

Abstract
Objective
The diagnosis of multiple sclerosis (MS) at disease onset is sometimes masqueraded by other diagnostic options resembling MS clinically or radiologically (NonMS). In the present study we utilized findings of large-scale Genome-Wide Association Studies (GWAS) to develop a blood gene expression-based classification tool to assist in diagnosis during the first demyelinating event.
Methods
We have merged knowledge of 110 MS susceptibility genes gained from MS GWAS studies together with our experimental results of differential blood gene expression profiling between 80 MS and 31 NonMS patients. Multiple classification algorithms were applied to this cohort to construct a diagnostic classifier that correctly distinguished between MS and NonMS patients. Accuracy of the classifier was tested on an additional independent group of 146 patients including 121 MS and 25 NonMS patients.
Results
We have constructed a 42 gene-transcript expression-based MS diagnostic classifier. The overall accuracy of the classifier, as tested on an independent patient population consisting of diagnostically challenging cases including NonMS patients with positive MRI findings, achieved a correct classification rate of 76.0 ± 3.5%.
Interpretation
The presented diagnostic classification tool complements the existing diagnostic McDonald criteria by assisting in the accurate exclusion of other neurological diseases at presentation of the first demyelinating event suggestive of MS.
Introduction
The diagnosis of multiple sclerosis (MS) is a challenging procedure and is currently based on the 2010 McDonald criteria requiring evidence of disease dissemination in time (DIT), dissemination in space (DIS) and “no better explanation” for symptoms.1 At first demyelinating symptomatology, clinical, imaging and laboratory findings may be similar between MS and other diseases that mimic MS (NonMS) including infectious, neoplastic, metabolic, vascular or idiopathic inflammatory demyelinating diseases.2–4 Therefore, a need exists to find accurate biomarkers and to develop a tool that can assist in the exclusion of NonMS diseases.
High throughput technology measuring simultaneous expression of thousands of genes or proteins has been shown to be potentially useful for diagnosis of MS.5–7 However, it is challenging to find biomarkers that not only have immunological and inflammatory properties, but are also specifically associated with MS pathogenesis. Large-scale genome-wide association studies (GWAS) examining single-nucleotide polymorphisms (SNP's) encompassing thousands of patients have been conducted and have established 110 MS-related SNP's,8–11 that contribute to MS susceptibility and pathophysiology. It has been previously demonstrated that SNPs could affect expression of nearby genes.12 Therefore, we have merged our findings of gene expression of MS susceptible loci genes with other highly differentially expressed genes (DEGs) between MS and NonMS patients to develop a blood gene expression-based diagnostic classification tool. This multi-gene classifier was tested on an independent cohort of patients during the first neurological event suggestive of MS demonstrating high accuracy and therefore could improve the diagnostic process in challenging cases.
Methods
Patients
We analyzed blood samples of patients selected according to the following criteria: (1) Age 18–60 years; (2) Free of steroid treatment for at least 30 days; (3) Patients with the first clinical demyelinating event suggestive of MS that upon presentation fulfilled DIT or DIS according to 2010 McDonald's criteria. After follow-up period of 5 years these patients were further divided into two groups: (a) patients that converted to relapsing remitting MS (RRMS) (Clinically Isolated Syndromes, CIS group); (b) patients that did not convert to MS and after extensive work-up a better explanation for the clinical and imaging findings was established (NonMS group). (4) Patients that at the time of sampling fulfilled McDonald 2010 criteria for RRMS and served for classifier training and testing.
Standard protocol approvals and patients consent
The study was approved by Sheba Medical Center Institutional Review and Ethical Board and all patients gave written informed consent. Demographic and clinical data were retrieved from Sheba Multiple Sclerosis Center computerized database.
Study design
Patient data set (n = 257) was divided into two subgroups: training set (n = 111) and test set (n = 146). The training set was used to construct blood gene expression-based classifier, while the independent test set allowed for subsequent testing of the classifier performance. Multiple classification algorithms implemented in Partek software (St. Louis, Missouri, USA) were applied to the training set subgroups to construct classifiers that can correctly distinguish between MS and NonMS patients. The classifier with the best performance on the training set was validated on the test set.
An additional internal cross-validation was performed by testing the classifier consistency on a subgroup of patients that were resampled within 0.8 ± 0.2 years from the initial blood sampling. The study flow chart is demonstrated in Figure1.
Figure 1.
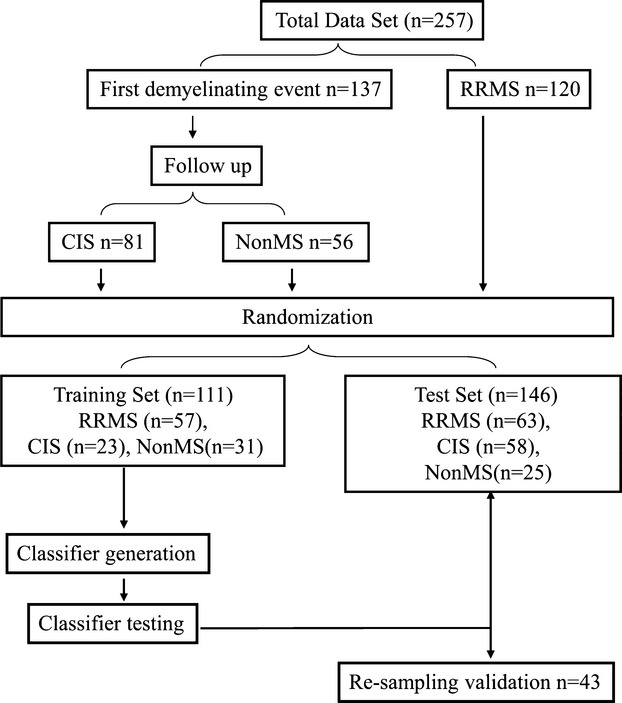
Flowchart of study design. Samples from 257 patients including 137 patients at first demyelinating event and 120 RRMS patients were subjected to gene expression microarray analysis and randomly divided into a training set (n = 111) and test set (147). training set was used for diagnostic classifier generation and then classifier performance was validated on independent test set. Resampling (n = 43) was done to demonstrate classifier consistency.
Microarray preparation
Total RNA from patients frozen peripheral blood mononuclear cells (PBMC) was extracted using Trizol (Invitrogen, Carlsbad, California, USA) and Phase-Look-Gel columns (Eppendorf, Hamburg, Germany) including a DNase digestion step. RNA quality was determined by BioRad Experion (Hercules, California, USA) automatic electrophoresis station. cDNA was synthesized from 3 μg total RNA using the One-Cycle cDNA Synthesis Kit, and in vitro transcription was performed with the GeneChip IVT Labeling Kit (Affymetrix, Inc., Santa Clara, CA). The biotin-labeled IVT-RNA was hybridized to HGU133A-2 arrays containing ∽22,000 gene transcripts corresponding to 14,500 well-annotated human genes, washed in a GeneChip Fluidics Station 450 and scanned on GeneArray-TM scanner (G2500A; Hewlett Packard Palo Alto, California, USA) according to standard Affymetrix Inc. protocol.
Data pretreatment, normalization, and statistical analysis
Following scanning of the arrays, the microarray raw data were initially normalized by R Bioconductor Packages13 as follows: (1) all arrays were normalized serially using a single sample microarray normalization approach designed for personal medicine workflows (SCAN Normalization)14; (2) an empirical bayes approach was used to address batch effect in the data as implemented in Combat SVA package.15,16
Partek Genomics Software (www.partek.com) was used for data analysis. All genes related to clinical and demographical confounders such as age, gender, immunomodulatory treatment with P < 0.01 were excluded from further analysis. Next, to select candidate biomarkers for the classifier we performed comparison gene expression analysis between MS and NonMS patients in the training set. Genes selected were comprised of: (1) DEGs within the entire transcriptome with P value <0.05 after Bonferroni correction for multiple comparisons and (2) DEGs with P value<0.05 from 207 gene transcripts corresponding to 110 loci found to be associated with MS in published GWAS (GWAS DEG).9,10,17,18 These genes were then used to build a support vector machine (SVM) classifier. The SVM was configured as cost based, with costs varied from 1 to 1001 in intervals of 100. The tolerance (termination criterion) was set at 0.001. The kernel for the SVM was a polynomial function, with gamma equal to the inverse of the number of evaluated probes. The optimal SVM was derived by shrinking centroids and 10-fold cross-validation. Principal Component Analysis (PCA) was used for visualization of results.
Functional analysis of genes included in diagnostic classifier
Biological functional analysis of the classifier genes was performed by Ingenuity Pathway Analysis (IPA) software (www.ingenuity.com) that links gene products with biological processes, molecular function and cellular components. Right-tailed Fisher's exact test was used to calculate a P value determining the probability that each biological function assigned to gene data set is not due to random chance. The P values obtained from Fisher's analysis were applied for Benjamini-Hochberg False Discovery Rate (FDR) multiple testing corrections to keep the overall error rate at P < 0.05.
Results
Patients
Blood samples were obtained from 137 patients that presented with symptomatology suggestive of MS at disease onset; 81 were diagnosed with CIS and 56 were diagnosed as NonMS patients. The CIS group included 50 females and 31 males, mean ± SE age 31.9 ± 1.2 years, the NonMS group included 43 females and 13 males, mean ± SE age 41.9 ± 1.5 years.
The group of 120 RRMS patients that served for the classifier training and testing included 80 females and 40 males, mean ± SE age 37.3 ± 1.0 years, disease duration 5.6 ± 0.5 years and Expanded Disability Status Scale (EDSS) 2.1 ± 0.1.
Demographic and clinical variables of the study patients in relation to experimental training set and test set groups are presented in Table1.
Table 1.
Demographical and clinical characteristics of patients.
| Group | N | Age average | F (M) | EDSS | |
|---|---|---|---|---|---|
| Training set | CIS | 23 | 31.6 ± 1.4* | 16 (7) | 1.4 ± 0.3 |
| RRMS | 57 | 36.0 ± 1.4* | 36 (12) | 2.1 ± 0.2 | |
| NonMS | 31 | 41.6 ± 2.3 | 24 (7) | NR | |
| Total | 111 | 36.6 ± 0.1 | 76 (35) | NR | |
| Test set | CIS | 58 | 32.2 ± 1.4* | 23 (35) | 1.4 ± 0.1 |
| RRMS | 63 | 38.4 ± 1.4* | 43 (20) | 2.2 ± 0.2 | |
| NonMS | 25 | 42.3 ± 1.7 | 19 (6) | NR | |
| Total | 146 | 36.6 ± 0.9 | 97 (49) | NR |
RRMS, relapsing remitting multiple sclerosis; NR, not relevant.
P < 0.05 as compare to NonMS group.
MS diagnostic classifier generation
The diagnostic classifier was generated based on the training set of samples from 111 patients including 80 MS and 31 NonMS. Gene expression analysis performed on these groups identified 29 DEG's with P < 0.05 after Bonferroni correction and 49 GWAS DEGs, resulting in 78 candidate genes for generating the diagnostic classifier. These 78 candidate biomarkers were used to construct and optimize classification accuracy of various SVMs. The most accurate SVM classifier used expression intensity of 42 gene – transcripts (Table S1) to correctly classify patients in the training set with a total correct rate of 94.6%, having a sensitivity of 95.0 ± 2.4% and a specificity of 94.0 ± 4.4%. Within the 42 transcripts chosen for the classifier, 18 were based on SNP's reported in the GWAS studies. PCA of the training set based on these 42 transcripts is shown in Figure2.
Figure 2.
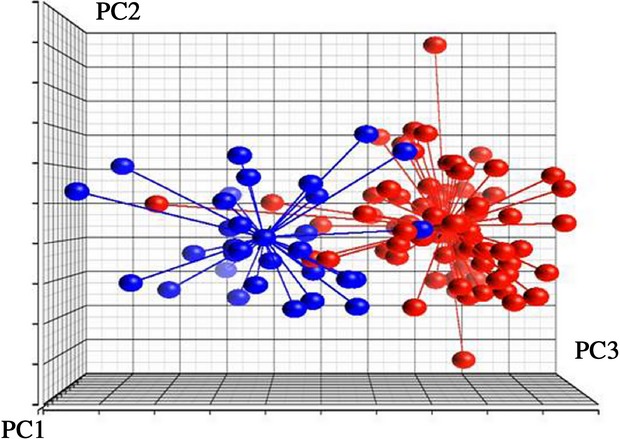
Principal component analysis (PCA) based on 42 gene-transcripts of the diagnostic classifier. This difference between MS and NonMS patients from training set is presented. Each dot represents patient sample principal components derived from expression of 42 diagnostic classifier gene-transcripts. The distance between any pair of points is related to the similarity between the two observations in high-dimensional (3D) space. Blue dots represent NonMS patients, Red dots represent MS patients.
Verification: classifier testing on the independent test set
The MS diagnostic classifier performance was verified on an independent test set, consisting of samples obtained from CIS (n = 58), RRMS (n = 63) and NonMS (n = 25) patients. First, the ability of the diagnostic classifier was tested in the group of RRMS patients and NonMS patients, demonstrating overall correct classification rate of 77.3 ± 4.5%, sensitivity of 78.0 ± 5.2% and specificity 76.0 ± 8.5% for the diagnosis of MS patients. Although RRMS patients already have an established diagnosis, this step was done to prove classifier ability to identify MS specific expression signature. Next, to assess clinical applicability, the classifier was applied to diagnostically relevant cohort of CIS and NonMS patients at the time of the first demyelinating event (n = 83). In this cohort the classifier demonstrated an overall accuracy of 74.7 ± 4.8%, sensitivity 74.0 ± 5.7%, and specificity 76.0 ± 8.5% for the diagnosis of MS. The overall accuracy of the classifier on the entire test set of independent patients (n = 146) was 76.0 ± 3.5%, Table2.
Table 2.
Summary of MS diagnostic classifier performance.
| Groups compared | Total accuracy | Sensitivity | Specificity |
|---|---|---|---|
| Test set CIS (n = 58) vs. NonMS (n = 25) | 74.7 ± 4.8% | 74.0 ± 5.7% | 76.0 ± 8.5% |
| Test set CIS EDSS > 0 (n = 45) vs. NonMS (n = 25) | 77.1 ± 5.0% | 78.0 ± 6.2% | 76.0 ± 8.5% |
| Test set RRMS (n = 63) vs. NonMS (n = 25) | 77.3 ± 4.5% | 78.0 ± 5.2% | 76.0 ± 8.5% |
| Test set all MS (n = 121) vs. NonMS (n = 25) | 76.0 ± 3.5% | 76.0 ± 3.9% | 76.0 ± 8.5% |
| Test set all MS EDSS > 0 (n = 103) vs. NonMS (n = 25) | 78.9 ± 3.6% | 80.0 ± 4.0% | 76.0 ± 8.5% |
RRMS, relapsing remitting multiple sclerosis.
Further analysis of classifier performance showed dependence of the classification accuracy on the initial clinical disability score of patients. Thus, including only patients with abnormal neurological findings (EDSS > 0) at the time of sampling, improved the classifier sensitivity to 78.0 ± 5.2% in the CIS patient group, and to 80.4 ± 4.0% overall (Table2).
An additional internal cross-validation test of the classifier validity was done by analysis of repeated samples. Consistency of the classifier to reproduce the same classification was tested in 43 patient samples retaken after an average time of 0.8 ± 0.2 years from the initial sampling. Twenty samples were retested from the test set group, and 23 samples were retested from the training set group. Consistency of the results was overall 81.4%. In the test set the consistency of results was 85.0% and in the training set the consistency was 78.0%.
As could be seen from Table1, in the training and test set the age of MS and NonMS patients was significantly different. To analyze the effect of age on classifier performance we examined the 42 gene-transcripts classifier performance in age-matched subgroups. This analysis demonstrated that, overall classifier ability to diagnose MS and NonMS patients was not affected by age. This is shown by the similar range of correct classification of 81.4 ± 7.5%, 72.2 ± 7.5%, 72.7 ± 6.7%, and 78.4 ± 6.7% in the respective age subgroups <25, 25–35, 35–45, and >45 years.
To assess the predictive contribution added by GWAS DEGs to DEG's we constructed two additional classifiers, the first based solely on GWAS DEGs genes and another based only on DEG's. Both classifiers performed inferiorly to the 42 gene-transcript classifier, with the GWAS DEGs classifier achieving an accuracy rate of 59.0 ± 4.6% with sensitivity 55.0 ± 5.2% and specificity 72.0 ± 9.0% and the only DEG's classifier demonstrating an accuracy of 76.0 ± 3.9% with sensitivity of 83.0 ± 3.9% but low level of specificity 53.0 ± 10.0%.
Functional analysis of diagnostic classifier genes
Functional analysis of the classifier genes showed enrichment of genes associated with different mechanisms of cell movement and migration (P value 3.21E-03 to 1.54E-05), immune cell trafficking including lymphocytes migration (P = 1.54E-05), cell movement of T lymphocytes (P = 6.79E-05), transmigration of T lymphocytes (P = 1.97E-04) and adhesion of immune cells (P = 1.49E-03), Table S2. These findings were further consistent with gene expression network analysis in which the classifier genes were shown to organize in four functional networks, including cell-mediated immune response and immune cell trafficking, similarly to the results of the enrichment analysis, Table S3. Additionally, the classifier genes were found to be upstream regulators of other molecules, including CXCR4 and JAK2 regulating CDH1. Furthermore, these regulators were related not only to downstream molecules, but they also could be interconnected between themselves, Figure3.
Figure 3.
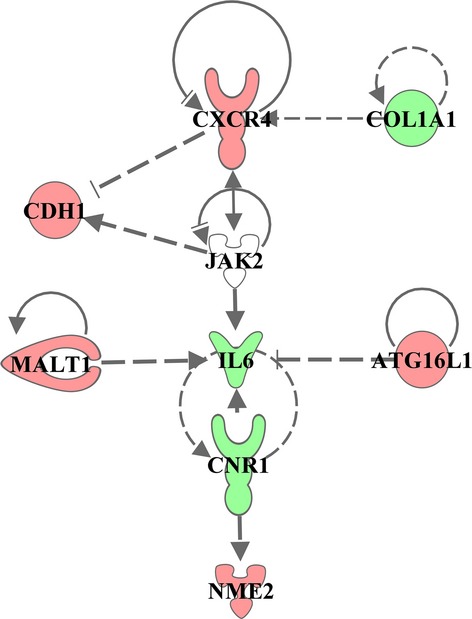
Functional regulatory network of classifier genes. Classifier gene network reconstructed based on literature-known relationships according to IPA software database. Each node in the regulation tree represents a regulating gene, arrows indicate literature confirmed regulatory interactions. Over-expressed genes are depicted in red, down-expressed in green.
Discussion
In this study, we present an innovative approach whereby we developed a blood gene expression-based diagnostic tool proved to be useful for excluding MS mimicking diseases in the early stage of the diagnostic process. For this purpose we have merged knowledge of MS susceptibility genes gained from MS GWAS studies together with our experimental results of differential gene expression profiling between MS and NonMS patients. This multimodal approach allowed us to utilize data acquired from very large scale studies to focus on a narrow list of candidate biomarkers that contribute to the pathological mechanisms operating in MS. While certain genetic associations have been consistently shown to contribute to the etiology of MS,11 the link between genome variability and disease phenotypes still remain poorly understood. To the best of our knowledge our study is the first to demonstrate the association between genomic variability and differential gene expression phenotypes of MS and other neurological diseases that mimic MS. SNP's may influence gene expression in several ways, including modulating transcription factor-binding sites, changing amino sequence of stop codons, affecting splicing patterns, altering proteins by changing single amino acids and modulating micro-RNA-binding sites activity.12,19 Although examining the specific mechanism by which each SNP affects its corresponding gene expression is beyond the scope of this study, overall we have shown significant differential expression of 49 MS susceptible loci associated transcripts, from which 18 were included in our diagnostic classifier.
Currently, the McDonald criteria used for MS diagnosis have specificity over 90% with sensitivity of 77% and accuracy of 86%.20–23 Notably, these high accuracy levels were achieved in multicenter trials conducted in tertiary MS centers, after exclusion of patients with other neurological diseases.1,22,24 Therefore, the relevance of the high accuracy of McDonald criteria has been questioned with regard to everyday clinical practice in which NonMS patients are not excluded prior to application of the criteria.22 A study examining the diagnostic performance of DIS criteria in such a cohort of difficult cases mimicking MS, showed that the sensitivity of the diagnosis decreased to 64%.22
Our diagnostic classifier was able to successfully discriminate between CIS converting to MS and NonMS patients during the early disease stage with sensitivity of 74.0% and specificity of 76.0%, therefore having potential use for clinical practice. This diagnostic classifier may assist to reach the correct diagnosis in patients with suspected MS at an early stage. Importantly, the accuracy rate of the classifier was tested on population that consisted of diagnostically challenging cases of NonMS patients having positive findings on MRI and fulfilling McDonald criteria for DIT or DIS.
Furthermore, the EDSS-dependent improvement of the classifier performance to reach a sensitivity of 78.0%, suggests that the expression of genes involved correlate with patients’ neurological findings. This suggests that genes included in the classifier represent disease activity profile rather than the constitutive gene expression profile of MS disease as we have previously reported.25,26
Functional enrichment analysis of the genes included in the classifier identified genes and functions with significant role in MS pathophysiology. The most significantly enriched functions are related to immune cell trafficking and adhesion. Specifically, the regulatory network including CXCR4, a master regulator having a known role as a stimulator of T- and B-cell proliferation and migration through the endothelium.27–30 CXCR4 connects to the downstream CD6 gene encoding the SRCR domain and binding site for activated cell adhesion molecules, and further continues the process of T-cell activation. It is of note that both CXCR4 and CD6 are associated with MS susceptibility loci. This process involving immune cells activation, proliferation, and adhesion appears to play a central role in the classifier network. Two additional important genes in the network include MALT1 and NME2. MALT1 is known to be involved in B- and T-cell receptor signaling, CD28 signaling in T Helper Cells and regulation of IL-2 Expression in Activated and Anergic T Lymphocytes.31,32 MALT1 also enhances BCL10-induced activation of NFKB and BCL10 cleavage leading to T-cell antigen receptor-induced integrin adhesion.33 NME2 is known to be involved in targeted activation of cell adhesion sites upon integrin engagement34 and is critical to the potassium channel KCa3.1 stability and the activation of CD4 + T cells.35
Molecular profiling technology, and specifically gene expression for diagnostic or prognostic purposes has been successfully applied in a number of different scenarios including Alzheimer, breast and lung cancer.36–39 These diagnostic assays were developed using established gene expression analysis approaches. The novelty of our study, however, is that we also apply knowledge gained in GWAS studies that although instrumental in understanding underlying genetics and pathogenesis of disease, have been somewhat limited in their clinical applicability in day to day practice.
Some limitations may apply to this work. First, although independent test and training sets were established for classification, all patients included in study were enrolled from a single MS center. Additional verification in a multi-center study design could better establish the clinical applicability of our findings. Second, the sample size was uneven between patient groups, with a relatively smaller sample size for the NonMS cohort. This issue should also be addressed in future studies.
In this study, we have focused on diagnostically challenging patients representing a true diagnostic dilemma, and present a novel MS diagnostic tool that can be used at the early disease stage to assist in accurate diagnostic decision making.
Acknowledgments
This study has not received any private or public funding.
Author Contributions
Michael Gurevich, Ph.D. – contributed to study design, analysis of data, and manuscript drafting and revising. Gadi Miron, M.D. – contributed to study design, analysis of data, and manuscript drafting and revising. Anat Achiron, M.D., Ph.D. – contributed to study design analysis of data and manuscript drafting and revising.
Conflict of Interest
Dr. Achiron, Dr. Gurevich and Dr. Miron has a patent US Prov 61/974,455 filed on 03/04/2014 pending.
Supporting Information
Table S1. Forty-two gene-transcripts of diagnostic classifier.
Table S2. Functional annotations of classifier genes.
Table S3. Focus molecules correspond to the number of genes included in functional network.
References
- Polman CH, Reingold SC, Banwell B, et al. Diagnostic criteria for multiple sclerosis: 2010 revisions to the McDonald criteria. Ann Neurol. 2011;69:292–302. doi: 10.1002/ana.22366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rolak LA, Fleming JO. The differential diagnosis of multiple sclerosis. Neurologist. 2007;13:57–72. doi: 10.1097/01.nrl.0000254705.39956.34. [DOI] [PubMed] [Google Scholar]
- Miller DH, Weinshenker BG, Filippi M, et al. Differential diagnosis of suspected multiple sclerosis: a consensus approach. Mult Scler. 2008;14:1157–1174. doi: 10.1177/1352458508096878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charil A, Yousry TA, Rovaris M, et al. MRI and the diagnosis of multiple sclerosis: expanding the concept of “no better explanation”. Lancet Neurol. 2006;5:841–852. doi: 10.1016/S1474-4422(06)70572-5. [DOI] [PubMed] [Google Scholar]
- Harris VK, Sadiq SA. Disease biomarkers in multiple sclerosis: potential for use in therapeutic decision making. Mol Diagn Ther. 2009;13:225–244. doi: 10.1007/BF03256329. [DOI] [PubMed] [Google Scholar]
- Tossberg JT, Crooke PS, Henderson MA, et al. Gene-expression signatures: biomarkers toward diagnosing multiple sclerosis. Genes Immun. 2012;13:146–154. doi: 10.1038/gene.2011.66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Comabella M, Montalban X. Body fluid biomarkers in multiple sclerosis. Lancet Neurol. 2014;13:113–126. doi: 10.1016/S1474-4422(13)70233-3. [DOI] [PubMed] [Google Scholar]
- Hoppenbrouwers IA, Hintzen RQ. Genetics of multiple sclerosis. Biochim Biophys Acta. 2011;1812:194–201. doi: 10.1016/j.bbadis.2010.09.017. [DOI] [PubMed] [Google Scholar]
- International Multiple Sclerosis Genetics C, Wellcome Trust Case Control C. Sawcer S, et al. Genetic risk and a primary role for cell-mediated immune mechanisms in multiple sclerosis. Nature. 2011;476:214–219. doi: 10.1038/nature10251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- International Multiple Sclerosis Genetics Consortium. Network-based multiple sclerosis pathway analysis with GWAS data from 15,000 cases and 30,000 controls. Am J Hum Genet. 2013;92:854–865. doi: 10.1016/j.ajhg.2013.04.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sawcer S, Franklin RJ, Ban M. Multiple sclerosis genetics. Lancet Neurol. 2014;13:700–709. doi: 10.1016/S1474-4422(14)70041-9. [DOI] [PubMed] [Google Scholar]
- Shastry BS. SNPs: impact on gene function and phenotype. Methods Mol Biol. 2009;578:3–22. doi: 10.1007/978-1-60327-411-1_1. [DOI] [PubMed] [Google Scholar]
- R development Core Team. R: a language and environment for statistical computing. Vienna, Austria: R foundation for Statistical Computing; 2013. [Google Scholar]
- Piccolo SR, Sun Y, Campbell JD, et al. A single-sample microarray normalization method to facilitate personalized-medicine workflows. Genomics. 2012;100:337–344. doi: 10.1016/j.ygeno.2012.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson WE, Li C, Rabinovic A. Adjusting batch effects in microarray expression data using empirical Bayes methods. Biostatistics. 2007;8:118–127. doi: 10.1093/biostatistics/kxj037. [DOI] [PubMed] [Google Scholar]
- Leek JT, Johnson WE, Parker HS, et al. The SVA package for removing batch effects and other unwanted variation in high-throughput experiments. Bioinformatics. 2012;28:882–883. doi: 10.1093/bioinformatics/bts034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arthur AT, Armati PJ, Bye C, et al. Genes implicated in multiple sclerosis pathogenesis from consilience of genotyping and expression profiles in relapse and remission. BMC Med Genet. 2008;9:17. doi: 10.1186/1471-2350-9-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patsopoulos NA, et al. Bayer Pharma MSGWG, Steering Committees of Studies Evaluating I-b. Genome-wide meta-analysis identifies novel multiple sclerosis susceptibility loci. Ann Neurol. 2011;70:897–912. doi: 10.1002/ana.22609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Z, Taylor JA. SNPinfo: integrating GWAS and candidate gene information into functional SNP selection for genetic association studies. Nucleic Acids Res. 2009;37(Web Server issue):W600–W605. doi: 10.1093/nar/gkp290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swanton JK, Rovira A, Tintore M, et al. MRI criteria for multiple sclerosis in patients presenting with clinically isolated syndromes: a multicentre retrospective study. Lancet Neurol. 2007;6:677–686. doi: 10.1016/S1474-4422(07)70176-X. [DOI] [PubMed] [Google Scholar]
- Swanton JK, Fernando K, Dalton CM, et al. Modification of MRI criteria for multiple sclerosis in patients with clinically isolated syndromes. J Neurol Neurosurg Psychiatry. 2006;77:830–833. doi: 10.1136/jnnp.2005.073247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nielsen JM, Uitdehaag BM, Korteweg T, et al. Performance of the Swanton multiple sclerosis criteria for dissemination in space. Mult Scler. 2010;16:985–987. doi: 10.1177/1352458510369244. [DOI] [PubMed] [Google Scholar]
- Gomez-Moreno M, Diaz-Sanchez M, Ramos-Gonzalez A. Application of the 2010 McDonald criteria for the diagnosis of multiple sclerosis in a Spanish cohort of patients with clinically isolated syndromes. Mult Scler. 2012;18:39–44. doi: 10.1177/1352458511417828. [DOI] [PubMed] [Google Scholar]
- Milo R, Miller A. Revised diagnostic criteria of multiple sclerosis. Autoimmun Rev. 2014;13:518–524. doi: 10.1016/j.autrev.2014.01.012. [DOI] [PubMed] [Google Scholar]
- Achiron A, Grotto I, Balicer R, et al. Microarray analysis identifies altered regulation of nuclear receptor family members in the pre-disease state of multiple sclerosis. Neurobiol Dis. 2010;38:201–209. doi: 10.1016/j.nbd.2009.12.029. [DOI] [PubMed] [Google Scholar]
- Achiron A, Feldman A, Gurevich M. Characterization of multiple sclerosis traits: nuclear receptors (NR) impaired apoptosis pathway and the role of 1-alpha 25-dihydroxyvitamin D3. J Neurol Sci. 2011;311:9–14. doi: 10.1016/j.jns.2011.06.038. [DOI] [PubMed] [Google Scholar]
- Banisadr G, Schwartz SR, Podojil JR, et al. Integrin/Chemokine receptor interactions in the pathogenesis of experimental autoimmune encephalomyelitis. J Neuroimmune Pharmacol. 2014;9:438–445. doi: 10.1007/s11481-014-9521-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franitza S, Grabovsky V, Wald O, et al. Differential usage of VLA-4 and CXCR4 by CD3 + CD56 + NKT cells and CD56 + CD16 + NK cells regulates their interaction with endothelial cells. Eur J Immunol. 2004;34:1333–1341. doi: 10.1002/eji.200324718. [DOI] [PubMed] [Google Scholar]
- Ganju RK, Brubaker SA, Meyer J, et al. The alpha-chemokine, stromal cell-derived factor-1alpha, binds to the transmembrane G-protein-coupled CXCR-4 receptor and activates multiple signal transduction pathways. J Biol Chem. 1998;273:23169–23175. doi: 10.1074/jbc.273.36.23169. [DOI] [PubMed] [Google Scholar]
- McCandless EE, Piccio L, Woerner BM, et al. Pathological expression of CXCL12 at the blood-brain barrier correlates with severity of multiple sclerosis. Am J Pathol. 2008;172:799–808. doi: 10.2353/ajpath.2008.070918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berger T, Saunders ME, Mak TW. Dissection of signaling in inflammation: three novel inflammatory regulators. Cold Spring Harb Symp Quant Biol. 2013;78:141–147. doi: 10.1101/sqb.2013.78.020107. [DOI] [PubMed] [Google Scholar]
- Fontan L, Melnick A. Molecular pathways: targeting MALT1 paracaspase activity in lymphoma. Clin Cancer Res. 2013;19:6662–6668. doi: 10.1158/1078-0432.CCR-12-3869. [DOI] [PubMed] [Google Scholar]
- Paul S, Schaefer BC. A new look at T cell receptor signaling to nuclear factor-kappaB. Trends Immunol. 2013;34:269–281. doi: 10.1016/j.it.2013.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fournier HN, Dupe-Manet S, Bouvard D, et al. Integrin cytoplasmic domain-associated protein 1alpha (ICAP-1alpha) interacts directly with the metastasis suppressor nm23-H2, and both proteins are targeted to newly formed cell adhesion sites upon integrin engagement. J Biol Chem. 2002;277:20895–20902. doi: 10.1074/jbc.M200200200. [DOI] [PubMed] [Google Scholar]
- Srivastava S, Li Z, Ko K, et al. Histidine phosphorylation of the potassium channel KCa3.1 by nucleoside diphosphate kinase B is required for activation of KCa3.1 and CD4 T cells. Mol Cell. 2006;24:665–675. doi: 10.1016/j.molcel.2006.11.012. [DOI] [PubMed] [Google Scholar]
- Spira A, Beane JE, Shah V, et al. Airway epithelial gene expression in the diagnostic evaluation of smokers with suspect lung cancer. Nat Med. 2007;13:361–366. doi: 10.1038/nm1556. [DOI] [PubMed] [Google Scholar]
- Gustafson AM, Soldi R, Anderlind C, et al. Airway PI3K pathway activation is an early and reversible event in lung cancer development. Sci Transl Med. 2010;2:26ra5. doi: 10.1126/scitranslmed.3000251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lunnon K, Sattlecker M, Furney SJ, et al. A blood gene expression marker of early Alzheimer's disease. J Alzheimers Dis. 2013;33:737–753. doi: 10.3233/JAD-2012-121363. [DOI] [PubMed] [Google Scholar]
- van de Vijver MJ, He YD, van't Veer LJ, et al. A gene-expression signature as a predictor of survival in breast cancer. N Engl J Med. 2002;347:1999–2009. doi: 10.1056/NEJMoa021967. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Forty-two gene-transcripts of diagnostic classifier.
Table S2. Functional annotations of classifier genes.
Table S3. Focus molecules correspond to the number of genes included in functional network.