

Abstract
Carbon monoxide (CO) is enzymatically generated in mammalian cells alongside the liberation of iron and the production of biliverdin and bilirubin. This occurs during the degradation of haem by haem oxygenase (HO) enzymes, a class of ubiquitous proteins consisting of constitutive and inducible isoforms. The constitutive HO2 is present in the gastrointestinal tract in neurons and interstitial cells of Cajal and CO released from these cells might contribute to intestinal inhibitory neurotransmission and/or to the control of intestinal smooth muscle cell membrane potential. On the other hand, increased expression of the inducible HO1 is now recognized as a beneficial response to oxidative stress and inflammation. Among the products of haem metabolism, CO appears to contribute primarily to the antioxidant and anti-inflammatory effects of the HO1 pathway explaining the studies conducted to exploit CO as a possible therapeutic agent. This article reviews the effects and, as far as known today, the mechanism(s) of action of CO administered either as CO gas or via CO-releasing molecules in acute gastrointestinal inflammation. We provide here a comprehensive overview on the effect of CO in experimental in vivo models of post-operative ileus, intestinal injury during sepsis and necrotizing enterocolitis. In addition, we will analyse the in vitro data obtained so far on the effect of CO on intestinal epithelial cell lines exposed to cytokines, considering the important role of the intestinal mucosa in the pathology of gastrointestinal inflammation.
Linked Articles
This article is part of a themed section on Pharmacology of the Gasotransmitters. To view the other articles in this section visit http://dx.doi.org/10.1111/bph.2015.172.issue-6
Keywords: carbon monoxide, CO-RMs, gastrointestinal, inflammation, post-operative ileus, sepsis, necrotizing enterocolitis, intestinal epithelial cells
The haem oxygenase (HO) pathway in the gastrointestinal (GI) tract
Physiological role of HO enzymes
Carbon monoxide (CO) is produced during the metabolism of haem by HO; this reaction also leads to the generation of ferrous iron (Fe2+) and biliverdin, the latter being subsequently reduced to bilirubin by biliverdin reductase. Originally thought of as a mechanism that only serves to dispose of haem, it is now clear that the products of haem metabolism have important functions under physiological and pathophysiological conditions. Three isoforms of HO have been identified (for nomenclature see Alexander et al., 2013). HO1 is the inducible isoform while HO2 is constitutive. In the GI tract, HO2 is present not only in enteric neurons (Miller et al., 2001; Colpaert et al., 2002), but also in non-neuronal cells, in particular interstitial cells of Cajal (Grozdanovic and Gossrau, 1996; Miller et al., 1998). A third isoform (HO3) has been reported but there are doubts whether HO3 has a functional role as it was never convincingly reported at the protein level (Hayashi et al., 2004; Wu and Wang, 2005). CO generated by HO enzymes binds preferentially and with high affinity to transition metals, in particular to the reduced form of haem iron (Fe2+) present in haemoproteins; it can thus be expected to bind to the iron in the haem group of soluble guanylate cyclase (sGC), the primary target of NO for GI smooth muscle relaxation. Still, CO is a weak activator of purified sGC in comparison to NO (Stone and Marletta, 1994; Friebe et al., 1996), but the short-lasting relaxations induced by bolus administration of CO in murine fundus and colon strips were abolished in strips of apo-sGC mice expressing haem-free sGC, suggesting that sGC is indeed essential for CO to cause relaxation in the GI tract (Cosyns et al., 2013). However, both sGC-dependent and sGC-independent mechanisms are involved in the effects induced by prolonged exposure of GI smooth muscle to CO (De Backer et al., 2008). CO has been proposed as another gaseous GI neurotransmitter but the evidence in favour of this is weaker than for NO. The possible role of CO as an inhibitory neurotransmitter in the GI tract or as an hyperpolarizing factor regulating GI smooth muscle membrane potential was very recently reviewed by Gibbons et al. (2013).
Protective effects of HO1 induction
HO1 is induced by a variety of physical and chemical stressors including haem, endotoxin, UV irradiation, heat shock and haemorrhagic shock, all sharing the ability to cause oxidative stress (Maines, 1997). Since the first report showing that HO1 induction crucially controls inflammation in a model of carrageenan-induced pleurisy in rats (Willis et al., 1996), evidence has accumulated that induction of HO1 is a fundamental adaptive response to oxidative stress and acute inflammation (Bauer et al., 2008), providing antioxidant, anti-inflammatory and cytoprotective effects. The homeostatic role of HO1 induction in mitigating the inflammatory response makes sense because reactive oxygen species (ROS) are indeed involved in the initiation and progression of inflammation (Mittal et al., 2014). Possible mechanisms include activation of toll-like receptors and the Nalp-3 inflammasome (Cannizzo et al., 2011) and formation of peroxynitrite by rapid combination of superoxide with NO (Mittal et al., 2014). In the GI tract, expression of HO1 is increased in many experimental models such as i.p. treatment with LPS (Otani et al., 2000), intracolonic administration of trinitrobenzene sulfonic acid (Wang et al., 2001) and surgical intestinal manipulation (De Backer et al., 2009). In patients with inflammatory bowel disease, it was found that macrophages and epithelial cells of colonic mucosa exhibit an increased expression of HO1 although levels of this protein decreased in the case of high inflammatory activity (Paul et al., 2005). Induction of HO1 by exogenously administered haemin reduces GI inflammation and tissue injury (Attuwaybi et al., 2004; Zhong et al., 2010; Yoriki et al., 2013). HO1 induction was also shown to mediate the protective effects of the amino acid glutamine in the GI tract (Uehara et al., 2005; Giriş et al., 2006; 2007) and to be a mechanism of action of 5-aminosalicylic acid, a common agent used for the treatment of inflammatory bowel disease (Whittle and Varga, 2010). This opens the possibility of exploiting the HO1 pathway as a therapeutic target for GI inflammation in a similar way to what has been proposed for the nervous and cardiovascular systems. A pilot clinical study using haemin to treat diabetic gastroparesis, which has been shown experimentally to be associated with insufficient HO1 expression in a particular set of macrophages (Choi et al., 2008; 2010), is ongoing (NCT01206582; http://clinicaltrials.gov).
Role of the by-products of HO1
The antioxidant and anti-inflammatory effects of HO1 induction are related to the breakdown of haem, which is known for its pro-oxidant and cytotoxic properties, and to a concerted action of the three products of haem metabolism, namely Fe2+, biliverdin and CO (Ryter and Choi, 2009). These emerging findings led gradually to the investigation of the biological effects and possible pharmacological application of each single by-product of HO1. First of all, free Fe2+ could be expected to act as a pro-oxidant through the generation of hydroxyl radicals in the Fenton reaction (Fe2+ + H2O2 → Fe3+ + OH· + OH-). However, induction of HO1 is accompanied by a concomitant up-regulation of ferritin (Nath et al., 1992; Ren et al., 2007), which sequesters Fe2+ and has antioxidant properties (Arosio et al., 2009). Additionally, an anti-apoptotic effect of heavy chain ferritin was shown in an ischaemia/reperfusion (I/R) model of the liver (Berberat et al., 2003). Biliverdin is quickly reduced by biliverdin reductase to bilirubin, which has been reported to act as potent intracellular antioxidant against lipid peroxidation (Stocker et al., 1987). Exogenous administration of nanomolar concentrations of bilirubin has been reported to restore post-ischaemic myocardial function and minimize infarct size similarly to induction of HO1 by haemin, suggesting a primary role of bilirubin in HO1-mediated tissue protection against reperfusion injury (Clark et al., 2000). Bilirubin also protects against I/R injury in the intestine, an effect that is associated with a decrease in intestinal lipid peroxidation products (Ceran et al., 2001; Hammerman et al., 2002). In a Dutch study, Caucasian patients with Crohn's disease are more likely to develop this pathology when not expressing the UDP-glucuronyltransferase 1A1*28 homozygous genotype, which is associated with Gilbert's syndrome and increased bilirubin levels (de Vries et al., 2012). Correspondingly, patients with Crohn's disease showed significantly lower levels of serum bilirubin compared with controls in a study in the Czech Republic (Leníček et al., 2014). Exogenous applications of bilirubin might thus be considered for the treatment of GI conditions where oxidative stress is involved. As biliverdin is rapidly reduced to bilirubin, exogenous administration of biliverdin was also attempted with success. In the GI tract, biliverdin attenuates transplantation-induced injury of the small bowel (Nakao et al., 2004) and ameliorates dextran sodium sulfate-induced colitis (Berberat et al., 2005). When oxidized by ROS, bilirubin is converted back to biliverdin, and a recycling loop between bilirubin and biliverdin via biliverdin reductase has been proposed (Wegiel and Otterbein, 2012) although there are also arguments against this antioxidant redox cycle (Jansen and Daiber, 2012). CO seems to contribute most significantly to the antioxidant, anti-inflammatory and cytoprotective effects of the HO1 pathway (Motterlini and Otterbein, 2010) in line with early reports that exogenous CO mimicked the protective effects of HO1 induction even under inhibition of HO1 activity (Sato et al., 2001). This explains the recent efforts to exploit the use of CO as a therapeutic agent to mimic the inherent beneficial actions of the HO1 pathway.
Mechanisms of action of CO
Although the exact molecular target of CO remains elusive, several mechanisms of action for this gaseous molecule have been proposed, mainly on the basis of data in cell lines and non-GI tissue. The protective effect of CO in acute GI inflammation during post-operative ileus (POI), sepsis and necrotizing enterocolitis (NEC) will be discussed in the respective sections.
Activation of sGC/p38 MAPK by CO
In the innate immune system, macrophages are one of the main players involved in the acute inflammatory response in reaction to pathogens, which is achieved by secretion of pro-/anti-inflammatory cytokines and pro-oxidant molecules. Interestingly, in a seminal paper by Otterbein and co-workers, it was shown that CO reduces the production of TNF-α and IL-1β and increases the synthesis of the anti-inflammatory IL-10 in LPS-stimulated macrophages; these effects do not involve activation of the sGC-cGMP pathway (Otterbein et al., 2000). In contrast, the inhibitory effect of CO on leukocyte adhesion in mesenteric venules and on neutrophil migration into the peritoneal cavity induced by carrageenan is counteracted by the sGC inhibitor 1H-[1,2,4]oxadiazolo[4,3-a]quinoxalin-1-one (ODQ) (Freitas et al., 2006). Similarly, the inhibitory effect of CO on apoptosis of neuroblastoma cells, induced by the inhibitor of mitochondrial complex I rotenone to mimic ischaemic respiratory arrest, is reversed by inhibition of sGC (Schallner et al., 2013). Notably, Otterbein and co-workers found that the inhibitory effect of CO on LPS-stimulated macrophages was related to further enhancement of p38 MAPK expression (Otterbein et al., 2000) and the involvement of p38 MAPK activation in the beneficial effects of CO has since been reported in many models in vivo (Otterbein et al., 2003a; Zhang et al., 2003; Kohmoto et al., 2007). However, CO is unlikely to activate p38 MAPK directly as it lacks a transition metal centre in the protein structure that would function as a binding site for the gaseous molecule. Thus, the p38 MAPK activation by CO might be the result of another upstream target. Interestingly, the inhibitory effect of CO on smooth muscle cell proliferation is linked to sGC-dependent activation of p38 MAPK (Otterbein et al., 2003b).
Interaction of the HO1/CO pathway with inducible NOS (iNOS)
An essential enzyme up-regulated in immune cells involved in the acute inflammatory response such as macrophages is iNOS. In LPS-stimulated macrophages, CO decreased nitrite (marker of NO production) levels without changing iNOS protein expression (Sawle et al., 2005), suggesting that CO might inhibit iNOS activity. However, in the same model, Tsoyi et al. (2009) reported that CO is able to reduce iNOS expression, and this was also observed in LPS-treated human umbilical vein endothelial cells (Sun et al., 2008).
Anti-/pro-oxidant effect of CO
In view of the contribution of ROS to the initiation and propagation of inflammation, inhibition of ROS production by CO might contribute to its anti-inflammatory action. NADPH oxidases (NOX) are a family of multicomponent enzymes solely devoted to the production of superoxide and macrophages contain the patriarch enzyme of the family, NOX2. Inhibition of NOX by CO, probably by binding to the haem-containing gp91phox/NOX2 unit, was shown to be involved in the anti-inflammatory actions of CO in LPS-stimulated macrophages (Nakahira et al., 2006) and its anti-proliferative actions in human airway smooth muscle cells (Taillé et al., 2005). The inhibitory effect of CO on vascular smooth muscle cell migration has been related to inhibition of NOX1 (Rodriguez et al., 2010). In contrast, binding of CO to cytochrome c oxidase (complex IV of the mitochondrial respiratory chain) was reported to result in a significant burst of mitochondrial ROS production and a concomitant conditioning of the cell with up-regulation of antioxidant and cytoprotective genes, which protects it from subsequent stress stimulation (Bilban et al., 2008). Moreover, in a caecal ligation and puncture model, CO induced a mild mitochondrial oxidative stress response in the heart which stimulated mitochondrial biogenesis and improved bioenergetics in association with a reduced inflammatory response (Lancel et al., 2009). This was confirmed in a recent study performed in rat isolated heart mitochondria (Lo Iacono et al., 2011) showing that low micromolar concentrations of CO, probably by partially binding to cytochrome c oxidase and/or other unidentified targets in the respiratory chain, increase oxygen consumption and mitochondrial hydrogen peroxide production when physiologically stimulating the electron transfer chain with pyruvate/malate, but significantly decrease the burst of ROS induced by stimulating the reverse electron flow in mitochondria, a phenomenon that appears to typify oxidative and inflammatory disease states (Chen et al., 2007).
Induction of HO1 by CO
The beneficial effects of CO might also be related, at least in some circumstances, to induction of HO1 providing a positive feedback loop (CO→ induction of HO1→ more CO→ further induction of HO1) and allowing the ROS scavenging properties of biliverdin/bilirubin to come into play (Rodella et al., 2006). In human endothelial cells, CO induces HO1 expression through a mechanism involving Nrf2 (nuclear factor erythroid 2-related factor) activation. This has been shown to be associated with upstream activation of PERK (protein kinase R-like endoplasmic reticulum kinase), one of the three endoplasmic reticulum stress sensors; this correlated with protection against endoplasmic reticulum stress-induced endothelial cell apoptosis (Kim et al., 2007).
Administration of CO as a therapeutic agent
CO gas
The recognition that HO1-derived CO serves as a signalling and cytoprotective intracellular mediator prompted scientists to devise strategies to deliver CO safely for therapeutic purposes. CO administered as gas at doses that do not compromise the oxygen carrying capacity of haemoglobin was firstly used in vivo to show that it protects against hyperoxic injury in rat lung (Otterbein et al., 1999). Since then, inhalation of CO gas at doses of 250 p.p.m. for a short period of time has shown efficacy in many animal models of disease, and devices for applying CO gas by inhalation in man have been developed (Motterlini and Otterbein, 2010). Using this protocol, Vanova et al. (2014) showed that the distribution of CO after 1 h CO inhalation is tissue specific and results in increased CO levels ranging from 32-fold in spleen to 2-fold in intestine.
CO in preservation solution
In transplantation surgery, CO has also been proposed to be used ex vivo as an adjuvant to preservation solution in which organs are normally stored prior to grafting. Indeed, cold storage of rat intestinal grafts in University of Wisconsin solution vigorously bubbled with 5% CO for 5 min before transplantation in recipient animals has been reported to markedly reduce the up-regulation of inflammatory mediators and improve graft blood flow and mucosal barrier function (Nakao et al., 2006b). Similar data have been obtained for rat kidney and lung grafts (Kohmoto et al., 2008; Nakao et al., 2008) and for renal transplantation in pigs (Yoshida et al., 2010). In contact with air, the CO-bubbled storage solution quickly loses CO by release into air, so that the CO-bubbled solution must be kept in a tightly sealed container without an air layer to keep the CO concentration constant (Kohmoto et al., 2008).
CO-releasing molecules (CO-RMs)
For this and other reasons, CO-RMs were developed to create the opportunity to deliver CO in a more practical, controllable and accurate way to the target site in comparison to CO gas inhalation. The most extensively studied CO-RMs today consist of two classes: (i) metal carbonyl complexes containing ruthenium, manganese or molybdenum which carry CO bound to metals and (ii) boranocarbonates that do not contain transition metals and release CO spontaneously based on pH changes. While the original CO-RMs had to be dissolved in organic solvents (Motterlini et al., 2002), water-soluble CO-RMs such as CORM-A1 and CORM-3 have been subsequently developed (Clark et al., 2003; Motterlini et al., 2005). In many animal models of inflammation, I/R injury and oxidative stress, these and other similar CO-RMs have been shown to have beneficial effects similar to CO gas (Motterlini, 2007; Motterlini and Otterbein, 2010). In the context of organ preservation, and similar to what has been described with CO gas above, addition of CORM-A1 and CORM-3 during kidney cold storage markedly increased glomerular filtration rate, vascular activity and energy metabolism at reperfusion (Sandouka et al., 2006). Despite the fact that these agents need to be optimized, and one has to keep in mind the possible toxicity originating from the molecule after release of CO especially when used chronically, CO-RMs offer a great opportunity in drug discovery as they differ from classical organic drugs and represent one of the few examples where transition metals are used as scaffolds to deliver the active principle. For instance, a metal within the CO-RMs scaffold can be viewed as the ‘Achilles heel’ in the development of these molecules to drugs due to the common perception that transition metals per se can catalyse cytotoxic reactions. However, a great portion of structural and enzymatic proteins in cells contain transition metals, and several metal-handling proteins that protect cells against potentially toxic metal-based reactions are known, suggesting that biological tissues may be able to tolerate low levels of metal carbonyls (Foresti and Motterlini, 2013). Interestingly, we are gradually learning that the transition metal in CO-RMs appears to be influential in transferring CO into the cells efficiently and limiting the potential toxic effects of free CO gas (Foresti and Motterlini, 2010; Michel et al., 2012).
Toxicity of exogenous CO
The interaction of CO with transition metals is central to the dichotomous nature (toxicity vs. beneficial effects) of this gaseous molecule. CO binds to haemoglobin with a 200–250 times higher affinity than oxygen, forming carbon monoxide-haemoglobin (COHb) that decreases the capacity of blood to deliver oxygen to the tissues. The basal COHb level in man is 0.1–1% in the absence of environmental contamination or smoking (Ryter and Choi, 2013). Toxicity symptoms can occur with COHb levels from 10% and above (Von Burg, 1999), although COHb may not be the only reliable biomarker for determining the toxicity of this gas (Foresti et al., 2008). In experimental studies with CO or CO-RMs, one thus usually attempts to keep COHb levels below 10%, levels that are also occasionally encountered in heavy smokers. Still, one can be reluctant to apply chronically CO or CO-RMs, for example, for treatment of inflammatory bowel disease, in doses inducing COHb levels of 5–10%. Deleterious effects to the heart were indeed shown for chronic ambient CO levels as low as 30 p.p.m. with peaks of 100 p.p.m. (Andre et al., 2010; Reboul et al., 2012). Short-term CO therapy should be devoid of such effects and must be expected to be easily acceptable for application in man. This review therefore considers the findings with CO gas and CO-RMs for conditions involving acute GI inflammation, that is, POI, septic ileus and NEC with particular attention on its role in the GI mucosa. Acute I/R injury of the gut will not be addressed here as this topic was very recently reviewed elsewhere (Liao et al., 2013).
CO in POI
Pathogenesis of POI
POI refers to the transient impairment of bowel motility after major abdominal surgery; it lasts 2–4 days until the first passage of flatus or stool combined with adequate oral intake for 24 h. Patients suffer from abdominal discomfort, nausea and vomiting. POI can delay post-operative recovery, induce morbidity and increase the length of hospital stay; the economic impact of POI has been estimated at $750 million per year (Doorly and Senagore, 2012). In the first hours after surgery, inhibition of GI motility is mainly due to activation of reflex inhibitory nervous pathways (Bauer and Boeckxstaens, 2004; Boeckxstaens and de Jonge, 2009). In a rat model of this early phase, it was reported that endogenous CO contributes to the retarded GI transit at 1.5 h after surgery as HO inhibitors reversed surgery-induced inhibition of transit (Korolkiewicz et al., 2004). The prolongation of GI motility suppression during POI is due to a complex immune-regulated inflammatory process in the intestinal muscularis, with a critical role for activation of the resident muscularis macrophage network (Kalff et al., 1999). The kinetically active substances NO (via iNOS) and prostaglandins (via COX-2) are released, as well as pro-inflammatory cytokines such as IL-1β, IL-6 and TNF-α, chemokines (MCP-1) and adhesion molecules (intercellular adhesion molecule-1; ICAM-1). This leads to additional recruitment of circulating leukocytes and further release of inflammatory mediators (Boeckxstaens and de Jonge, 2009).
Effect of CO on POI
The preclinical studies investigating the influence of CO on the inflammatory phase of POI by manipulating the small intestine after laparotomy and measuring transit 24 h after surgery are summarized in Table 1.
Table 1.
Effects of CO on GI motility and on inflammatory parameters in the muscle layer in preclinical models of post-operative ileus
| Reference | 1 | 2 | 2 | 3 | 4 | |||||
| Species | Mouse | Rat | Pig | Mouse | Mouse | |||||
| CO source | Inhalation | Inhalation | Inhalation | i.p. | i.p. | |||||
| 250 p.p.m. | 250 p.p.m. | 250 p.p.m. | Ringer's lactate, 100% CO bubbled at the end of surgery | CORM-3 40 mg·kg−1 | ||||||
| 1 h before to 24 h after surgery | 3 h before surgery | 3 h before surgery | 3 and 1 h before surgery | |||||||
| Blood COHb | – | – | 6% | 8% | 2.3% | |||||
| POI | +CO | POI | +CO | POI | +CO | POI | +CO | POI | +CO | |
| GI transit (24 h) | ↓ | ↑ | ↓ | ↑ | ↑ | ↓ | ↑ | ↓ | ↑ | |
| In vitro muscle activity | ↓ | ↑ | – | – | – | ↓ | ↑ | |||
| In vitro betanechol | ↓ | ↑ | – | ↓ | ↑ | – | – | |||
| sGC involvement in CO effect | – | – | – | Yes | – | |||||
| p38 MAPK phosphorylation | – | – | – | ↑ | = | ↑ | ↑ | |||
| ERK MAPK phosphorylation | – | – | – | ↑ | ↓ | ↑ | ↓ | |||
| JNK MAPK phosphorylation | – | – | – | ↑ | ↓ | ↑ | = | |||
| iNOS mRNA | ↑ | ↓ | – | – | ↑ | ↓ | – | |||
| Activity | – | – | – | – | ↑ | ↓ | ||||
| ROS | – | – | – | – | ↑ | ↓ | ||||
| HO1 mRNA | ↑ | ↑ | – | – | ↑ | = | – | |||
| Protein | – | – | – | – | ↑ | ↑ | ||||
| HO total activity | – | – | – | – | ↑ | ↑ | ||||
| MPO (infiltrating leukocytes) | ↑ | = | – | ↑ | = | ↑ | ↓ | ↑ | ↓ | |
| IL-1β mRNA | ↑ | ↓ | – | – | ↑ | ↓ | – | |||
| IL-6 mRNA (protein in 4) | ↑ | = | – | – | ↑ | = | ↑ | ↓ | ||
| IL-10 mRNA (protein in 4) | ↑ | ↑ | – | – | ↑ | ↑ | ↑ | ↑ | ||
| COX-2 mRNA | ↑ | = | – | – | ↑ | ↓ | – | |||
| ICAM-1 mRNA (protein in 4) | – | – | – | ↑ | ↓ | ↑ | ↓ | |||
| MCP-1 mRNA (protein in 4) | – | – | – | ↑ | = | ↑ | ↓ | |||
| NF-κB DNA binding activity | – | – | – | ↑ | ↓ | – | ||||
CO gas
Moore et al. (2003) reported that inhaled CO (250 p.p.m. for 1 h before and for 24 h after surgery) in mice significantly improved the prolongation of transit induced by surgery, corresponding to improvement of the surgery-induced suppression of spontaneous and betanechol-induced activity in small intestinal circular muscle strips obtained 24 h after surgery. No negative effect of inhaled CO, induced by its possible GI relaxant effect, was observed, as CO inhalation in control non-operated animals did not significantly influence transit. Surgery increased iNOS mRNA expression in the small intestinal muscular layer at 3 and 6 h, and nitrite release in the culture medium of muscular material obtained 24 h after surgery; these effects were suppressed by CO inhalation, suggesting that CO might act by inhibiting iNOS gene expression as well as iNOS activity. Surgery also increased the mRNA expression of pro-inflammatory IL-1β, IL-6 and COX-2; CO reduced IL-1β expression and the anti-inflammatory IL-10 and HO1 further enhanced IL-10 and HO1 expression. This supports the possibility that the beneficial effects of CO are at least partially due to increased HO1 expression. Interestingly, the increase in muscular leukocyte infiltration induced by surgery was not suppressed by CO.
The same group (Moore et al., 2005) showed that inhalation of 250 p.p.m. CO for 3 h before surgery in rats was sufficient to fully reverse surgery-induced retardation in transit, indicating a more feasible protocol for application in humans than prolonged inhalation. They also showed that 250 p.p.m. for 3 h before surgery is effective in pigs receiving repetitive opioid analgesia to mimic the human condition, whereby the GI inhibitory effects of opioids also contribute to ileus. Blood COHb levels reached 6% just before surgery and progressively declined thereafter. Although CO inhalation suppressed the increase in total blood white cell count at 4 h after surgery, it did not suppress muscular leukocyte infiltration at 24 h, once again suggesting that CO by inhalation is not able to inhibit additional leukocyte recruitment during the pathogenesis of POI.
CO in i.p. solution
Following the report that the intestinal grafts performance is enhanced by their preservation in solutions saturated with CO (Nakao et al., 2006b), Nakao et al. (2006a) showed that an i.p. injection of Ringer's lactate solution bubbled with 100% CO for 15 min just before closure of the abdomen prevents postsurgical ileus in mice. COHb levels reached almost 8% at 5 min after administration of CO but decreased to less than 4% within 30 min. This study showed that treatment with solutions containing CO affects surgery-induced mRNA expression of pro- and anti-inflammatory parameters as reported by Moore et al. (2003) for inhaled CO. Importantly, i.p. injection of CO suppressed muscular leukocyte infiltration. The study further showed that surgery induced activation of MAPK p38, ERK and JNK, and the transcription factor NF-κB, and that CO suppressed the activation of all these signalling proteins except p38. The majority of the effects of CO were diminished by pretreatment with the sGC inhibitor ODQ, suggesting activation of sGC by CO early in its protective pathway.
CO-RMs
An i.p. injection of the ‘fast’ CO-releaser CORM-3 at 3 and 1 h before surgery in mice (40 mg·kg−1)was found to increase blood COHb levels to 2.3% after 10 min from injection; this was associated with partial protection against retarded transit, while the inactive compound i-CORM-3 was not effective (De Backer et al., 2009). A similar effect was obtained with equimolecular amounts of the ‘slow’ CO-releaser CORM-A1 but COHb levels in this case increased to 8.4% at 20 min after injection. Similar to data obtained with CO in i.p. solution, the i.p. administration of CORM-3 suppressed muscular leukocyte infiltration at 6 and 24 h after surgery, decreased ERK activation but increased MAPK p38 activation. Surgery induced a progressive increase in protein expression of HO1 and in total HO activity; this was further enhanced by CORM-3 at 1–6 h after surgery in a MAPK p38-dependent manner. Finally, the study showed that CORM-3 suppresses the progressive increase in muscular oxidative stress levels induced by surgery, illustrating that the anti-oxidative properties of CO might also contribute to its beneficial effect in POI. In contrast to previous studies, De Backer et al. (2009) also investigated the role of the mucosal layer. Surgery did not induce pronounced expression of pro-inflammatory cytokines in the mucosa, but led to an early mucosal oxidative stress burst 1 h after surgery, which might contribute to intestinal epithelial barrier dysfunction during POI (Snoek et al., 2012). CORM-3 reduced this early mucosal oxidative burst in a fully HO1-dependent way, as shown with the HO inhibitor chromium mesoporphyrin, while the anti-inflammatory/anti-oxidative effects of CORM-3 in the muscularis were only partially HO1-dependent.
Cells involved in the protective effect of CO in POI
None of the studies described investigated the type of cells that are targeted by CO in promoting its beneficial effects in POI. In view of the crucial role of resident macrophages in the muscle layer in the pathogenesis of POI, the mechanisms and effects of CO reported in LPS-stimulated macrophages are rather instructive (see Mechanisms of Action: Otterbein et al., 2000; Sawle et al., 2005; Nakahira et al., 2006; Tsoyi et al., 2009). Although very recently contested (Gomez-Pinilla et al., 2014), activation of peritoneal mast cells is also considered to play an important role in the inflammatory cascade during POI (de Jonge et al., 2004; The et al., 2008; Boeckxstaens and de Jonge, 2009). The compound 48/80 or antigen-induced release of histamine from rat and guinea pig mast cells is inhibited by an incubation medium saturated with CO or by addition of a CO-RM; this is associated with calcium sequestration through a cGMP-dependent mechanism (Di Bello et al., 1998; Ndisang et al., 1999; Vannacci et al., 2004). Whether this mechanism plays any role in the protective effect of CO in POI needs further investigation.
Perspectives
Thus, all studies so far show that CO is able to protect against POI but i.p. administration of CO seems to have a more pronounced anti-inflammatory effect than inhalation. The results obtained with one specific CO-RM cannot be automatically extrapolated to all CO-RMs available especially with regard to safety. A scheme of the possible mechanisms of action of CO in POI is shown in Figure 1. The first clinical trial in patients requiring colonic resection is ongoing with a protocol consisting of CO inhalation for 1 h before and 1 h after resection (NCT01050712; http://clinicaltrials.gov).
Figure 1.
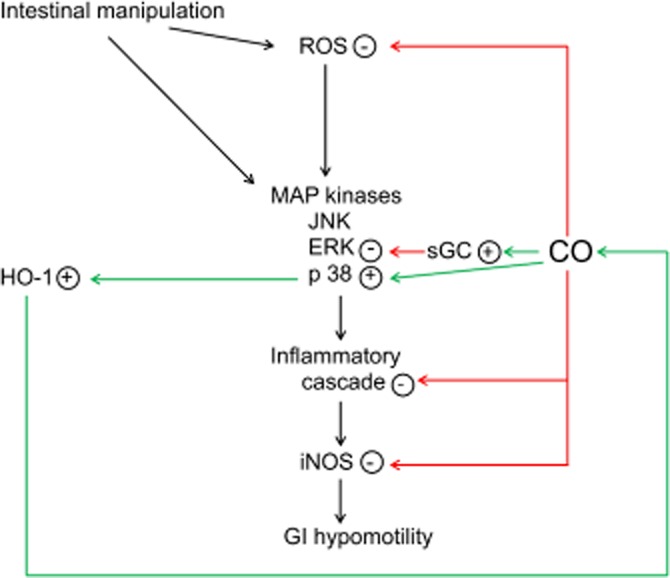
Graphical representation of the possible mechanisms of action of CO in POI. Intestinal manipulation induces oxidative stress and an inflammatory cascade, leading to increased expression/activity of iNOS and GI hypomotility via iNOS-derived NO. CO reduces the inflammatory cascade and iNOS expression/activity (Moore et al., 2003; Nakao et al., 2006a; De Backer et al., 2009). Contributing mechanisms can be inhibition of ROS production (De Backer et al., 2009), suppression of ERK MAPK phosphorylation (De Backer et al., 2009) in a sGC-dependent way (Nakao et al., 2006a) and induction of HO1 (Moore et al., 2003) via enhanced p38 MAPK phosphorylation (De Backer et al., 2009).
CO and the intestine in sepsis
Sepsis is a frequent cause of mortality in intensive care. It is defined as a systemic inflammatory response to infection, which can progress to severe sepsis with multiple organ failure and further to septic shock with acute circulatory failure and refractory hypotension. Sepsis is driven by a complex cascade of events, initiated by bacteria-derived molecules such as LPS (endotoxin) with subsequent formation of inflammatory cytokines, ROS and nitrogen species, I/R and mitochondrial dysfunction (Cinel and Opal, 2009). Animal models of sepsis consist of administration of LPS or bacteria, or caecal ligation and puncture (De Winter and De Man, 2010). In these models, CO either inhaled as a gas or supplied as CO-RMs has been shown to increase survival and to decrease inflammation and tissue damage in critical organs involved in septic multiple organ failure including the lung, liver, heart and kidney (Sarady et al., 2004; Hoetzel et al., 2007; Cepinskas et al., 2008; Lancel et al., 2009; Mizuguchi et al., 2009; Lee et al., 2014). In most studies, CO is applied before or just after administering the septic stimulus, which is clinically irrelevant in patients with established sepsis, but a recent study showed that inhalation of CO in a therapeutic mode (i.e. 2.5 h after starting the LPS infusion) still provides some degree of protection (Koulouras et al., 2011).
The role of the intestine in the pathogenesis of sepsis
While the beneficial effects of CO in the critical organs during sepsis can certainly be attributed to a local anti-inflammatory and antioxidant action, the therapeutic action of CO might also affect the intestine. It is surprising that experimental studies on CO in sepsis rarely include intestinal readouts as the intestine is very vulnerable during systemic inflammation and can trigger and perpetuate sepsis (Clark and Coopersmith, 2007). Conditions such as major trauma, extensive burns and hypovolemia lead to I/R of the intestine, triggering an intestinal inflammatory reaction and intestinal epithelial barrier dysfunction (Magnotti et al., 1998; de Haan et al., 2009; Flessas et al., 2011). Pro-inflammatory mediators produced in the intestine also reach the systemic circulation, promoting systemic inflammation. Dysfunction of the epithelial barrier allows translocation of live bacteria and/or their components such as LPS through the intestinal mucosa, further reaching the circulation via the intestinal lymph nodes. The intestinal events are accompanied by severe impairment of intestinal motility (septic ileus), promoting bacterial colonization of the normally sterile small intestine and stomach; this will facilitate bacterial translocation and predisposes to pneumonia by aspiration of gastric content (Hassoun et al., 2001; Deitch, 2002; Balzan et al., 2007). The mechanism of sepsis-induced ileus, as assessed by injecting LPS, shows similarity with that of the inflammatory phase of POI (Eskandari et al., 1997), although differences in the degree of particular responses were observed when comparing animals with a similar degree of transit retardation, induced either by surgical intestinal manipulation or by i.p. injection of LPS. The POI model induced a greater inflammatory response and a greater degree of leukocyte infiltration (Schmidt et al., 2012). In experimental sepsis, HO1 is induced counteracting intestinal tissue injury (Fujii et al., 2003) and additional HO1 induction by pretreatment of animals with haemin 24 h before administration of LPS protected against the retardation of transit and the intestinal circular muscle dysmotility (Bortscher et al., 2012).
Effect of CO on the intestine in sepsis
Studies concentrating on the effect of CO on the intestine during experimental sepsis are summarized in Table 2. The intestinal readouts involve whole tissue homogenates of small intestinal fragments without separation of mucosal or muscular layers. Liu et al. (2007) reported that CO inhalation at 250 p.p.m. from 1 to 4 h after injection of LPS in rats ameliorates intestinal injury. This effect was associated with significant suppression of the LPS-induced intestinal cell apoptosis. Although inhalation of CO gas induced an increase in arterial COHb levels to 6.9%, no significant differences in the partial arterial oxygen pressure and arterial oxygen saturation were observed. Injection of LPS increased intestinal HO1 mRNA expression, which was further augmented following CO inhalation. CO inhalation reduced the LPS-induced increase in intestinal ICAM-1 and platelet activator factor expression as well as leukocyte infiltration and lipid peroxidation, so CO might exert its protective effect via anti-inflammatory, anti-oxidative and anti-apoptotic actions. Similar protective effects were obtained when continuously perfusing 2 L·min−1 of 250 p.p.m. CO (from CO gas compressed at 250 p.p.m. with balanced air in a cylinder) into the peritoneal cavity (through an inlet in the hypogastric right region with an outlet in the epigastric left section) from 1 to 7 h after i.v. administration of LPS (Liu et al., 2010a). Parameters were measured after 1, 3 and 6 h of CO treatment and illustrated that the beneficial effect of CO was maintained for the whole perfusion period. As measured at 3 h after LPS challenge, p38 MAPK phosphorylation was increased by LPS and further enhanced by CO treatment. The group of Liu et al. (2010b) then also investigated the effect of a single i.p. injection of 2 mL·kg−1 of 250 p.p.m. CO 1 h after LPS treatment. At 1 and 3 h after injection of CO, the LPS-modified parameters were beneficially influenced by CO but no longer at 6 h. In addition to their previous studies, intestinal levels of superoxide dismutase activity and IL-10 protein were measured; both markers were suppressed by LPS but partially reversed by CO, with IL-10 being still elevated 6 h after CO treatment. Dal-Secco et al. (2010) confirmed in mice the importance of ICAM-1 for LPS-induced neutrophil migration. The increased expression of ICAM-1 on mesenteric venular endothelium observed 2 h after an i.p. injection of LPS was significantly reduced by treating the animals with the CO donor dimanganese decacarbonyl (3 mg·kg−1 s.c.) 30 min before LPS challenge. The effect of the CO donor was prevented by ODQ, illustrating that its effect depends on sGC activation. Recently, Wang et al. (2012) studied the mouse small intestine 24 h after caecal ligation and puncture. The intestine showed increased lipid peroxidation, leukocyte infiltration (myeloperoxidase activity), and TNF-α, IL-1β, ICAM-1 and iNOS protein levels, the latter corresponding to increased NO production. All these markers of inflammation were attenuated by CORM-2 (8 mg·kg−1) injected i.v. immediately after the caecal ligation procedure. Histological analysis revealed hydropic degeneration and granulocyte infiltration by the septic procedure; while CORM-2 decreased granulocyte infiltration, it did not improve the hydropic degeneration.
Table 2.
Effects of CO on intestinal tissue integrity and on inflammatory parameters in preclinical models of sepsis
| Reference | 1 | 2 | 3 | 4 | ||||
| Species | Rat | Rat | Rat | Mouse | ||||
| Model | LPS injection i.v. | LPS injection i.v. | LPS injection i.v. | CLP | ||||
| CO source | Inhalation | i.p. | i.p. | i.v. | ||||
| 250 p.p.m. | perfusion | 2 mL·kg−1 | CORM-2 | |||||
| from 1 to 4 h after LPS challenge | 2 L·min−1 | 250 p.p.m. CO | 8 mg·kg−1 | |||||
| 250 p.p.m. CO from 1 to 7 h after LPS challenge | 1 h after LPS challenge | immediately after CLP | ||||||
| Blood COHb | 6.9% | 7.0% | 2.6% | – | ||||
| LPS | +CO | LPS | +CO | LPS | +CO | CLP | +CO | |
| Intestinal tissue damage | ↑ | ↓ | ↑ | ↓ | ↑ | ↓ | ↑ | = |
| Intestinal cell apoptosis | ↑ | ↓ | ↑ | ↓ | ↑ | ↓ | – | |
| p38 MAPK phosphorylation | – | ↑ | ↑ | ↑ | ↑ | – | ||
| iNOS protein | – | – | – | ↑ | ↓ | |||
| NO production | – | – | – | ↑ | ↓ | |||
| ROS | ↑ | ↓ | ↑ | ↓ | ↑ | ↓ | ↑ | ↓ |
| SOD activity | – | – | ↓ | ↑ | – | |||
| HO1 mRNA | ↑ | ↑ | – | – | – | |||
| MPO (infiltrating leukocytes) | ↑ | ↓ | ↑ | ↓ | ↑ | ↓ | ↑ | ↓ |
| TNF-α protein | – | – | – | ↑ | ↓ | |||
| IL-1β protein | – | – | – | ↑ | ↓ | |||
| IL-10 protein | – | – | ↓ | ↑ | – | |||
| ICAM-1 protein | ↑ | ↓ | ↑ | ↓ | ↑ | ↓ | ↑ | ↓ |
| PAF-1 protein | ↑ | ↓ | ↑ | ↓ | ↑ | ↓ | – | |
Effect of CO on the intestine in conditions predisposing to sepsis
Experimental studies also confirmed that conditions clinically predisposing to the development of sepsis such as haemorrhagic shock and burn injury induce intestinal damage and inflammation. The effects of CO gas inhalation or CO-RMs on the intestine in these conditions are summarized in Table 3, and show similar results to the sepsis models per se. In a mouse model of haemorrhagic shock (90–150 min at an arterial pressure of 25 mmHg by blood withdrawal followed by resuscitation with the shed blood), inhalation of 250 p.p.m. CO from the onset of shock until intestinal harvest 4 h after resuscitation prevented the shock-induced intestinal damage (oedema, loss of epithelial and mucosal architecture with areas of focal inflammatory cell influx; Zuckerbraun et al., 2005a). Two studies investigated the intestinal consequences 24 h after a 15% total body surface full-thickness thermal injury (Sun et al., 2007; Liu et al., 2008). An i.v. treatment with CORM-2 immediately after burn injury had the same beneficial effect on pro- and anti-inflammatory mediators as observed in sepsis models. Only one of the two studies included histological analysis. Although CORM-2 treatment decreased burn-induced granulocyte infiltration, it did not improve hydropic degeneration (oedema and sloughing of the villous tips; Sun et al., 2007). Finally, Scott et al. (2009) reported that even remote I/R from the intestine (hindlimb) increases leukocyte recruitment, as measured by intravital microscopy of the submucosal postcapillary venules, and intestinal TNF-α, ICAM-1 and ROS levels. Inhalation of 250 p.p.m. CO for 3 h from the start of hindlimb reperfusion completely prevented the increase in intestinal leukocyte recruitment and TNF-α and ICAM-1 expression, but not the increase in ROS levels; this suggests that CO is mainly active through its anti-inflammatory properties in this model.
Table 3.
Effects of CO on intestinal tissue integrity and on inflammatory parameters in preclinical models of conditions predisposing to sepsis
| Reference | 1 | 2 | 3 | 4 | ||||
| Species | Mouse | Mouse | Mouse | Mouse | ||||
| Model | Haemorrhagia | Burn injury | Burn injury | Hindlimb I/R | ||||
| CO source | Inhalation | i.v. | i.v. | Inhalation | ||||
| 250 p.p.m. | CORM-2 | CORM-2 | 250 p.p.m. | |||||
| from onset of shock | 8 mg·kg−1 | 8 mg·kg−1 | concomitant with reperfusion | |||||
| immediately after burn injury | immediately after burn injury | |||||||
| Blood COHb | – | – | – | 8.4% | ||||
| Haemorrhage | + CO | Burn | +CO | Burn | +CO | I/R | + CO | |
| Intestinal tissue damage | ↑ | ↓ | ↑ | = | – | – | ||
| iNOS protein | – | – | ↑ | ↓ | – | |||
| NO production | – | – | ↑ | ↓ | – | |||
| ROS | – | – | ↑ | ↓ | ↑ | = | ||
| HO1 protein | – | ↑ | ↑ | – | – | |||
| MPO (infiltrating leukocytes) | – | ↑ | ↓ | – | – | |||
| TNF-α protein | – | – | ↑ | ↓ | ↑ | ↓ | ||
| IL-1β protein | – | – | ↑ | ↓ | = | = | ||
| ICAM-1 protein | – | ↑ | ↓ | – | ↑ | ↓ | ||
| NF-κB DNA binding activity | – | ↑ | ↓ | – | – | |||
Perspectives
The studies summarized in Tables 2 and 3 support the idea that the beneficial effect of CO in sepsis might be due, at least in part, to prevention or recovery of intestinal inflammation and tissue damage, therefore, preventing the triggering role of the intestine in sepsis. Although none of the studies in Tables 2 and 3 measured gut barrier function, it is expected that the anti-inflammatory and antioxidant effects of CO on the intestine will also decrease the intestinal barrier dysfunction in sepsis. Additionally, the enhancement of host bacterial clearance (Chung et al., 2008; Onyiah et al., 2013) and direct bacteriostatic/bactericidal effects by CO and CO-RMs (Desmard et al., 2012) might contribute to their beneficial effect in sepsis. Still, when healthy volunteers were subjected to CO gas inhalation (500 p.p.m. CO for 1 h) just before i.v. injection of LPS (2 ng·kg−1), increasing COHb levels to 7%, no changes in plasma neutrophil counts and cytokine levels were observed compared with LPS alone (Mayr et al., 2005). However, Takaki et al. (2010) reported that arterial CO levels and monocyte HO1 protein expression are increased in patients in intensive care, with a significantly more pronounced increase in patients with severe sepsis/septic shock than in non-septic patients. In the septic patients, there was a positive correlation between survival and CO/HO1 levels. These findings further support the importance of trials with CO treatment in septic patients, especially if the beneficial effects of acute CO treatment is confirmed in humans in the ongoing clinical study in colonic surgery patients mentioned above. However, this will first require additional preclinical work to select the optimal protocol and procedures of CO delivery (concentration of CO, selection of CO-RM).
CO in NEC
Pathogenesis of NEC
NEC is an important cause of death in premature infants. It is estimated that up to 12% infants with a birth weight of less than 1500 g will develop the disease, and 30% of them will not survive as overwhelming sepsis can occur when the intestinal mucosal barrier breaks down (Gephart et al., 2012). The clinical signs are often non-specific but when developing, diffuse areas of bowel necrosis can occur with the possibility of perforation, necessitating emergency surgery. The pathogenesis of NEC is not fully understood but NEC does not occur until at least 8–10 days postpartum when bacteria have colonized the gut. The intestinal immaturity with inadequate intestinal barrier function and an excessive inflammatory response towards the post-natal microbial colonization of the intestine predisposes to NEC (Neu and Walker, 2011). As tissues resected from infants with NEC are obtained at later severe stages of the disease, they cannot reveal early pathogenesis. Animal models have therefore been developed, usually combining enteral feeding and hypothermic or hypoxic stress in the preterm or early postterm period, to identify the mediators of GI inflammation in NEC (Sodhi et al., 2008). A unifying pathogenetic hypothesis was recently put forward (De Plaen, 2013). Following the introduction of food, there is a proliferation of the intestinal bacteria. These bacteria and their products adhere to the epithelium, breach the immature intestinal mucosal barrier and activate lamina propria immunocytes. These produce inflammatory agents that attract further inflammatory cells, in particular neutrophils, and inflict further damage to the intestinal barrier with bacterial translocation and mucosal necrosis. The way to sepsis and shock is then open.
Role of HO1/effect of CO in NEC
In line with observations that intestinal specimens of children with NEC show increased expression of HO1 protein (Zuckerbraun et al., 2005b), two recent experimental studies suggest that the impaired ability of the intestine to react with HO1 expressed in the immature intestine contributes to the development of NEC. Exposure of newborn rats (day 1) to hypercapnia followed by re-oxygenation induced gut barrier failure (increased peroral FITC-dextran uptake) and increased intestinal iNOS expression and caspase-3 activity, but not so in weanling rats (15 days). In the latter case, the procedure induced the expression of HO1 protein in the GI tract within 24 h, while HO1 protein was only increased at 72 h in newborn rats. Pretreatment of newborn rats with haemin induced earlier expression of HO1 and attenuated gut barrier failure and apoptosis (Pietzcker et al., 2012). Using 7-day-old heterozygous HO1 (+/-) mice, Schulz et al. (2013) showed that a partial lack of HO1 promotes the development of NEC induced by combining oral gavage of formula food and exposure to hypoxia; the enterocolitis incidence was higher and the survival rate lower than in wild-type mice. Whether either one or both of the HO1 products, CO and biliverdin-bilirubin, prevent enterocolitis was not investigated in the studies of Pietzcker et al. (2012) and Schulz et al. (2013). Zuckerbraun et al. (2005b) showed that exposure to 250 p.p.m. CO for 1 h per day in neonatal rats exposed to hypoxia and formula feeding from day 1 to 3 decreased terminal ileal inflammation and reduced mortality on day 4. The beneficial effects of CO were associated with attenuation of the hypoxia/formula-induced iNOS expression and protein nitration in terminal ileum mucosa. CO also prevented the increase in serum nitrite, TNF-α and IL-1β levels, suggesting that CO also has a systemic effect on inflammatory cells.
Perspectives
CO might thus be considered as a therapeutic agent in human NEC. As systematic exposure of premature infants to CO will not be acceptable, additional experimental studies are required to investigate whether CO can still be effective when the signs and symptoms of suspected NEC occur, and whether the protective effects can also be obtained with CO-RMs.
CO in intestinal epithelial cells
From the above sections, it is clear that the intestinal epithelial cell layer plays a crucial role in acute GI inflammation. This underlines the importance of studies with intestinal epithelial cell lines; publications on the protective effect of CO in intestinal epithelial cell lines are summarized in Table 4. The intestinal epithelial cell lines studied are from murine, rat and human origin; the rodent cell lines (MODE-Κ, YAMC and IEC-6) are derived from non-cancerous GI tissues while the human cell lines (Caco-2, HT-29 and DLD-1) are from tumorous tissue sources. The pro-inflammatory cytokine TNF-α is an important inflammatory mediator in GI inflammation (Holtmann et al., 2002) explaining why studies so far investigating the effect of CO or CO-RMs involved exposing monolayers of enterocytes to either TNF-α alone or in combination with other pro-inflammatory cytokines such as IL-1β and IFN-γ (cytokine mix). All these studies involve acute measurements where the cells were exposed to TNF-α or cytokine mix from 30 min to maximally 24 h followed by cellular and transcriptional readouts.
Table 4.
Effects of CO on intestinal epithelial cell lines
| Cell type | Inflammatory trigger | CO source | Effect of inflammatory markers/overall outcome | Reference |
|---|---|---|---|---|
| Human colonic epithelial cell line, DLD-1 | CM | 250–400 p.p.m. CO | ↓ iNOS mRNA expression | 1 |
| Rat small intestinal epithelial cell line, IEC-6 | CM | 250 p.p.m. CO | ↓ iNOS protein expression | 2 |
| ↓ Nitrite production | ||||
| TNF-α plus actinomycin D | 250 p.p.m. CO | ↓ Cell death (apoptosis) | ||
| LPS/hypoxia | 250 p.p.m. CO | ↓ Cell death | ||
| ↓ iNOS protein expression | ||||
| ↓ Transcriptional activation of iNOS promoter | ||||
| Human colonic epithelial cell line, HT-29 | TNF-α | CORM-2 (5–20 μM) | ↓ IL-8 mRNA expression | 3 |
| ↓ ICAM-1 and COX-2 protein expression | ||||
| Human colonic epithelial cell line, Caco-2 | CM | CORM-2 (50–150 μM) | ↓ Nitrite production | 4 |
| ↓ iNOS mRNA expression | ||||
| ↓ IL-8 protein expression | ||||
| ↓ Transcriptional activation of NF-κB | ||||
| ↓ IκBα phosphorylation | ||||
| ↓ MAPK (p38, ERK 1/2 and JNK 1/2) phosphorylation | ||||
| Young adult mouse colonic epithelial cell line, YAMC | No trigger* | CORM-2 (1–10 μM) | ↑ Wound repair of cell monolayer | 5 |
| Young adult mouse colonic epithelial cell line, YAMC | TNF-α | CORM-2 (10 μM) | ↓ KC mRNA expression | 6 |
| ↓ KC production | ||||
| ↓ NF-κB activity | ||||
| Mouse duodenal epithelial cell line, MODE-K | TNF-α plus cycloheximide | CORM-A1 (100 μM) | ↓ Cell death (apoptosis) | 7 |
| ↑ GSH levels | ||||
| ↓ ROS production | ||||
| ↓ Caspase-3/7 activity |
CO treatment protects against intestinal epithelial cell death induced by inflammatory cytokines, as shown with CO gas in rat and CORM-A1 in mouse small intestinal epithelial cells (Zuckerbraun et al., 2005b; Babu et al., 2012). With regard to the mechanism of its protective effect, none of the studies have investigated the involvement of sGC and only the study by Megias et al. (2007) examined a possible contribution of p38 MAPK. In fact, CORM-2 treatment reduced the cytokine-induced increase in phosphorylation of MAPK p38, similar to its effect on ERK 1/2 and JNK 1/2 phosphorylation. This is in contrast with many in vivo studies on acute GI inflammation reporting that CO further enhances inflammation-induced p38 MAPK phosphorylation (see Tables 1 and 2). An up-regulation of iNOS and sustained overproduction of NO have been associated with intestinal mucosal damage and barrier dysfunction of the gut in acute GI inflammation. Hence, several studies investigated the effect of CO on iNOS expression/NO production in intestinal epithelial cell lines. Consistently, CO treatment reduced the cytokine-induced expression of iNOS at the mRNA or protein level, correlating with suppression of nitrite production (Dijkstra et al., 2004; Zuckerbraun et al., 2005b; Megias et al., 2007). No evidence for a selective effect of CO on iNOS activity has been obtained (Sawle et al., 2005). In a study with the rat small intestinal epithelial cell line IEC-6 (Zuckerbraun et al., 2005b), CO also inhibited LPS- and/or hypoxia (1% oxygen)-induced increase in iNOS protein expression, which was corroborated by the decrease in the transcriptional activation of the iNOS promoter. The effect of CO on iNOS in intestinal epithelial cells might be related to its upstream influence on the transcription factors involved in inflammation-induced iNOS expression. In the elaborate study of Megias et al. (2007) in human Caco-2 cells, CORM-2 indeed reduced the transcriptional activation of NF-κB triggered by a cytokine mixture. Moreover, IκBα phosphorylation after cytokine challenge was also prevented by CORM-2, thus implying that CO might directly influence NF-κB activation. Interestingly, this study also reported that CORM-2 reduced the increased expression of IL-8. IL-8 is the best characterized member of the neutrophil chemoattractant family (Rot and von Andrian, 2004). It is rapidly up-regulated in human colonic epithelial cell lines in response to stimulation with bacteria, TNF-α or IL-1β and mimics acute mucosal inflammatory conditions (Eckmann et al., 1993; Jung et al., 1995; Gewirtz et al., 2001). Also increased IL-8 mRNA expression has been observed in the intestinal epithelial layer from infants with NEC (Nadler et al., 2001). Moreover, IL-8 expression is transcriptionally regulated by MAPK phosphorylation and NF-κB activation in intestinal epithelial cells (Berin et al., 2002). Rodents lack IL-8, but express keratinocyte chemoattractant (KC), a functionally related homologue of IL-8 in humans. This KC derived from epithelial cells has been reported to be involved in the pathophysiology of neutrophil-dependent animal models of intestinal inflammation (Li et al., 2004; Liu et al., 2010c). CORM-2 inhibited KC gene expression and protein production along with inhibition of NF-κB transcriptional activation in TNF-α-stimulated young adult mouse colonic epithelial cells (Takagi et al., 2011).
The anti-inflammatory effects of CO/CO-RMs required HO1 induction in other isolated cell systems such as hepatocytes (Zuckerbraun et al., 2003), macrophages (Sawle et al., 2005), microglia (Min et al., 2006) and endothelial cells (Kim et al., 2007); however, treatment with CO-RMs does not influence HO1 protein expression in human colonic Caco-2 cells (Megias et al., 2007) and murine small intestinal epithelial MODE-K cells (Babu et al., 2012), and thus no evidence for a positive feedback loop CO/HO1/CO was obtained. In another human colonic epithelial cell line, HT-29, Lee et al. (2007) reported that bilirubin per se had a similar inhibitory effect as CORM-2 on TNF-α-induced IL-8, ICAM-1 and COX-2 expression, and the effect was additive when CORM and bilirubin were incubated together, suggesting that pharmacological induction of HO1 might lead to more pronounced beneficial effects than exogenous CO. However, the HO1 inducer cobalt protoporphyrin decreased TNF-α plus actinomycin D-induced apoptotic cell death of IEC-6 cells only to the same extent as exogenous CO gas (Zuckerbraun et al., 2005b). Only one study investigated the influence of CO on ROS production in intestinal epithelial cells (Babu et al., 2012). The increased ROS production and the decreased levels of cellular reduced GSH induced by TNF-α/cycloheximide in MODE-Κ cells were partially prevented by treatment with CORM-A1. This correlated with diminished caspase-3/7 activity, apoptosis and cell death, suggesting that modulation of ROS/oxidative stress might be involved in the anti-apoptotic and cytoprotective effects of CORM-A1.
During acute GI inflammation associated with oxidative stress, impairment of the epithelial surface barrier with accompanying erosion demands rapid resealing of this barrier to maintain homeostasis. The whole process of rapid resealing of superficial wounds involving migration of the epithelial cells adjacent to the injured surface to the site of mucosal wound defect, differentiation and reorganization of the cytoskeleton with ultimate closure of the denuded area to establish the continuity of surface epithelium is termed ‘epithelial restitution’. Epithelial restitution is a critical process which does not involve epithelial cell proliferation, but contraction of the cytoplasmic processes of underlying myofibrolasts (Moore et al., 1989). Uchiyama et al. (2010) provided evidence in YAMC cells that CO can promote colonic mucosal restitution. Myofibroblast-conditioned medium enhanced the wound repair of a manually denuded monolayer of YAMC cells to a more pronounced extent when the myofibroblasts were treated with CORM-2. This beneficial effect of CORM-2 on epithelial cell restitution was abolished by treating the myofibroblasts with siRNA for fibroblast growth factor (FGF)15. CORM-2 increased FGF15 mRNA and protein expression probably by down-regulating microRNA miR-710.
All studies in intestinal epithelial cell lines thus support the hypothesis that CO can reduce acute mucosal inflammation and damage, and even stimulate intestinal epithelial cell restitution. The beneficial effect of CO in in vivo models of sepsis and NEC might thus at least be partially due to its effects at the mucosal level. It is thus advisable that intestinal inflammation and the effect of CO in these models are studied separately in the mucosal versus the muscular layer of the GI tract to further elucidate the mechanism of action in these conditions.
Conclusion
In preclinical in vivo models of acute GI inflammation during POI after abdominal surgery, sepsis and NEC, treatment with CO gas or CO-RMs suppresses the intestinal infiltration of leukocytes and the expression of inflammatory mediators, leading to improved GI transit (POI) and decreased intestinal damage (sepsis, NEC). This beneficial effect is confirmed in intestinal epithelial cell lines exposed to cytokines. The mechanism of action of CO in acute GI inflammation has not been profoundly examined, but enhanced p38 MAPK phosphorylation, suppressed iNOS expression and decreased production of ROS are most consistently reported. However, the beneficial effects of CO in humans affected by acute GI inflammation still need to be confirmed. The most straightforward indication to attempt CO for acute GI inflammation in humans, as administration can be started in a preventive way before the planned operation starts, is abdominal surgery, and that is exactly where a clinical trial with inhaled CO is now ongoing. Even though this trial will lead to positive results with CO, the application of CO in humans with sepsis or NEC will require more extensive preclinical work to determine the optimal route of CO administration. For instance, what is the maximal tolerable and beneficial dose of CO gas inhalation? Which CO-RM should be used and what is the dose and the best route of administration? When is the best time to start CO treatment once sepsis or NEC has developed? How long should the CO gas inhalation be applied for? Are single or repetitive administrations of CO-RM or CO gas needed? Is the residual molecule formed after CO release from CO-RMs safe or toxic? All these and similar important issues are required to be properly addressed in animal studies before moving on to humans.
Acknowledgments
This work was supported by COST Action BM1005 (European Network on Gasotransmitters).
Glossary
- CO
carbon monoxide
- COHb
carbon monoxide-haemoglobin
- CO-RM
CO-releasing molecule
- ER
endoplasmic reticulum
- FGF
fibroblast growth factor
- GI
gastrointestinal
- HO
haem oxygenase
- ICAM-1
intercellular adhesion molecule-1
- iNOS
inducible NOS
- I/R
ischaemia/reperfusion
- KC
keratinocyte chemoattractant
- NEC
necrotizing enterocolitis
- NOX
NADPH oxidase
- Nrf2
nuclear factor erythroid 2-related factor
- ODQ
1H-[1,2,4]oxadiazolo[4,3-a]quinoxalin-1-one
- PERK
protein kinase R-like endoplasmic reticulum kinase
- POI
post-operative ileus
- ROS
reactive oxygen species
- sGC
soluble guanylate cyclase
Conflict of interest
No conflict of interest.
References
- Alexander SPH, Benson HE, Faccenda E, Pawson AJ, Sharman JL, Spedding M, Peters JA, Harmar AJ CGTP Collaborators. The Concise Guide to PHARMACOLOGY 2013/14: Enzymes. Br J Pharmacol. 2013;170:1797–1867. doi: 10.1111/bph.12451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andre L, Boissière J, Reboul C, Perrier R, Zalvidea S, Meyer G, et al. Carbon monoxide pollution promotes cardiac remodeling and ventricular arrhythmia in healthy rats. Am J Respir Crit Care Med. 2010;181:587–595. doi: 10.1164/rccm.200905-0794OC. [DOI] [PubMed] [Google Scholar]
- Arosio P, Ingrassia R, Cavadini P. Ferritins: a family of molecules for iron storage, antioxidation and more. Biochim Biophys Acta. 2009;1790:589–599. doi: 10.1016/j.bbagen.2008.09.004. [DOI] [PubMed] [Google Scholar]
- Attuwaybi BO, Kozar RA, Moore-Olufemi SD, Sato N, Hassoun HT, Weisbrodt NW, et al. Heme oxygenase-1 induction by hemin protects against gut ischemia/reperfusion injury. J Surg Res. 2004;118:53–57. doi: 10.1016/j.jss.2004.01.010. [DOI] [PubMed] [Google Scholar]
- Babu D, Soenen SJ, Raemdonck K, Leclercq G, De Backer O, Motterlini R, et al. TNF-alpha/cycloheximide-induced oxidative stress and apoptosis in murine intestinal epithelial MODE-K cells. Curr Pharm Des. 2012;18:4414–4425. doi: 10.2174/138161212802481291. [DOI] [PubMed] [Google Scholar]
- Balzan S, de Almeida Quadros C, de Cleva R, Zilberstein B, Cecconello I. Bacterial translocation: overview of mechanisms and clinical impact. J Gastroenterol Hepatol. 2007;22:464–471. doi: 10.1111/j.1440-1746.2007.04933.x. [DOI] [PubMed] [Google Scholar]
- Bauer AJ, Boeckxstaens GE. Mechanisms of postoperative ileus. Neurogastroenterol Motil. 2004;16(Suppl. 2):54–60. doi: 10.1111/j.1743-3150.2004.00558.x. [DOI] [PubMed] [Google Scholar]
- Bauer M, Huse K, Settmacher U, Claus RA. The heme oxygenase–carbon monoxide system: regulation and role in stress response and organ failure. Intensive Care Med. 2008;34:640–648. doi: 10.1007/s00134-008-1010-2. [DOI] [PubMed] [Google Scholar]
- Berberat PO, Katori M, Kaczmarek E, Anselmo D, Lassman C, Ke B, et al. Heavy chain ferritin acts as an anti-apoptotic gene that protects livers from ischemia-reperfusion injury. FASEB J. 2003;17:1724–1726. doi: 10.1096/fj.03-0229fje. [DOI] [PubMed] [Google Scholar]
- Berberat PO, A-Rahim YI, Yamashita K, Warny MM, Csizmadia E, Robson SC, et al. Heme oxygenase-1-generated biliverdin ameliorates experimental murine colitis. Inflamm Bowel Dis. 2005;11:350–359. doi: 10.1097/01.mib.0000164017.06538.8a. [DOI] [PubMed] [Google Scholar]
- Berin MC, Darfeuille-Michaud A, Egan LJ, Miyamoto Y, Kagnoff MF. Role of EHEC O157:H7 virulence factors in the activation of intestinal epithelial cell NF-kappaB and MAP kinase pathways and the upregulated expression of interleukin 8. Cell Microbiol. 2002;4:635–648. doi: 10.1046/j.1462-5822.2002.00218.x. [DOI] [PubMed] [Google Scholar]
- Bilban M, Haschemi A, Wegiel B, Chin BY, Wagner O, Otterbein LE. Heme oxygenase and carbon monoxide initiate homeostatic signaling. J Mol Med. 2008;86:267–279. doi: 10.1007/s00109-007-0276-0. [DOI] [PubMed] [Google Scholar]
- Boeckxstaens GE, de Jonge WJ. Neuroimmune mechanisms in postoperative ileus. Gut. 2009;58:1300–1311. doi: 10.1136/gut.2008.169250. [DOI] [PubMed] [Google Scholar]
- Bortscher S, Chang J, Vilz TO, Schäfer N, Sommer N, Wehner S, et al. Hemin induction of HO-1 protects against LPS-induced septic ileus. J Surg Res. 2012;178:866–873. doi: 10.1016/j.jss.2012.07.064. [DOI] [PubMed] [Google Scholar]
- Cannizzo ES, Clement CC, Sahu R, Follo C, Santambrogio L. Oxidative stress, inflamm-aging and immunosenescence. J Proteomics. 2011;74:2313–2323. doi: 10.1016/j.jprot.2011.06.005. [DOI] [PubMed] [Google Scholar]
- Cepinskas G, Katada K, Bihari A, Potter RF. Carbon monoxide liberated from carbon monoxide-releasing molecule CORM-2 attenuates inflammation in the liver of septic mice. Am J Physiol Gastrointest Liver Physiol. 2008;294:G184–G191. doi: 10.1152/ajpgi.00348.2007. [DOI] [PubMed] [Google Scholar]
- Ceran C, Sönmez K, Türkyllmaz Z, Demirogullarl B, Dursun A, Düzgün E, et al. Effect of bilirubin in ischemia/reperfusion injury on rat small intestine. J Pediatr Surg. 2001;36:1764–1767. doi: 10.1053/jpsu.2001.28816. [DOI] [PubMed] [Google Scholar]
- Chen YR, Chen CL, Pfeiffer DR, Zweier JL. Mitochondrial complex II in the post-ischemic heart: oxidative injury and the role of protein S-glutathionylation. J Biol Chem. 2007;282:32640–32654. doi: 10.1074/jbc.M702294200. [DOI] [PubMed] [Google Scholar]
- Choi KM, Gibbons SJ, Nguyen TV, Stoltz GJ, Lurken MS, Ordog T, et al. Heme oxygenase-1 protects interstitial cells of Cajal from oxidative stress and reverses diabetic gastroparesis. Gastroenterology. 2008;135:2055–2064. doi: 10.1053/j.gastro.2008.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi KM, Kashyap PC, Dutta N, Stoltz GJ, Ordog T, Shea Donohue T, et al. CD206-positive M2 macrophages that express heme oxygenase-1 protect against diabetic gastroparesis in mice. Gastroenterology. 2010;138:2399–2409. doi: 10.1053/j.gastro.2010.02.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung SW, Liu X, Macias AA, Baron RM, Perrella MA. Heme oxygenase-1-derived carbon monoxide enhances the host defense response to microbial sepsis in mice. J Clin Invest. 2008;118:239–247. doi: 10.1172/JCI32730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cinel I, Opal SM. Molecular biology of inflammation and sepsis: a primer. Crit Care Med. 2009;37:291–304. doi: 10.1097/CCM.0b013e31819267fb. [DOI] [PubMed] [Google Scholar]
- Clark JA, Coopersmith CM. Intestinal crosstalk – a new paradigm for understanding the gut as the ‘motor’ of critical illness. Shock. 2007;28:384–393. doi: 10.1097/shk.0b013e31805569df. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark JE, Foresti R, Sarathchandra P, Kaur H, Green CJ, Motterlini R. Heme oxygenase-1-derived bilirubin ameliorates postischemic myocardial dysfunction. Am J Physiol Heart Circ Physiol. 2000;278:H643–H651. doi: 10.1152/ajpheart.2000.278.2.H643. [DOI] [PubMed] [Google Scholar]
- Clark JE, Naughton P, Shurey S, Green CJ, Johnson TR, Mann BE, et al. Cardioprotective actions by a water-soluble carbon monoxide-releasing molecule. Circ Res. 2003;93:e2–e8. doi: 10.1161/01.RES.0000084381.86567.08. [DOI] [PubMed] [Google Scholar]
- Colpaert EE, Timmermans JP, Lefebvre RA. Immunohistochemical localization of the antioxidant enzymes biliverdin reductase and heme oxygenase-2 in human and pig gastric fundus. Free Radic Biol Med. 2002;32:630–637. doi: 10.1016/s0891-5849(02)00754-2. [DOI] [PubMed] [Google Scholar]
- Cosyns SM, Dhaese I, Thoonen R, Buys ES, Vral A, Brouckaert P, et al. Heme deficiency of soluble guanylate cyclase induces gastroparesis. Neurogastroenterol Motil. 2013;25:e339–e352. doi: 10.1111/nmo.12120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dal-Secco D, Freitas A, Abreu MA, Garlet TP, Rossi MA, Ferreira SH, et al. Reduction of ICAM-1 expression by carbon monoxide via soluble guanylate cyclase activation accounts for modulation of neutrophil migration. Naunyn Schmiedebergs Arch Pharmacol. 2010;381:483–493. doi: 10.1007/s00210-010-0500-2. [DOI] [PubMed] [Google Scholar]
- De Backer O, Elinck E, Sips P, Buys E, Brouckaert P, Lefebvre RA. Role of the soluble guanylyl cyclase α1/α2 subunits in the relaxant effect of CO and CORM-2 in murine gastric fundus. Naunyn Schmiedebergs Arch Pharmacol. 2008;378:493–502. doi: 10.1007/s00210-008-0315-6. [DOI] [PubMed] [Google Scholar]
- De Backer O, Elinck E, Blanckaert B, Leybaert L, Motterlini R, Lefebvre RA. Water-soluble CO-releasing molecules reduce the development of postoperative ileus via modulation of MAPK/HO-1 signalling and reduction of oxidative stress. Gut. 2009;58:347–356. doi: 10.1136/gut.2008.155481. [DOI] [PubMed] [Google Scholar]
- De Plaen IG. Inflammatory signaling in necrotizing enterocolitis. Clin Perinatol. 2013;40:109–124. doi: 10.1016/j.clp.2012.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Winter BY, De Man JG. Interplay between inflammation, immune system and neuronal pathways: effect on gastrointestinal motility. World J Gastroenterol. 2010;16:5523–5535. doi: 10.3748/wjg.v16.i44.5523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deitch EA. Bacterial translocation or lymphatic drainage of toxic products from the gut: what is important in human beings? Surgery. 2002;131:241–244. doi: 10.1067/msy.2002.116408. [DOI] [PubMed] [Google Scholar]
- Desmard M, Foresti R, Morin D, Dagoussat M, Berdeaux A, Denamur E, et al. Differential antibacterial activity against Pseudomonas aeruginosa by carbon monoxide-releasing molecules. Antioxid Redox Signal. 2012;16:153–163. doi: 10.1089/ars.2011.3959. [DOI] [PubMed] [Google Scholar]
- Di Bello MG, Berni L, Gai P, Mirabella C, Ndisang JF, Masini E, et al. A regulatory role for carbon monoxide in mast cell function. Inflamm Res. 1998;47(Suppl. 1):S7–S8. doi: 10.1007/s000110050238. [DOI] [PubMed] [Google Scholar]
- Dijkstra G, Blokzijl H, Bok L, Homan M, van Goor H, Faber KN, et al. Opposite effect of oxidative stress on inducible nitric oxide synthase and haem oxygenase-1 expression in intestinal inflammation: anti-inflammatory effect of carbon monoxide. J Pathol. 2004;204:296–303. doi: 10.1002/path.1656. [DOI] [PubMed] [Google Scholar]
- Doorly MG, Senagore AJ. Pathogenesis and clinical and economic consequences of postoperative ileus. Surg Clin North Am. 2012;92:259–272. doi: 10.1016/j.suc.2012.01.010. [DOI] [PubMed] [Google Scholar]
- Eckmann L, Jung HC, Schürer-Maly C, Panja A, Morzycka-Wroblewska E, Kagnoff MF. Differential cytokine expression by human intestinal epithelial cell lines: regulated expression of interleukin 8. Gastroenterology. 1993;105:1689–1697. doi: 10.1016/0016-5085(93)91064-o. [DOI] [PubMed] [Google Scholar]
- Eskandari MK, Kalff JC, Billiar TR, Lee KK, Bauer AJ. Lipopolysaccharide activates the muscularis macrophage network and suppresses circular smooth muscle activity. Am J Physiol. 1997;273:G727–G734. doi: 10.1152/ajpgi.1997.273.3.G727. [DOI] [PubMed] [Google Scholar]
- Flessas II, Papalois AE, Toutouzas K, Zagouri F, Zografos GC. Effects of lazaroids on intestinal ischemia and reperfusion injury in experimental models. J Surg Res. 2011;166:265–274. doi: 10.1016/j.jss.2010.08.031. [DOI] [PubMed] [Google Scholar]
- Foresti R, Motterlini R. Interaction of carbon monoxide with transition metals: evolutionary insights into drug target discovery. Curr Drug Targets. 2010;11:1595–1604. doi: 10.2174/1389450111009011595. [DOI] [PubMed] [Google Scholar]
- Foresti R, Motterlini R. CO-releasing molecules: avoiding toxicity and exploiting the beneficial effects of CO for the treatment of cardiovascular disorders. Future Med Chem. 2013;5:367–369. doi: 10.4155/fmc.13.10. [DOI] [PubMed] [Google Scholar]
- Foresti R, Bani-Hani MG, Motterlini R. Use of carbon monoxide as a therapeutic agent: promises and challenges. Intensive Care Med. 2008;34:649–658. doi: 10.1007/s00134-008-1011-1. [DOI] [PubMed] [Google Scholar]
- Freitas A, Alves-Filho JC, Secco DD, Neto AF, Ferreira SH, Barja-Fidalgo C, et al. Heme oxygenase/carbon monoxide-biliverdin pathway down regulates neutrophil rolling, adhesion and migration in acute inflammation. Br J Pharmacol. 2006;149:345–354. doi: 10.1038/sj.bjp.0706882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friebe A, Schultz G, Koesling D. Sensitizing soluble guanylyl cyclase to become a highly CO-sensitive enzyme. EMBO J. 1996;15:6863–6868. [PMC free article] [PubMed] [Google Scholar]
- Fujii H, Takahashi T, Nakahira K, Uehara K, Shimizu H, Matsumi M, et al. Protective role of heme oxygenase-1 in the intestinal tissue injury in an experimental model of sepsis. Crit Care Med. 2003;31:893–902. doi: 10.1097/01.CCM.0000050442.54044.06. [DOI] [PubMed] [Google Scholar]
- Gephart SM, McGrath JM, Effken JA, Halpern MD. Necrotizing enterocolitis risk: state of the science. Adv Neonatal Care. 2012;12:77–87. doi: 10.1097/ANC.0b013e31824cee94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gewirtz AT, Simon PO, Jr, Schmitt CK, Taylor LJ, Hagedorn CH, O'Brien AD, et al. Salmonella typhimurium translocates flagellin across intestinal epithelia, inducing a proinflammatory response. J Clin Invest. 2001;107:99–109. doi: 10.1172/JCI10501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibbons SJ, Verhulst PJ, Bharucha A, Farrugia G. Review article: carbon monoxide in gastrointestinal physiology and its potential therapeutics. Aliment Pharmacol Ther. 2013;38:689–702. doi: 10.1111/apt.12467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giriş M, Erbil Y, Oztezcan S, Olgaç V, Barbaros U, Deveci U, et al. The effect of heme oxygenase-1 induction by glutamine on radiation-induced intestinal damage: the effect of heme oxygenase-1 on radiation enteritis. Am J Surg. 2006;191:503–509. doi: 10.1016/j.amjsurg.2005.11.004. [DOI] [PubMed] [Google Scholar]
- Giriş M, Erbil Y, Doğru-Abbasoğlu S, Yanik BT, Aliş H, Olgaç V, et al. The effect of heme oxygenase-1 induction by glutamine on TNBS-induced colitis. Int J Colorectal Dis. 2007;22:591–599. doi: 10.1007/s00384-006-0238-y. [DOI] [PubMed] [Google Scholar]
- Gomez-Pinilla PJ, Farro G, Di Giovangiulio M, Stakenborg N, Némethova A, de Vries A, et al. Mast cells play no role in the pathogenesis of postoperative ileus induced by intestinal manipulation. PLoS ONE. 2014;9:e85304. doi: 10.1371/journal.pone.0085304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grozdanovic Z, Gossrau R. Expression of heme oxygenase-2 (HO-2)-like immunoreactivity in rat tissues. Acta Histochem. 1996;98:203–214. doi: 10.1016/S0065-1281(96)80040-7. [DOI] [PubMed] [Google Scholar]
- de Haan JJ, Lubbers T, Derikx JP, Relja B, Henrich D, Greve JW, et al. Rapid development of intestinal cell damage following severe trauma: a prospective observational cohort study. Crit Care. 2009;13:R86. doi: 10.1186/cc7910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammerman C, Goldschmidt D, Caplan MS, Kaplan M, Bromiker R, Eidelman AI, et al. Protective effect of bilirubin in ischemia-reperfusion injury in the rat intestine. J Pediatr Gastroenterol Nutr. 2002;35:344–349. doi: 10.1097/00005176-200209000-00020. [DOI] [PubMed] [Google Scholar]
- Hassoun HT, Kone BC, Mercer DW, Moody FG, Weisbrodt NW, Moore FA. Post-injury multiple organ failure: the role of the gut. Shock. 2001;15:1–10. doi: 10.1097/00024382-200115010-00001. [DOI] [PubMed] [Google Scholar]
- Hayashi S, Omata Y, Sakamoto H, Higashimoto Y, Hara T, Sagara Y, et al. Characterization of rat heme oxygenase-3 gene. Implication of processed pseudogenes derived from heme oxygenase-2 gene. Gene. 2004;336:241–250. doi: 10.1016/j.gene.2004.04.002. [DOI] [PubMed] [Google Scholar]
- Hoetzel A, Dolinay T, Schmidt R, Choi AM, Ryter SW. Carbon monoxide in sepsis. Antioxid Redox Signal. 2007;9:2013–2026. doi: 10.1089/ars.2007.1762. [DOI] [PubMed] [Google Scholar]
- Holtmann MH, Schütz M, Galle PR, Neurath MF. Functional relevance of soluble TNF-alpha, transmembrane TNF-alpha and TNF-signal transduction in gastrointestinal diseases with special reference to inflammatory bowel diseases. Z Gastroenterol. 2002;40:587–600. doi: 10.1055/s-2002-33418. [DOI] [PubMed] [Google Scholar]
- Jansen T, Daiber A. Direct antioxidant properties of bilirubin and biliverdin. Is there a role for biliverdin reductase? Front Pharmacol. 2012;3:30. doi: 10.3389/fphar.2012.00030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Jonge WJ, The FO, van der Coelen D, Bennink RJ, Reitsma PH, van Deventer SJ, et al. Mast cell degranulation during abdominal surgery initiates postoperative ileus in mice. Gastroenterology. 2004;127:535–545. doi: 10.1053/j.gastro.2004.04.017. [DOI] [PubMed] [Google Scholar]
- Jung HC, Eckmann L, Yang SK, Panja A, Fierer J, Morzycka-Wroblewska E, et al. A distinct array of proinflammatory cytokines is expressed in human colon epithelial cells in response to bacterial invasion. J Clin Invest. 1995;95:55–65. doi: 10.1172/JCI117676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalff JC, Buchholz BM, Eskandari MK, Hierholzer C, Schraut WH, Simmons RL, et al. Biphasic response to gut manipulation and temporal correlation of cellular infiltrates and muscle dysfunction in rat. Surgery. 1999;126:498–509. [PubMed] [Google Scholar]
- Kim KM, Pae HO, Zheng M, Park R, Kim YM, Chung HT. Carbon monoxide induces heme oxygenase-1 via activation of protein kinase R-like endoplasmic reticulum kinase and inhibits endothelial cell apoptosis triggered by endoplasmic reticulum stress. Circ Res. 2007;101:919–927. doi: 10.1161/CIRCRESAHA.107.154781. [DOI] [PubMed] [Google Scholar]
- Kohmoto J, Nakao A, Stolz DB, Kaizu T, Tsung A, Ikeda A, et al. Carbon monoxide protects rat lung transplants from ischemia-reperfusion injury via a mechanism involving p38 MAPK pathway. Am J Transplant. 2007;7:2279–2290. doi: 10.1111/j.1600-6143.2007.01940.x. [DOI] [PubMed] [Google Scholar]
- Kohmoto J, Nakao A, Sugimoto R, Wang Y, Zhan J, Ueda H, et al. Carbon monoxide-saturated preservation solution protects lung grafts from ischemia-reperfusion injury. J Thorac Cardiovasc Surg. 2008;136:1067–1075. doi: 10.1016/j.jtcvs.2008.06.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korolkiewicz RP, Sein-Anand J, Ruczyński J, Rekowski P, Bieniaszewski L, Chodorowski Z, et al. The role and interactions of nitric oxide (NO), carbon monoxide (CO), and prostanoids in the pathogenesis of postoperative ileus in rats. J Gastrointest Surg. 2004;8:346–357. doi: 10.1016/j.gassur.2003.11.004. [DOI] [PubMed] [Google Scholar]
- Koulouras VP, Li R, Chen L, Hedenstierna GG. Effects of inhaled carbon monoxide and glucocorticoids in porcine endotoxin sepsis. Int J Clin Exp Med. 2011;4:53–66. [PMC free article] [PubMed] [Google Scholar]
- Lancel S, Hassoun SM, Favory R, Decoster B, Motterlini R, Neviere R. Carbon monoxide rescues mice from lethal sepsis by supporting mitochondrial energetic metabolism and activating mitochondrial biogenesis. J Pharmacol Exp Ther. 2009;329:641–648. doi: 10.1124/jpet.108.148049. [DOI] [PubMed] [Google Scholar]
- Lee S, Lee SJ, Coronata AA, Fredenburgh LE, Chung SW, Perrella MA, et al. Carbon monoxide confers protection in sepsis by enhancing beclin 1-dependent autophagy and phagocytosis. Antioxid Redox Signal. 2014;20:432–442. doi: 10.1089/ars.2013.5368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee SH, Sohn DH, Jin XY, Kim SW, Choi SC, Seo GS. 2′,4′,6′-tris(methoxymethoxy) chalcone protects against trinitrobenzene sulfonic acid-induced colitis and blocks tumor necrosis factor-alpha-induced intestinal epithelial inflammation via heme oxygenase 1-dependent and independent pathways. Biochem Pharmacol. 2007;74:870–880. doi: 10.1016/j.bcp.2007.06.034. [DOI] [PubMed] [Google Scholar]
- Leníček M, Duricová D, Hradsky O, Dušátková P, Jirásková A, Lukáš M, et al. The relationship between serum bilirubin and Crohn's disease. Inflamm Bowel Dis. 2014;20:481–487. doi: 10.1097/01.MIB.0000440817.84251.98. [DOI] [PubMed] [Google Scholar]
- Li N, Liboni K, Fang MZ, Samuelson D, Lewis P, Patel R, et al. Glutamine decreases lipopolysaccharide-induced intestinal inflammation in infant rats. Am J Physiol Gastrointest Liver Physiol. 2004;286:G914–G921. doi: 10.1152/ajpgi.00493.2003. [DOI] [PubMed] [Google Scholar]
- Liao YF, Zhu W, Li DP, Zhu X. Heme oxygenase-1 and gut ischemia/reperfusion injury: a short review. World J Gastroenterol. 2013;19:3555–3561. doi: 10.3748/wjg.v19.i23.3555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu DM, Sun BW, Sun ZW, Jin Q, Sun Y, Chen X. Suppression of inflammatory cytokine production and oxidative stress by CO-releasing molecules-liberated CO in the small intestine of thermally-injured mice. Acta Pharmacol Sin. 2008;29:838–846. doi: 10.1111/j.1745-7254.2008.00816.x. [DOI] [PubMed] [Google Scholar]
- Liu SH, Xu XR, Ma K, Xu B. Protection of carbon monoxide inhalation on lipopolysaccharide-induced multiple organ injury in rats. Chin Med Sci J. 2007;22:169–176. [PubMed] [Google Scholar]
- Liu SH, Ma K, Xu B, Xu XR. Protection of carbon monoxide intraperitoneal administration from rat intestine injury induced by lipopolysaccharide. Chin Med J (Engl) 2010a;123:1039–1046. [PubMed] [Google Scholar]
- Liu SH, Ma K, Xu XR, Xu B. A single dose of carbon monoxide intraperitoneal administration protects rat intestine from injury induced by lipopolysaccharide. Cell Stress Chaperones. 2010b;15:717–727. doi: 10.1007/s12192-010-0183-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Fatheree NY, Mangalat N, Rhoads JM. Human-derived probiotic Lactobacillus reuteri strains differentially reduce intestinal inflammation. Am J Physiol Gastrointest Liver Physiol. 2010c;299:G1087–G1096. doi: 10.1152/ajpgi.00124.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lo Iacono L, Boczkowski J, Zini R, Salouage I, Berdeaux A, Motterlini R, et al. A carbon monoxide-releasing molecule (CORM-3) uncouples mitochondrial respiration and modulates the production of reactive oxygen species. Free Radic Biol Med. 2011;50:1556–1564. doi: 10.1016/j.freeradbiomed.2011.02.033. [DOI] [PubMed] [Google Scholar]
- Magnotti LJ, Upperman JS, Xu DZ, Lu Q, Deitch EA. Gut-derived mesenteric lymph but not portal blood increases endothelial cell permeability and promotes lung injury after hemorrhagic shock. Ann Surg. 1998;228:518–527. doi: 10.1097/00000658-199810000-00008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maines MD. The heme oxygenase system: a regulator of second messenger gases. Annu Rev Pharmacol Toxicol. 1997;37:517–554. doi: 10.1146/annurev.pharmtox.37.1.517. [DOI] [PubMed] [Google Scholar]
- Mayr FB, Spiel A, Leitner J, Marsik C, Germann P, Ullrich R, et al. Effects of carbon monoxide inhalation during experimental endotoxemia in humans. Am J Respir Crit Care Med. 2005;171:354–360. doi: 10.1164/rccm.200404-446OC. [DOI] [PubMed] [Google Scholar]
- Megias J, Busserolles J, Alcaraz MJ. The carbon monoxide-releasing molecule CORM-2 inhibits the inflammatory response induced by cytokines in Caco-2 cells. Br J Pharmacol. 2007;150:977–986. doi: 10.1038/sj.bjp.0707184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michel BW, Lippert AR, Chang CJ. A reaction-based fluorescent probe for selective imaging of carbon monoxide in living cells using a palladium-mediated carbonylation. J Am Chem Soc. 2012;134:15668–15671. doi: 10.1021/ja307017b. [DOI] [PubMed] [Google Scholar]
- Miller SM, Farrugia G, Schmalz PF, Ermilov LG, Maines MD, Szurszewski JH. Heme oxygenase 2 is present in interstitial cell networks of the mouse small intestine. Gastroenterology. 1998;114:239–244. doi: 10.1016/s0016-5085(98)70473-1. [DOI] [PubMed] [Google Scholar]
- Miller SM, Reed D, Sarr MG, Farrugia G, Szurszewski JH. Haem oxygenase in enteric nervous system of human stomach and jejunum and co-localization with nitric oxide synthase. Neurogastroenterol Motil. 2001;13:121–131. doi: 10.1046/j.1365-2982.2001.00255.x. [DOI] [PubMed] [Google Scholar]
- Min KJ, Yang MS, Kim SU, Jou I, Joe EH. Astrocytes induce heme oxygenase-1 expression in microglia: a feasible mechanism for preventing excessive brain inflammation. J Neurosci. 2006;26:1880–1887. doi: 10.1523/JNEUROSCI.3696-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mittal M, Siddiqui MR, Tran K, Reddy SP, Malik AB. Reactive oxygen species in inflammation and tissue injury. Antioxid Redox Signal. 2014;20:1126–1167. doi: 10.1089/ars.2012.5149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizuguchi S, Stephen J, Bihari R, Markovic N, Suehiro S, Capretta A, et al. CORM-3-derived CO modulates polymorphonuclear leukocyte migration across the vascular endothelium by reducing levels of cell surface-bound elastase. Am J Physiol Heart Circ Physiol. 2009;297:H920–H929. doi: 10.1152/ajpheart.00305.2009. [DOI] [PubMed] [Google Scholar]
- Moore BA, Otterbein LE, Türler A, Choi AM, Bauer AJ. Inhaled carbon monoxide suppresses the development of postoperative ileus in the murine small intestine. Gastroenterology. 2003;124:377–391. doi: 10.1053/gast.2003.50060. [DOI] [PubMed] [Google Scholar]
- Moore BA, Overhaus M, Whitcomb J, Ifedigbo E, Choi AM, Otterbein LE, et al. Brief inhalation of low-dose carbon monoxide protects rodents and swine from postoperative ileus. Crit Care Med. 2005;33:1317–1326. doi: 10.1097/01.ccm.0000166349.76514.40. [DOI] [PubMed] [Google Scholar]
- Moore R, Carlson S, Madara JL. Villus contraction aids repair of intestinal epithelium after injury. Am J Physiol. 1989;257:G274–G283. doi: 10.1152/ajpgi.1989.257.2.G274. [DOI] [PubMed] [Google Scholar]
- Motterlini R. Carbon monoxide-releasing molecules (CO-RMs): vasodilatory, anti-ischaemic and anti-inflammatory activities. Biochem Soc Trans. 2007;35:1142–1146. doi: 10.1042/BST0351142. [DOI] [PubMed] [Google Scholar]
- Motterlini R, Otterbein LE. The therapeutic potential of carbon monoxide. Nat Rev Drug Discov. 2010;9:728–743. doi: 10.1038/nrd3228. [DOI] [PubMed] [Google Scholar]
- Motterlini R, Clark JE, Foresti R, Sarathchandra P, Mann BE, Green CJ. Carbon monoxide-releasing molecules: characterization of biochemical and vascular activities. Circ Res. 2002;90:E17–E24. doi: 10.1161/hh0202.104530. [DOI] [PubMed] [Google Scholar]
- Motterlini R, Sawle P, Hammad J, Bains S, Alberto R, Foresti R, et al. CORM-A1: a new pharmacologically active carbon monoxide-releasing molecule. FASEB J. 2005;19:284–286. doi: 10.1096/fj.04-2169fje. [DOI] [PubMed] [Google Scholar]
- Nadler EP, Stanford A, Zhang XR, Schall LC, Alber SM, Watkins SC, et al. Intestinal cytokine gene expression in infants with acute necrotizing enterocolitis: interleukin-11 mRNA expression inversely correlates with extent of disease. J Pediatr Surg. 2001;36:1122–1129. doi: 10.1053/jpsu.2001.25726. [DOI] [PubMed] [Google Scholar]
- Nakahira K, Kim HP, Geng XH, Nakao A, Wang X, Murase N, et al. Carbon monoxide differentially inhibits TLR signaling pathways by regulating ROS-induced trafficking of TLRs to lipid rafts. J Exp Med. 2006;203:2377–2389. doi: 10.1084/jem.20060845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakao A, Otterbein LE, Overhaus M, Sarady JK, Tsung A, Kimizuka K, et al. Biliverdin protects the functional integrity of a transplanted syngeneic small bowel. Gastroenterology. 2004;127:595–606. doi: 10.1053/j.gastro.2004.05.059. [DOI] [PubMed] [Google Scholar]
- Nakao A, Schmidt J, Harada T, Tsung A, Stoffels B, Cruz RJ, Jr, et al. A single intraperitoneal dose of carbon monoxide-saturated Ringer's lactate solution ameliorates postoperative ileus in mice. J Pharmacol Exp Ther. 2006a;319:1265–1275. doi: 10.1124/jpet.106.108654. [DOI] [PubMed] [Google Scholar]
- Nakao A, Toyokawa H, Tsung A, Nalesnik MA, Stolz DB, Kohmoto J, et al. Ex vivo application of carbon monoxide in University of Wisconsin solution to prevent intestinal cold ischemia/reperfusion injury. Am J Transplant. 2006b;6:2243–2255. doi: 10.1111/j.1600-6143.2006.01465.x. [DOI] [PubMed] [Google Scholar]
- Nakao A, Faleo G, Shimizu H, Nakahira K, Kohmoto J, Sugimoto R, et al. Ex vivo carbon monoxide prevents cytochrome P450 degradation and ischemia/reperfusion injury of kidney grafts. Kidney Int. 2008;74:1009–1016. doi: 10.1038/ki.2008.342. [DOI] [PubMed] [Google Scholar]
- Nath KA, Balla G, Vercellotti GM, Balla J, Jacob HS, Levitt MD, et al. Induction of heme oxygenase is a rapid, protective response in rhabdomyolysis in the rat. J Clin Invest. 1992;90:267–270. doi: 10.1172/JCI115847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ndisang JF, Gai P, Berni L, Mirabella C, Baronti R, Mannaioni PF, et al. Modulation of the immunological response of guinea pig mast cells by carbon monoxide. Immunopharmacology. 1999;43:65–73. doi: 10.1016/s0162-3109(99)00045-4. [DOI] [PubMed] [Google Scholar]
- Neu J, Walker WA. Necrotizing enterocolitis. N Engl J Med. 2011;364:255–264. doi: 10.1056/NEJMra1005408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Onyiah JC, Sheikh SZ, Maharshak N, Steinbach EC, Russo SM, Kobayashi T, et al. Carbon monoxide and heme oxygenase-1 prevent intestinal inflammation in mice by promoting bacterial clearance. Gastroenterology. 2013;144:789–798. doi: 10.1053/j.gastro.2012.12.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otani K, Shimizu S, Chijiiwa K, Morisaki T, Yamaguchi T, Yamaguchi K, et al. Administration of bacterial lipopolysaccharide to rats induces heme oxygenase-1 and formation of antioxidant bilirubin in the intestinal mucosa. Dig Dis Sci. 2000;45:2313–2319. doi: 10.1023/a:1005626622203. [DOI] [PubMed] [Google Scholar]
- Otterbein LE, Mantell LL, Choi AM. Carbon monoxide provides protection against hyperoxic lung injury. Am J Physiol Lung Cell Mol Physiol. 1999;276:L688–L694. doi: 10.1152/ajplung.1999.276.4.L688. [DOI] [PubMed] [Google Scholar]
- Otterbein LE, Bach FH, Alam J, Soares M, Tao Lu H, Wysk M, et al. Carbon monoxide has anti-inflammatory effects involving the mitogen-activated protein kinase pathway. Nat Med. 2000;6:422–428. doi: 10.1038/74680. [DOI] [PubMed] [Google Scholar]
- Otterbein LE, Otterbein SL, Ifedigbo E, Liu F, Morse DE, Fearns C, et al. MKK3 mitogen-activated protein kinase pathway mediates carbon monoxide-induced protection against oxidant-induced lung injury. Am J Pathol. 2003a;163:2555–2563. doi: 10.1016/S0002-9440(10)63610-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otterbein LE, Zuckerbraun BS, Haga M, Liu F, Song R, Usheva A, et al. Carbon monoxide suppresses arteriosclerotic lesions associated with chronic graft rejection and with balloon injury. Nat Med. 2003b;9:183–190. doi: 10.1038/nm817. [DOI] [PubMed] [Google Scholar]
- Paul G, Bataille F, Obermeier F, Bock J, Klebl F, Strauch U, et al. Analysis of intestinal haem-oxygenase-1 (HO-1) in clinical and experimental colitis. Clin Exp Immunol. 2005;140:547–555. doi: 10.1111/j.1365-2249.2005.02775.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pietzcker J, Kluthea C, Klinghammer K, Bührer C, Rüstow B, Guthmann F. Developmental delay in hypoxia-induced HO-1 expression predisposes to gut injury. J Perinat Med. 2012;40:191–197. doi: 10.1515/jpm.2011.117. [DOI] [PubMed] [Google Scholar]
- Reboul C, Thireau J, Meyer G, André L, Obert P, Cazorla O, et al. Carbon monoxide exposure in the urban environment: an insidious foe for the heart? Respir Physiol Neurobiol. 2012;184:204–212. doi: 10.1016/j.resp.2012.06.010. [DOI] [PubMed] [Google Scholar]
- Ren H, Leib SL, Ferriero DM, Täuber MG, Christen S. Induction of haem oxygenase-1 causes cortical non-haem iron increase in experimental pneumococcal meningitis: evidence that concomitant ferritin up-regulation prevents iron-induced oxidative damage. J Neurochem. 2007;100:532–544. doi: 10.1111/j.1471-4159.2006.04230.x. [DOI] [PubMed] [Google Scholar]
- Rodella L, Lamon BD, Rezzani R, Sangras B, Goodman AI, Falck JR, et al. Carbon monoxide and biliverdin prevent endothelial cell sloughing in rats with type I diabetes. Free Radic Biol Med. 2006;40:2198–2205. doi: 10.1016/j.freeradbiomed.2006.02.018. [DOI] [PubMed] [Google Scholar]
- Rodriguez AI, Gangopadhyay A, Kelley EE, Pagano PJ, Zuckerbraun BS, Bauer PM. HO-1 and CO decrease platelet-derived growth factor-induced vascular smooth muscle cell migration via inhibition of Nox1. Arterioscler Thromb Vasc Biol. 2010;30:98–104. doi: 10.1161/ATVBAHA.109.197822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rot A, von Andrian UH. Chemokines in innate and adaptive host defense: basic chemokinese grammar for immune cells. Annu Rev Immunol. 2004;22:891–928. doi: 10.1146/annurev.immunol.22.012703.104543. [DOI] [PubMed] [Google Scholar]
- Ryter SW, Choi AM. Heme oxygenase-1/carbon monoxide: from metabolism to molecular therapy. Am J Respir Cell Mol Biol. 2009;41:251–260. doi: 10.1165/rcmb.2009-0170TR. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryter SW, Choi AM. Carbon monoxide: present and future indications for a medical gas. Korean J Intern Med. 2013;28:123–140. doi: 10.3904/kjim.2013.28.2.123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sandouka A, Fuller BJ, Mann BE, Green CJ, Foresti R, Motterlini R. Treatment with CO-RMs during cold storage improves renal function at reperfusion. Kidney Int. 2006;69:239–247. doi: 10.1038/sj.ki.5000016. [DOI] [PubMed] [Google Scholar]
- Sarady JK, Zuckerbraun BS, Bilban M, Wagner O, Usheva A, Liu F, et al. Carbon monoxide protection against endotoxic shock involves reciprocal effects on iNOS in the lung and liver. FASEB J. 2004;18:854–856. doi: 10.1096/fj.03-0643fje. [DOI] [PubMed] [Google Scholar]
- Sato K, Balla J, Otterbein L, Smith RN, Brouard S, Lin Y, et al. Carbon monoxide generated by heme oxygenase-1 suppresses the rejection of mouse-to-rat cardiac transplants. J Immunol. 2001;166:4185–4194. doi: 10.4049/jimmunol.166.6.4185. [DOI] [PubMed] [Google Scholar]
- Sawle P, Foresti R, Mann BE, Johnson TR, Green CJ, Motterlini R. Carbon monoxide-releasing molecules (CO-RMs) attenuate the inflammatory response elicited by lipopolysaccharide in RAW264.7 murine macrophages. Br J Pharmacol. 2005;145:800–810. doi: 10.1038/sj.bjp.0706241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schallner N, Romão CC, Biermann J, Lagrèze WA, Otterbein LE, Buerkle H, et al. Carbon monoxide abrogates ischemic insult to neuronal cells via the soluble guanylate cyclase-cGMP pathway. PLoS ONE. 2013;8:e60672. doi: 10.1371/journal.pone.0060672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt J, Stoffels B, Chanthaphavong RS, Buchholz BM, Nakao A, Bauer AJ. Differential molecular and cellular immune mechanisms of postoperative and LPS-induced ileus in mice and rats. Cytokine. 2012;59:49–58. doi: 10.1016/j.cyto.2012.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schulz S, Wong RJ, Jang KY, Kalish F, Chisholm KM, Zhao H, et al. Heme oxygenase-1 deficiency promotes the development of necrotizing enterocolitis-like intestinal injury in a newborn mouse model. Am J Physiol Gastrointest Liver Physiol. 2013;304:G991–G1001. doi: 10.1152/ajpgi.00363.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scott JR, Cukiernik MA, Ott MC, Bihari A, Badhwar A, Gray DK, et al. Low-dose inhaled carbon monoxide attenuates the remote intestinal inflammatory response elicited by hindlimb ischemia-reperfusion. Am J Physiol Gastrointest Liver Physiol. 2009;296:G9–G14. doi: 10.1152/ajpgi.90243.2008. [DOI] [PubMed] [Google Scholar]
- Snoek SA, Dhawan S, van Bree SH, Cailotto C, van Diest SA, Duarte JM, et al. Mast cells trigger epithelial barrier dysfunction, bacterial translocation and postoperative ileus in a mouse model. Neurogastroenterol Motil. 2012;24:172–184. doi: 10.1111/j.1365-2982.2011.01820.x. [DOI] [PubMed] [Google Scholar]
- Sodhi C, Richardson W, Gribar S, Hackam DJ. The development of animal models for the study of necrotizing enterocolitis. Dis Model Mech. 2008;1:94–98. doi: 10.1242/dmm.000315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stocker R, Yamamoto Y, McDonagh AF, Glazer AN, Ames BN. Bilirubin is an antioxidant of possible physiological importance. Science. 1987;235:1043–1046. doi: 10.1126/science.3029864. [DOI] [PubMed] [Google Scholar]
- Stone JR, Marletta MA. Soluble guanylate cyclase from bovine lung: activation with nitric oxide and carbon monoxide and spectral characterization of the ferrous and ferric states. Biochemistry. 1994;33:5636–5640. doi: 10.1021/bi00184a036. [DOI] [PubMed] [Google Scholar]
- Sun B, Zou X, Chen Y, Zhang P, Shi G. Preconditioning of carbon monoxide releasing molecule-derived CO attenuates LPS-induced activation of HUVEC. Int J Biol Sci. 2008;4:270–278. doi: 10.7150/ijbs.4.270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun BW, Jin Q, Sun Y, Sun ZW, Chen X, Chen ZY, et al. Carbon liberated from CO-releasing molecules attenuates leukocyte infiltration in the small intestine of thermally injured mice. World J Gastroenterol. 2007;13:6183–6190. doi: 10.3748/wjg.v13.i46.6183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taillé C, El-Benna J, Lanone S, Boczkowski J, Motterlini R. Mitochondrial respiratory chain and NAD(P)H oxidase are targets for the antiproliferative effect of carbon monoxide in human airway smooth muscle. J Biol Chem. 2005;280:25350–25360. doi: 10.1074/jbc.M503512200. [DOI] [PubMed] [Google Scholar]
- Takagi T, Naito Y, Uchiyama K, Suzuki T, Hirata I, Mizushima K, et al. Carbon monoxide liberated from carbon monoxide-releasing molecule exerts an anti-inflammatory effect on dextran sulfate sodium-induced colitis in mice. Dig Dis Sci. 2011;56:1663–1671. doi: 10.1007/s10620-010-1484-y. [DOI] [PubMed] [Google Scholar]
- Takaki S, Takeyama N, Kajita Y, Yabuki T, Noguchi H, Miki Y, et al. Beneficial effects of the heme oxygenase-1/.carbon monoxide system in patients with severe sepsis/septic shock. Intensive Care Med. 2010;36:42–48. doi: 10.1007/s00134-009-1575-4. [DOI] [PubMed] [Google Scholar]
- The FO, Bennink RJ, Ankum WM, Buist MR, Busch OR, Gouma DJ, et al. Intestinal handling-induced mast cell activation and inflammation in human postoperative ileus. Gut. 2008;57:33–40. doi: 10.1136/gut.2007.120238. [DOI] [PubMed] [Google Scholar]
- Tsoyi K, Ha YM, Kim YM, Lee YS, Kim HJ, Kim HJ, et al. Activation of PPAR-gamma by carbon monoxide from CORM-2 leads to the inhibition of iNOS but not COX-2 expression in LPS-stimulated macrophages. Inflammation. 2009;32:364–371. doi: 10.1007/s10753-009-9144-0. [DOI] [PubMed] [Google Scholar]
- Uchiyama K, Naito Y, Takagi T, Mizushima K, Hayashi N, Harusato A, et al. Carbon monoxide enhance colonic epithelial restitution via FGF15 derived from colonic myofibroblasts. Biochem Biophys Res Commun. 2010;391:1122–1126. doi: 10.1016/j.bbrc.2009.12.035. [DOI] [PubMed] [Google Scholar]
- Uehara K, Takahashi T, Fujii H, Shimizu H, Omori E, Matsumi M, et al. The lower intestinal tract-specific induction of heme oxygenase-1 by glutamine protects against endotoxemic intestinal injury. Crit Care Med. 2005;33:381–390. doi: 10.1097/01.ccm.0000153407.14237.7f. [DOI] [PubMed] [Google Scholar]
- Vannacci A, Di Felice A, Giannini L, Marzocca C, Pierpaoli S, Zagli G, et al. The effect of a carbon monoxide-releasing molecule on the immunological activation of guinea-pig mast cells and human basophils. Inflamm Res. 2004;53(Suppl. 1):S9–S10. doi: 10.1007/s00011-003-0303-8. [DOI] [PubMed] [Google Scholar]
- Vanova K, Suk J, Petr T, Cerny D, Slanar O, Vreman HJ, et al. Protective effects of inhaled carbon monoxide in endotoxin-induced cholestasis is dependent on its kinetics. Biochimie. 2014;97:173–180. doi: 10.1016/j.biochi.2013.10.009. [DOI] [PubMed] [Google Scholar]
- Von Burg R. Carbon Monoxide. J Appl Toxicol. 1999;19:379–386. doi: 10.1002/(sici)1099-1263(199909/10)19:5<379::aid-jat563>3.0.co;2-8. [DOI] [PubMed] [Google Scholar]
- de Vries HS, te Morsche RH, Jenniskens K, Peters WH, de Jong DJ. A functional polymorphism in UGT1A1 related to hyperbilirubinemia is associated with a decreased risk for Crohn's disease. J Crohn's Colitis. 2012;6:597–602. doi: 10.1016/j.crohns.2011.11.010. [DOI] [PubMed] [Google Scholar]
- Wang WP, Guo X, Koo MW, Wong BC, Lam SK, Ye YN, et al. Protective role of heme oxygenase-1 on trinitrobenzene sulfonic acid-induced colitis in rats. Am J Physiol Gastrointest Liver Physiol. 2001;281:G586–G594. doi: 10.1152/ajpgi.2001.281.2.G586. [DOI] [PubMed] [Google Scholar]
- Wang X, Cao J, Sun BW, Liu DD, Liang F, Gao L. Exogenous carbon monoxide attenuates inflammatory responses in the small intestine of septic mice. World J Gastroenterol. 2012;18:5719–5728. doi: 10.3748/wjg.v18.i40.5719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wegiel B, Otterbein LE. Go green: the anti-inflammatory effects of biliverdin reductase. Front Pharmacol. 2012;3:47. doi: 10.3389/fphar.2012.00047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whittle BJ, Varga C. New light on the anti-colitic actions of therapeutic aminosalicylates: the role of heme oxygenase. Pharmacol Rep. 2010;62:548–556. doi: 10.1016/s1734-1140(10)70312-1. [DOI] [PubMed] [Google Scholar]
- Willis D, Moore AR, Frederick R, Willoughby DA. Heme oxygenase: a novel target for the modulation of the inflammatory response. Nat Med. 1996;2:87–90. doi: 10.1038/nm0196-87. [DOI] [PubMed] [Google Scholar]
- Wu L, Wang R. Carbon monoxide: endogenous production, physiological functions, and pharmacological applications. Pharmacol Rev. 2005;57:585–630. doi: 10.1124/pr.57.4.3. [DOI] [PubMed] [Google Scholar]
- Yoriki H, Naito Y, Takagi T, Mizusima K, Hirai Y, Harusato A, et al. Hemin ameliorates indomethacin-induced small intestinal injury in mice through the induction of heme oxygenase-1. J Gastroenterol Hepatol. 2013;28:632–638. doi: 10.1111/jgh.12074. [DOI] [PubMed] [Google Scholar]
- Yoshida J, Ozaki KS, Nalesnik MA, Ueki S, Castillo-Rama M, Faleo G, et al. Ex vivo application of carbon monoxide in UW solution prevents transplant-induced renal ischemia/reperfusion injury in pigs. Am J Transplant. 2010;10:763–772. doi: 10.1111/j.1600-6143.2010.03040.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Shan P, Otterbein LE, Alam J, Flavell RA, Davis RJ, et al. Carbon monoxide inhibition of apoptosis during ischemia-reperfusion lung injury is dependent on the p38 mitogen-activated protein kinase pathway and involves caspase 3. J Biol Chem. 2003;278:1248–1258. doi: 10.1074/jbc.M208419200. [DOI] [PubMed] [Google Scholar]
- Zhong W, Xia Z, Hinrichs D, Rosenbaum JT, Wegmann KW, Meyrowitz J, et al. Hemin exerts multiple protective mechanisms and attenuates dextran sulfate sodium-induced colitis. J Pediatr Gastroenterol Nutr. 2010;50:132–139. doi: 10.1097/MPG.0b013e3181c61591. [DOI] [PubMed] [Google Scholar]
- Zuckerbraun BS, Billiar TR, Otterbein SL, Kim PK, Liu F, Choi AM, et al. Carbon monoxide protects against liver failure through nitric oxide-induced heme oxygenase 1. J Exp Med. 2003;198:1707–1716. doi: 10.1084/jem.20031003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuckerbraun BS, McCloskey CA, Gallo D, Liu F, Ifedigbo E, Otterbein LE, et al. Carbon monoxide prevents multiple organ injury in a model of hemorrhagic shock and resuscitation. Shock. 2005a;23:527–532. [PubMed] [Google Scholar]
- Zuckerbraun BS, Otterbein LE, Boyle P, Jaffe R, Upperman J, Zamora R, et al. Carbon monoxide protects against the development of experimental necrotizing enterocolitis. Am J Physiol Gastrointest Liver Physiol. 2005b;289:G607–G613. doi: 10.1152/ajpgi.00055.2005. [DOI] [PubMed] [Google Scholar]