

Abstract
Although the physiological regulatory function of the gasotransmitter NO (a diatomic free radical) was discovered decades ago, NO is still in the frontline research in biomedicine. NO has been implicated in a variety of physiological and pathological processes; therefore, pharmacological modulation of NO levels in various tissues may have significant therapeutic value. NO is generated by NOS in most of cell types and by non-enzymatic reactions. Measurement of NO is technically difficult due to its rapid chemical reactions with a wide range of molecules, such as, for example, free radicals, metals, thiols, etc. Therefore, there are still several contradictory findings on the role of NO in different biological processes. In this review, we briefly discuss the major techniques suitable for measurement of NO (electron paramagnetic resonance, electrochemistry, fluorometry) and its derivatives in biological samples (nitrite/nitrate, NOS, cGMP, nitrosothiols) and discuss the advantages and disadvantages of each method. We conclude that to obtain a meaningful insight into the role of NO and NO modulator compounds in physiological or pathological processes, concomitant assessment of NO synthesis, NO content, as well as molecular targets and reaction products of NO is recommended.
Linked Articles
This article is part of a themed section on Pharmacology of the Gasotransmitters. To view the other articles in this section visit http://dx.doi.org/10.1111/bph.2015.172.issue-6
Table of Links
| TARGETS | LIGANDS |
|---|---|
| Arginase | cGMP |
| Endothelial NOS | Hydrogen peroxide (H2O2) |
| Inducible NOS | L-arginine |
| Neuronal NOS | L-citrulline |
| L-arginine | NADPH |
| Soluble guanylyl cyclase (sGC) | Tetrahydrobiopterin (sapropterin) |
| NO |
This Table lists key protein targets and ligands in this document, which are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Pawson et al., 2014) and are permanently archived in the Concise Guide to PHARMACOLOGY 2013/14 (Alexander et al., 2013).
Introduction
Although the physiological regulatory function of the gasotransmitter NO was discovered decades ago, NO is still in the frontline research as shown by the continuously increasing number of annual hits for ‘nitric oxide’ in the PubMed database. The increasing interest in NO in biomedical research is due to several facts: (i) NO is an ubiquitous free radial molecule found in most of cells of all tissues intracellularly as well as in the extracellular fluids; (ii) NO is involved in a variety of physiological and pathological processes; and (iii) utilization of gaseous NO and some NO donor molecules for human therapy has entered into clinical therapy and the development of further NO-related therapies are promising (see for review, Pacher et al., 2007). However, the chemical reactions of NO with other free radicals and various small and macromolecules have raised many new questions regarding the involvement of NO in different cellular or intercellular/interorgan signalling pathways. Moreover, measurement of NO is technically difficult, due to its rapid chemical reactions with a wide range of biomolecules and its very short half-life of approximately a few seconds. Therefore, there are still contradictory findings on the role of NO in several biological processes as reviewed previously by Ferdinandy and Schulz (2003) and Schulz et al. (2004). Correct application of the different NO measurement techniques are essential to gain more knowledge on the physiology and pathology of NO.
In this review, we describe the major techniques used most frequently in the literature for measurement of NO in biological samples and discuss the advantages and disadvantages of each method.
Synthesis and major metabolic pathways of NO
NO is a diatomic hydrophobic gas that can permeate various cellular membranes and other hydrophobic structures, and thus it has a high diffusion capacity in a physiological environment. NO can be produced in biological systems by both enzymatic and non-enzymatic reactions. In mammals, NO is biosynthesized endogenously by various isoforms of NOS using the substrates L-arginine and molecular oxygen (Figure 1). NOS catalyses the oxidation of the terminal guanidino nitrogen of L-arginine to produce NO and L-citrulline. Several cofactors are required for the reaction including NADPH, flavin adenine dinucleotide, flavin mononucleotide, haem, tetrahydrobiopterin and calmodulin. Insufficient availability of L-arginine and some of the cofactors (i.e. tetrahydrobiopterin), as well as S-glutathionylation of certain NOS isoforms may lead to reduced NO formation and uncoupling of NOS resulting in superoxide production (Forstermann and Li, 2011; Zweier et al., 2011). Three distinct isoforms of NOS have been described in mammals: neuronal NOS (nNOS, NOS-1), endothelial NOS (eNOS, NOS-3) and inducible NOS (iNOS, NOS-2) (Knowles et al., 1989; Mayer et al., 1989; Mulsch et al., 1989; Palacios et al., 1989; Palmer and Moncada, 1989; Stuehr et al., 1989; Moncada et al., 1997; Forstermann and Sessa, 2012). Moreover, the existence of a putative mitochondrial NOS (mtNOS) has also been suggested (Zaobornyj and Ghafourifar, 2012). The constitutively expressed isoforms nNOS and eNOS are Ca2+-dependent, whereas iNOS is Ca2+-independent (Moncada et al., 1991). The biological effects of NO and the activities of NOS isoenzymes are further regulated by compartmentalization (Villanueva and Giulivi, 2010) as well as transcriptional, post-transcriptional and post-translational modulations such as for instance phosphorylation, S-nitrosation, interaction with modulatory proteins (e.g. calmodulin, HSP90, caveolin, etc.), dimerization, inhibition by endogenous methyl-arginines, etc. (as reviewed in detail elsewhere, Zhou and Zhu, 2009; Pautz et al., 2010; Qian and Fulton, 2013).
Figure 1.
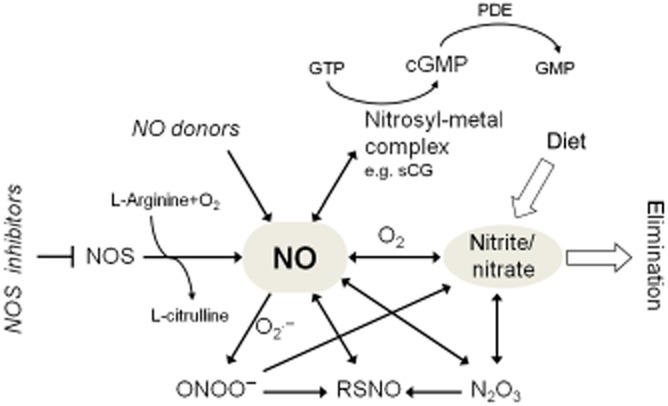
Major metabolic pathways of NO.
It has been clarified that NOS-independent reduction of dietary or endogenous sources of nitrate and nitrite are important contributors for the production of NO in mammalian tissues (Lundberg et al., 2008; Rassaf et al., 2014) (Figure 1). Nitrate found in significant amounts in certain vegetables (e.g. lettuce, spinach and beetroot, etc.), needs to be initially reduced to nitrite by nitrate reductase enzymes of bacteria in the gastrointestinal tract. Nitrite – also found in certain dietary sources – can be reduced to NO by several pathways and conditions including low pH, ascorbic acid, haemoglobin, myoglobin, polyphenols and xanthine oxidoreductase (Lundberg et al., 2008; Rassaf et al., 2014). The formation of NO by these pathways may become especially important during hypoxia when pH becomes acidic and oxygen-dependent NOS activities are limited.
NO has a short biological half-life (estimated to be a few seconds) due to its rapid reaction with a variety of molecules (Figure 1). Although the chemistry of NO is well-established in a test tube, the exact biochemistry of NO is still far from clear (Hill et al., 2010). The biologically relevant reactions of NO have been reviewed elsewhere in more detail (Gow, 2006; Bryan and Grisham, 2007; Habib and Ali, 2011; Tennyson and Lippard, 2011). The major pathway for the metabolism of NO is its oxidation to nitrite and nitrate eventually followed by their urinary excretion. NO in the presence of molecular oxygen is oxidized to nitrogen-dioxide (NO2), which by reacting with another NO molecule forms N2O3, an intermedier that participates in nitrosation reactions. N2O3 may be decomposed to nitrite and a one-electron reduction of NO2 may also lead to nitrite formation. Nitrite has a half-life of a few minutes in the circulation as it can be further oxidized to the more stable nitrate by certain oxyhaemoproteins such as oxygenated haemoglobin or myoglobin. Alternatively, NO may directly react with oxyhaemoproteins to form nitrate (Bryan and Grisham, 2007). In addition to its reaction with oxygen, NO rapidly reacts with superoxide to yield peroxynitrite, a short-lived oxidant, nitrating and nitrosating agent (Pacher et al., 2007; Radi, 2013).
Important molecular targets of NO are transition metal ions. NO binds to transition metal ions to form nitrosyl-metal ion complexes. The nitrosyl-Fe2+ adduct – such as in haem – is particularly stable, as the binding of the nitrosyl ligand to Fe2+ is very strong. Relevant examples of proteins in which the formation of nitrosyl-metal complexes affect biological function include soluble GC (sGC, see later), haemoglobin, cytochromes, etc (Toledo and Augusto, 2012). NO – following oxidation to N2O3 – plays an important role in the formation of S-nitrosothiols (RSNOs) via S-nitrosation of small molecular weight thiols and thiol-containing proteins (Broniowska and Hogg, 2012). This type of reaction is often incorrectly referred to as ‘S-nitrosylation’ (i.e. direct addition of NO to a reactant) in the literature [for more details on NO chemistry and terminology see a recent review by Heinrich et al., (2013)]. Nevertheless, NO may also react directly with thiyl radicals formed after oxidation of thiols to produce RSNOs. Enzyme-dependent and -independent S-nitrosation, transnitrosation and denitrosation are potential post-translational modifications that may regulate biological function of several proteins (Lima et al., 2010; Stamler and Hess, 2010; Gould et al., 2013; Maron et al., 2013). RSNOs may play an important role in endogenous transport and storage of NO as well as in NO-related cell signalling.
Analytical tools for the assessment of NO: what to consider before selection?
In general, NO measurement techniques can be classified as direct (the target of measurement is NO itself) and indirect methods. Most of the NO measurement techniques in the literature are indirect ones that are measurements of NOS activities, activation of molecular targets of NO, such as GC-derived cGMP, or products of reactions of NO, such as RSNOs or nitrite/nitrate. The major analytical tools for the detection of these analytes are spectroscopic or electrochemical methods. The spectroscopic methods include colorimetric, fluorometry, luminometry and electron spin resonance spectroscopy (ESR). These analytical tools have been extensively reviewed previously (Hetrick and Schoenfisch, 2009; Coneski and Schoenfisch, 2012). The sensitivity and specificity of these techniques for NO varies a lot, and they cannot provide an insight into the in situ NO levels in biological systems. Direct NO measurement techniques that are more specific for NO such as, for example, ESR after in vivo or ex vivo spin trapping or NO-specific biosensors are less frequently used.
Choosing the most appropriate method for measurement of NO in biological systems is not easy. Therefore, we suggest consideration of two additional aspects before planning experiments. Firstly, for the measurement of NO, a variety of different commercial products are available on the market, most of which can be purchased in ready-to-use formats. However, these kits and/or instruments that operate with different background principles are developed for various scientific, industrial and environmental application purposes. Secondly, the specificity and cross-reactivity of the NO sensors with NO derivatives (e.g. reactive nitrogen species such as peroxynitrite) and other non-NO-related molecules (e.g. reactive oxygen species) still remain a major challenge in NO research (Rodriguez-Rodriguez and Simonsen, 2012; Woolley et al., 2013). This is mainly due to the small size of the NO molecule and its complex chemical nature in biological systems. Therefore, before scientific use, careful consideration of various aspects including background assay principle, specificity, sensitivity, advantages, disadvantages and financials (see Table 1) is strongly recommended to choose the most proper analytical technique(s), which best fits the study objectives. As an aid to finding the most appropriate method(s) for measurement of NO in a particular study, a detailed questionnaire can be constructed and answered as suggested by Wardman (2007). Moreover, we suggest the use of NO donors for positive control and inhibitors of NO formation as negative controls to complete the data obtained either by direct or indirect assays to understand the exact role of NO in biological systems.
Table 1.
Specificity and major advantages and disadvantages of techniques of NO assessment with a brief summary
| Technique | Sensitivity | Advantages | Disadvantages |
|---|---|---|---|
| EPR | 0.05–0.4 nMa | Direct, considered as a ‘stand alone’ assay; the most specific method, due to NO-specific spin traps | Semiquantitative; expensive, not particularly common instrumentation; complex evaluation requires significant expertise |
| NO spin trapping followed by spectrometry in magnetic field | |||
| Electrochemistry | 0.3–10 nMb | Direct; continuous; real-time, portable; no chemical contamination of the sample | Difficult to calibrate; influenced by temperature and ambient electrical noise; uncertain specificity (NO-specific membrane or layer); sensitive to electrode tip position |
| amperometry or voltammetry using NO-specific electrode | |||
| Fluorometry | 0.6–8 nMc | Direct; sensitive (detection limit in nM range); can be two- or three-dimensional | Uncertain specificity (NO-specific fluorescent dyes); semiquantitative |
| spectrometry or imaging of fluorophore-labelled NO | |||
| Griess assay | 500 nMd | Cheap, fast; commercially available ready-to-use kits | Indirect, indicative of NO oxidative products; only measures nitrite (nitrate should be reduced); interferes with dietary and environmental nitrite and nitrate |
| diazotization assay measures nitrite by photometry | |||
| NOS activity | n.a. | Measures enzymatic production of NO; sensitive; specific | Indirect; non-enzymatic NO formation is not considered; measures active NOS protein under optimized in vitro conditions, does not reflect in situ NO synthesis |
| biochemical enzyme activity assay | |||
| RSNO | n.a. | Important NO target besides cGMP; represents downstream NO signalling | Indirect; represents downstream NO signalling; does not reflect actual NO levels |
| detection of nitrosated proteins/peptides | |||
| cGMP assays | n.a. | Most known cellular target of NO is sGC resulting in cGMP formation; represents downstream NO signalling | Indirect, assesses effect of NO on sGC; represents downstream NO signalling; does not consider NO-independent cGMP formation |
| measurement of cGMP level | |||
| NO donors and NOS inhibitors | n.a. | Use of them completes other assays; helps to understand the role of NO | Alone does not reflect NO production |
In the present review, we focus on the most important possibilities to determine NO or its derivatives in biological matrices. We discuss background assay principles, specificities, sensitivities, advantages/disadvantages and possible limitations.
Direct methods to estimate NO
Electron paramagnetic resonance spectroscopy
As NO is a free radical, hence of paramagnetic nature, electron paramagnetic resonance spectroscopy (EPR; also referred to as ESR) is considered to be the most appropriate tool for the direct detection of NO. The main advantage of EPR compared with other NO detection techniques is that it only detects paramagnetic molecules, and the EPR spectrum is a unique fingerprint of the chemical and electronic structure around the unpaired electron. EPR measures the transitions induced between the Zeeman levels of a paramagnetic molecule via its interaction with a static magnetic field and an oscillating electromagnetic field, most commonly in the X microwave frequency band, around 9–9.5 GHz. To take an EPR spectrum, the microwave frequency is held constant and the static magnetic field is swept (despite its name) across the desired range. The equation for resonance absorption is ΔE = hv = gβB, in which h is Planck's constant; ν is the frequency of the microwaves; β is the Bohr magneton, a physical constant; B is the external magnetic field; and g is the g-value, or the Zeeman splitting factor (Weil and Bolton, 2007). The g-value is a characteristics of the paramagnetic molecule [in case of NO in liquid g = 2.035, (Hogg, 2010)]. A further specific property of EPR spectra originates from the coupling of the electron spin with the surrounding nuclear magnetic spins, measured by the so-called hyperfine coupling constant, A, in mT.
The paramagnetic NO can be measured directly by EPR irrespective of the optical appearance of the sample; however, due to the rapid relaxation of its excited electron spin state to the ground state (Maples et al., 1991) and the high reactivity of NO, spin-trapping techniques have been developed. Spin traps are compounds that interact with the less stable radicals, producing a more stable adduct, which can be then detected by EPR (Janzen, 1984; Tosaki et al., 1996; Berliner and Fujii, 2004; Villamena and Zweier, 2004).
As natural spin traps, the iron centre of haemoglobin and other haem-proteins may interact with NO with high affinity forming nitrosyl derivatives, which are paramagnetic and exhibit a characteristic EPR spectrum (Greenberg et al., 1990; Henry et al., 1991; Eriksson, 1994; Katayama et al., 2001). The spectra of the haemoglobin-NO is sensitive to many factors such as tertiary and quaternary structures of the protein, concentration of O2, pH, degree of hydration, temperature, etc. (Sanches, 1988; Hall and Buettner, 1996). The detection limit of haemoglobin-NO has been reported to be ∼200 nM, but the basal level of NO in blood appears to be below this limit (Piknova et al., 2005).
There are several classes of chemical spin traps for the detection of NO like the (i) nitroxide spin traps (Arroyo and Forray, 1991a; Arroyo and Kohno, 1991b) including nitrones (e.g. DMPO, 5,5-dimethyl-pyrroline-N-oxide and its related spin trap, DEPMPO, 5-diethoxyphosphoryl-5-methyl-1-pyrroline-N-oxide; PBN, α-phenyl-N-tert-butyl-nitrone; and POBN, α(4-pyridyl-1-oxide)-N-tert-butyl-nitrone) and nitroso compounds (e.g. MNP, 2-methyl-2-nitrosopropane and DBNBs, 3,5-dibromo-4-nitrosobenzene); (ii) the NO cheletropic traps (double carbon-centred biradical equivalents; Korth et al., 1992); and (iii) the nitronyl nitroxides (e.g. carboxy-PTIO, [2-(4-carboxyphenyl)-4,4,5,5-tetramethylimidazoline-1-oxide] (Katayama et al., 2001; Hawkins and Davies, 2014). However, the spin traps mentioned earlier have several limitations, such as instability at certain pH and low sensitivity or selectivity (Katayama et al., 2001; Hawkins and Davies, 2014).
The efficiency of NO spin trapping can be sharply increased using various transition metal complexes (see, e.g. Archer, 1993; Henry et al., 1993). Thus, the most popular and most extensively used NO spin traps are the iron (II) dithiocarbamate (DTC) type of complexes (Kleschyov et al., 2007; Hong et al., 2009; Hogg, 2010). Iron complexed with N,N-diethyl-dithiocarmabate [DETC, (Mordvintcev et al., 1991; Mülsch et al., 1992)] is hydrophobic, whereas iron complexed with N-methyl-D-glucamine dithiocarmabate (MGD; Komarov et al., 1993) is hydrophilic; hence, these iron complexes trap NO in different environments. One of the major advantages of using these iron complexes as spin traps is that they react with NO extremely rapidly, that is NO binds to dithiocarbamate complexes with a rate constant of 1–5 × 108 M−1·s−1 (Hogg, 2010). Fe2+(DTC)2 are suitable spin traps for NO in vivo and in real-time measurements because the rate constant of the formation of NO-Fe2+(DTC)2 adduct is much larger than that with other NO spin traps (Vanin et al., 2000; Nagano and Yoshimura, 2002), and also because of the high solubility of NO in membranes (Nedeianu et al., 2004).
Whereas DETC is soluble in aqueous media, it is ferrous and the mononitrosyl ferrous complexes precipitate at neutral pH (Csont et al., 2003). Therefore, when injected in animals, it is recommended to inject Fe2+ separately from DETC. The reagents are inexpensive, commercially available and are typically used in vivo [100 mM DETC, 20 mM Fe(II), usually at a ratio of 1 iron : 5 ligand (Berliner and Fujii, 2004)]. The Fe2+(DETC)2 is distributed throughout the body, as observed by the detection of the NO-Fe2+(DETC)2 complexes in different organs in vitro and in vivo in animal models [see, e.g. Csont et al. (1998); Fejer et al. (2005); for a review, see Hong et al. (2009)], while Fe2+(MGD)2 is suitable for ex vivo spin trapping in different tissue samples and cell cultures (Csonka et al., 1999; Radak et al., 1999; Csont et al., 2010), including human tissue samples (Radak et al., 1999).
NO interacts with high affinity with these Fe2+(DTC)2 complexes forming stable nitrosyl iron-dithiocarbamate complexes [according to Vanin et al. (2000) the predominant binding of iron to dithiocarbamate ligands takes place only after its binding to NO, as nitrosylated iron manifests much higher affinity for these ligands that for any non-thiol compounds (Vanin and Poltorakov, 2009)]. At ambient temperature, the solution of NO-Fe2+(DTC)2 is characterized by the isotropic EPR signal at a g-value of 2.035 and triplet hyperfine structure (not shown) and a spectrum with axial symmetry in the frozen state (Figure 2A). The triplet (three-line) splitting originates from the hyperfine interaction of an unpaired electron with the 14N nucleus of the NO ligand [solution spectra are characterized in Nedeianu and Pali (2002)]. DETC penetrates the cell wall and binds not only the free intracellular Fe2+, but also endogenous Cu2+ forming a Cu2+(DETC)2 complex, characterized by a four-line EPR spectrum that overlaps with the NO-Fe2+(DTC)2 spectrum at low temperature (Suzuki et al., 1997; see also Figure 2B).
Figure 2.
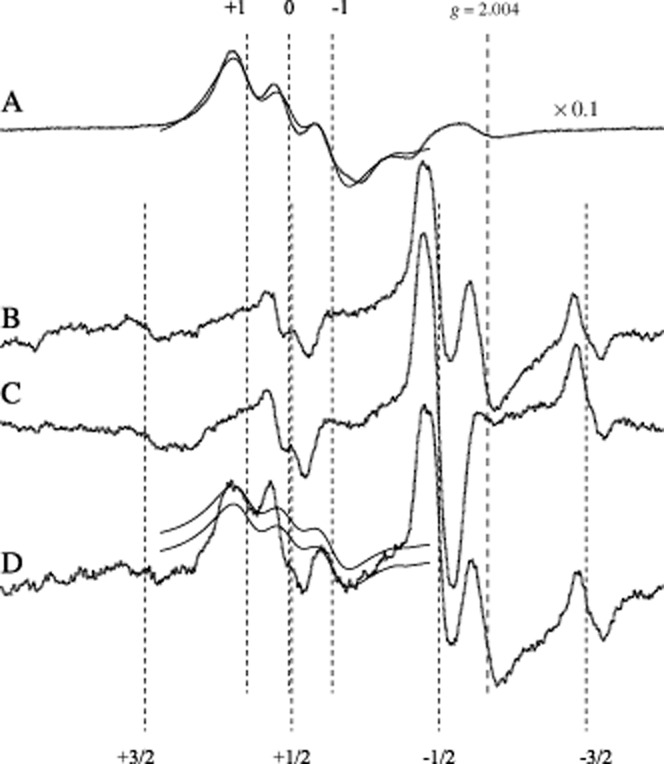
X-band EPR spectra of NO-Fe2+(DETC)2 complex in left ventricular tissue samples of rat hearts. Trace A: dotted line, positive control (NO donor sodium nitroprusside), with gain reduced by a factor of 10 compared with curves B–D; solid line, sum of Lorentzians fitted to the NO signal. Trace B: background spectrum, Cu2+(DETC)2. Trace C: negative control (NOS inhibitor NG-nitro-L-arginine). Trace D: increased NO production in nitroglycerine-tolerant animals. The solid lines on trace D are the fitted spectrum from trace A, but scaled down to match the +1 and −1 hyperfine lines, upper and lower solid lines, respectively, of the NO spectral component of the spectrum. The +1, 0, −1 numbers indicate the hyperfine lines of NO-Fe2+(DETC)2 triplet (perpendicular orientation). The +3/2, +1/2, −1/2, −3/2 numbers indicate the hyperfine lines of Cu2+(DETC)2 (perpendicular orientation). Sample temperature was 160 K, central field and scan range are 335.6 and 34.0 mT respectively. The g = 2.004 position of the g-value was determined using a g standard.
Figure was modified with permission from Csont et al. (1998).
Specimens in a quartz tube with a small sample volume (<100 μL) are measurable in an X-band EPR spectrometer. However, larger biological samples (e.g. tissues, organs and live animals) cannot be measured with a conventional X-band spectrometer due to the high dielectric loss of water at such frequencies, and the small size of the EPR cavity resonator. EPR spectroscopy at lower frequencies, the L-band (0.4–1.6 GHz) or S-band (1.6–4 GHz), can be used for in vivo EPR imaging (Nagano and Yoshimura, 2002; Fujii and Berliner, 2004). As the NO-Fe2+(DTC)2 complexes are still reactive free radicals, freezing of the samples until and during measurement is required. Measurements are done typically below 200 K. The detection limit of NO by the DTC-based spin trapping EPR method is 0.05 nM in biological samples (Mülsch et al., 1992; Khoo et al., 2004). We found the detection limit for NO released from an NO donor to be 0.4 nM in solution under anaerobic conditions (Nedeianu et al., 2004). However, precise and absolute quantification of EPR detection of NO in biological samples is not reliable due to an uneven distribution of the spin trap complex Fe2+(DTC)2 in the aqueous and the lipid phase of biological samples (Ferdinandy et al., 1997). In addition, as the concentration of the Fe2+(DTC)2 complexes cannot be predicted from the concentration of Fe2+ and DTC administered, and as the trapping efficiency of the spin trap in the given environment is not known, absolute quantitation of NO is not a realistic objective in practice. Hence, in the vast majority of studies, relative changes in the NO level are reported as NO signal intensity in arbitrary units.
Typical EPR spectra are shown in Figure 2 for NO trapped with Fe2+(DETC)2 (traces A, C and D), together with the most common compromising signal Cu2+(DETC)2 (trace B) and the spectra of control samples. The EPR spectrum is proportional with the first derivative of the microwave absorption (Y-axis, measured in arbitrary units) as function of the magnetic field (B, X-axis). The second integral of the EPR spectrum (the area under the absorption spectrum) is proportional to the number of spins present in the active volume of the resonator. Therefore, in this case, the second integrals can be used for relative quantitation of NO. In Csont et al. (1998) for instance, analysis of NO content was performed with double integration of all spectra, after subtracting the background signal of Cu2+(DETC)2. However, in most studies, the background signal also changes significantly from sample to sample, even if the same test tube is used for the sample and the background signals. As the second integral approach is very sensitive to the baseline, an alternative approach is followed: the use of a positive control, which is the same tissue, but loaded with an NO donor and the spin trap. A positive control yields an NO triplet that has much higher intensity than the background signal, hence, it can be analysed and fitted easily [this technique was used in Fejer et al. (2005)]. A fit of a simulated NO signal to the positive control is also shown in Figure 2 trace A, solid line. A simple approach to evaluate NO spectra is to measure the peak-to-peak amplitude of the +1 and/or the −1 NO peak and compare them with that of a positive control signal (Radak et al., 1999; Csont et al., 2010).
In summary, EPR measurement of NO spin-trapped with Fe2+(DTC)2 provides a very sensitive and one of the most specific methods for direct detection of NO both in vivo and in vitro. The EPR spectrum is characteristic for the nitrosyl-adduct and it can be recorded under various conditions; however, the limitation of this method is that complex evaluation of the results requires significant expertise.
Electrochemical assays using microelectrodes specific for NO
There are a lot of different electrochemical assays available for NO measurement. They all integrate the advantages of electrochemistry: small size, continuous (often in situ) and fast measurements, no chemical contamination of the samples (thus samples can be used for other assays). However, large differences may occur among the different electrodes in selectivity. Selectivity is controlled by the voltage applied between the electrodes (∼860 mV) and the NO-selective layer around or on the surface of the electrode.
The majority of electrodes on the market dedicated to measure NO uses micro ion electrodes measuring simply nitrite and/or nitrate. For the advantage and disadvantage of nitrite and nitrate measurement versus direct NO measurement, see succeeding sections. Compared with the classical Griess spectrophotometric assay, which is the gold standard of nitrite/nitrate determination, use of electrodes has several advantages: (i) requires smaller amount of samples; (ii) wide range of samples can be used including turbid, opaque or even non-liquid samples for example cell cultures, tissues and tissue homogenates; (iii) per sample cost savings over Griess; (iv) avoid interferences during measurements from other components for example NADPH and antioxidants; and (v) minimize sample preparation steps.
To date, among the several electrochemical techniques that have been shown to be useful for the direct measurement of NO, amperometric detection of NO is the most popular technique sensitive enough to detect relevant concentrations of NO in real-time and in vivo (Serpe and Zhang, 2007; Davies and Zhang, 2008; Yap et al., 2013). Generally, this technique involves applying a fixed (poise) voltage potential to a working electrode versus a reference electrode, and monitoring the redox current produced by the oxidation of NO (Serpe and Zhang, 2007). This technique has proven to be very useful for NO detection due to its fast response time of less than a few seconds, and its acceptable sensitivity. In amperometry, both electrodes are encased within a protective Faraday-shielded stainless steel sleeve. The tip of the sleeve is covered with an NO-selective membrane, and the sleeve itself contains electrolyte (Davies and Zhang, 2008). NO diffuses across the gas-permeable/NO-selective membrane (e.g. nitrocellulose, chroloprene, silicon, Teflon, graphene, Nafion, cellulose acetate and polycarbazole) and ultimately is oxidized at the working electrode surface producing a redox current (Serpe and Zhang, 2007; Wang and Hu, 2009). The selectivity of the membranes for NO is still debatable, and after NO diffuses through the membrane, there is no more qualitative control on the analytical signal. The amount of NO oxidized is proportional to the current flow between the working and reference electrodes, which is measured by an NO meter. The redox current generated by the oxidation of NO in biological systems is extremely small, typically in the range of 1–10 pA corresponding to an approximately 10−8–10−9 M concentration range (Table 2014).
In addition to amperometry, voltammetry techniques may provide an alternative electrochemical approach to detect NO; however, they are less frequently used. These techniques typically employ a classical three-electrode configuration consisting of a working electrode, reference electrode and a counter electrode.
Besides the classic Clark-type electrodes mentioned earlier, where selectivity is insured by a passive NO-selective membrane or layer around the electrode, a new generation of NO electrodes have been recently developed (Wang and Hu, 2009; Bedioui et al., 2010; Jiang et al., 2013; Yap et al., 2013). Instead of membranes believed to ensure NO-selectivity, an NO-specific active scavenger layer is electropolymerized onto the surface of a glassy carbon electrode that can chemically react directly with NO. Among these materials, the electropolymerized film of metalloporphyrins has been used most extensively (Diab et al., 2003; Li et al., 2009). Electric signals form only after the direct chemical link between NO and the electropolymerized layers of the electrode; thus, electrocatalytic oxidation of NO enhance NO specificity of the electrode significantly. Parallel with advancements in nanotechnology (e.g. Santos et al., 2013), recently there has been significant progress in the development of these electrodes. Hence, they have great potential as a future technology suitable for direct NO measurements in biological samples.
Even if electrochemical techniques offer great promise for the measurement of NO production, some disadvantages should be considered. The tip of the electrode is used to detect the signal; therefore, the location of the electrode is critical as small changes in its position can dramatically influence spatial information. Theoretically, this problem can be eliminated using ultramicroelectrode sensor arrays in which each electrode, or groups of electrodes, are individually addressable, and used for mapping the analyte to achieve spatio–temporal analysis (Quinton et al., 2011; Griveau and Bedioui, 2013). As NO dissolves well in lipids, the distance of the electrode from different membranes may also influence signal intensity. The diameter of the electrodes, which varies from the micrometre to the millimetre range, also influences the sensitivity of the electrode as the surface of the electrode (determined by diameter and length) is proportional to the NO signal detected. To get reliable results using electrochemical assays, the preparation of standard solutions to calibrate the electrode is also a critical step in the measurement, and all the following options are rather problematic: (i) application of gaseous NO to prepare a standard solution (concentration can be calculated from the dissolving coefficient, and deoxygenation of the standard is important to prevent oxidation of NO); (ii) known concentration of NO donors; or (iii) NO produced by chemical reactions (Serpe and Zhang, 2007). Temperature may affect sensitivity of the electrode by influencing the partial pressure of dissolved NO, the permeability of the coatings and the conductivities of various sensor components. Therefore, a careful temperature control is recommended during calibration and experiments. External electrical noise sources (e.g. magnetic stirrers, fluorescent lights, MRI machines, electric motors, computers, pumps and other electrical instruments) may couple into the sensor signal path electromagnetically and impose undesirable signals on the output record. Because NO signal intensity is in the pA range, it is important to ground and shield the system properly (Serpe and Zhang, 2007).
Fluorometry
Fluorescence assays are sensitive techniques for detection of NO. The assay principle is that NO and/or its oxidative derivatives react with a non-fluorescent compound forming a fluorescent product. Fluorescent compounds (frequently called probes or dyes) greatly vary in their specificity for NO and applicability in biological systems. In order to detect the spatial and temporal dimensions of NO generation and accumulation in living cells, the fluorescent compounds must pass several strict criteria. The probe should be small, cell or membrane permeable, non-toxic, water soluble, photostable, have excitation/emission wavelengths in the visible range to minimize damage of biological samples, well-separated excitation/emission wavelengths, possesses a large extinction coefficient and quantum yield, and have a large signal-to-noise ratio and linear response. In addition, it is highly desirable that the molecular probe is selective for NO among competing reactive nitrogen species. If oxidized forms of NO such as nitrite and nitrate, and also reactive oxygen species, such as superoxide, hydrogen peroxide, and peroxynitrite, do not react with the probes to give a fluorescent product, then in the absence of NO fluorescent dye is not formed. Once the fluorescent dye is formed, it can be detected by any instrument suitable for detection of fluorescence, including flow cytometers, microscopes, fluorescent microplate readers and fluorometers. Fluorescence microscopy can provide two- or even three-dimensional imaging, and high spatial–temporal resolution; therefore, in biological samples, it is the most informative detection technique despite limited quantification.
There are two main classes of different fluorescent probes specifically designed for NO measurement: organic-based and metal-based sensors. In both instances, the goal is to alter the fluorescent properties of the probe by specific reaction with NO. Organic probes employ fluorophores with pendant functional groups that serve to quench their fluorescence until restored by a specific reaction with NO. Metal-based probes take advantage of the high reactivity of NO with transition metals to form metal-nitrosyl complexes (McQuade and Lippard, 2010b).
The first fluorescent probe developed for the measurement of NO was diaminonaphthalene (DAN) (Ji and Hollocher, 1988). However, Ji and Hollocher (1988) suggested that chemical nitrosation of DAN requires O2, therefore, DAN directly measures oxidative derivatives of NO (N2O3). Later, Misko et al. developed an assay using DAN to detect nitrite in biological samples (Misko et al., 1993). Another popular organic-based fluorescein probe, diaminofluorescein (DAF), was developed more than 15 years ago (Kojima et al., 1998a,b). DAF is essentially non-fluorescent until it reacts with NO (or more precisely N2O3) to form a fluorescent benzotriazole. Besides specificity, cell membrane permeability was improved by developing its diacetate derivative, which passively diffuses across cellular membranes. Once inside cells, it is deacetylated by intracellular esterases to become membrane-impermeable. These probes have been extensively used for estimation of NO production in a variety of biological samples. However, it turned out that DAF-2 can be enzymatically converted into a variety of highly fluorescent derivatives both intra- and extracellularly, of which only a minor part appeared NO-dependent (Roychowdhury et al., 2002). Thus, DAF-fluorescence does not necessarily indicate NO·production; therefore, it is not generally accepted as an NO-specific probe. The presence or absence of oxygen (i.e. in ischaemia or reperfusion) can also influence fluorescence through formation of N2O3 (Rumer et al., 2012). Despite these limitations, further applications of DAF were developed, that is a high-throughput technique based on DAF-fluorescence capable of detecting RSNOs (Doctor et al., 2005). Other frequently used organic NO fluorophores are o-diaminorhodamines (DARs; typically DAR-2 and DAR-4M AM), o-diamino-boron-dipyrromethenes (DAMBOs; typically DAMBO and DAMBO-CO2Et), and o-phenylenediamine cyanines (DACs) (Nagano, 2009; Zhang et al., 2014). The currently used organic fluorescent probes are reviewed in (Nagano, 2009; 2010; Woolley et al., 2013).
As organic probes are not suitable for direct detection of NO, metal-based probes were also developed. As transition metals can reversibly form bonds with NO, many researchers have focused on metal–ligand constructs where the ligand contains a fluorophore. (McQuade and Lippard, 2010a). To date, NO sensing has been accomplished using a wide array of metal ions, including Co(II), Fe(II), Ru(II), Rh(II) and Cu(II) (Hong et al., 2009; Hu et al., 2011). One of the most promising of metal-based sensors for direct, cellular detection of NO is a copper-based fluorescent probe CuFL (Lim et al., 2006; McQuade and Lippard, 2010b).
Although, the use of fluorescent techniques is a promising tool to detect NO, it has several disadvantages. The optimal dilution buffer and working concentration of the fluorescent dye, and optimal loading concentration, time and temperature are needed to be determined empirically, which can vary among different labs. Moreover, quantification of NO is nearly impossible. It is desirable to use the lowest dye concentration yielding fluorescence signals with adequate signal-to-noise ratios. Moreover, close excitation/emission wavelengths, cytotoxicity, strong auto-fluorescence, small extinction coefficient and insufficient solubility in neutral buffers, pH dependence of the fluorescence intensity are mentioned as further disadvantages of this method. Some further limitations still exist to affect sensitive imaging of NO, such as poor photostability (e.g. DAFs, DACs and transition metal complex probes), small changes of fluorescence quantum yield after reaction with NO (e.g. DACs and transition metal complex probes), fast leakage from cells (e.g. DAFs and DARs) and possible fluorescence interference from biological matrix (e.g. DAFs, DAMBOs and DARs) (Zhang et al., 2014).
A great deal of improvement has been made to fluorescent NO probes in recent years, mostly in the area of sensitivity and selectivity (e.g. Nagano, 2010; Rodriguez-Rodriguez and Simonsen, 2012; Woolley et al., 2013; Alam et al., 2014; Zhang et al., 2014). Although these improvements have allowed for more accurate measurements of NO, the specificity of fluorophores for NO is still a very important issue and needs to be further validated in biological systems.
Indirect methods to estimate NO
Determination of nitrate/nitrite
Due to its short half-life, the direct measurement of NO is extremely challenging in a complex biological environment. As NO is rapidly metabolized to nitrite and nitrate in the presence of oxygen (Figure 1), the determination of both nitrite and nitrate (termed NOx) is commonly used to estimate total NO production. A number of analytical techniques have been developed to determine nitrite and nitrate in biological samples including the Griess colorimetric assay, fluorometry, flow or sequential injection analysis with visible absorbance, chemiluminescence and electrochemical detection (Moorcroft et al., 2001; Bryan and Grisham, 2007). Chromatographic methods including GC–MS, capillary electrophoresis, and HPLC have also been developed using a variety of detection systems.
A major general disadvantage of nitrite/nitrate determination to estimate tissue NO level is that dietary intake of nitrite (e.g. cured meat) and nitrate (e.g. vegetables) are rather significant, which markedly influences plasma NOx level (Rassaf et al., 2014). In addition, environmental conditions such as nitrite/nitrate pollution in chemical reagents, cell culture media, and even plastic labware may interfere with nitrite/nitrate analysis.
Nitrite and nitrate can be measured from a variety of biological fluids. A relatively simple, long-known colorimetric assay based on the Griess reaction is probably used most extensively for assaying NOx. The Griess reaction – also called diazotization assay – is based on the conversion of nitrite to a purple-coloured azo-dye that can be spectrophotometrically assayed at a wavelength of ∼540 nm. The most widely used reagents required for the reaction are sulphanilamide and N-naphthyl-ethylenediamine (Moorcroft et al., 2001; Sun et al., 2003; Bryan and Grisham, 2007; Hetrick and Schoenfisch, 2009). Advantages of this method include a strong literature background, numerous commercially available reagent kits and wide availability of infrastructure. However, prior to this reaction, nitrate needs to be reduced either chemically or enzymatically to nitrite in order to determine NOx. Chemical reduction is not specific for the nitrate–nitrite conversion and nitrite may be reduced further to NO thereby leading to underestimation of nitrite (Sun et al., 2003). Enzymatic reduction of nitrate by nitrate reductase requires NADPH; however, excess NADPH interferes with the subsequent Griess reaction and thus limits sensitivity of the assay. Moreover, the high protein content of cell lysates and plasma may interfere with the nitrate reduction or the Griess reaction; therefore, deproteinization of samples is highly recommended. As the detection limit is ∼0.5 μM of nitrite/nitrate, this assay is not suitable for measuring physiological amounts of NO (Sun et al., 2003; Hetrick and Schoenfisch, 2009).
The other most widely used approach for the analysis of nitrite and nitrate is based on electrochemical detection. For general advantages and disadvantages of the electrochemical approach, see previous sections.
Estimation of NO formation by measuring NOS activity
Measurement of NOS activity may provide an alternative solution for investigating NO metabolism both in vivo and in vitro (Figure 1). NOS activity can be determined by measuring either L-citrulline or the NO metabolites nitrite/nitrate. The classical citrulline assay is based on the conversion of 3H- or 14C-labelled L-arginine to L-citrulline, where after removal of unreacted L-arginine with resin, radioactivity of the end-product L-citrulline is proportional to NOS activity. Radioactivity is usually measured by liquid scintillation counting. This method is suitable for distinguishing Ca2+-dependent and Ca2+-independent NOS activities. It is relatively sensitive (Hevel and Marletta, 1994) and after improvement by Giraldez and Zweier (1998) it became suitable for detection of low levels of NOS activity found in certain tissues (e.g. heart).
NOS enzyme activity measured in vitro conditions strongly depends on the availability of exogenously administered cofactors such as tetrahydrobiopterin (also known as sapropterin) and NADPH. A radiochemical HPLC-based citrulline assay was developed for the measurement of NOS activity in intact tissue samples (de Bono et al., 2007; Crabtree et al., 2009a,b), thereby avoiding the influence of the measurements by exogenously administered cofactors.
Other commonly used assays for measurement of NOS activity are based on detection of nitrite/nitrate generated from NO. These assays are less specific, but more simple and cost effective compared with the citrulline assay, and are also suitable for high-throughput analysis. Another method to improve specificity and sensitivity of NOS activity assays based on nitrate/nitrite determination involves a GC–MS based measurement of 15N-labelled nitrite/nitrate converted from L-[guanidino-15N]-arginine-derived 15NO (Tsikas, 2008; Shin and Fung, 2011).
NOS activity assays indicate the amount of enzymatically active NOS protein in a biological system under optimized in vitro conditions; however, they do not necessarily reflect in vivo NO production and actual NO concentration. Care should be taken to avoid interference by other enzymes affecting L-arginine metabolism (e.g. arginase) and non-enzymatic NO formation should be also considered. Specificity of the assays should be ensured by application of inhibitors of NOS isoforms in parallel measurements.
Detection of RSNOs in biological systems
Protein RSNOs derived from NO and/or its derivatives (Figure 1) play an important role in NO signalling in both physiological and pathological conditions. Therefore, measurement of RSNOs contributes to the understanding of NO biochemistry; however, it does not directly reflect actual NO levels.
To date, no universal direct method exists to identify protein RSNOs specifically. The most commonly used methods for the detection of S-nitrosated proteins are (i) Saville reaction (simplest and least expensive) in which mercury replaces a nitrosyl from a thiol group to form nitrite followed by Griess assay (Saville, 1958); (ii) biotin switch (suitable to detect low μM concentrations of biotinylated proteins without requiring special instrumentation) (Jaffrey and Snyder, 2001; Forrester et al., 2009); and (iii) chemiluminesence-based assays (Basu et al., 2008). A DAF-fluorescence-based high-throughput technique measuring RSNOs in the low nanomolar range was also described (Doctor et al., 2005). Mass spectrometry detection became the gold standard for directly studying low-level RSNOs in a physiologically relevant context (Barglow et al., 2011); however, it is expensive and requires 15N-labelled internal standards (Gow et al., 2007). The methods mentioned earlier suffer from lengthy preparative protocols and selectivity issues. Most recently, triarylphosphines and cyclization reactions with phosphines have been addressed to be the milestones of direct detection of RSNOs; however, these methods require further improvements (Bechtold and King, 2012).
Estimation of NO formation by measuring cGMP
In biological systems, the most known target of NO is sGC that is responsible for the synthesis of cGMP (Figure 1). The connection between NO and cGMP in most of the tissues is so close that cGMP is frequently used as a surrogate for the rate of NO synthesis both in vitro and in vivo (Tsikas, 2008); however, the relationship between tissue NO and cGMP level in the heart tissue seems more complicated (Csont et al., 1998).
There are several available methods to measure cGMP in a variety of biological samples including tissue lysate, blood, urine and culture supernatants. RIAs are commercially available, with high specificity and sensitivity (pM range) (Steiner et al., 1972; Evgenov et al., 2004). The method is based on the competitive binding of cGMP in the sample and a radioiodinated derivative of cGMP ([125I] cGMP) to a highly specific antibody. After separation of antibody-bound cGMP from free cGMP, 125I is determined by a γ-counter.
A similar immune-detection approach applying non-radioactive reagents is used in commercially available elisa assays with colorimetric detection of optical densities by a plate reader. Another method for cGMP quantification is liquid chromatography coupled to tandem mass spectrometry (LC-MS) characterized by complicated sample preparation and high sensitivity and selectivity (Lorenzetti et al., 2007; Martens-Lobenhoffer et al., 2010).
All these methods mentioned earlier are expensive and labour intensive. Moreover, these are end point assays, not suitable for real-time measurements. However, real-time monitoring of cGMP based on homogenous time-resolved fluorescence has been recently described (van Mastbergen et al., 2012). In addition, NO reporter assays were also developed allowing real-time detection of NO synthesis within living cells (Wunder et al., 2005; 2007).
Although determination of cGMP is frequently used to demonstrate NO production in biological systems, cGMP level is modified by several other factors including activities of particulate GC and phosphodiesterases, sampling time, subcellular localization and NO-independent regulation of sGC (Lucas et al., 2000; Bender and Beavo, 2006; Francis et al., 2010).
Conclusions
A wide range of analytical methods are available for the detection of NO in biological samples; however, each method has certain advantages and limitations. Therefore, the use of direct NO detection methods as first choice should be considered. In addition, a series of methods to follow NO synthesis, NO content, molecular targets and reaction products of NO are recommended to get meaningful insights into the role of NO in a certain physiological or pathological process. Moreover, application of NO donors and NOS inhibitors to provide appropriate positive and negative controls is also recommended to overcome limitations of individual methodologies.
Acknowledgments
We thank partial financial support from the Hungarian National Science Fund (OTKA K101633, OTKA ANN 107803, OKTA K109737, PD106001), TÁMOP-4.2.2.A-11/1/KONV-2012-0035. F. P. holds a Szentágothai Professorship of the National Excellence Program of Hungary (TÁMOP 4.2.4.A/2-11-1-2012-0001). This paper was supported by the János Bolyai Research Scholarship of the Hungarian Academy of Sciences.
Glossary
- DAC
o-phenylenediamine cyanine
- DAF
diaminofluorescein
- DAMBO
diamino-boron-dipyrromethene
- DAN
diaminonaphthalene
- DAR
o-diaminorhodamine
- DETC
N,N-diethyl-dithiocarmabate
- DTC
dithiocarbamate
- EPR
electron paramagnetic resonance spectroscopy
- ESR
electron spin resonance spectroscopy
- MGD
N-methyl-D-glucamine dithiocarmabate
- RSNO
S-nitrosothiols
- sGC
soluble GC
Author contributions
C. C. did the study concept and design; wrote the following sections: ‘Fluorometry’, ‘Electrochemical assays using microelectrodes specific for NO’, ‘Electron paramagnetic resonance spectroscopy’, and ‘Analytical tools for the assessment of NO: what to consider before selection?’, Table 2014, Figure 1; and revised the paper. T. P. wrote the sections ‘Electron paramagnetic resonance spectroscopy’ and Figure 2. P. B. wrote the sections ‘Estimation of NO formation by measuring NOS activity’ and ‘Detection of RSNOs in biological systems’. A. G. wrote the section ‘Estimation of NO formation by measuring cGMP’. P. F. wrote the ‘Introduction’, and consulted, proofread and edited the paper to improve the language. T. C. did the study concept and design; wrote the following sections: ‘Synthesis and major metabolic pathways of NO’, ‘Determination of nitrate/nitrite’, ‘Analytical tools for the assessment of NO: what to consider before selection?’, ‘Conclusion’ and ‘Summary’ edited and revised the paper; and did the final approval.
Conflict of interest
None.
References
- Alam R, Mistri T, Mondal P, Das D, Mandal SK, Khuda-Bukhsh AR, et al. A novel copper(II) complex as a nitric oxide turn-on fluorosensor: intracellular applications and DFT calculation. Dalton Trans. 2014;43:2566–2576. doi: 10.1039/c3dt52521j. [DOI] [PubMed] [Google Scholar]
- Alexander SPH, Benson HE, Faccenda E, Pawson AJ, Sharman JL, Spedding M, et al. The Concise Guide to PHARMACOLOGY 2013/14:Enzymes. Br J Pharmacol. 2013;170:1797–1867. doi: 10.1111/bph.12451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Archer S. Measurement of nitric oxide in biological models. FASEB J. 1993;7:349–360. doi: 10.1096/fasebj.7.2.8440411. [DOI] [PubMed] [Google Scholar]
- Arroyo CM, Forray C. Activation of cyclic GMP formation in mouse neuroblastoma cells by a labile nitroxyl radical. An electron paramagnetic resonance/spin trapping study. Eur J Pharmacol. 1991a;208:157–161. doi: 10.1016/0922-4106(91)90066-q. [DOI] [PubMed] [Google Scholar]
- Arroyo CM, Kohno M. Difficulties encountered in the detection of nitric oxide (NO) by spin trapping techniques. A cautionary note. Free Radic Res Commun. 1991b;14:145–155. doi: 10.3109/10715769109094127. [DOI] [PubMed] [Google Scholar]
- Barglow KT, Knutson CG, Wishnok JS, Tannenbaum SR, Marletta MA. Site-specific and redox-controlled S-nitrosation of thioredoxin. Proc Natl Acad Sci U S A. 2011;108:E600–E606. doi: 10.1073/pnas.1110736108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basu S, Wang X, Gladwin MT, Kim-Shapiro DB. Chemiluminescent detection of S-nitrosated proteins: comparison of tri-iodide, copper/CO/cysteine, and modified copper/cysteine methods. Methods Enzymol. 2008;440:137–156. doi: 10.1016/S0076-6879(07)00808-7. [DOI] [PubMed] [Google Scholar]
- Bechtold E, King SB. Chemical methods for the direct detection and labeling of S-nitrosothiols. Antioxid Redox Signal. 2012;17:981–991. doi: 10.1089/ars.2012.4570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bedioui F, Quinton D, Griveau S, Nyokong T. Designing molecular materials and strategies for the electrochemical detection of nitric oxide, superoxide and peroxynitrite in biological systems. Phys Chem Chem Phys. 2010;12:9976–9988. doi: 10.1039/c0cp00271b. [DOI] [PubMed] [Google Scholar]
- Bender AT, Beavo JA. Cyclic nucleotide phosphodiesterases: molecular regulation to clinical use. Pharmacol Rev. 2006;58:488–520. doi: 10.1124/pr.58.3.5. [DOI] [PubMed] [Google Scholar]
- Berliner LJ, Fujii H. In vivo spin trapping of nitric oxide. Antioxid Redox Signal. 2004;6:649–656. doi: 10.1089/152308604773934413. [DOI] [PubMed] [Google Scholar]
- de Bono JP, Warrick N, Bendall JK, Channon KM, Alp NJ. Radiochemical HPLC detection of arginine metabolism: measurement of nitric oxide synthesis and arginase activity in vascular tissue. Nitric Oxide. 2007;16:1–9. doi: 10.1016/j.niox.2006.03.008. [DOI] [PubMed] [Google Scholar]
- Broniowska KA, Hogg N. The chemical biology of S-nitrosothiols. Antioxid Redox Signal. 2012;17:969–980. doi: 10.1089/ars.2012.4590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bryan NS, Grisham MB. Methods to detect nitric oxide and its metabolites in biological samples. Free Radic Biol Med. 2007;43:645–657. doi: 10.1016/j.freeradbiomed.2007.04.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coneski PN, Schoenfisch MH. Nitric oxide release: part III. Measurement and reporting. Chem Soc Rev. 2012;41:3753–3758. doi: 10.1039/c2cs15271a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crabtree MJ, Tatham AL, Al-Wakeel Y, Warrick N, Hale AB, Cai S, et al. Quantitative regulation of intracellular endothelial nitric-oxide synthase (eNOS) coupling by both tetrahydrobiopterin-eNOS stoichiometry and biopterin redox status: insights from cells with tet-regulated GTP cyclohydrolase I expression. J Biol Chem. 2009a;284:1136–1144. doi: 10.1074/jbc.M805403200. [DOI] [PubMed] [Google Scholar]
- Crabtree MJ, Tatham AL, Hale AB, Alp NJ, Channon KM. Critical role for tetrahydrobiopterin recycling by dihydrofolate reductase in regulation of endothelial nitric-oxide synthase coupling: relative importance of the de novo biopterin synthesis versus salvage pathways. J Biol Chem. 2009b;284:28128–28136. doi: 10.1074/jbc.M109.041483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Csonka C, Szilvassy Z, Fulop F, Pali T, Blasig IE, Tosaki A, et al. Classic preconditioning decreases the harmful accumulation of nitric oxide during ischemia and reperfusion in rat hearts. Circulation. 1999;100:2260–2266. doi: 10.1161/01.cir.100.22.2260. [DOI] [PubMed] [Google Scholar]
- Csont T, Pali T, Szilvassy Z, Ferdinandy P. Lack of correlation between myocardial nitric oxide and cyclic guanosine monophosphate content in both nitrate-tolerant and -nontolerant rats. Biochem Pharmacol. 1998;56:1139–1144. doi: 10.1016/s0006-2952(98)00167-1. [DOI] [PubMed] [Google Scholar]
- Csont T, Csonka C, Kovacs P, Jancso G, Ferdinandy P. Capsaicin-sensitive sensory neurons regulate myocardial nitric oxide and cGMP signaling. Eur J Pharmacol. 2003;476:107–113. doi: 10.1016/s0014-2999(03)02117-4. [DOI] [PubMed] [Google Scholar]
- Csont T, Gorbe A, Bereczki E, Szunyog A, Aypar E, Toth ME, et al. Biglycan protects cardiomyocytes against hypoxia/reoxygenation injury: role of nitric oxide. J Mol Cell Cardiol. 2010;48:649–652. doi: 10.1016/j.yjmcc.2010.01.013. [DOI] [PubMed] [Google Scholar]
- Davies IR, Zhang X. Nitric oxide selective electrodes. Methods Enzymol. 2008;436:63–95. doi: 10.1016/S0076-6879(08)36005-4. [DOI] [PubMed] [Google Scholar]
- Diab N, Oni J, Schulte A, Radtke I, Blochl A, Schuhmann W. Pyrrole functionalised metalloporphyrins as electrocatalysts for the oxidation of nitric oxide. Talanta. 2003;61:43–51. doi: 10.1016/S0039-9140(03)00358-8. [DOI] [PubMed] [Google Scholar]
- Doctor A, Platt R, Sheram ML, Eischeid A, McMahon T, Maxey T, et al. Hemoglobin conformation couples erythrocyte S-nitrosothiol content to O2 gradients. Proc Natl Acad Sci U S A. 2005;102:5709–5714. doi: 10.1073/pnas.0407490102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eriksson LE. Binding of nitric oxide to intact human erythrocytes as monitored by electron paramagnetic resonance. Biochem Biophys Res Commun. 1994;203:176–181. doi: 10.1006/bbrc.1994.2165. [DOI] [PubMed] [Google Scholar]
- Evgenov OV, Ichinose F, Evgenov NV, Gnoth MJ, Falkowski GE, Chang Y, et al. Soluble guanylate cyclase activator reverses acute pulmonary hypertension and augments the pulmonary vasodilator response to inhaled nitric oxide in awake lambs. Circulation. 2004;110:2253–2259. doi: 10.1161/01.CIR.0000144469.01521.8A. [DOI] [PubMed] [Google Scholar]
- Fejer G, Szalay K, Gyory I, Fejes M, Kusz E, Nedieanu S, et al. Adenovirus infection dramatically augments lipopolysaccharide-induced TNF production and sensitizes to lethal shock. J Immunol. 2005;175:1498–1506. doi: 10.4049/jimmunol.175.3.1498. [DOI] [PubMed] [Google Scholar]
- Ferdinandy P, Schulz R. Nitric oxide, superoxide, and peroxynitrite in myocardial ischaemia-reperfusion injury and preconditioning. Br J Pharmacol. 2003;138:532–543. doi: 10.1038/sj.bjp.0705080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferdinandy P, Szilvassy Z, Horvath LI, Csont T, Csonka C, Nagy E, et al. Loss of pacing-induced preconditioning in rat hearts: role of nitric oxide and cholesterol-enriched diet. J Mol Cell Cardiol. 1997;29:3321–3333. doi: 10.1006/jmcc.1997.0557. [DOI] [PubMed] [Google Scholar]
- Forrester MT, Foster MW, Benhar M, Stamler JS. Detection of protein S-nitrosylation with the biotin-switch technique. Free Radic Biol Med. 2009;46:119–126. doi: 10.1016/j.freeradbiomed.2008.09.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forstermann U, Li H. Therapeutic effect of enhancing endothelial nitric oxide synthase (eNOS) expression and preventing eNOS uncoupling. Br J Pharmacol. 2011;164:213–223. doi: 10.1111/j.1476-5381.2010.01196.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forstermann U, Sessa WC. Nitric oxide synthases: regulation and function. Eur Heart J. 2012;33:829–837. doi: 10.1093/eurheartj/ehr304. 837a–837d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Francis SH, Busch JL, Corbin JD, Sibley D. cGMP-dependent protein kinases and cGMP phosphodiesterases in nitric oxide and cGMP action. Pharmacol Rev. 2010;62:525–563. doi: 10.1124/pr.110.002907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujii H, Berliner LJ. Detection of bioradicals by in vivo L-band electron spin resonance spectrometry. NMR Biomed. 2004;17:311–318. doi: 10.1002/nbm.898. [DOI] [PubMed] [Google Scholar]
- Giraldez RR, Zweier JL. An improved assay for measurement of nitric oxide synthase activity in biological tissues. Anal Biochem. 1998;261:29–35. doi: 10.1006/abio.1998.2721. [DOI] [PubMed] [Google Scholar]
- Gould N, Doulias PT, Tenopoulou M, Raju K, Ischiropoulos H. Regulation of protein function and signaling by reversible cysteine S-nitrosylation. J Biol Chem. 2013;288:26473–26479. doi: 10.1074/jbc.R113.460261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gow A, Doctor A, Mannick J, Gaston B. S-nitrosothiol measurements in biological systems. J Chromatogr B Analyt Technol Biomed Life Sci. 2007;851:140–151. doi: 10.1016/j.jchromb.2007.01.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gow AJ. The biological chemistry of nitric oxide as it pertains to the extrapulmonary effects of inhaled nitric oxide. Proc Am Thorac Soc. 2006;3:150–152. doi: 10.1513/pats.200506-058BG. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greenberg SS, Wilcox DE, Rubanyi GM. Endothelium-derived relaxing factor released from canine femoral artery by acetylcholine cannot be identified as free nitric oxide by electron paramagnetic resonance spectroscopy. Circ Res. 1990;67:1446–1452. doi: 10.1161/01.res.67.6.1446. [DOI] [PubMed] [Google Scholar]
- Griveau S, Bedioui F. Overview of significant examples of electrochemical sensor arrays designed for detection of nitric oxide and relevant species in a biological environment. Anal Bioanal Chem. 2013;405:3475–3488. doi: 10.1007/s00216-012-6671-6. [DOI] [PubMed] [Google Scholar]
- Habib S, Ali A. Biochemistry of nitric oxide. Indian J Clin Biochem. 2011;26:3–17. doi: 10.1007/s12291-011-0108-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall DM, Buettner GR. In vivo spin trapping of nitric oxide by heme: electron paramagnetic resonance detection ex vivo. Methods Enzymol. 1996;268:188–192. doi: 10.1016/s0076-6879(96)68020-3. [DOI] [PubMed] [Google Scholar]
- Hawkins CL, Davies MJ. Detection and characterisation of radicals in biological materials using EPR methodology. Biochim Biophys Acta. 2014;1840:708–721. doi: 10.1016/j.bbagen.2013.03.034. [DOI] [PubMed] [Google Scholar]
- Heinrich TA, da Silva RS, Miranda KM, Switzer CH, Wink DA, Fukuto JM. Biological nitric oxide signalling: chemistry and terminology. Br J Pharmacol. 2013;169:1417–1429. doi: 10.1111/bph.12217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henry Y, Ducrocq C, Drapier JC, Servent D, Pellat C, Guissani A. Nitric oxide, a biological effector. Electron paramagnetic resonance detection of nitrosyl–iron–protein complexes in whole cells. Eur Biophys J. 1991;20:1–15. doi: 10.1007/BF00183275. [DOI] [PubMed] [Google Scholar]
- Henry Y, Lepoivre M, Drapier JC, Ducrocq C, Boucher JL, Guissani A. EPR characterization of molecular targets for NO in mammalian cells and organelles. FASEB J. 1993;7:1124–1134. doi: 10.1096/fasebj.7.12.8397130. [DOI] [PubMed] [Google Scholar]
- Hetrick EM, Schoenfisch MH. Analytical chemistry of nitric oxide. Annu Rev Anal Chem (Palo Alto Calif) 2009;2:409–433. doi: 10.1146/annurev-anchem-060908-155146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hevel JM, Marletta MA. Nitric-oxide synthase assays. Methods Enzymol. 1994;233:250–258. doi: 10.1016/s0076-6879(94)33028-x. [DOI] [PubMed] [Google Scholar]
- Hill BG, Dranka BP, Bailey SM, Lancaster JR, Jr, Darley-Usmar VM. What part of NO don't you understand? Some answers to the cardinal questions in nitric oxide biology. J Biol Chem. 2010;285:19699–19704. doi: 10.1074/jbc.R110.101618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hogg N. Detection of nitric oxide by electron paramagnetic resonance spectroscopy. Free Radic Biol Med. 2010;49:122–129. doi: 10.1016/j.freeradbiomed.2010.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong H, Sun J, Cai W. Multimodality imaging of nitric oxide and nitric oxide synthases. Free Radic Biol Med. 2009;47:684–698. doi: 10.1016/j.freeradbiomed.2009.06.011. [DOI] [PubMed] [Google Scholar]
- Hu X, Wang J, Zhu X, Dong D, Zhang X, Wu S, et al. A copper(II) rhodamine complex with a tripodal ligand as a highly selective fluorescence imaging agent for nitric oxide. Chem Commun (Camb) 2011;47:11507–11509. doi: 10.1039/c1cc14032a. [DOI] [PubMed] [Google Scholar]
- Jaffrey SR, Snyder SH. The biotin switch method for the detection of S-nitrosylated proteins. Sci STKE. 2001;2001:pl1. doi: 10.1126/stke.2001.86.pl1. [DOI] [PubMed] [Google Scholar]
- Janzen EG. Spin trapping. Methods Enzymol. 1984;105:188–198. doi: 10.1016/s0076-6879(84)05025-4. [DOI] [PubMed] [Google Scholar]
- Ji XB, Hollocher TC. Mechanism for nitrosation of 2,3-diaminonaphthalene by Escherichia coli: enzymatic production of NO followed by O2-dependent chemical nitrosation. Appl Environ Microbiol. 1988;54:1791–1794. doi: 10.1128/aem.54.7.1791-1794.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang S, Cheng R, Wang X, Xue T, Liu Y, Nel A, et al. Real-time electrical detection of nitric oxide in biological systems with sub-nanomolar sensitivity. Nat Commun. 2013;4:2225. doi: 10.1038/ncomms3225. [DOI] [PubMed] [Google Scholar]
- Katayama Y, Soh N, Maeda M. A new strategy for the design of molecular probes for investigating endogenous nitric oxide using an EPR or fluorescent technique. Chemphyschem. 2001;2:655–661. doi: 10.1002/1439-7641(20011119)2:11<655::AID-CPHC655>3.0.CO;2-S. [DOI] [PubMed] [Google Scholar]
- Khoo JP, Alp NJ, Bendall JK, Kawashima S, Yokoyama M, Zhang YH, et al. EPR quantification of vascular nitric oxide production in genetically modified mouse models. Nitric Oxide. 2004;10:156–161. doi: 10.1016/j.niox.2004.04.003. [DOI] [PubMed] [Google Scholar]
- Kleschyov AL, Wenzel P, Munzel T. Electron paramagnetic resonance (EPR) spin trapping of biological nitric oxide. J Chromatogr B Analyt Technol Biomed Life Sci. 2007;851:12–20. doi: 10.1016/j.jchromb.2006.10.006. [DOI] [PubMed] [Google Scholar]
- Knowles RG, Palacios M, Palmer RM, Moncada S. Formation of nitric oxide from L-arginine in the central nervous system: a transduction mechanism for stimulation of the soluble guanylate cyclase. Proc Natl Acad Sci U S A. 1989;86:5159–5162. doi: 10.1073/pnas.86.13.5159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kojima H, Nakatsubo N, Kikuchi K, Kawahara S, Kirino Y, Nagoshi H, et al. Detection and imaging of nitric oxide with novel fluorescent indicators: diaminofluoresceins. Anal Chem. 1998a;70:2446–2453. doi: 10.1021/ac9801723. [DOI] [PubMed] [Google Scholar]
- Kojima H, Sakurai K, Kikuchi K, Kawahara S, Kirino Y, Nagoshi H, et al. Development of a fluorescent indicator for nitric oxide based on the fluorescein chromophore. Chem Pharm Bull (Tokyo) 1998b;46:373–375. doi: 10.1248/cpb.46.373. [DOI] [PubMed] [Google Scholar]
- Komarov A, Mattson D, Jones MM, Singh PK, Lai CS. In vivo spin trapping of nitric oxide in mice. Biochem Biophys Res Commun. 1993;195:1191–1198. doi: 10.1006/bbrc.1993.2170. [DOI] [PubMed] [Google Scholar]
- Korth H-G, Ingold KU, Sustmann R, de Groot H, Sies H. Tetramethyl-Ortho-quinodimethane, first member of a family of custom-tailored cheletropic spin traps for nitric oxide. Angew Chem Int Ed Engl. 1992;31:891–893. [Google Scholar]
- Li CZ, Alwarappan S, Zhang W, Scafa N, Zhang X. Metallo protoporphyrin functionalized microelectrodes for electrocatalytic sensing of nitric oxide. Am J Biomed Sci. 2009;1:274–282. doi: 10.5099/aj090300274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim MH, Xu D, Lippard SJ. Visualization of nitric oxide in living cells by a copper-based fluorescent probe. Nat Chem Biol. 2006;2:375–380. doi: 10.1038/nchembio794. [DOI] [PubMed] [Google Scholar]
- Lima B, Forrester MT, Hess DT, Stamler JS. S-nitrosylation in cardiovascular signaling. Circ Res. 2010;106:633–646. doi: 10.1161/CIRCRESAHA.109.207381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lorenzetti R, Lilla S, Donato JL, de Nucci G. Simultaneous quantification of GMP, AMP, cyclic GMP and cyclic AMP by liquid chromatography coupled to tandem mass spectrometry. J Chromatogr B Analyt Technol Biomed Life Sci. 2007;859:37–41. doi: 10.1016/j.jchromb.2007.09.008. [DOI] [PubMed] [Google Scholar]
- Lucas KA, Pitari GM, Kazerounian S, Ruiz-Stewart I, Park J, Schulz S, et al. Guanylyl cyclases and signaling by cyclic GMP. Pharmacol Rev. 2000;52:375–414. [PubMed] [Google Scholar]
- Lundberg JO, Weitzberg E, Gladwin MT. The nitrate-nitrite-nitric oxide pathway in physiology and therapeutics. Nat Rev Drug Discov. 2008;7:156–167. doi: 10.1038/nrd2466. [DOI] [PubMed] [Google Scholar]
- Maples KR, Sandstrom T, Su YF, Henderson RF. The nitric oxide/heme protein complex as a biologic marker of exposure to nitrogen dioxide in humans, rats, and in vitro models. Am J Respir Cell Mol Biol. 1991;4:538–543. doi: 10.1165/ajrcmb/4.6.538. [DOI] [PubMed] [Google Scholar]
- Maron BA, Tang SS, Loscalzo J. S-nitrosothiols and the S-nitrosoproteome of the cardiovascular system. Antioxid Redox Signal. 2013;18:270–287. doi: 10.1089/ars.2012.4744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martens-Lobenhoffer J, Dautz C, Bode-Boger SM. Improved method for the determination of cyclic guanosine monophosphate (cGMP) in human plasma by LC-MS/MS. J Chromatogr B Analyt Technol Biomed Life Sci. 2010;878:487–491. doi: 10.1016/j.jchromb.2009.12.009. [DOI] [PubMed] [Google Scholar]
- van Mastbergen J, Jolas T, Allegra L, Page CP. The mechanism of action of doxofylline is unrelated to HDAC inhibition, PDE inhibition or adenosine receptor antagonism. Pulm Pharmacol Ther. 2012;25:55–61. doi: 10.1016/j.pupt.2011.10.007. [DOI] [PubMed] [Google Scholar]
- Mayer B, Schmidt K, Humbert P, Bohme E. Biosynthesis of endothelium-derived relaxing factor: a cytosolic enzyme in porcine aortic endothelial cells Ca2+-dependently converts L-arginine into an activator of soluble guanylyl cyclase. Biochem Biophys Res Commun. 1989;164:678–685. doi: 10.1016/0006-291x(89)91513-1. [DOI] [PubMed] [Google Scholar]
- McQuade LE, Lippard SJ. Fluorescence-based nitric oxide sensing by Cu(II) complexes that can be trapped in living cells. Inorg Chem. 2010a;49:7464–7471. doi: 10.1021/ic100802q. [DOI] [PubMed] [Google Scholar]
- McQuade LE, Lippard SJ. Fluorescent probes to investigate nitric oxide and other reactive nitrogen species in biology (truncated form: fluorescent probes of reactive nitrogen species) Curr Opin Chem Biol. 2010b;14:43–49. doi: 10.1016/j.cbpa.2009.10.004. [DOI] [PubMed] [Google Scholar]
- Misko TP, Schilling RJ, Salvemini D, Moore WM, Currie MG. A fluorometric assay for the measurement of nitrite in biological samples. Anal Biochem. 1993;214:11–16. doi: 10.1006/abio.1993.1449. [DOI] [PubMed] [Google Scholar]
- Moncada S, Palmer RM, Higgs EA. Nitric oxide: physiology, pathophysiology, and pharmacology. Pharmacol Rev. 1991;43:109–142. [PubMed] [Google Scholar]
- Moncada S, Higgs A, Furchgott R. International Union of Pharmacology Nomenclature in Nitric Oxide Research. Pharmacol Rev. 1997;49:137–142. [PubMed] [Google Scholar]
- Moorcroft MJ, Davis J, Compton RG. Detection and determination of nitrate and nitrite: a review. Talanta. 2001;54:785–803. doi: 10.1016/s0039-9140(01)00323-x. [DOI] [PubMed] [Google Scholar]
- Mordvintcev P, Mulsch A, Busse R, Vanin A. On-line detection of nitric oxide formation in liquid aqueous phase by electron paramagnetic resonance spectroscopy. Anal Biochem. 1991;199:142–146. doi: 10.1016/0003-2697(91)90282-x. [DOI] [PubMed] [Google Scholar]
- Mulsch A, Bassenge E, Busse R. Nitric oxide synthesis in endothelial cytosol: evidence for a calcium-dependent and a calcium-independent mechanism. Naunyn Schmiedebergs Arch Pharmacol. 1989;340(6 Pt 2):767–770. doi: 10.1007/BF00169688. [DOI] [PubMed] [Google Scholar]
- Mülsch A, Mordvintcev P, Vanin A. Quantification of nitric oxide in biological samples by electron spin resonance spectroscopy. Neuroprotocols. 1992;1:165–173. [Google Scholar]
- Nagano T. Bioimaging probes for reactive oxygen species and reactive nitrogen species. J Clin Biochem Nutr. 2009;45:111–124. doi: 10.3164/jcbn.R09-66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagano T. Development of fluorescent probes for bioimaging applications. Proc Jpn Acad Ser B Phys Biol Sci. 2010;86:837–847. doi: 10.2183/pjab.86.837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagano T, Yoshimura T. Bioimaging of nitric oxide. Chem Rev. 2002;102:1235–1270. doi: 10.1021/cr010152s. [DOI] [PubMed] [Google Scholar]
- Nedeianu S, Pali T. EPR spectroscopy of common nitric oxide – spin trap complexes. Cell Mol Biol Lett. 2002;7:142–143. [PubMed] [Google Scholar]
- Nedeianu S, Pali T, Marsh D. Membrane penetration of nitric oxide and its donor S-nitroso-N-acetylpenicillamine: a spin-label electron paramagnetic resonance spectroscopic study. Biochim Biophys Acta. 2004;1661:135–143. doi: 10.1016/j.bbamem.2003.12.008. [DOI] [PubMed] [Google Scholar]
- Pacher P, Beckman JS, Liaudet L. Nitric oxide and peroxynitrite in health and disease. Physiol Rev. 2007;87:315–424. doi: 10.1152/physrev.00029.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palacios M, Knowles RG, Palmer RM, Moncada S. Nitric oxide from L-arginine stimulates the soluble guanylate cyclase in adrenal glands. Biochem Biophys Res Commun. 1989;165:802–809. doi: 10.1016/s0006-291x(89)80037-3. [DOI] [PubMed] [Google Scholar]
- Palmer RM, Moncada S. A novel citrulline-forming enzyme implicated in the formation of nitric oxide by vascular endothelial cells. Biochem Biophys Res Commun. 1989;158:348–352. doi: 10.1016/s0006-291x(89)80219-0. [DOI] [PubMed] [Google Scholar]
- Pautz A, Art J, Hahn S, Nowag S, Voss C, Kleinert H. Regulation of the expression of inducible nitric oxide synthase. Nitric Oxide. 2010;23:75–93. doi: 10.1016/j.niox.2010.04.007. [DOI] [PubMed] [Google Scholar]
- Pawson AJ, Sharman JL, Benson HE, Faccenda E, Alexander SP, Buneman OP, et al. NC-IUPHAR. The IUPHAR/BPS Guide to PHARMACOLOGY: an expert-driven knowledgebase of drug targets and their ligands. Nucl Acids Res. 2014;42(Database Issue):D1098–D1106. doi: 10.1093/nar/gkt1143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piknova B, Gladwin MT, Schechter AN, Hogg N. Electron paramagnetic resonance analysis of nitrosylhemoglobin in humans during NO inhalation. J Biol Chem. 2005;280:40583–40588. doi: 10.1074/jbc.M506292200. [DOI] [PubMed] [Google Scholar]
- Qian J, Fulton D. Post-translational regulation of endothelial nitric oxide synthase in vascular endothelium. Front Physiol. 2013;4:347. doi: 10.3389/fphys.2013.00347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quinton D, Girard A, Thi Kim LT, Raimbault V, Griscom L, Razan F, et al. On-chip multi-electrochemical sensor array platform for simultaneous screening of nitric oxide and peroxynitrite. Lab Chip. 2011;11:1342–1350. doi: 10.1039/c0lc00585a. [DOI] [PubMed] [Google Scholar]
- Radak Z, Pucsok J, Mecseki S, Csont T, Ferdinandy P. Muscle soreness-induced reduction in force generation is accompanied by increased nitric oxide content and DNA damage in human skeletal muscle. Free Radic Biol Med. 1999;26:1059–1063. doi: 10.1016/s0891-5849(98)00309-8. [DOI] [PubMed] [Google Scholar]
- Radi R. Peroxynitrite, a stealthy biological oxidant. J Biol Chem. 2013;288:26464–26472. doi: 10.1074/jbc.R113.472936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rassaf T, Ferdinandy P, Schulz R. Nitrite in organ protection. Br J Pharmacol. 2014;171:1–11. doi: 10.1111/bph.12291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez-Rodriguez R, Simonsen U. Measurement of nitric oxide and reactive oxygen species in the vascular wall. Curr Anal Chem. 2012;8:485–494. [Google Scholar]
- Roychowdhury S, Luthe A, Keilhoff G, Wolf G, Horn TF. Oxidative stress in glial cultures: detection by DAF-2 fluorescence used as a tool to measure peroxynitrite rather than nitric oxide. Glia. 2002;38:103–114. doi: 10.1002/glia.10024. [DOI] [PubMed] [Google Scholar]
- Rumer S, Krischke M, Fekete A, Mueller MJ, Kaiser WM. DAF-fluorescence without NO: elicitor treated tobacco cells produce fluorescing DAF-derivatives not related to DAF-2 triazol. Nitric Oxide. 2012;27:123–135. doi: 10.1016/j.niox.2012.05.007. [DOI] [PubMed] [Google Scholar]
- Sanches R. Dehydration effects on the heme environment of nitric oxide hemoglobin. Biochim Biophys Acta. 1988;955:310–314. doi: 10.1016/0167-4838(88)90209-9. [DOI] [PubMed] [Google Scholar]
- Santos RM, Rodrigues MS, Laranjinha J, Barbosa RM. Biomimetic sensor based on hemin/carbon nanotubes/chitosan modified microelectrode for nitric oxide measurement in the brain. Biosens Bioelectron. 2013;44:152–159. doi: 10.1016/j.bios.2013.01.015. [DOI] [PubMed] [Google Scholar]
- Saville B. A scheme for the colorimetric determination of microgram amount of thiols. Analyst. 1958;83:670–672. [Google Scholar]
- Schulz R, Kelm M, Heusch G. Nitric oxide in myocardial ischemia/reperfusion injury. Cardiovasc Res. 2004;61:402–413. doi: 10.1016/j.cardiores.2003.09.019. [DOI] [PubMed] [Google Scholar]
- Serpe MJ, Zhang X. The principles, development and application of microelectrodes for the in vivo determination of nitric oxide. In: Michael AC, Borland LM, editors. Electrochemical Methods for Neuroscience. edn. Boca Raton, FL: CRC Press; 2007. Chapter 21. [PubMed] [Google Scholar]
- Shin S, Fung HL. Evaluation of an LC-MS/MS assay for 15N-nitrite for cellular studies of L-arginine action. J Pharm Biomed Anal. 2011;56:1127–1131. doi: 10.1016/j.jpba.2011.08.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stamler JS, Hess DT. Nascent nitrosylases. Nat Cell Biol. 2010;12:1024–1026. doi: 10.1038/ncb1110-1024. [DOI] [PubMed] [Google Scholar]
- Steiner AL, Parker CW, Kipnis DM. Radioimmunoassay for cyclic nucleotides. I. Preparation of antibodies and iodinated cyclic nucleotides. J Biol Chem. 1972;247:1106–1113. [PubMed] [Google Scholar]
- Stuehr DJ, Kwon NS, Gross SS, Thiel BA, Levi R, Nathan CF. Synthesis of nitrogen oxides from L-arginine by macrophage cytosol: requirement for inducible and constitutive components. Biochem Biophys Res Commun. 1989;161:420–426. doi: 10.1016/0006-291x(89)92615-6. [DOI] [PubMed] [Google Scholar]
- Sun J, Zhang XJ, Broderick M, Fein H. Measurement of nitric oxide production in biological systems by using Griess reaction assay. Sensors. 2003;3:276–284. [Google Scholar]
- Suzuki Y, Fujii S, Tominaga T, Yoshimoto T, Yoshimura T, Kamada H. The origin of an EPR signal observed in dithiocarbamate-loaded tissues. Copper(II)-dithiocarbamate complexes account for the narrow hyperfine lines. Biochim Biophys Acta. 1997;1335:242–245. doi: 10.1016/s0304-4165(97)00027-5. [DOI] [PubMed] [Google Scholar]
- Tennyson AG, Lippard SJ. Generation, translocation, and action of nitric oxide in living systems. Chem Biol. 2011;18:1211–1220. doi: 10.1016/j.chembiol.2011.09.009. [DOI] [PubMed] [Google Scholar]
- Toledo JC, Jr, Augusto O. Connecting the chemical and biological properties of nitric oxide. Chem Res Toxicol. 2012;25:975–989. doi: 10.1021/tx300042g. [DOI] [PubMed] [Google Scholar]
- Tosaki A, Pali T, Droy-Lefaix MT. Effects of Ginkgo biloba extract and preconditioning on the diabetic rat myocardium. Diabetologia. 1996;39:1255–1262. doi: 10.1007/s001250050567. [DOI] [PubMed] [Google Scholar]
- Tsikas D. A critical review and discussion of analytical methods in the L-arginine/nitric oxide area of basic and clinical research. Anal Biochem. 2008;379:139–163. doi: 10.1016/j.ab.2008.04.018. [DOI] [PubMed] [Google Scholar]
- Vanin A, Poltorakov A. NO spin trapping in biological systems. Front Biosci (Landmark Ed) 2009;14:4427–4435. doi: 10.2741/3538. [DOI] [PubMed] [Google Scholar]
- Vanin AF, Liu X, Samouilov A, Stukan RA, Zweier JL. Redox properties of iron-dithiocarbamates and their nitrosyl derivatives: implications for their use as traps of nitric oxide in biological systems. Biochim Biophys Acta. 2000;1474:365–377. doi: 10.1016/s0304-4165(00)00033-7. [DOI] [PubMed] [Google Scholar]
- Villamena FA, Zweier JL. Detection of reactive oxygen and nitrogen species by EPR spin trapping. Antioxid Redox Signal. 2004;6:619–629. doi: 10.1089/152308604773934387. [DOI] [PubMed] [Google Scholar]
- Villanueva C, Giulivi C. Subcellular and cellular locations of nitric oxide synthase isoforms as determinants of health and disease. Free Radic Biol Med. 2010;49:307–316. doi: 10.1016/j.freeradbiomed.2010.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Hu S. Nitric oxide sensor based on poly (p-phenylenevinylene) derivative modified electrode and its application in rat heart. Bioelectrochemistry. 2009;74:301–305. doi: 10.1016/j.bioelechem.2008.11.002. [DOI] [PubMed] [Google Scholar]
- Wardman P. Fluorescent and luminescent probes for measurement of oxidative and nitrosative species in cells and tissues: progress, pitfalls, and prospects. Free Radic Biol Med. 2007;43:995–1022. doi: 10.1016/j.freeradbiomed.2007.06.026. [DOI] [PubMed] [Google Scholar]
- Weil JA, Bolton JR. Electron Paramagnetic Resonance: Elementary Theory and Practical Applications. 2nd edn. Hoboken, NJ: John Wiley & Sons; 2007. [Google Scholar]
- Woolley JF, Stanicka J, Cotter TG. Recent advances in reactive oxygen species measurement in biological systems. Trends Biochem Sci. 2013;38:556–565. doi: 10.1016/j.tibs.2013.08.009. [DOI] [PubMed] [Google Scholar]
- Wunder F, Stasch JP, Hutter J, Alonso-Alija C, Huser J, Lohrmann E. A cell-based cGMP assay useful for ultra-high-throughput screening and identification of modulators of the nitric oxide/cGMP pathway. Anal Biochem. 2005;339:104–112. doi: 10.1016/j.ab.2004.12.025. [DOI] [PubMed] [Google Scholar]
- Wunder F, Buehler G, Huser J, Mundt S, Bechem M, Kalthof B. A cell-based nitric oxide reporter assay useful for the identification and characterization of modulators of the nitric oxide/guanosine 3′,5′-cyclic monophosphate pathway. Anal Biochem. 2007;363:219–227. doi: 10.1016/j.ab.2007.02.001. [DOI] [PubMed] [Google Scholar]
- Yap CM, Xu GQ, Ang SG. Amperometric nitric oxide sensor based on nanoporous platinum phthalocyanine modified electrodes. Anal Chem. 2013;85:107–113. doi: 10.1021/ac302081h. [DOI] [PubMed] [Google Scholar]
- Zaobornyj T, Ghafourifar P. Strategic localization of heart mitochondrial NOS: a review of the evidence. Am J Physiol Heart Circ Physiol. 2012;303:H1283–H1293. doi: 10.1152/ajpheart.00674.2011. [DOI] [PubMed] [Google Scholar]
- Zhang HX, Chen JB, Guo XF, Wang H, Zhang HS. Highly sensitive low-background fluorescent probes for imaging of nitric oxide in cells and tissues. Anal Chem. 2014;86:3115–3123. doi: 10.1021/ac4041718. [DOI] [PubMed] [Google Scholar]
- Zhou L, Zhu DY. Neuronal nitric oxide synthase: structure, subcellular localization, regulation, and clinical implications. Nitric Oxide. 2009;20:223–230. doi: 10.1016/j.niox.2009.03.001. [DOI] [PubMed] [Google Scholar]
- Zweier JL, Chen CA, Druhan LJ. S-glutathionylation reshapes our understanding of endothelial nitric oxide synthase uncoupling and nitric oxide/reactive oxygen species-mediated signaling. Antioxid Redox Signal. 2011;14:1769–1775. doi: 10.1089/ars.2011.3904. [DOI] [PMC free article] [PubMed] [Google Scholar]