neutrophils and inflamed colon
Exposure of rat neutrophils to sulfide for 7 min resulted in a concentration-dependent inhibition of MPO activity, with statistically significant effects at >25 µM added sulfide (Figure 9A). When a similar experiment was performed using homogenized colonic tissue from rats with colitis, inhibition of MPO activity was also observed (Figure 9B). However, in this experiment, a significant inhibition was obtained at higher (>100 µM) sulfide concentrations. As discussed recently, sample maceration and cell lysis affect sulfide-producing and -consuming reactions and hence the actual sulfide levels are expected to vary (Vitvitsky et al., 2012). Therefore, sulfide concentrations in these systems after 7 min incubation were measured using the MB method to be able to assess approximate sulfide levels in the assay solutions. Table 2 shows the measured sulfide concentrations in the incubated biological samples. It is important to note that this method measures the content of the so called ‘acid labile sulfide pool’ that is expected to be much larger than the actual free sulfide concentrations (Shen et al., 2012; Nagy et al., 2014). Based on a rough estimation of the actual sulfide concentrations in the MPO activity assay solutions (by correcting for the endogenous sulfide concentrations and the 300-fold dilution) and taking into account the fully reversible nature of MPO inhibition by sulfide (see Sulfide inhibition of MPO is reversible section), the measured statistically significant inhibition of MPO activity is likely to occur at low or sub-µM sulfide concentrations, as in the isolated enzyme experiments (see Table 2 and Figure 9).
Figure 9.
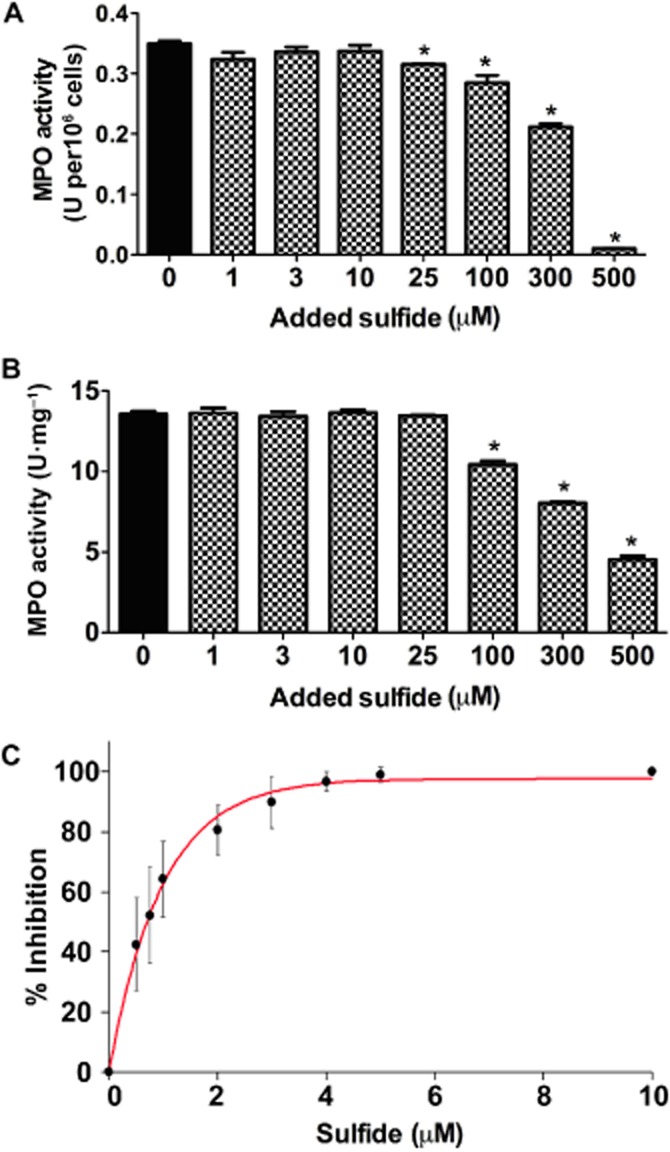
Inhibition of MPO peroxidase activity in (A) rat neutrophil lysates (B) rat inflamed colon homogenates and (C) human neutrophil lysates. (A, B) MPO activity was measured after a 7 min exposure to the indicated amount of added sulfide. However, free sulfide concentrations are expected to be considerably lower based on the measured (by the MB method) ‘acid labile’ sulfide levels in the samples (see Table 2). Furthermore, a 300-fold dilution of the incubated samples preceded the activity measurements (10-fold in HTAB followed by 30-fold in the assay buffer) and therefore, the observed significant inhibition of MPO activity is likely to be in the presence of sub- or low µM sulfide. One unit of MPO activity was defined as that degrading 1 µmol H2O2 min-1 at 25°C. Values are expressed as units of MPO activity per mg tissue or per 106 neutrophils. Each experiment had a sample size of three to five, and they were repeated three times. *P < 0.05, significant effect of sulfide; one-way anova followed by Dunnett's test. (C) Percentage of inhibition of MPO peroxidase activity as a function of added sulfide concentration in human neutrophil lysates, where sulfide was added after dilution to assay conditions. In the absence of added sulfide 100% MPO activity corresponds to 0.27 ± 0.05 unit per 106 cells (for unit definition, see earlier). Data shown are means ± SD of six independent experiments.
Sulfide inhibition of MPO in human neutrophil homogenates
To confirm the reversible nature of sulfide inhibition of MPO activity in biological samples, we measured the effects of sulfide on MPO peroxidase activity in human neutrophil homogenates, where sulfide was added to the diluted homogenate samples just before the activity measurements (Figure 9C). The observed IC50 value of ∼1 µM is similar to that measured for the purified enzyme, which together with the rat neutrophil and tissue homogenate experiments suggest that MPO inhibition will indeed be reversible and dependent on the actual available sulfide levels in a biological environment.
Discussion and conclusions
Interactions of sulfide with ferric MPO
We have found several lines of evidence for a favourable interaction between ferric MPO and sulfide. Our data indicate that sulfide coordination to the Fe(III) centre results in formation of a low-spin MPOFe(III)–hydrosulfido complex that is subsequently reduced to the ferrous counterpart (Scheme 11). The latter reduction step is consistent with previous results showing that sulfide efficiently reduces iron centres of porphyrin systems to the corresponding ferrous–H2S complexes (Pavlik et al., 2010). Furthermore, in haem proteins, Nicholl's and López-Garriga's groups proposed (albeit with different vertebrate proteins such as cytochrome c oxidase, catalase, haemoglobin or myoglobin) that an initial fast coordination process to the ferric centre is followed by a slower reduction to the ferrous form at higher sulfide concentrations (see Scheme 1; Nicholls and Kim, 1981; 1982; Pietri et al., 2009; 2011). Both reactions were found to be reversible, where the rates of ligand binding and release (i.e. k1, k-1) and the concomitant redox reactions (i.e. k2 and k-2) depend on the polarity of the distal side residues at the active site (Pietri et al., 2009; 2011; Nagy et al., 2014). The observed relatively good isosbestic point in the UV-vis and ECD spectra at low sulfide levels and the lack of a simple two-state transition at higher sulfide, together with the formation of similar products in the reactions of ferric and ferrous MPO with sulfide (Figure 2), confirm that sulfide binding to ferric MPO is most likely followed by MPOFe(III) reduction in consecutive reversible reactions (see Scheme 1).
Figure 11. Scheme.
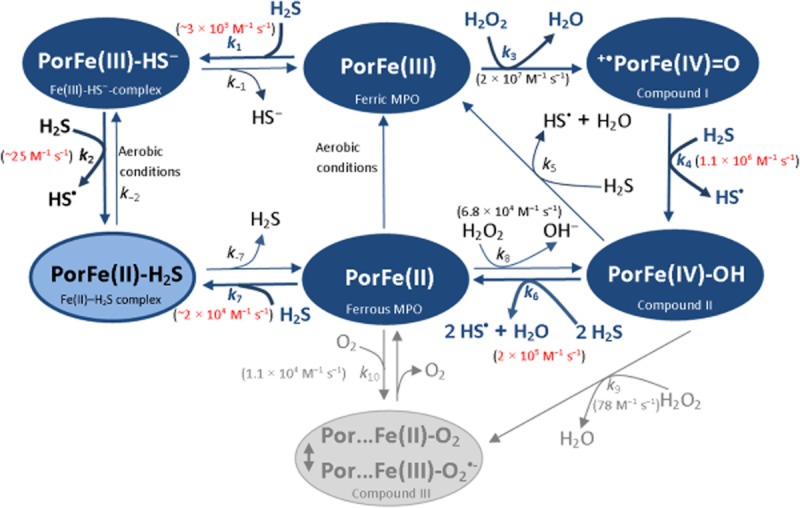
Proposed model for the interactions of sulfide with MPO catalysis. The rate constants for the reactions of sulfide with the different enzyme forms (highlighted with red) are measured values from this study. The model is consistent with all of our experimental data.
Competitive versus non-competitive inhibition of MPO catalysis
Sulfide binding to native MPO suggests competitive inhibition of the reaction of ferric MPO with H2O2 (Scheme 1). However, similar IC50 values were obtained for sulfide inhibition over a wide peroxide concentration range (5–60 µM). Additionally, it is important to note that the reactions of compounds I and II with sulfide will be negligible under the conditions of the peroxidase assay performed in this work. Although the rate constants of compounds I and II with TMB (i.e. 3.6 × 106 and 9.4 × 105 M−1 s−1, respectively) are in the same order of magnitude as with sulfide, even at the largest applied sulfide concentration there was an at least 100-fold excess of TMB over sulfide. Consequently, less than 1% of the peroxide will be consumed for sulfide oxidation under the applied conditions. This is confirmed by the fact that we have observed similar IC50 values at 0.5 and 2 mM TMB (data not shown) suggesting saturating TMB concentrations. Hence, because sulfide inhibition of MPO activity is fully reversible and that the reactions of compounds I and II with sulfide in the peroxidase assay are kinetically not competent, our data strongly suggest dead-end inhibition, via reversible slow equilibria for reactions 1 and 2 (Scheme 1). Although the second-order rate constant for the reaction of ferric MPO with peroxide is almost an order of magnitude faster than the rate constants of compounds I and II with TMB (Marquez and Dunford, 1997), because of the applied large excess of TMB over peroxide in the peroxidase assay (at least a 20-fold excess) the dominating redox state of the enzyme is expected to be the ferric form. Hence the apparent non–competitive-like inhibition of sulfide versus peroxide can be explained by the relatively slow dissociation of the MPOFe(II/III)–sulfide complexes.
Based on this proposed model, a similar effect of sulfide on the initial phase of the chlorination cycle is likely. Our data indicate that MPO-generated oxidants are efficiently captured by sulfide making it difficult to assess the effect of sulfide on the chlorination activity (see Supporting Information Figure S5). Nevertheless, an important conclusion that can be drawn from our attempts on the chlorination activity measurements in the presence of sulfide is that sulfide oxidation by MPO-generated oxidants reactivates the enzyme (see Supporting Information Figure S5). This observation is also consistent with Figure 4 showing that sulfide oxidation products have no effect on the enzymic activity.
Sulfide is a potential substrate for MPO
The large rate constants we obtained for the reactions of sulfide with compounds I and II suggest that sulfide could serve as a substrate for MPO.
Although the second-order rate constant for compound I is comparable with the ones that were measured for the reactions with proposed biological substrates of MPO (such as Cl−, SCN−, Br− or Tyr), because of the relatively small physiological free sulfide levels it is unlikely that sulfide will be able to efficiently compete for compound I in vivo. However, administered sulfide at sites of inflammation could potentiate this reaction.
During enzymic turnover, compound II is generally the least reactive enzyme form, and therefore, it represents the dominating redox state of MPO during its catalytic cycle in vivo. Good peroxidase substrates reduce compound II directly to the ferric enzyme. However, with aliphatic and aromatic thiols (Burner et al., 1999) and sulfide, this reaction is very slow. By contrast, sulfide efficiently mediates the two-electron reduction of compound II to ferrous MPO and the relatively large second-order rate constant obtained for this reaction substantiates its feasibility during MPO catalysis in several physiological conditions (see later).
Our kinetic analysis of the reactions of sulfide with the different enzyme forms allowed us to propose a model for MPO-catalysed sulfide oxidation (Scheme 1), which includes rapid compound I reduction to compound II (k4 = 1.0 × 106 M−1 s−1) followed by reduction of compound II to ferrous MPO (k5 = 2.0 × 105 M−1 s−1). The latter converts to the corresponding MPOFe(II)–H2S complex in its reaction with sulfide (k7 ∼ 2.0 × 104 M−1 s−1).
Importantly, the reactions of sulfide with either MPOFe(III), MPOFe(II), compound I or compound II all result in the formation of the ferrous–MPO–H2S complex, which therefore acts as a sink (shown in light blue in Scheme 1) because it traps MPO in an inactive form when sulfide is present.
Although we could not trap the spectral signatures of a classical MPO compound III species [which could form as a result of the reaction of MPOFe(II) with O2 (k10 in Scheme 1) and having absorbance maxima at 449 and 625 nm] in any of our reactions, we cannot fully exclude it as a potential intermediate species during catalytic turnover.
The slow oxygen-mediated re-oxidation of the MPOFe(II)–H2S to the MPOFe(III)–HS– complex apparently represents the rate limiting step of the catalytic cycle in the presence of sulfide, which is also in favour of our proposed model (Scheme 1). Previous studies that investigated the interactions of sulfide with LPO and catalase proposed similar models (Nicholls, 1961; Nakamura et al., 1984). In the LPO study, it was possible to distinguish between the LPOFe(II)–H2S (with a sharp band at 638 nm) and LPOFe(III)–HS− complexes (absorbance bands at 505 and 727 nm) spectrophotometrically and for both catalase and LPO, similar to our observations, a rate-determining oxidation of the MPOFe(II)–H2S to the MPOFe(III)–HS− complex by oxygen was proposed.
Possible physiological effects of sulfide–MPO interactions
The published free sulfide levels in biological systems (see Nagy et al., 2014) together with the observed favourable interactions of MPO with sulfide predicts that substantial amounts of circulating and endothelium-bound MPO are likely to be in a low-spin–sulfide complex form and thus inhibited under normal physiological conditions. Under these conditions, the steady-state levels of H2O2 are also likely to be very low (Halliwell et al., 2000). However, our results also suggest that consumption of sulfide as a result of inflammation-induced oxidative stress (during which local concentrations of peroxide are likely to temporarily increase by orders of magnitude) can reactivate sulfide-bound MPO, which could exacerbate inflammation-induced damage. However, administered sulfide could restitute MPO inhibition at sites of inflammation or oxidative stress, which may explain some of the observed anti-inflammatory or protective effects. In addition, it is possible that the large amounts of sulfide that are released by bowel bacteria (Flannigan et al., 2011) may represent a protective mechanism against the inflammatory process initiated by the innate immune system, through alleviating MPO-induced oxidative stress.
It is important to emphasize that the kinetic properties of the relevant reactions in the conditions mentioned earlier will have a large effect on the biological outcome. For example, a sulfide-buffering role was proposed (Nagy et al., 2014) for the large biomolecule-bound sulfide pools that were detected in most biological systems beside the relatively low free sulfide levels (in the µM to nM range). However, the nature and chemical properties, including the rate of sulfide release from the different biomolecule–sulfide complexes (representing the buffers) show large variations. Therefore, the efficiency of consumed sulfide replacement (i.e. buffering) will most likely be system-specific with a potential regulatory role. On the other hand, the reversible inhibition of MPO by sulfide could serve as a model for sulfide modulation of endogenous oxidant production and consumption rates, which in light of the well documented redox regulation of cellular signalling could represent an alternative element of sulfide's regulatory role.
Furthermore, oxidation of sulfide by chlorinating oxidants result in polysulfide formation (Nagy and Winterbourn, 2010), which were shown to trigger protein sulfhydration (Greiner et al., 2013). This could affect, among others, the inflammatory process in a different way, for example by modulating the activities of NF-κB or NF-κB repressing factor (Sen et al., 2012; Koike et al., 2013). Hence, the observation that MPO inhibition is via coordination chemical interaction of sulfide rather than its oxidation products and that the oxidant products of MPO can alter the relative amounts of reduced versus oxidized sulfide forms (that will have different biological functions) could serve as a prime example of a fine-tuned signalling system.
Acknowledgments
This work was supported by an FP7-PEOPLE-2010-RG Marie Curie International Reintegration Grant (Grant No. PIRG08-GA-2010-277006), The Hungarian National Science Foundation (OTKA; Grant No. K 109843) and by the Austrian Science Foundation (FWF project P20664 and doctoral programme BioToP W1224). An STSM from COST Action BM1203 supported P. Z. to carry out the stopped-flow and ECD experiments in Vienna. COST Action BM 1005 is acknowledged for covering travel costs for N. P., Kyle Flannigan is acknowledged for technical assistance in measuring sulfide levels for Table 2, and Monika Soudi for her assistance in the preparation of ferrous MPO and studies of its reaction with sulfide. We are grateful for Krisztina Ballagó, Dorottya Garai and Judit Barancsiné Szűcs for their assistance with the isolation of human neutrophils and some steady-state kinetic measurements. Professor Anthony Kettle is acknowledged for valuable advice.
Glossary
- cw
continuous wave
- DMPO
5,5-dimethyl-1-pyrroline-N-oxide
- DTNB
5,5'-dithiobis-(2-nitrobenzoic acid)
- DTPA
diethylenetriaminpenta-acetic acid
- ECD
electronic circular dichroism
- EPR
electron paramagnetic resonance
- HTAB
hexadecyltrimethylammonium bromide
- LPO
lactoperoxidase
- MB
methylene blue
- MPO
myeloperoxidase
- Nrf
NFκB repressing factor
- TMB
3,3',5,5'-tetramethylbenzidine
Author contributions
Z. P. was responsible for the majority of the experimental work and he was involved in data analyses and contributed to the writing of the paper. P. G. F. contributed to the stopped-flow and ECD experiments, data analyses and writing of the paper. A. N. carried out, or assisted to most of the steady-state enzyme kinetic experiments. C. J. performed the experiments in Figure 8C&D and contributed to the stopped-flow results. K. F. P. was responsible for the EPR results. M. M. and K. J. performed the rat colon homogenate and rat neutrophil experiments on Figure 9A&B. J. L. W. contributed to the design and interpretation of the experiments in Figure 9A&B and Table 2. C. O. contributed to the design and interpretation of experimental results, and to the editing of the paper. P. N. conceived the study and had overall responsibility for the project; he contributed to the design and interpretation of experimental results and wrote the paper.
Conflict of interest
The authors declare that there are no conflicts of interests.
Supporting Information
Appendix S1 Supporting methods.
Figure S1 Sulfide binding to ferric myeloperoxidase observed by electronic circular dichroism (ECD) and UV-vis spectroscopies. (A, B) Spectral changes of haem ECD spectra (A) and haem Soret absorption (B) during the titration of 7 µM MPO in 5 mM phosphate buffer at pH = 7.4 with different sulfide concentrations (3, 6.5, 10.5, 14, 17.5, 28, 38, 52, 70, 104, 139, 193, 275, 408 and 571 µM). (C) Plot of the measured differences in absorbance values at 429 nm upon the titration of ferric MPO with sulfide compared with the pure ferric MPO spectrum. The red line represents the rectangular hyperbola fit to equation f = a × x/(b + x). Inset to Figure C shows the isosbestic point at 444 nm during the spectral titration (at <60 µM sulfide).
Figure S2 Inhibition of MPO peroxidase activity. (A) A similar curve was obtained for sulfide-mediated percentage of inhibition of the peroxidase activity using the guaiacol assay as with the TMB method (compared with Figure 3A of the main text). (B) The obtained kinetic traces exhibit a sulfide-dependent induction period indicating interference with the photometric detection: in the absence of sulfide (dark blue line), or in the presence of 0.25 µM (green line), 0.5 µM (red line), 1.0 µM (black line) and 4.0 µM (purple line) sulfide. (C) 1.0 µM sulfide (red line) inhibits H2O2 consumption by ∼50% compared with when no sulfide was added (purple line) as measured by a peroxide selective electrode, which confirms direct inhibition by sulfide on the enzymic activity. Green line, representing the control sample, was recorded under similar conditions to the red line with the exception that MPO was not present. For experimental details see Supporting Information Appendix S1.
Figure S3 Simulation of compound I and compound II reduction with sulfide. (A) Simulated spectra of the different enzyme forms obtained by singular value decomposition analysis of the polychromatic data that were collected for the reduction of compound I (A) or compound II (C) by 20 µM sulfide. Calculated concentration distribution diagram of redox intermediates during the course of the reaction of compound I (B) or compound II (D) with 20 µM sulfide. Reaction conditions are similar as in Figure 5 (for A&B) or Figure 6 (for C&D).
Figure S4 Sulfide dependency of the obtained pseudo first-order rate constants for the rate determining reformation of MPO–sulfide complexes [which we propose is a mixture of MPOFe(III)-hydrosulfido and MPOFe(II)–H2S complexes]. Rate constants were obtained by an exponential fit of the slowest phase of the kinetic traces that were recorded at 430 nm in aerated solutions for the reactions of compound I (blue points) and compound II (red points) with sulfide. Conditions: 2 µM compound I or compound II were mixed with the corresponding concentrations of sulfide in 100 mM phosphate buffer, pH 7.4, and 25°C in a sequential-mixing stopped-flow experiment (for details, see the Methods section in the main text).
Figure S5 Effect of sulfide on the halogenation activity of MPO. Kinetic curves indicate that sulfide consumption by MPO-generated glycine chloramine during the assay results in full recovery of the enzymic activity, which has potential biological implications (see the Discussion and conclusions section in the main text). Kinetic curves are representative of traces that were recorded in the absence of sulfide (blue line), in the presence of 1 µM (red line), or 4 µM (green line) sulfide. For experimental details see Supporting Information Appendix S1.
References
- Aldib I, Soubhye J, Zouaoui Boudjeltia K, Vanhaeverbeek M, Rousseau A, Furtmüller PG, et al. Evaluation of new scaffolds of myeloperoxidase inhibitors by rational design combined with high-throughput virtual screening. J Med Chem. 2012;55:7208–7218. doi: 10.1021/jm3007245. [DOI] [PubMed] [Google Scholar]
- Alexander SPH, Benson HE, Faccenda E, Pawson AJ, Sharman JL, Spedding M, et al. The Concise Guide to PHARMACOLOGY 2013/14: Enzymes. Br J Pharmacol. 2013;170:1797–1867. doi: 10.1111/bph.12451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bir SC, Kevil CG. Sulfane sustains vascular health: insights into cystathionine gamma-lyase function. Circulation. 2013;127:2472–2474. doi: 10.1161/CIRCULATIONAHA.113.003489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyum A. Isolation of mononuclear cells and granulocytes from human blood – isolation of mononuclear cells by one centrifugation and of granulocytes by combining centrifugation and sedimentation at L G. Scand J Clin Lab Invest Suppl. 1968;21:77–89. [PubMed] [Google Scholar]
- Bradley PP, Priebat DA, Christensen RD, Rothstein G. Measurement of cutaneous inflammation: estimation of neutrophil content with an enzyme marker. J Invest Dermatol. 1982;78:206–209. doi: 10.1111/1523-1747.ep12506462. [DOI] [PubMed] [Google Scholar]
- Burner U, Jantschko W, Obinger C. Kinetics of oxidation of aliphatic and aromatic thiols by myeloperoxidase compounds I and II. FEBS Lett. 1999;443:290–296. doi: 10.1016/s0014-5793(98)01727-x. [DOI] [PubMed] [Google Scholar]
- Calvert JW, Jha S, Gundewar S, Elrod JW, Ramachandran A, Pattillo CB, et al. Hydrogen sulfide mediates cardioprotection through Nrf2 signaling. Circ Res. 2009;105:365–374. doi: 10.1161/CIRCRESAHA.109.199919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Dalen CJ, Whitehouse MW, Winterbourn CC, Kettle AJ. Thiocyanate and chloride as competing substrates for myeloperoxidase. Biochem J. 1997;327(Pt 2):487–492. doi: 10.1042/bj3270487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elrod JW, Calvert JW, Morrison J, Doeller JE, Kraus DW, Tao L, et al. Hydrogen sulfide attenuates myocardial ischemia-reperfusion injury by preservation of mitochondrial function. Proc Natl Acad Sci USA. 2007;104:15560–15565. doi: 10.1073/pnas.0705891104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- English DR, Hendrickson DN, Suslick KS, Eigenbrot CW, Scheidt WR. Low-spin five-coordinate ferric porphyrin complex: [5, 10, 15, 20-tetrakis (4-methoxyphenyl) porphyrinato] (hydrosulfido) iron (III) J Am Chem Soc. 1984;106:7258–7259. [Google Scholar]
- Flannigan KL, McCoy KD, Wallace JL. Eukaryotic and prokaryotic contributions to colonic hydrogen sulfide synthesis. Am J Physiol. 2011;301:G188–G193. doi: 10.1152/ajpgi.00105.2011. [DOI] [PubMed] [Google Scholar]
- Flannigan KL, Ferraz JGP, Wang R, Wallace JL. Enhanced synthesis and diminished degradation of hydrogen sulfide in experimental colitis: a site-specific, pro-resolution mechanism. PLoS ONE. 2013;8:e71962. doi: 10.1371/journal.pone.0071962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forbes LV, Furtmüller PG, Khalilova I, Turner R, Obinger C, Kettle AJ. Isoniazid as a substrate and inhibitor of myeloperoxidase: identification of amine adducts and the influence of superoxide dismutase on their formation. Biochem Pharmacol. 2012;84:949–960. doi: 10.1016/j.bcp.2012.07.020. [DOI] [PubMed] [Google Scholar]
- Gong QH, Shi XR, Hong ZY, Pan LL, Liu XH, Zhu YZ. A new hope for neurodegeneration: possible role of hydrogen sulfide. J Alzheimers Dis. 2011;24(Suppl. 2):173–182. doi: 10.3233/JAD-2011-110128. [DOI] [PubMed] [Google Scholar]
- Greiner R, Palinkas Z, Basell K, Becher D, Antelmann H, Nagy P, et al. Polysulfides link H2S to protein thiol oxidation. Antioxid Redox Signal. 2013;19:1749–1765. doi: 10.1089/ars.2012.5041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halliwell B, Clement MV, Longa LH. Hydrogen peroxide in the human body. FEBS Lett. 2000;486:10–13. doi: 10.1016/s0014-5793(00)02197-9. [DOI] [PubMed] [Google Scholar]
- Hegde A, Bhatia M. Hydrogen sulfide in inflammation: friend or foe? Inflamm Allergy Drug Targets. 2011;10:118–122. doi: 10.2174/187152811794776268. [DOI] [PubMed] [Google Scholar]
- Heinecke JW. Mechanisms of oxidative damage of low density lipoprotein in human atherosclerosis. Curr Opin Lipidol. 1997;8:268–274. doi: 10.1097/00041433-199710000-00005. [DOI] [PubMed] [Google Scholar]
- Jantschko W, Furtmüller PG, Zederbauer M, Jakopitsch C, Obinger C. Kinetics of oxygen binding to ferrous myeloperoxidase. Arch Biochem Biophys. 2004;426:91–97. doi: 10.1016/j.abb.2004.03.019. [DOI] [PubMed] [Google Scholar]
- Johansson MW, Patarroyo M, Oberg F, Siegbahn A, Nilsson K. Myeloperoxidase mediates cell adhesion via the alpha M beta 2 integrin (Mac-1, CD11b/CD18) J Cell Sci. 1997;110(Pt 9):1133–1139. doi: 10.1242/jcs.110.9.1133. [DOI] [PubMed] [Google Scholar]
- Josephy PD, Eling T, Mason RP. The horseradish peroxidase-catalyzed oxidation of 3,5,3',5'-tetramethylbenzidine. Free radical and charge-transfer complex intermediates. J Biol Chem. 1982;257:3669–3675. [PubMed] [Google Scholar]
- Kettle AJ, Gedye CA, Winterbourn CC. Mechanism of inactivation of myeloperoxidase by 4-aminobenzoic acid hydrazide. Biochem J. 1997;321:503–508. doi: 10.1042/bj3210503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kilkenny C, Browne W, Cuthill IC, Emerson M, Altman DG. Animal research: reporting in vivo experiments: the ARRIVE guidelines. Br J Pharmacol. 2010;160:1577–1579. doi: 10.1111/j.1476-5381.2010.00872.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimura H. Physiological role of hydrogen sulfide and polysulfide in the central nervous system. Neurochem Int. 2013;63:492–497. doi: 10.1016/j.neuint.2013.09.003. [DOI] [PubMed] [Google Scholar]
- Kimura H. Production and physiological effects of hydrogen sulfide. Antioxid Redox Signal. 2014;20:783–793. doi: 10.1089/ars.2013.5309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimura Y, Kimura H. Hydrogen sulfide protects neurons from oxidative stress. FASEB J. 2004;18:1165–1167. doi: 10.1096/fj.04-1815fje. [DOI] [PubMed] [Google Scholar]
- Kimura Y, Mikami Y, Osumi K, Tsugane M, Oka J, Kimura H. Polysulfides are possible H2S-derived signaling molecules in rat brain. FASEB J. 2013;27:2451–2457. doi: 10.1096/fj.12-226415. [DOI] [PubMed] [Google Scholar]
- King AL, Lefer DJ. Cytoprotective actions of hydrogen sulfide in ischaemia-reperfusion injury. Exp Physiol. 2011;96:840–846. doi: 10.1113/expphysiol.2011.059725. [DOI] [PubMed] [Google Scholar]
- Klebanoff SJ. Myeloperoxidase–halide–hydrogen peroxide antibacterial system. J Bacteriol. 1968;95:2131–2138. doi: 10.1128/jb.95.6.2131-2138.1968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klebanoff SJ. Myeloperoxidase: friend and foe. J Leukoc Biol. 2005;77:598–625. doi: 10.1189/jlb.1204697. [DOI] [PubMed] [Google Scholar]
- Klebanoff SJ, Kettle AJ, Rosen H, Winterbourn CC, Nauseef WM. Myeloperoxidase: a front-line defender against phagocytosed microorganisms. J Leukoc Biol. 2013;93:185–198. doi: 10.1189/jlb.0712349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koike S, Ogasawara Y, Shibuya N, Kimura H, Ishii K. Polysulfide exerts a protective effect against cytotoxicity caused by t-buthylhydroperoxide through Nrf2 signaling in neuroblastoma cells. FEBS Lett. 2013;587:3548–3555. doi: 10.1016/j.febslet.2013.09.013. [DOI] [PubMed] [Google Scholar]
- Kraus DW, Wittenberg JB, Lu JF, Peisach J. Hemoglobins of the Lucina pectinata/bacteria symbiosis. II. An electron paramagnetic resonance and optical spectral study of the ferric proteins. J Biol Chem. 1990;265:16054–16059. [PubMed] [Google Scholar]
- Laggner H, Muellner MK, Schreier S, Sturm B, Hermann M, Exner M, et al. Hydrogen sulphide: a novel physiological inhibitor of LDL atherogenic modification by HOC1. Free Radical Res. 2007;41:741–747. doi: 10.1080/10715760701263265. [DOI] [PubMed] [Google Scholar]
- Mani S, Li H, Untereiner A, Wu L, Yang G, Austin RC, et al. Decreased endogenous production of hydrogen sulfide accelerates atherosclerosis. Circulation. 2013;127:2523–2534. doi: 10.1161/CIRCULATIONAHA.113.002208. [DOI] [PubMed] [Google Scholar]
- Marquez LA, Dunford HB. Mechanism of the oxidation of 3,5,3',5'-tetramethylbenzidine by myeloperoxidase determined by transient- and steady-state kinetics. Biochemistry. 1997;36:9349–9355. doi: 10.1021/bi970595j. [DOI] [PubMed] [Google Scholar]
- McGrath J, Drummond G, McLachlan E, Kilkenny C, Wainwright C. Guidelines for reporting experiments involving animals: the ARRIVE guidelines. Br J Pharmacol. 2010;160:1573–1576. doi: 10.1111/j.1476-5381.2010.00873.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miljkovic JL, Kenkel I, Ivanović-Burmazović I, Filipovic MR. Generation of HNO and HSNO from nitrite by heme-iron-catalyzed metabolism with H2S. Angew Chem Int Ed. 2013;52:12061–12064. doi: 10.1002/anie.201305669. [DOI] [PubMed] [Google Scholar]
- Nagy P, Winterbourn CC. Rapid reaction of hydrogen sulfide with the neutrophil oxidant hypochlorous acid to generate polysulfides. Chem Res Toxicol. 2010;23:1541–1543. doi: 10.1021/tx100266a. [DOI] [PubMed] [Google Scholar]
- Nagy P, Palinkas Z, Nagy A, Budai B, Toth I, Vasas A. Chemical aspects of hydrogen sulfide measurements in physiological samples. Biochim Biophys Acta. 2014;1840:876–891. doi: 10.1016/j.bbagen.2013.05.037. [DOI] [PubMed] [Google Scholar]
- Nakamura S, Nakamura M, Yamazaki I, Morrison M. Reactions of ferryl lactoperoxidase (compound-Ii) with sulfide and sulfhydryl compounds. J Biol Chem. 1984;259:7080–7085. [PubMed] [Google Scholar]
- Nashef AS, Osuga DT, Feeney RE. Determination of hydrogen sulfide with 5,5'-dithiobis-(2-nitrobenzoic acid), N-ethylmaleimide, and parachloromercuribenzoate. Anal Biochem. 1977;79:394–405. doi: 10.1016/0003-2697(77)90413-4. [DOI] [PubMed] [Google Scholar]
- Nicholls P. The formation and properties of sulphmyoglobin and sulphcatalase. Biochem J. 1961;81:374–383. doi: 10.1042/bj0810374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicholls P, Kim JK. Oxidation of sulphide by cytochrome aa3. Biochim Biophys Acta. 1981;637:312–320. doi: 10.1016/0005-2728(81)90170-5. [DOI] [PubMed] [Google Scholar]
- Nicholls P, Kim JK. Sulphide as an inhibitor and electron donor for the cytochrome c oxidase system. Can J Biochem. 1982;60:613–623. doi: 10.1139/o82-076. [DOI] [PubMed] [Google Scholar]
- Nicholls SJ, Hazen SL. Myeloperoxidase and cardiovascular disease. Arterioscler Thromb Vasc Biol. 2005;25:1102–1111. doi: 10.1161/01.ATV.0000163262.83456.6d. [DOI] [PubMed] [Google Scholar]
- Nussbaum C, Klinke A, Adam M, Baldus S, Sperandio M. Myeloperoxidase: a leukocyte-derived protagonist of inflammation and cardiovascular disease. Antioxid Redox Signal. 2013;18:692–713. doi: 10.1089/ars.2012.4783. [DOI] [PubMed] [Google Scholar]
- Paul BD, Snyder SH. H2S signalling through protein sulfhydration and beyond. Nat Rev Mol Cell Biol. 2012;13:499–507. doi: 10.1038/nrm3391. [DOI] [PubMed] [Google Scholar]
- Pavlik JW, Noll BC, Oliver AG, Schulz CE, Scheidt WR. Hydrosulfide (HS–) coordination in iron porphyrinates. Inorg Chem. 2010;49:1017–1026. doi: 10.1021/ic901853p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peisach J, Blumberg WE, Ogawa S, Rachmilewitz EA, Oltzik R. The effects of protein conformation on the heme symmetry in high spin ferric heme proteins as studied by electron paramagnetic resonance. J Biol Chem. 1971;246:3342–3355. [PubMed] [Google Scholar]
- Pietri R, Lewis A, Leon RG, Casabona G, Kiger L, Yeh SR, et al. Factors controlling the reactivity of hydrogen sulfide with hemeproteins. Biochemistry. 2009;48:4881–4894. doi: 10.1021/bi801738j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pietri R, Roman-Morales E, Lopez-Garriga J. Hydrogen sulfide and hemeproteins: knowledge and mysteries. Antioxid Redox Signal. 2011;15:393–404. doi: 10.1089/ars.2010.3698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rivers JR, Badiei A, Bhatia M. Hydrogen sulfide as a therapeutic target for inflammation. Expert Opin Ther Targets. 2012;16:439–449. doi: 10.1517/14728222.2012.673591. [DOI] [PubMed] [Google Scholar]
- Sen N, Paul BD, Gadalla MM, Mustafa AK, Sen T, Xu R, et al. Hydrogen sulfide-linked sulfhydration of NF-kappaB mediates its antiapoptotic actions. Mol Cell. 2012;45:13–24. doi: 10.1016/j.molcel.2011.10.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen X, Peter EA, Bir S, Wang R, Kevil CG. Analytical measurement of discrete hydrogen sulfide pools in biological specimens. Free Radic Biol Med. 2012;52:2276–2283. doi: 10.1016/j.freeradbiomed.2012.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shibuya N, Tanaka M, Yoshida M, Ogasawara Y, Togawa T, Ishii K, et al. 3-Mercaptopyruvate sulfurtransferase produces hydrogen sulfide and bound sulfane sulfur in the brain. Antioxid Redox Signal. 2009;11:703–714. doi: 10.1089/ars.2008.2253. [DOI] [PubMed] [Google Scholar]
- Singh S, Banerjee R. PLP-dependent H2S biogenesis. Biochim Biophys Acta. 2011;1814:1518–1527. doi: 10.1016/j.bbapap.2011.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soubhye J, Prevost M, Van Antwerpen P, Zouaoui Boudjeltia K, Rousseau A, Furtmüller PG, et al. Structure-based design, synthesis, and pharmacological evaluation of 3-(aminoalkyl)-5-fluoroindoles as myeloperoxidase inhibitors. J Med Chem. 2010;53:8747–8759. doi: 10.1021/jm1009988. [DOI] [PubMed] [Google Scholar]
- Soubhye J, Aldib I, Elfving B, Gelbcke M, Furtmüller PG, Podrecca M, et al. Design, synthesis, and structure-activity relationship studies of novel 3-alkylindole derivatives as selective and highly potent myeloperoxidase inhibitors. J Med Chem. 2013;56:3943–3958. doi: 10.1021/jm4001538. [DOI] [PubMed] [Google Scholar]
- Stamp LK, Khalilova I, Tarr JM, Senthilmohan R, Turner R, Haigh RC, et al. Myeloperoxidase and oxidative stress in rheumatoid arthritis. Rheumatology. 2012;51:1796–1803. doi: 10.1093/rheumatology/kes193. [DOI] [PubMed] [Google Scholar]
- Stipanuk MH, Beck PW. Characterization of the enzymic capacity for cysteine desulfhydration in liver and kidney of the rat. Biochem J. 1982;206:267–277. doi: 10.1042/bj2060267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stoll S, Schweiger AJ. EasySpin, a comprehensive software package for spectral simulation and analysis in EPR. J Magn Reson. 2006;178:42–55. doi: 10.1016/j.jmr.2005.08.013. [DOI] [PubMed] [Google Scholar]
- Szabo C. Hydrogen sulphide and its therapeutic potential. Nat Rev Drug Discov. 2007;6:917–935. doi: 10.1038/nrd2425. [DOI] [PubMed] [Google Scholar]
- Tiden AK, Sjogren T, Svensson M, Bernlind A, Senthilmohan R, Auchere F, et al. 2-Thioxanthines are mechanism-based inactivators of myeloperoxidase that block oxidative stress during inflammation. J Biol Chem. 2011;286:37578–37589. doi: 10.1074/jbc.M111.266981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vitvitsky V, Kabil O, Banerjee R. High turnover rates for hydrogen sulfide allow for rapid regulation of its tissue concentrations. Antioxid Redox Signal. 2012;17:22–31. doi: 10.1089/ars.2011.4310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallace JL, Ferraz JG, Muscara MN. Hydrogen sulfide: an endogenous mediator of resolution of inflammation and injury. Antioxid Redox Signal. 2012;17:58–67. doi: 10.1089/ars.2011.4351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang JG, Mahmud SA, Nguyen J, Slungaard A. Thiocyanate-dependent induction of endothelial cell adhesion molecule expression by phagocyte peroxidases: a novel HOSCN-specific oxidant mechanism to amplify inflammation. J Immunol. 2006;177:8714–8722. doi: 10.4049/jimmunol.177.12.8714. [DOI] [PubMed] [Google Scholar]
- Wang R. Physiological implications of hydrogen sulfide: a whiff exploration that blossomed. Physiol Rev. 2012;92:791–896. doi: 10.1152/physrev.00017.2011. [DOI] [PubMed] [Google Scholar]
- Wever R, Van Gelder BF, Dervartanian DV. Biochemical and biophysical studies on cytochrome c oxidase XX. Reaction with sulphide. Biochim Biophys Acta. 1975;387:189–193. doi: 10.1016/0005-2728(75)90102-4. [DOI] [PubMed] [Google Scholar]
- Whiteman M, Winyard PG. Hydrogen sulfide and inflammation: the good, the bad, the ugly and the promising. Expert Rev Clin Pharmacol. 2011;4:13–32. doi: 10.1586/ecp.10.134. [DOI] [PubMed] [Google Scholar]
- Whiteman M, Haigh R, Tarr JM, Gooding KM, Shore AC, Winyard PG. Detection of hydrogen sulfide in plasma and knee-joint synovial fluid from rheumatoid arthritis patients: relation to clinical and laboratory measures of inflammation. Ann N Y Acad Sci. 2010;1203:146–150. doi: 10.1111/j.1749-6632.2010.05556.x. [DOI] [PubMed] [Google Scholar]
- Winterbourn CC, Kettle AJ. Redox reactions and microbial killing in the neutrophil phagosome. Antioxid Redox Signal. 2013;18:642–660. doi: 10.1089/ars.2012.4827. [DOI] [PubMed] [Google Scholar]
- Yang G, Zhao K, Ju Y, Mani S, Cao Q, Puukila S, et al. Hydrogen sulfide protects against cellular senescence via S-sulfhydration of Keap1 and activation of Nrf2. Antioxid Redox Signal. 2013;18:1906–1919. doi: 10.1089/ars.2012.4645. [DOI] [PubMed] [Google Scholar]
- Yap YW, Whiteman M, Cheung NS. Chlorinative stress: an under appreciated mediator of neurodegeneration? Cell Signal. 2007;19:219–228. doi: 10.1016/j.cellsig.2006.06.013. [DOI] [PubMed] [Google Scholar]
- Zanardo RC, Brancaleone V, Distrutti E, Fiorucci S, Cirino G, Wallace JL. Hydrogen sulfide is an endogenous modulator of leukocyte-mediated inflammation. FASEB J. 2006;20:2118–2120. doi: 10.1096/fj.06-6270fje. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix S1 Supporting methods.
Figure S1 Sulfide binding to ferric myeloperoxidase observed by electronic circular dichroism (ECD) and UV-vis spectroscopies. (A, B) Spectral changes of haem ECD spectra (A) and haem Soret absorption (B) during the titration of 7 µM MPO in 5 mM phosphate buffer at pH = 7.4 with different sulfide concentrations (3, 6.5, 10.5, 14, 17.5, 28, 38, 52, 70, 104, 139, 193, 275, 408 and 571 µM). (C) Plot of the measured differences in absorbance values at 429 nm upon the titration of ferric MPO with sulfide compared with the pure ferric MPO spectrum. The red line represents the rectangular hyperbola fit to equation f = a × x/(b + x). Inset to Figure C shows the isosbestic point at 444 nm during the spectral titration (at <60 µM sulfide).
Figure S2 Inhibition of MPO peroxidase activity. (A) A similar curve was obtained for sulfide-mediated percentage of inhibition of the peroxidase activity using the guaiacol assay as with the TMB method (compared with Figure 3A of the main text). (B) The obtained kinetic traces exhibit a sulfide-dependent induction period indicating interference with the photometric detection: in the absence of sulfide (dark blue line), or in the presence of 0.25 µM (green line), 0.5 µM (red line), 1.0 µM (black line) and 4.0 µM (purple line) sulfide. (C) 1.0 µM sulfide (red line) inhibits H2O2 consumption by ∼50% compared with when no sulfide was added (purple line) as measured by a peroxide selective electrode, which confirms direct inhibition by sulfide on the enzymic activity. Green line, representing the control sample, was recorded under similar conditions to the red line with the exception that MPO was not present. For experimental details see Supporting Information Appendix S1.
Figure S3 Simulation of compound I and compound II reduction with sulfide. (A) Simulated spectra of the different enzyme forms obtained by singular value decomposition analysis of the polychromatic data that were collected for the reduction of compound I (A) or compound II (C) by 20 µM sulfide. Calculated concentration distribution diagram of redox intermediates during the course of the reaction of compound I (B) or compound II (D) with 20 µM sulfide. Reaction conditions are similar as in Figure 5 (for A&B) or Figure 6 (for C&D).
Figure S4 Sulfide dependency of the obtained pseudo first-order rate constants for the rate determining reformation of MPO–sulfide complexes [which we propose is a mixture of MPOFe(III)-hydrosulfido and MPOFe(II)–H2S complexes]. Rate constants were obtained by an exponential fit of the slowest phase of the kinetic traces that were recorded at 430 nm in aerated solutions for the reactions of compound I (blue points) and compound II (red points) with sulfide. Conditions: 2 µM compound I or compound II were mixed with the corresponding concentrations of sulfide in 100 mM phosphate buffer, pH 7.4, and 25°C in a sequential-mixing stopped-flow experiment (for details, see the Methods section in the main text).
Figure S5 Effect of sulfide on the halogenation activity of MPO. Kinetic curves indicate that sulfide consumption by MPO-generated glycine chloramine during the assay results in full recovery of the enzymic activity, which has potential biological implications (see the Discussion and conclusions section in the main text). Kinetic curves are representative of traces that were recorded in the absence of sulfide (blue line), in the presence of 1 µM (red line), or 4 µM (green line) sulfide. For experimental details see Supporting Information Appendix S1.