

Abstract
Background and Purpose
Whether NO, carbon monoxide (CO) and hydrogen sulfide (H2S) compensate for each other when one or more is depleted is unclear. Inhibiting NOS causes hypertension and kidney injury. Both global depletion of H2S by cystathionine γ-lyase (CSE) gene deletion and low levels of exogenous H2S cause hypertension. Inhibiting CO-producing enzyme haeme oxygenase-1 (HO-1) makes rodents hypersensitive to hypertensive stimuli. We hypothesized that combined inhibition of NOS and HO-1 exacerbates hypertension and renal injury, but how combined inhibition of NOS and CSE affect hypertension and renal injury was unclear.
Experimental Approach
Rats were treated with inhibitors of NOS (L-nitroarginine; LNNA), CSE (DL-propargylglycine; PAG), or HO-1 (tin protoporphyrin; SnPP) singly for 1 or 4 weeks or in combinations for 4 weeks.
Key Results
LNNA always reduced NO, decreased H2S and increased CO after 4 weeks. PAG abolished H2S, always enhanced CO and reduced NO, but not when used in combination with other inhibitors. SnPP always increased NO, enhanced H2S and inhibited CO after 1 week. Rats treated with LNNA, but not PAG and SnPP, rapidly developed hypertension followed by renal dysfunction. LNNA-induced hypertension was ameliorated and renal dysfunction prevented by all additional treatments. Renal HO-1 expression was increased by LNNA in injured tubules and increased in all tubules by all other treatments.
Conclusions and Implications
The amelioration of LNNA-induced hypertension and renal injury by additional inhibition of H2S and/or CO-producing enzymes appeared to be associated with secondary increases in renal CO or NO production.
Linked Articles
This article is part of a themed section on Pharmacology of the Gasotransmitters. To view the other articles in this section visit http://dx.doi.org/10.1111/bph.2015.172.issue-6
Keywords: nitric oxide, hydrogen sulfide, carbon monoxide, renal, hypertension, cystathionine γ-lyase, haeme oxygenase-1
Introduction
The family of biologically active gases, NO, carbon monoxide (CO) and hydrogen sulfide (H2S) exert important, often overlapping homeostatic functions in the reno-vascular system (Li et al., 2009). BP regulation is strongly dependent on renal NO availability (Rajapakse and Mattson, 2013), and hypertension and chronic kidney disease (CKD) are associated with decreased NO levels in the kidney (Baylis and Vallance, 1996; Baylis, 2008). Prolonged pharmacological inhibition of NOS invariably results in hypertension, and renal and cardiac injury (Zatz and de Nucci, 1991; Baylis et al., 1992; Verhagen et al., 1998). In addition, NO, CO and H2S also induce vasodilatation (Li et al., 2009). Indeed, mice with deletion of the key enzymes in NO and H2S production, endothelial NOS (eNOS) and cystathionine γ-lyase (CSE), both display hypertension (Ortiz and Garvin, 2003; Yang et al., 2008). Mice with deletion of haeme oxygenase-1 (HO-1) are not hypertensive but, as in eNOS knockout mice (Whiting et al., 2013) their sensitivity to angiotensin-induced hypertension and to renal injury is increased (Agarwal and Nick, 2000; Wiesel et al., 2001). At present, it is unclear whether these three gases compensate for each other (Gadalla and Snyder, 2010), in other words, how production and function of the other two gaseous vasodilators is adapted to depletion of the third.
Depletion of NO by non-selective NOS inhibition is an established model of hypertension and CKD (Verhagen et al., 1998; 2001; Zatz and Baylis, 1998; van Koppen et al., 2013). In view of the key role of NO availability in the pathogenesis of hypertension (Rajapakse and Mattson, 2013), we investigated the renal transcriptome of chronically (21 days) NO-depleted rats that had developed hypertension and renal injury, and found that expression of the H2S-producing enzyme CSE was decreased while the CO-producing enzyme HO-1 was increased (Wesseling et al., 2007). At an early stage (4 days), when hypertension and renal injury had not yet developed, gene expression of these enzymes was not affected. Because our previous observations were limited to gene expression, the first questions of the current study were whether prolonged NO depletion indeed decreases renal H2S production and increases renal CO production and whether depletion of H2S or CO affects the renal production of the other two gases. Furthermore, we explored, in different combinations, how simultaneous depletion of two gases would affect renal production of the third.
HO-1 knockout mice had normal BP and no renal injury, but the kidney was much more sensitive to nephrotoxins (e.g. cisplatin; Shiraishi et al., 2000). Although inhibition of HO-1 did not induce hypertension in normotensive rats (Ushiyama et al., 2002; Chok et al., 2009; Chandrashekar et al., 2012), it further increased BP (Ushiyama et al., 2002; Chandrashekar et al., 2012) and induced renal injury in hypertensive rats (Chandrashekar et al., 2012). At low concentrations, H2S acts as a vasoconstrictor by inhibiting production and availability of NO (Liu et al., 2011). However, deletion of the H2S-producing enzymes CSE or cystathionine β-synthase (CBS) impaired endothelial function (Lentz et al., 2000; Yang et al., 2008), and caused age-dependent mild hypertension, although kidney function was preserved (Yang et al., 2008). In rats, combined inhibition of CSE and CBS was required to induce hypertension (Roy et al., 2012). Thus, based on these and our previous observations of decreased CSE expression in NO-depleted rats we hypothesized that combined inhibition of NOS and HO-1 would exacerbate hypertension and renal injury, while combined inhibition of NOS and CSE would have uncertain effects on hypertension and renal injury.
To answer the first question and to understand the effect of combined inhibition on the third gas, on BP and on renal function, we first observed the effects of single inhibition. In the present study two time points in the treatment were chosen based on our previous experience with non-specific NOS inhibition (Attia et al., 2001; Wesseling et al., 2007) for two reasons: firstly, to check whether the effect of short-term single inhibition (1 week) on gases remained stable after prolonged single inhibition (4 weeks) and, secondly, to differentiate the effect of intact (1 week) and diminished (4 weeks) renal function on gases. This was followed by combined inhibition for 4 weeks.
Methods
Animal experiments
Male 5 week-old, Sprague Dawley rats (Harlan-Olac, Blackthorn, Bicester, Oxon, UK) were acclimatized, at 22°C, 60% humidity, exposed to 12-h light/dark cycle, tamed and trained for BP measurement. At 8 weeks of age, administration of the different inhibitors was started (see Drugs section) after a baseline urine collection and BP measurement. In the first phase (single inhibition), the rats were treated for 1 or 4 weeks. In the second phase (combined inhibition), the rats were treated for 4 weeks. An overview of the animal experiment is shown in Figure 1. All groups contained six rats. Rats had free access to drinking water and standard chow. Systolic BP measurements were performed using a tail cuff (LE 5002 Storage Pressure Meter; PanLab, Barcelona, Spain); an independent technician, blinded to the treatments as the rats were numbered, repeatedly measured each rat until at least five good measurements were achieved. For the 24 h urine collection, rats were placed in metabolic cages without chow, but with free access to glucose-containing water (2% w·v−1). Urine was collected in antibiotics to prevent formation of NO metabolites and frozen after collection. We found no difference in drinking water intake between rats on different treatments (∼25 mL per rat day−1). Only the rats on L-nitroarginine (LNNA; see Drugs section) started to drink more water after 4 weeks of treatment because of renal dysfunction. After 1 or 4 weeks the rats were anaesthetized with Nembutal (pentobarbital injection i.p., 5.5 mg 100 g−1 BW), an aortic blood sample was drawn and kidneys were excised. Blood plasma and parts of the renal cortex were snap frozen and stored at −80°C. Half of one kidney was stored in 4% formaldehyde for histology. The Animal Ethical Committee of the University of Utrecht approved the protocol. All studies involving animals are reported in accordance with the ARRIVE guidelines for reporting experiments involving animals (Kilkenny et al., 2010; McGrath et al., 2010).
Figure 1.
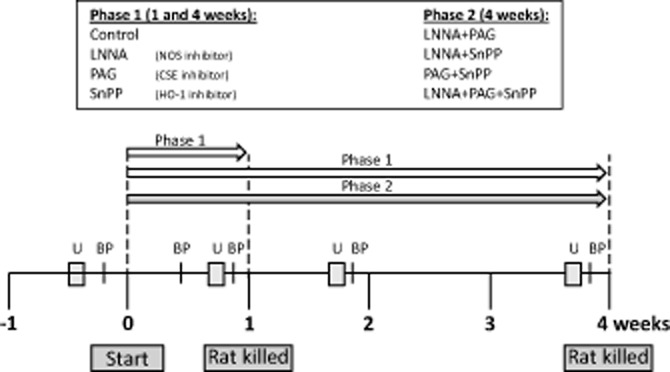
Flow sheet for the study design. The experimental set-up from −1 to 4 weeks of treatment with the inhibitors that started at week 0 is depicted. The durations of phases 1 and 2 are shown by white and grey arrows respectively. Inhibitors were LNNA (a NOS inhibitor), PAG (H2S-producing enzyme inhibitor) and SnPP (HO-1 inhibitor). U = 24 h urine collection.
Drugs
Drugs used were NOS inhibitor LNNA (Sigma-Aldrich, Zwijndrecht, the Netherlands), irreversible H2S-synthesizing enzyme CSE inhibitor DL-propargylglycine (PAG; Sigma-Aldrich) and HO-1 inhibitor Sn(IV) protoporphyrin IX dichloride (tin protoporphyrin, SnPP; Frontier Scientific, Newark, NJ, USA). LNNA was dissolved in drinking water (125 mg·L−1). PAG was dissolved in saline (30 mg·mL−1) for daily i.p. injection (37.5 mg·kg−1 BW). SnPP was dissolved in 1 M NaOH and diluted fourfold with saline (final concentration 75 mg·mL−1). The acidity of SnPP solution was checked (∼pH 7.4) before weekly s.c. injection (50 μmol·kg−1 BW). The dose of PAG used in the study was effective in abolishing H2S production in kidney and liver (data not shown). Thus, we had no reason to increase the dose of PAG. This decision was supported by reports that normal rats with inhibited CSE had no change in BP (Roy et al., 2012; Issa et al., 2013). Inhibition of CSE by D-propargylglycine (D-PAG) was found to be nephrotoxic (Maekawa et al., 2005). This was caused by a metabolite of D-PAG produced by D-amino-acid oxidase (Konno et al., 2000). However, PAG had no toxicity, and as mentioned earlier, no renal injury occurs when only PAG is administered (Roy et al., 2012). PAG is able to effectively inhibit CSE by covalent binding to it, which changes the active structure of CSE•pyridoxal-5′-phosphate (PLP) complex; in the CSE•PLP•PAG complex PLP was not affected (Sun et al., 2009). PAG also is able to react with PLP forming an electrophile (Washtien and Abeles, 1977) causing it to irreversibly react, for example, with the active lysine site. Thus, please note that PAG, although highly specific for CSE, might affect other PLP-dependent enzymes (e.g. CBS). Reported doses of SnPP are often twofold lower than that used in the present study. The absence of a change in BP induced by inhibiting HO-1 activity was supported by a gene-deletion study (Wiesel et al., 2001). Protoporphyrins are known for their phototoxicity, so we avoided high doses. A report was found where 30 μmol·kg−1·BW was effective in inhibiting HO-1 activity (bilirubin-based) and additional SnPP (50 μmol·kg−1·BW) did not further reduce HO-1 activity (Chandrashekar et al., 2012). Thus we decided, based on literature and toxicity data, to use a dose of 50 μmol·kg−1·BW SnPP. Please note that metallo-based protoporphyrins that act as inhibitors of HO-1 (e.g. ZnPP) can also inhibit soluble guanylate cyclase (Christodoulides et al., 1995) and haeme-regulated enzymes such as the cytochrome P450 enzymes (Trakshel et al., 1992).
Renal NO level
Urine nitrate + nitrite (NOx) metabolites, reflecting renal NO availability, were determined in urine using a colourimetric Assay (Cayman Chemical; Ann Arbor, CA, USA).
H2S production in kidney
Renal H2S production was measured in a closed 25 mL Erlenmeyer. A cryotube (1.8 mL), with a rolled up filter (Whatman, Rotterdam, the Netherlands; grade 3 MM, size 2.5 cm) filled with 500 μL trapping solution [3 M NaOH and 1% ZnAc (w·v−1 pH 6); 15:1, respectively], was placed in the Erlenmeyer with the opening at the top. Next, a mixture of potassium phosphate (50 mM; pH 7.4), L-cysteine (10 mM), PLP (2 mM) and renal homogenate (1.5% w·v−1 potassium phosphate pH 6.8) was added at the bottom; avoiding contact between mixture and filter. After plugging and flushing the Erlenmeyer with nitrogen, the contents were incubated at 37°C under slight shaking for 90 min. After injecting 500 μL trichloroacetic acid (50% w·v−1) carefully into the mixture through a needle inserted in the rubber plug (to inactivate the enzymes and to acidify the mixture causing expulsion of remaining dissolved H2S), the incubation at 37°C was prolonged for 60 min. The contents of the cryotube (filter plus trapping solution) were transferred to a tube containing 3.5 mL millipore H2O. Consecutively, 500 μL N,N-dimethyl-p-phenylenediamine sulfate salt (20 mM in 7.2 M HCl) and FeCl3 (30 mM in 1.2 M HCl) were added and after 30 min in the dark the absorbance of the solution was measured at 670 nm. The amount of H2S produced was calculated using a dilution range of NaHS.
CO production in kidney
The HO-1 activity is measured using Ferrozine, based on methods described by Reed et al. and modified by us for measurement in tissue (Huber et al., 2009; Reed et al., 2010). All solutions and steps were ice-cold or performed on ice. The same Tris buffer was used throughout (100 mM; pH 7.4). The renal cortex samples were homogenized in Tris buffer (20% w·v−1) using an ultra turrax, rested for 15 min and the homogenate was then centrifuged at 1500 g for 15 min. After separation of the cystolic fraction, the pellet was resuspended in the same amount of Tris buffer containing detergent Triton X-100 (1% w·v−1). After 15 min of resting followed by centrifugation at 1500 g for 15 min the supernatant (membranous fraction) was used for HO-1 activity measurements. A dilution range of samples (20.0–0.1%) was made with Tris buffer containing Triton X-100. Ferrozine (10 mM) dissolved in Tris buffer and hemin (12 mM) dissolved first in 1/5th of end-volume 0.1 M NaOH followed by adding 4/5th of end-volume Tris buffer were freshly made and kept in the dark. A reaction solution of ferrozine and hemin (500 and 300 μM, respectively) in Tris buffer was incubated at 37°C for at least 30 min in the dark for removal of any residual ferrous ions and placed on ice and kept in the dark. A 96-well plate was placed on an ice-cold platform and each well was filled with 25 μL of homogenate, 25 μL of Tris buffer and 50 μL of reaction solution. The 96-well plate was put in a plate reader, heated at 37°C and the absorbance in each well was real-time measured at 560 nm. A negative control containing only reaction solution was used to normalize the sample dilution curves. Dilution range of FeCl2 dissolved in Tris buffer containing triton X-100 was also included in each 96-well plates and the curves were all straight (r2 = 0.998 ± 0.001) with slope of 0.0022 (= adding 455 μmol Fe2+ increased absorbance by one). In the samples, the slope at the start of the curve represents the enzymatic activity of HO-1. In the dilution range of a sample, the homogenized concentration that had the highest activity was selected for calculation of enzyme activity.
HO-1 – Western blot and immunohistochemistry
For western blots, lysed renal samples in Laemmli buffer and 1 mM DTT were loaded and run in 12% acrylamide gel and then transferred to a nitrocellulose membrane. After being blocked with BSA (5% w·v−1) the membrane blots were incubated with polyclonal rabbit antibody HO-1 (MBL International, Woburn, MA, USA; SR-895; 1:1000) overnight at 4°C followed by incubation with secondary anti-rabbit antibody HRP for 1 h at room temperature (RT). After an incubation with ECL solution, digital images were analysed on a ChemiDoc XRS + molecular imager (Bio-Rad Laboratories; Veenendaal, the Netherlands) using Imagelab software (version 4.0.1).
For the immunohistochemistry, paraffin-embedded slides were deparaffinized using xylene and ethanol, and after blocking endogenous peroxidases with H2O2 (1.5%), boiled in citric buffer (10 mM trisodium citrate dehydrate; pH 6.0) for 20 min. The slides were incubated with polyclonal rabbit antibody HO-1 (MBL International; 1:100) for 1 h at 4°C followed by incubation with Goat anti-Rabbit poly-HRP BrightVision for 1 h at RT. Next, NovaRed solution (Vector) was applied for 10 min at RT and stained with haematoxylin for 30 s.
Renal function and injury
In urine, total protein levels were measured using the Bradford method (Bio-Rad Laboratories). Plasma, urea was determined using a kit (DiaSys Diagnostic Systems; Holzheim, Germany). Glomerulosclerosis was scored on periodic acid schiff-stained paraffin-embedded slides as described (Attia et al., 2002; Koeners et al., 2008).
Statistics
Results are expressed as mean ± SEM. Data were compared using repeated measurement one- or two-way anova where appropriate followed by the Student–Newman–Keuls (SNK) post hoc test. For terminal data one-way anova was applied followed by the SNK post hoc test. P < 0.05 was considered significant.
Results
Single inhibition – effect of 1 week of enzyme inhibition
After 1 week, NO metabolites in urine were reduced by LNNA (as expected) and by PAG, but were increased by SnPP (all P < 0.05; Figure 2A). The production of renal H2S was not affected by LNNA but was completely inhibited by PAG (0.1% vs. 100% in control; P < 0.001) and strongly induced by SnPP (P < 0.001; Figure 2B). The production of renal CO was not affected by LNNA, but was strongly stimulated by PAG (1890% vs. control; P < 0.001) and diminished by SnPP (54% vs. control; P < 0.05; Figure 2C). For clarification an overview of interaction between gases is shown (Figure 2D); it shows that restricting production of renal H2S or CO triggered responses from other gases, while inhibition of NOS did not. All rats receiving treatments displayed normal behaviour and no physical abnormalities. After 1 week, SBP was elevated by LNNA (P < 0.01; Table 1) but not affected by other inhibitors. No difference in heart rate was found between groups (data not shown). Plasma urea, proteinuria and body weight (268 ± 6 g) were at control levels in all groups. Renal histology did not reveal any change in glomerulosclerosis score (data not shown).
Figure 2.

Urine NOx, renal H2S and CO production in rats treated with a single inhibitor for 1 and 4 weeks. NO metabolites in urine (A), renal H2S production (B), renal CO production (C) and overview of interaction between gases (D). The pattern of each treatment group is shown in legend in panel A. To clearly show the effects of chronic inhibition on NO metabolites, urine NOx (μmol 100 g−1 BW day−1) was compared with baseline of the same treatment group set at 100%. The average level of urine NOx at baseline day 0 was 2.02 ± 0.46 μmol 100 g−1 BW day−1 at which point there were no significant differences between treatment groups. The average level of H2S or CO production in the control after 1 week was 7.6 ± 1.1 μmol·mg−1 tissue min−1 and 10.3 ± 1.1 μmol·mg−1 tissue min−1 respectively. The interaction between gases showed how the production of each gas responded to inhibition of each other gas after 1 and 4 weeks (panels A to C) and may not reflect the situation when the production of each gas is stimulated. The only change in the interaction between gases after 4 weeks of single inhibition is represented by the dotted arrows. Note that in the interaction between gases, effects are solely based on the effect of inhibition of one of the gas-producing enzymes. #P < 0.05, ##P < 0.01 versus control of the same treatment time.
Table 1.
Systolic BP, proteinuria and plasma urea in rats after 1 week of a single treatment
| Group | Systolic BP (mmHg) | Proteinuria (mg day−1) | Plasma urea (mmol L−1) |
|---|---|---|---|
| Control | 140 ± 14 | 17 ± 2 | 6.3 ± 0.6 |
| LNNA | 173 ± 12# | 16 ± 3 | 6.3 ± 0.4 |
| PAG | 135 ± 3 | 36 ± 2 | 8.5 ± 0.2 |
| SnPP | 119 ± 3 | 10 ± 2 | 4.1 ± 0.3 |
Mean ± SEM, n = 6 per group.
P < 0.01 versus control.
Single inhibition – effect of 4 weeks of enzyme inhibition
After 4 weeks of inhibition NO metabolites in urine remained effectively reduced by LNNA and PAG and were increased by SnPP (all P < 0.001; Figure 2A). Note that NO metabolites were more reduced by LNNA after 4 w as compared with after 1 w. Renal H2S production was reduced by LNNA (49% vs. control; P < 0.001; Figure 2B) and continued to be completely inhibited by PAG (1% vs. control; P < 0.001), but was at control levels during SnPP. Renal CO production was increased in LNNA and PAG-treated rats (all P < 0.001; Figure 2C) and was at control level in SnPP-treated rats. The overview of interaction between gases differed from 1 w single intervention in that prolonged NO depletion caused reduction and induction in H2S and CO production, respectively (Figure 2D), while inhibition of HO-1 (note that CO production was at control level) no longer enhanced H2S production.
All rats receiving treatments displayed normal behaviour and no physical abnormalities, except for LNNA rats in the fourth week (ruffled fur and weight loss). Systolic BP in LNNA-treated rats continued to increase over time (223 ± 10 vs. 137 ± 3 mmHg; P < 0.001; Figure 3A). No difference in heart rate was found among groups (data not shown). After 4 weeks, LNNA caused proteinuria (P < 0.001; Figure 3B), higher plasma urea (P < 0.05, Figure 3C), lower body weight (305 ± 15 vs. 341 ± 11 g; P < 0.05) and renal injury as assessed by glomerulosclerosis (P < 0.001; Figure 3D). PAG and SnPP had no effect on BP, renal function or injury.
Figure 3.
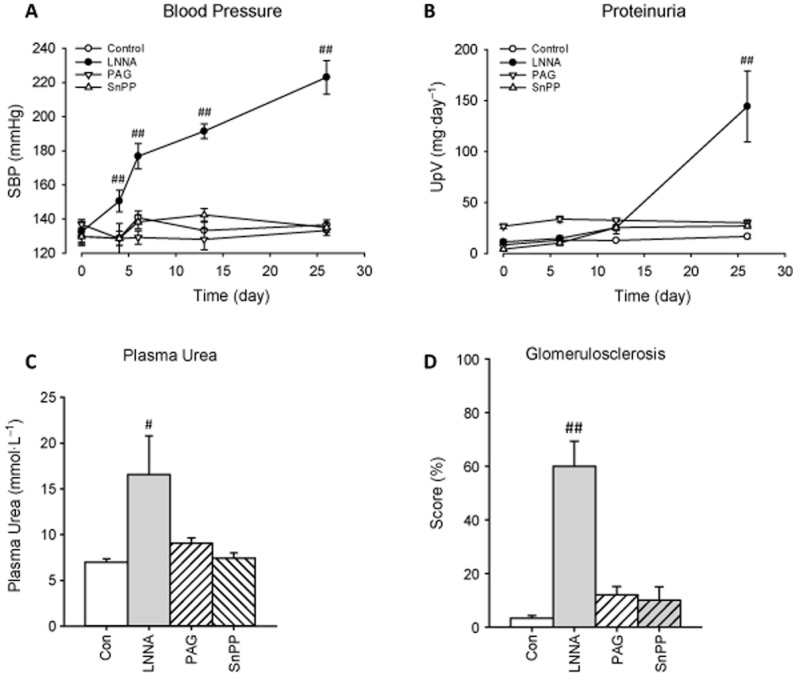
Systolic BP, renal function and renal injury in rats after 4 weeks of a single inhibitor. Systolic BP, proteinuria and plasma urea, and glomerulosclerosis (sum of partial and total glomerulosclerosis) are shown in panels A–D respectively. #P < 0.05, ##P < 0.01 versus control.
Single inhibition – renal HO-1 expression
Western blot on renal cortex samples revealed increased protein expression of HO-1 by LNNA only after 4 weeks and by PAG and SnPP at 1 and 4 weeks (all P < 0.001; Figure 4A). The IHC staining of HO-1 was almost absent in the renal cortex of both control groups and in 1 week LNNA rats, and was strongly positive in all other treated rats (Figure 4B). In all rats, the location of HO-1 was primarily tubular. The pattern of HO-1 staining in 4 week LNNA rats was characteristic of marked proteinuria (leaking glomeruli), while in other treated rats the HO-1 was uniformly spread throughout the proximal tubules, but not in glomeruli.
Figure 4.
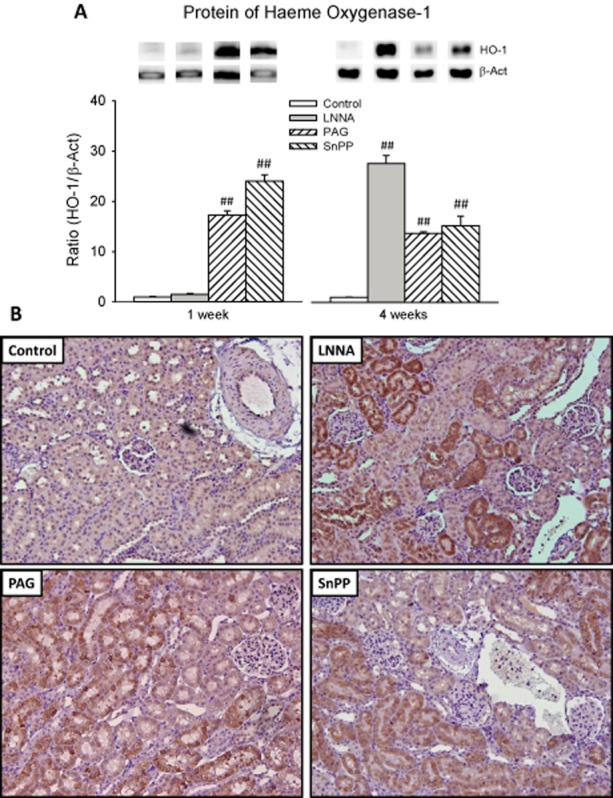
Renal HO-1 in rats, at 1 or 4 weeks, treated with a single inhibitor and interaction between gases. HO-1 protein was quantified using Western blots (A). Immunohistochemistry was applied to assess intrarenal protein expression and location of HO-1 (B) of rats treated for 4 weeks. Staining of HO-1 is shown by the brown colour. Staining of the 4 week control is representative of 1 week control and 1 week LNNA rats. Staining of 4 week PAG and 4 week SnPP is representative of 1 week PAG and 1 week SnPP, respectively (data not shown). ##P < 0.01 versus control (same week).
Combined inhibition – effect of 4 weeks combined inhibitors
NO metabolites in urine that were markedly diminished by LNNA (Figure 2A) were unaffected by additional PAG (P < 0.001 vs. controls) and were increased to an intermediate level between LNNA and controls by additional SnPP (P < 0.05). This intermediate level of LNNA + SnPP was not altered by adding PAG. NO metabolites in PAG + SnPP-treated rats were at control level (Figure 5A). The H2S production remained effectively inhibited in all PAG groups (P < 0.001) and was enhanced in the LNNA + SnPP group (P < 0.001; Figure 5B). HO-1 activity was enhanced by LNNA + PAG (P < 0.01; Figure 5C), but was at control levels in all SnPP groups. The results are compatible with the interaction among gases (Figure 2D).
Figure 5.
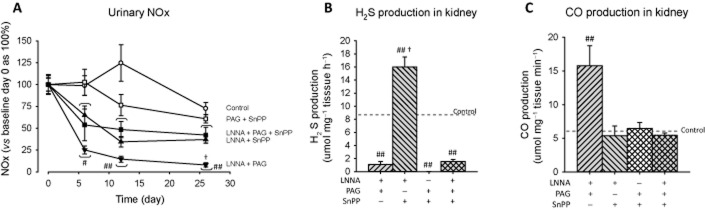
Urine NOx, renal H2S and CO production in rats treated with multiple inhibitors. NO metabolites in urine (A), renal H2S production (B), renal CO production (C). To clearly show the effects of chronic inhibition on NO metabolites, urine NOx (μmol 100 g−1 BW day−1) was compared with baseline of the same treatment group set at 100%. In (B), the horizontal dotted line represents the control level as shown in Figure 2B. #P < 0.05, ##P < 0.01 versus control (same week) and versus baseline day 0 (same inhibitor). †P < 0.01 versus SnPP + LNNA + PAG.
All rats receiving combined treatments displayed normal behaviour and no physical abnormalities. All combinations including LNNA resulted in hypertension (P < 0.001; Figure 6A). SnPP + PAG-treated rats remained normotensive. Note that SBP in rats treated with only LNNA (Figure 3A) was higher than in all other groups (P < 0.01). Adding PAG to LNNA stabilized SBP after 1 week and after 4 weeks SBP was midway between LNNA and control (P < 0.001). Adding SnPP to LNNA (with or without PAG) caused a slightly lower SBP than LNNA alone. Only addition of PAG to LNNA resulted in lower heart rate (315 ± 24 vs. 362 ± 13 bpm; P < 0.05). Proteinuria in LNNA rats was prevented by all additional inhibitors (Figure 6B), plasma urea was similar in all groups with the exception of SnPP + LNNA + PAG (9.8 ± 0.8 vs. 7.0 ± 0.4 mmol·L−1; P < 0.05, Figure 6C), body weight was at control level and histology revealed no renal injury (data not shown).
Figure 6.
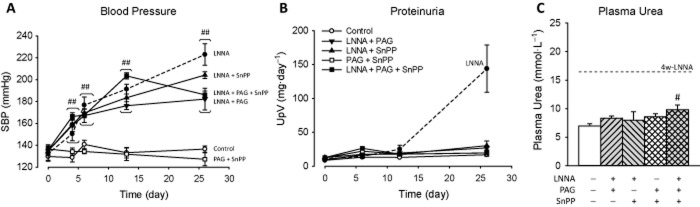
Systolic BP (SBP) and renal function and injury in rats after 4 weeks of multiple inhibitors. SBP, proteinuria and plasma urea are shown in panels A–C respectively. Data for LNNA only (shown in Figure 4) are represented by dotted lines. Significant differences between groups with multiple inhibitors including LNNA and LNNA only are mentioned in the results. #P < 0.05, ##P < 0.01 versus control (same week).
Combined inhibition – renal HO-1 expression
Renal cortex protein expression and tubular staining of HO-1 was induced by all inhibitors, and most for LNNA + SnPP (Figure 7).
Figure 7.
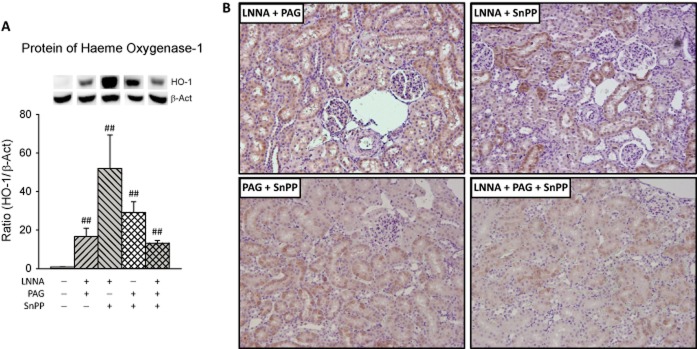
Renal HO-1 in rats with multiple inhibitors. Protein (A) expression and intrarenal location (B) were quantified using western blots and immunohistochemistry respectively. For details see legend to Figure 4. ##P < 0.01 versus control.
Discussion and conclusions
The three gases NO, H2S and CO are involved in many physiological processes (Nakao et al., 2008; Tripatara et al., 2008; Jadhav et al., 2009). There are indications, for various pairs of these three gases, that they interact and that mutual adaptation occurs when one of the gases diminishes or increases (Rodriguez et al., 2004; Botros and Navar, 2006; Rong-na et al., 2011). However, a complete overview of the reciprocal relations between three gases in the kidney has not yet been published. Here, the impact loss of one or more gases, through inhibition of gas-producing enzymes, on the production of other gases in the kidney and the effect on BP, renal function and renal injury were investigated.
Single inhibition
In the present study, the renal activity of CSE and HO-1 was measured in vitro. This may not reflect the actual activities in vivo. Nevertheless, higher activities measured in vitro suggest that the kidney can generate higher amounts of gases when needed. H2S depletion had consistent effects on the other gas-producing enzymes: at both time points NO metabolites were reduced and CO production was strongly increased. Synergy between H2S and NO has been observed previously. For example, exogenously elevated H2S activated NO production (Meng et al., 2013) and vice versa (Zhao et al., 2003) in non-renal tissues. Also H2S and NO were mutually required for control of vascular function (Coletta et al., 2012). However, in the present study, non-mutuality appears to exist in the kidneys; depleting H2S reduced NO metabolites, but H2S was not reduced when NO was depleted. In the literature, interaction between H2S and CO varies. Generally, endogenous H2S and CO were shown to inhibit each other's production (Jin et al., 2006), but recently, exogenous H2S was shown to increase HO-1 expression in kidney cells (D'Araio et al., 2014). This suggests a bell-shaped biological response of HO-1 to H2S (Szabo and Papapetropoulos, 2011; Modis et al., 2013). Nevertheless, the present study clearly showed that inhibition of CSE or HO-1 enhanced CO or H2S production respectively. High levels of CO have been shown to inhibit NOS activity and NO generation (Thorup et al., 1999). One may keep in mind that HO-1 controls the availability of cellular haeme (Quan et al., 2001) an essential co-factor for NOS (Andrew and Mayer, 1999) and CBS (Singh et al., 2007) that produces substrate for CSE. Thus, the increase in H2S and NO metabolites during SnPP exposure was probably due to reduced CO binding to the haeme co-factor of CBS and NO synthases respectively. Inhibiting H2S or CO-producing enzymes had no effect on BP and renal function. These data confirmed other studies that also reported absence of BP change in normal rats after inhibiting CSE (Roy et al., 2012; Issa et al., 2013) or HO-1 (Ushiyama et al., 2002; Wesseling et al., 2007; Chok et al., 2009; Chandrashekar et al., 2012) in different experimental settings. Generally, inhibition of CSE or HO-1 in the absence of pre-existent injury did not damage tissues, but made the tissues more sensitive to induction of injury. In perspective to the present data, PAG and SnPP did not appear to decrease renal function and BP remained normal. Importantly, BP and renal function and structure were not affected by practically abolishing renal H2S production. Thus, within the time frame of our study, the secondary decrease in NO production and increase in CO production during H2S depletion did not have functional consequences.
BP was already increased by NO depletion after 4 days and continued to increase. This was accompanied by renal injury and loss of function (as assessed by proteinuria, glomerulosclerosis and uraemia) after 4 weeks of NO depletion. Whether the drop in H2S and boost in CO production induced by LNNA merely associate with or are causally involved in the hypertension and diminished renal function was unclear. In the present study, renal protein expression of HO-1 was strongly induced by LNNA after 4 weeks only when kidneys are dysfunctional, but by PAG and SnPP at both time points. In NO-depleted rats, proteinuria correlated strongly with HO-1 gene expression (Wesseling et al., 2007). In rats with glomerular proteinuria tubular, HO-1 expression was increased (Datta et al., 2006). In the presence of proteinuria induced by NO depletion some nephrons invariably show more injury than others (Verhagen et al., 1999; Ying et al., 2003). The pattern of HO-1 staining in 4 week LNNA rats was characteristic of marked proteinuria (scattered protein leak in some glomeruli), while after PAG or SnPP HO-1 staining was uniformly spread throughout the proximal tubules. In all cases, the location of HO-1 was primarily tubular. A large body of literature supports protective actions of HO-1 (Nath, 2013), and proximal tubular HO-1 expression may reflect a stress response to injury (Pedraza-Chaverri et al., 2006). Interestingly, inhibition of HO-1 prior to ischaemia reperfusion increased renal expression of HO-1 and provided protection independent of enzyme activity (Kaizu et al., 2003). Moreover, in the current study, the direct measurement of CO production correlated with increased HO-1 expression in LNNA- or PAG-treated rats and established the efficiency of HO-1 inhibition by SnPP despite strong protein induction of HO-1. In LNNA-treated rats, we do not know if increased HO-1 expression was a response to local injury, reduction in H2S production or both.
Combined inhibition
Remarkably little information is available on the effects of combined inhibition of the production of these gases in extrarenal tissues, let alone in the kidney. Effects of exogenously elevated gas(es) are more widely published. Interaction between NOS or CSE inhibition was addressed on cardiac function in metabolic syndrome (Rong-na et al., 2011), where inhibiting CSE or NOS resulted in increased NO or H2S respectively. Also there was no study on combined inhibition of CSE and HO-1, but CBS and HO-1 were explored in myocardium; after left coronary ligation both H2S and CO production increased and inhibition of CBS or HO-1 increased CO or H2S, respectively, notably without worsening myocardial injury. Only when both CBS and HO-1 were inhibited did myocardial injury worsen (Zhu et al., 2008). Crosstalk between H2S and CO was explored in the carotid body where hypoxia decreased CO and increased H2S production and subsequently inhibiting CO production also increased H2S production (Prabhakar, 2012). Cirrhosis in rats blunted the hypoxic pressor response and elevated HO-1 and this response was partially restored by inhibiting HO-1. Cirrhotic rats treated with NOS inhibitor showed both a normal HO-1 expression and hypoxic pressor response (Carter et al., 2002). It appears from the examples of crosstalk that interaction between gases found in the present study is tissue-specific.
The production of H2S was diminished in all PAG groups and the CO production was at control level in all SnPP groups, but the effects of treatments on NO metabolites was complex. The level of NO metabolites in LNNA or LNNA + SnPP rats was not affected by additional H2S depletion. This suggested that the reducing effect of PAG on NO metabolites, observed during single inhibition, must have been indirect. Interestingly, all PAG-treated rats, with or without other inhibitors, had enhanced HO-1 expression. Note that because of HO-1 inhibition by SnPP in other combined treatment groups only LNNA + PAG rats had increased renal CO production. This was specifically caused by PAG, because after 1 week in LNNA rats when renal function was normal HO-1 expression was unaltered. As mentioned, high levels of CO had been shown to inhibit NOS activity and NO generation (Thorup et al., 1999) by binding to co-factor haeme. Combining these observations suggests that PAG treatment increased CO production by inducing HO-1, and CO in turn reduced NO metabolites by inhibiting NOS. Thus, in the kidneys, the effect of CSE on NOS appears to run via HO-1. Indeed, in macrophages, the inhibitory effect of H2S on NOS activity was dependent on active and intact HO-1 (Oh et al., 2006). Furthermore, excretion of NO metabolites in LNNA + SnPP groups was higher than in the LNNA group without other inhibitors. LNNA treatment for 4 weeks reduced NO metabolites to lower levels than after 1 week, and this probably was caused by increased renal CO production. Thus increased CO production together with NO depletion cumulatively reduced NO metabolites in LNNA rats after 4 weeks. In short, the intermediate level of NO metabolites in LNNA + SnPP groups as compared with the LNNA group without other inhibitors was caused by keeping CO production at control levels (despite strong induction of HO-1).
BP of LNNA rats was lower when co-treated with PAG, SnPP or both. For co-treatment with PAG this may be expected because endogenous H2S can act as a constrictor (Liu et al., 2011). However, the BP-lowering effect of SnPP in combination with LNNA was counter-intuitive. In one study, enzyme expression in the kidney in relation to BP was evaluated. Hypertensive rats had several times higher renal HO-1 expression than normotensive rats and inhibiting HO-1 increased BP in hypertensive but not in normotensive rats (Ushiyama et al., 2002). Spontaneously, hypertensive rats treated with hemin decreased (Ndisang et al., 2010) or normalized BP (Wang et al., 2006). Thus there is a suggestion that the BP-lowering effect of depleting H2S in LNNA rats was caused by increased renal CO production. Studies on interaction of NOS and HO-1 inhibition on BP were not consistent. For example, HO-1 inhibition increased BP in NO-depleted rats (Ushiyama et al., 2002), or failed to trigger a pressor response when NOS was impaired (Polizio et al., 2011). In the present study, all SnPP groups had higher level of NO metabolites than singly inhibited LNNA-treated rats. Arginine analogues dose-dependently increase BP and by inference there is an inverse association between NO level and BP (Verhagen et al., 1998). This suggested that the BP-lowering effect of SnPP in LNNA rats was caused by increased NO. BP in rats where all three systems were inhibited was also lower than in singly inhibited LNNA rats, and this might also be related to a higher level of NO metabolites. In short, hypertension in LNNA rats was milder with additional inhibitor(s). Possibly initiation of renal injury in this model requires a certain threshold of hypertension or longer treatment time when hypertension is milder. Although NO, H2S and CO share many common properties (Li et al., 2009), regulation of BP, and secondary to this, maintenance of normal renal function, appear primarily dependent on functional NOS.
HO-1 protein expression was enhanced by all combined treatments. Although strong induction of renal HO-1 is often associated with renal injury, there are several examples where increased renal expression of HO-1 might be protective. In rats with unilateral ureter obstruction (Correa-Costa et al., 2010), 5/6 subtotal nephrectomy (Liu et al., 2004) or diabetes (Ptilovanciv et al., 2013) co-treatment with hemin ameliorated renal dysfunction. In all these rats HO–1-stained positive in tubular cells. This suggests that strong induction of renal HO-1 in the absence of pre-existent injury might provide additional protection against hypertension and possibly also against detrimental actions of reduced NO availability.
In the present study, we observed complex relations among NO, H2S and CO in the kidney. In short, decreased NO had no effect on other gases, while reduction in CO increased both NO metabolites and H2S production, and that reduction in H2S reduced NO metabolites by enhancing CO production (Figure 8). Moreover, the increased CO production in the combined study detected with PAG and LNNA suggested that when sufficient NO and H2S are produced CO is redundant while when NO and H2S productions are inhibited CO was augmented as a compensatory mechanism. In addition, from the single and combined inhibition studies, it appears that H2S is a stronger regulator of CO than NO. So, it appears that CO is able to modulate NO production and has a ying-yang relationship with H2S but there appear to be no direct interactions between NO and H2S in the kidney. Altogether, based on the present study, CO appears to be the connecting player between NO and H2S in the kidney. The antihypertensive and possibly the renoprotective effect of PAG or SnPP in NO-depleted rats merits further study of the effects of PAG and SnPP in other models of hypertension and renal injury.
Figure 8.
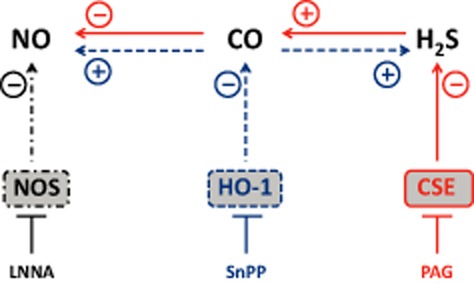
Complete interaction between gases bases on single and combined inhibition. The effect of an inhibitor on the direct product of the targeted enzyme and the secondary effects on the other gases are represented by three series of arrows: LNNA/NOS, black dot-dash arrows; SnPP/HO-1, blue dashed arrows; PAG/CSE, red continuous arrows. These line types also surround the enzymes.
Acknowledgments
We thank Adele v. Dijk, Krista den Ouden, Chantal Tilburgs, Nel Willekes and Irene Huisman for their laboratory expertise and Paula Martens for her excellent care of animals. This study was financially supported by Dutch Kidney Foundation National (C09.2332).
Glossary
- CKD
chronic kidney disease
- CSE
cystathionine γ-lyase
- FGS
focal glomerulosclerosis
- HO-1
haeme oxygenase-1
- LNNA
L-nitroarginine
- PAG
DL-propargylglycine
- PLP
pyridoxal-5′-phosphate
- SD
Sprague Dawley (rats)
- SnPP
Sn(IV) protoporphyrin IX dichloride
Conflict of interest
None.
References
- Agarwal A, Nick HS. Renal response to tissue injury: lessons from heme oxygenase-1 GeneAblation and expression. J Am Soc Nephrol. 2000;11:965–973. doi: 10.1681/ASN.V115965. [DOI] [PubMed] [Google Scholar]
- Andrew PJ, Mayer B. Enzymatic function of nitric oxide synthases. Cardiovasc Res. 1999;43:521–531. doi: 10.1016/s0008-6363(99)00115-7. [DOI] [PubMed] [Google Scholar]
- Attia DM, Verhagen AM, Stroes ES, van Faassen EE, Grone HJ, De Kimpe SJ, et al. Vitamin E alleviates renal injury, but not hypertension, during chronic nitric oxide synthase inhibition in rats. J Am Soc Nephrol. 2001;12:2585–2593. doi: 10.1681/ASN.V12122585. [DOI] [PubMed] [Google Scholar]
- Attia DM, Ni ZN, Boer P, Attia MA, Goldschmeding R, Koomans HA, et al. Proteinuria is preceded by decreased nitric oxide synthesis and prevented by a NO donor in cholesterol-fed rats. Kidney Int. 2002;61:1776–1787. doi: 10.1046/j.1523-1755.2002.00313.x. [DOI] [PubMed] [Google Scholar]
- Baylis C. Nitric oxide deficiency in chronic kidney disease. Am J Physiol Renal Physiol. 2008;294:F1–F9. doi: 10.1152/ajprenal.00424.2007. [DOI] [PubMed] [Google Scholar]
- Baylis C, Vallance P. Nitric oxide and blood pressure: effects of nitric oxide deficiency. Curr Opin Nephrol Hypertens. 1996;5:80–88. doi: 10.1097/00041552-199601000-00014. [DOI] [PubMed] [Google Scholar]
- Baylis C, Mitruka B, Deng A. Chronic blockade of nitric oxide synthesis in the rat produces systemic hypertension and glomerular damage. J Clin Invest. 1992;90:278–281. doi: 10.1172/JCI115849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Botros FT, Navar LG. Interaction between endogenously produced carbon monoxide and nitric oxide in regulation of renal afferent arterioles. Am J Physiol Heart Circ Physiol. 2006;291:H2772–H2778. doi: 10.1152/ajpheart.00528.2006. [DOI] [PubMed] [Google Scholar]
- Carter EP, Hartsfield CL, Miyazono M, Jakkula M, Morris KG, Jr, McMurtry IF. Regulation of heme oxygenase-1 by nitric oxide during hepatopulmonary syndrome. Am J Physiol Lung Cell Mol Physiol. 2002;283:L346–L353. doi: 10.1152/ajplung.00385.2001. [DOI] [PubMed] [Google Scholar]
- Chandrashekar K, Lopez-Ruiz A, Juncos R, Nath K, Stec DE, Vera T, et al. The modulatory role of heme oxygenase on subpressor angiotensin ii-induced hypertension and renal injury. Int J Hypertens. 2012;2012:392890. doi: 10.1155/2012/392890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chok MK, Ferlicot S, Conti M, Almolki A, Durrbach A, Loric S, et al. Renoprotective potency of heme oxygenase-1 induction in rat renal ischemia-reperfusion. Inflamm Allergy Drug Targets. 2009;8:252–259. doi: 10.2174/187152809789352186. [DOI] [PubMed] [Google Scholar]
- Christodoulides N, Durante W, Kroll MH, Schafer AI. Vascular smooth muscle cell heme oxygenases generate guanylyl cyclase-stimulatory carbon monoxide. Circulation. 1995;91:2306–2309. doi: 10.1161/01.cir.91.9.2306. [DOI] [PubMed] [Google Scholar]
- Coletta C, Papapetropoulos A, Erdelyi K, Olah G, Modis K, Panopoulos P, et al. Hydrogen sulfide and nitric oxide are mutually dependent in the regulation of angiogenesis and endothelium-dependent vasorelaxation. Proc Natl Acad Sci U S A. 2012;109:9161–9166. doi: 10.1073/pnas.1202916109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Correa-Costa M, Semedo P, Monteiro AP, Silva RC, Pereira RL, Goncalves GM, et al. Induction of heme oxygenase-1 can halt and even reverse renal tubule-interstitial fibrosis. PLoS ONE. 2010;5:e14298. doi: 10.1371/journal.pone.0014298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D'Araio E, Shaw N, Millward A, Demaine A, Whiteman M, Hodgkinson A. Hydrogen sulfide induces heme oxygenase-1 in human kidney cells. Acta Diabetol. 2014;51:155–157. doi: 10.1007/s00592-013-0501-y. [DOI] [PubMed] [Google Scholar]
- Datta PK, Reddy S, Sharma M, Lianos EA. Differential nephron HO-1 expression following glomerular epithelial cell injury. Nephron Exp Nephrol. 2006;103:e131–e138. doi: 10.1159/000092544. [DOI] [PubMed] [Google Scholar]
- Gadalla MM, Snyder SH. Hydrogen sulfide as a gasotransmitter. J Neurochem. 2010;113:14–26. doi: 10.1111/j.1471-4159.2010.06580.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huber WJ, 3rd, Marohnic CC, Peters M, Alam J, Reed JR, Masters BS, et al. Measurement of membrane-bound human heme oxygenase-1 activity using a chemically defined assay system. Drug Metab Dispos. 2009;37:857–864. doi: 10.1124/dmd.108.025023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Issa K, Kimmoun A, Collin S, Ganster F, Fremont-Orlowski S, Asfar P, et al. Compared effects of inhibition and exogenous administration of hydrogen sulphide in ischaemia-reperfusion injury. Crit Care. 2013;17:R129. doi: 10.1186/cc12808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jadhav A, Torlakovic E, Ndisang JF. Hemin therapy attenuates kidney injury in deoxycorticosterone acetate-salt hypertensive rats. Am J Physiol Renal Physiol. 2009;296:F521–F534. doi: 10.1152/ajprenal.00510.2007. [DOI] [PubMed] [Google Scholar]
- Jin HF, Du JB, Li XH, Wang YF, Liang YF, Tang CS. Interaction between hydrogen sulfide/cystathionine gamma-lyase and carbon monoxide/heme oxygenase pathways in aortic smooth muscle cells. Acta Pharmacol Sin. 2006;27:1561–1566. doi: 10.1111/j.1745-7254.2006.00425.x. [DOI] [PubMed] [Google Scholar]
- Kaizu T, Tamaki T, Tanaka M, Uchida Y, Tsuchihashi S, Kawamura A, et al. Preconditioning with tin-protoporphyrin IX attenuates ischemia/reperfusion injury in the rat kidney. Kidney Int. 2003;63:1393–1403. doi: 10.1046/j.1523-1755.2003.00882.x. [DOI] [PubMed] [Google Scholar]
- Kilkenny C, Browne W, Cuthill IC, Emerson M, Altman DG. Animal research: reporting in vivo experiments: the ARRIVE guidelines. Br J Pharmacol. 2010;160:1577–1579. doi: 10.1111/j.1476-5381.2010.00872.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koeners MP, Braam B, van der Giezen DM, Goldschmeding R, Joles JA. A perinatal nitric oxide donor increases renal vascular resistance and ameliorates hypertension and glomerular injury in adult fawn-hooded hypertensive rats. Am J Physiol Regul Integr Comp Physiol. 2008;294:R1847–R1855. doi: 10.1152/ajpregu.00073.2008. [DOI] [PubMed] [Google Scholar]
- Konno R, Ikeda M, Yamaguchi K, Ueda Y, Niwa A. Nephrotoxicity of D-proparglyglycine in mice. Arch Toxicol. 2000;74:473–479. doi: 10.1007/s002040000156. [DOI] [PubMed] [Google Scholar]
- van Koppen A, Verhaar MC, Bongartz LG, Joles JA. 5/6th nephrectomy in combination with high salt diet and nitric oxide synthase inhibition to induce chronic kidney disease in the Lewis rat. J Vis Exp. 2013;77:e50398. doi: 10.3791/50398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lentz SR, Erger RA, Dayal S, Maeda N, Malinow MR, Heistad DD, et al. Folate dependence of hyperhomocysteinemia and vascular dysfunction in cystathionine beta-synthase-deficient mice. Am J Physiol Heart Circ Physiol. 2000;279:H970–H975. doi: 10.1152/ajpheart.2000.279.3.H970. [DOI] [PubMed] [Google Scholar]
- Li L, Hsu A, Moore PK. Actions and interactions of nitric oxide, carbon monoxide and hydrogen sulphide in the cardiovascular system and in inflammation – a tale of three gases! Pharmacol Ther. 2009;123:386–400. doi: 10.1016/j.pharmthera.2009.05.005. [DOI] [PubMed] [Google Scholar]
- Liu S, Shi L, Liu X. Effect of heme oxygenase-1 inducer hemin on chronic renal failure rats. J Huazhong Univ Sci Technolog Med Sci. 2004;24:250–253. doi: 10.1007/BF02832004. [DOI] [PubMed] [Google Scholar]
- Liu YH, Yan CD, Bian JS. Hydrogen sulfide: a novel signaling molecule in the vascular system. J Cardiovasc Pharmacol. 2011;58:560–569. doi: 10.1097/FJC.0b013e31820eb7a1. [DOI] [PubMed] [Google Scholar]
- Maekawa M, Okamura T, Kasai N, Hori Y, Summer KH, Konno R. D-amino-acid oxidase is involved in D-serine-induced nephrotoxicity. Chem Res Toxicol. 2005;18:1678–1682. doi: 10.1021/tx0500326. [DOI] [PubMed] [Google Scholar]
- McGrath J, Drummond G, McLachlan E, Kilkenny C, Wainwright C. Guidelines for reporting experiments involving animals: the ARRIVE guidelines. Br J Pharmacol. 2010;160:1573–1576. doi: 10.1111/j.1476-5381.2010.00873.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meng J, Ganesan Adaikan P, Srilatha B. Hydrogen sulfide promotes nitric oxide production in corpus cavernosum by enhancing expression of endothelial nitric oxide synthase. Int J Impot Res. 2013;25:86–90. doi: 10.1038/ijir.2012.39. [DOI] [PubMed] [Google Scholar]
- Modis K, Wolanska K, Vozdek R. Hydrogen sulfide in cell signaling, signal transduction, cellular bioenergetics and physiology in C. elegans. Gen Physiol Biophys. 2013;32:1–22. doi: 10.4149/gpb_2013001. [DOI] [PubMed] [Google Scholar]
- Nakao A, Faleo G, Shimizu H, Nakahira K, Kohmoto J, Sugimoto R, et al. Ex vivo carbon monoxide prevents cytochrome P450 degradation and ischemia/reperfusion injury of kidney grafts. Kidney Int. 2008;74:1009–1016. doi: 10.1038/ki.2008.342. [DOI] [PubMed] [Google Scholar]
- Nath KA. The role of renal research in demonstrating the protective properties of heme oxygenase-1. Kidney Int. 2013;84:3–6. doi: 10.1038/ki.2013.88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ndisang JF, Lane N, Syed N, Jadhav A. Up-regulating the heme oxygenase system with hemin improves insulin sensitivity and glucose metabolism in adult spontaneously hypertensive rats. Endocrinology. 2010;151:549–560. doi: 10.1210/en.2009-0471. [DOI] [PubMed] [Google Scholar]
- Oh GS, Pae HO, Lee BS, Kim BN, Kim JM, Kim HR, et al. Hydrogen sulfide inhibits nitric oxide production and nuclear factor-kappaB via heme oxygenase-1 expression in RAW264.7 macrophages stimulated with lipopolysaccharide. Free Radic Biol Med. 2006;41:106–119. doi: 10.1016/j.freeradbiomed.2006.03.021. [DOI] [PubMed] [Google Scholar]
- Ortiz PA, Garvin JL. Cardiovascular and renal control in NOS-deficient mouse models. Am J Physiol Regul Integr Comp Physiol. 2003;284:R628–R638. doi: 10.1152/ajpregu.00401.2002. [DOI] [PubMed] [Google Scholar]
- Pedraza-Chaverri J, Murali NS, Croatt AJ, Alam J, Grande JP, Nath KA. Proteinuria as a determinant of renal expression of heme oxygenase-1: studies in models of glomerular and tubular proteinuria in the rat. Am J Physiol Renal Physiol. 2006;290:F196–F204. doi: 10.1152/ajprenal.00230.2005. [DOI] [PubMed] [Google Scholar]
- Polizio AH, Santa-Cruz DM, Balestrasse KB, Gironacci MM, Bertera FM, Hocht C, et al. Heme oxygenase-1 overexpression fails to attenuate hypertension when the nitric oxide synthase system is not fully operative. Pharmacology. 2011;87:341–349. doi: 10.1159/000327939. [DOI] [PubMed] [Google Scholar]
- Prabhakar NR. Carbon monoxide (CO) and hydrogen sulfide (H(2)S) in hypoxic sensing by the carotid body. Respir Physiol Neurobiol. 2012;184:165–169. doi: 10.1016/j.resp.2012.05.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ptilovanciv EO, Fernandes GS, Teixeira LC, Reis LA, Pessoa EA, Convento MB, et al. Heme oxygenase 1 improves glucoses metabolism and kidney histological alterations in diabetic rats. Diabetol Metab Syndr. 2013;5:3. doi: 10.1186/1758-5996-5-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quan S, Yang L, Abraham NG, Kappas A. Regulation of human heme oxygenase in endothelial cells by using sense and antisense retroviral constructs. Proc Natl Acad Sci U S A. 2001;98:12203–12208. doi: 10.1073/pnas.211399398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajapakse NW, Mattson DL. Role of cellular L-arginine uptake and nitric oxide production on renal blood flow and arterial pressure regulation. Curr Opin Nephrol Hypertens. 2013;22:45–50. doi: 10.1097/MNH.0b013e32835a6ff7. [DOI] [PubMed] [Google Scholar]
- Reed JR, Huber WJ, 3rd, Backes WL. Human heme oxygenase-1 efficiently catabolizes heme in the absence of biliverdin reductase. Drug Metab Dispos. 2010;38:2060–2066. doi: 10.1124/dmd.110.034777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez F, Lamon BD, Gong W, Kemp R, Nasjletti A. Nitric oxide synthesis inhibition promotes renal production of carbon monoxide. Hypertension. 2004;43:347–351. doi: 10.1161/01.HYP.0000111721.97169.97. [DOI] [PubMed] [Google Scholar]
- Rong-na L, Xiang-jun Z, Yu-han C, Ling-qiao L, Gang H. Interaction between hydrogen sulfide and nitric oxide on cardiac protection in rats with metabolic syndrome. Zhongguo Yi Xue Ke Xue Yuan Xue Bao. 2011;33:25–32. doi: 10.3881/j.issn.1000-503X.2011.01.007. [DOI] [PubMed] [Google Scholar]
- Roy A, Khan AH, Islam MT, Prieto MC, Majid DS. Interdependency of cystathione gamma-lyase and cystathione beta-synthase in hydrogen sulfide-induced blood pressure regulation in rats. Am J Hypertens. 2012;25:74–81. doi: 10.1038/ajh.2011.149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shiraishi F, Curtis LM, Truong L, Poss K, Visner GA, Madsen K, et al. Heme oxygenase-1 gene ablation or expression modulates cisplatin-induced renal tubular apoptosis. Am J Physiol Renal Physiol. 2000;278:F726–F736. doi: 10.1152/ajprenal.2000.278.5.F726. [DOI] [PubMed] [Google Scholar]
- Singh S, Madzelan P, Banerjee R. Properties of an unusual heme cofactor in PLP-dependent cystathionine beta-synthase. Nat Prod Rep. 2007;24:631–639. doi: 10.1039/b604182p. [DOI] [PubMed] [Google Scholar]
- Sun Q, Collins R, Huang S, Holmberg-Schiavone L, Anand GS, Tan CH, et al. Structural basis for the inhibition mechanism of human cystathionine gamma-lyase, an enzyme responsible for the production of H(2)S. J Biol Chem. 2009;284:3076–3085. doi: 10.1074/jbc.M805459200. [DOI] [PubMed] [Google Scholar]
- Szabo C, Papapetropoulos A. Hydrogen sulphide and angiogenesis: mechanisms and applications. Br J Pharmacol. 2011;164:853–865. doi: 10.1111/j.1476-5381.2010.01191.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thorup C, Jones CL, Gross SS, Moore LC, Goligorsky MS. Carbon monoxide induces vasodilation and nitric oxide release but suppresses endothelial NOS. Am J Physiol. 1999;277(6):F882–F889. doi: 10.1152/ajprenal.1999.277.6.F882. Pt 2): [DOI] [PubMed] [Google Scholar]
- Trakshel GM, Sluss PM, Maines MD. Comparative effects of tin- and zinc-protoporphyrin on steroidogenesis: tin-protoporphyrin is a potent inhibitor of cytochrome P-450-dependent activities in the rat adrenals. Pediatr Res. 1992;31:196–201. doi: 10.1203/00006450-199202000-00022. [DOI] [PubMed] [Google Scholar]
- Tripatara P, Patel NS, Collino M, Gallicchio M, Kieswich J, Castiglia S, et al. Generation of endogenous hydrogen sulfide by cystathionine gamma-lyase limits renal ischemia/reperfusion injury and dysfunction. Lab Invest. 2008;88:1038–1048. doi: 10.1038/labinvest.2008.73. [DOI] [PubMed] [Google Scholar]
- Ushiyama M, Morita T, Katayama S. Carbon monoxide regulates blood pressure cooperatively with nitric oxide in hypertensive rats. Heart Vessels. 2002;16:189–195. doi: 10.1007/s003800200020. [DOI] [PubMed] [Google Scholar]
- Verhagen AM, Rabelink TJ, Braam B, Opgenorth TJ, Grone HJ, Koomans HA, et al. Endothelin A receptor blockade alleviates hypertension and renal lesions associated with chronic nitric oxide synthase inhibition. J Am Soc Nephrol. 1998;9:755–762. doi: 10.1681/ASN.V95755. [DOI] [PubMed] [Google Scholar]
- Verhagen AM, Braam B, Boer P, Grone HJ, Koomans HA, Joles JA. Losartan-sensitive renal damage caused by chronic NOS inhibition does not involve increased renal angiotensin II concentrations. Kidney Int. 1999;56:222–231. doi: 10.1046/j.1523-1755.1999.00542.x. [DOI] [PubMed] [Google Scholar]
- Verhagen AM, Koomans HA, Joles JA. Predisposition of spontaneously hypertensive rats to develop renal injury during nitric oxide synthase inhibition. Eur J Pharmacol. 2001;411:175–180. doi: 10.1016/s0014-2999(00)00900-6. [DOI] [PubMed] [Google Scholar]
- Wang R, Shamloul R, Wang X, Meng Q, Wu L. Sustained normalization of high blood pressure in spontaneously hypertensive rats by implanted hemin pump. Hypertension. 2006;48:685–692. doi: 10.1161/01.HYP.0000239673.80332.2f. [DOI] [PubMed] [Google Scholar]
- Washtien W, Abeles RH. Mechanism of inactivation of gamma-cystathionase by the acetylenic substrate analogue propargylglycine. Biochemistry. 1977;16:2485–2491. doi: 10.1021/bi00630a026. [DOI] [PubMed] [Google Scholar]
- Wesseling S, Joles JA, van Goor H, Bluyssen HA, Kemmeren P, Holstege FC, et al. Transcriptome-based identification of pro- and antioxidative gene expression in kidney cortex of nitric oxide-depleted rats. Physiol Genomics. 2007;28:158–167. doi: 10.1152/physiolgenomics.00077.2006. [DOI] [PubMed] [Google Scholar]
- Whiting C, Castillo A, Haque MZ, Majid DS. Protective role of the endothelial isoform of nitric oxide synthase in angiotensin II induced inflammatory responses in the kidney. Am J Physiol Renal Physiol. 2013;305:F1031–F1041. doi: 10.1152/ajprenal.00024.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiesel P, Patel AP, Carvajal IM, Wang ZY, Pellacani A, Maemura K, et al. Exacerbation of chronic renovascular hypertension and acute renal failure in heme oxygenase-1-deficient mice. Circ Res. 2001;88:1088–1094. doi: 10.1161/hh1001.091521. [DOI] [PubMed] [Google Scholar]
- Yang G, Wu L, Jiang B, Yang W, Qi J, Cao K, et al. H2S as a physiologic vasorelaxant: hypertension in mice with deletion of cystathionine gamma-lyase. Science. 2008;322:587–590. doi: 10.1126/science.1162667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ying L, Flamant M, Vandermeersch S, Boffa JJ, Chatziantoniou C, Dussaule JC, et al. Renal effects of omapatrilat and captopril in salt-loaded, nitric oxide-deficient rats. Hypertension. 2003;42:937–944. doi: 10.1161/01.HYP.0000099240.89890.94. [DOI] [PubMed] [Google Scholar]
- Zatz R, Baylis C. Chronic nitric oxide inhibition model six years on. Hypertension. 1998;32:958–964. doi: 10.1161/01.hyp.32.6.958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zatz R, de Nucci G. Effects of acute nitric oxide inhibition on rat glomerular microcirculation. Am J Physiol. 1991;261(2):F360–F363. doi: 10.1152/ajprenal.1991.261.2.F360. Pt 2): [DOI] [PubMed] [Google Scholar]
- Zhao W, Ndisang JF, Wang R. Modulation of endogenous production of H2S in rat tissues. Can J Physiol Pharmacol. 2003;81:848–853. doi: 10.1139/y03-077. [DOI] [PubMed] [Google Scholar]
- Zhu JC, Shao JL, Ma H, Wang JK. Interaction between endogenous cystathionine synthase/hydrogen sulfide and heme oxygenase-1/.carbon monoxide systems during myocardial ischemic-reperfusion: experiment with rats] Zhonghua Yi Xue Za Zhi. 2008;88:3222–3225. [PubMed] [Google Scholar]