

Abstract
Disruption of the functional protein balance in living cells activates protective quality control systems to repair damaged proteins or sequester potentially cytotoxic misfolded proteins into aggregates. The established model based on Saccharomyces cerevisiae indicates that aggregating proteins in the cytosol of eukaryotic cells partition between cytosolic juxtanuclear (JUNQ) and peripheral deposits. Substrate ubiquitination acts as the sorting principle determining JUNQ deposition and subsequent degradation. Here, we show that JUNQ unexpectedly resides inside the nucleus, defining a new intranuclear quality control compartment, INQ, for the deposition of both nuclear and cytosolic misfolded proteins, irrespective of ubiquitination. Deposition of misfolded cytosolic proteins at INQ involves chaperone-assisted nuclear import via nuclear pores. The compartment-specific aggregases, Btn2 (nuclear) and Hsp42 (cytosolic), direct protein deposition to nuclear INQ and cytosolic (CytoQ) sites, respectively. Intriguingly, Btn2 is transiently induced by both protein folding stress and DNA replication stress, with DNA surveillance proteins accumulating at INQ. Our data therefore reveal a bipartite, inter-compartmental protein quality control system linked to DNA surveillance via INQ and Btn2.
Keywords: chaperones, protein aggregation, protein disaggregation, proteostasis, ubiquitin–proteasome system
Introduction
In any organism, physiological health and life span depend critically on maintenance of protein homeostasis (proteostasis) under adverse conditions, such as exposure to heat or intrinsic perturbation of the proteome (Morimoto, 2011). Proteostasis is achieved by an efficient and adaptive protein quality control system that detects non-functional and potentially harmful misfolded proteins and promotes their refolding by chaperones and proteolytic degradation by the ubiquitin–proteasome system (UPS) or autophagy (Chen et al, 2011; Hartl et al, 2011; Rosenbaum & Gardner, 2011). Stress levels exceeding the capacity of this system result in increased protein aggregation, which is associated with aging and pathophysiologies. Aggregation, however, also sequesters out potentially toxic mis-folded proteins, which is cytoprotective and facilitates subsequent quality control activities (Cohen et al, 2006; Douglas et al, 2008; Kaganovich et al, 2008). Asymmetric aggregate segregation at cell division produces rejuvenated cytoplasm devoid of aggregates in daughter cells (Rujano et al, 2006; Nystrom & Liu, 2014).
Although the local concentration of misfolded proteins drives aggregation initially, diffusion within cellular compartments allows aggregate scattering and is observed in the cytosol of eukaryotic cells including Saccharomyces cerevisiae cells under conditional protein folding stress (Specht et al, 2011; Zhou et al, 2011). With persistent folding stress however, protein aggregation exhibits marked spatial organization. Spatially distinct, specific aggregate deposition sites are differentially occupied by misfolded protein species (Tyedmers et al, 2010; Sontag et al, 2014).
Mammalian cells contain deposits of various types, including perinuclear deposits destined for autophagy, termed ‘aggresomes’ and ‘aggresome-like induced structures (ALIS)’ (Johnston et al, 1998; Lelouard et al, 2002; Szeto et al, 2006). These are located at the microtubule-organizing center (MTOC) and form via retrograde transport of misfolded proteins along microtubules mediated by an energy-dependent machinery (Johnston et al, 1998; Garcia-Mata et al, 1999; Kawaguchi et al, 2003).
In contrast, in S. cerevisiae, cytosolic misfolded proteins are sequestered into various deposition sites. Peripheral aggregates (also termed ‘stress foci’ or ‘Q-bodies’) (Kaganovich et al, 2008; Specht et al, 2011; Spokoini et al, 2012; Escusa-Toret et al, 2013) appear scattered throughout the cytosol. Here, we refer to these aggregates as CytoQ, allowing for unification and simplification of the current diverse nomenclature. But distinct and specific aggregation sites are also discernable. One is associated with the nucleus (juxtanuclear quality control compartment, JUNQ), and up to now considered to coincide with an apparent indentation at the surface of the nucleus. Another is found adjacent to the vacuole and contains stably associated misfolded proteins (immobile protein deposit, IPOD) (Kaganovich et al, 2008; Specht et al, 2011; Zhou et al, 2011; Spokoini et al, 2012).
Targeting of cytosolic misfolded proteins to the nuclear-associated site (JUNQ) requires the heat-shock protein Btn2 (Malinovska et al, 2012). The cytosolic targeting function of Btn2 is thought to rely on interaction with the J-domain protein Sis1, an Hsp70 co-chaperone which directly binds substrates. Formation of CytoQs during physiological heat stress, however, requires the small heat-shock protein Hsp42 (Specht et al, 2011) which localizes exclusively to CytoQ, consistent with its specific role in aggregate formation (Specht et al, 2011).
The principles regulating sorting and distribution of aggregation-prone proteins are unclear. The fate of aggregated proteins and the precise roles of Btn2 and Hsp42 also are poorly understood. Ubiquitination was suggested to sort sequestered proteins to proteasomal degradation via the nuclear-associated site previously termed ‘JUNQ’ (Kaganovich et al, 2008). However, the central protein disaggregase in yeast, the Hsp104-Hsp70 bi-chaperone, also associates with this deposit (Specht et al, 2011). This suggests that sequestration of misfolded protein precedes the triage decisions determining proteasomal degradation versus chaperone-assisted refolding. Another unexplored issue is the nature of deposits for misfolding proteins residing in the nucleus and the relationship between cytosolic and putative nuclear deposits.
Here, we show that nuclear deposition of misfolded proteins is integral to cytosolic aggregation function. We define an inter-compartmental organization of aggregated protein deposits, involving nuclear import of misfolded proteins and compartment-specific Hsp42 and Btn2 aggregases, which is linked to cellular DNA surveillance responses.
Results
JUNQ is an intranuclear quality control compartment (INQ)
To identify the deposition site(s) for misfolded nuclear proteins and to relate this site to those for cytosolic proteins, we simultaneously monitored aggregation of two model substrates, cytosolic mCherry-VHL and nuclear GFP-luciferase-DM-NLS, coexpressed in S. cerevisiae wild-type (wt) cells (Fig1A). VHL (von Hippel–Lindau protein) is a heterologous protein that misfolds in the yeast cytosol due to the absence of partner proteins required for stabilization (McClellan et al, 2005). Luciferase-DM-NLS (R188Q/R261Q) is a hyperlabile variant of firefly luciferase that misfolds already at physiological temperatures (Gupta et al, 2011) and is targeted to the nucleus by fusion to the SV40 nuclear localization sequence (NLS). Misfolding and aggregation of both proteins was triggered by mild temperature upshift from 30°C to 37°C with simultaneous inhibition of the proteasome by the addition of MG132. These were the conditions used in the original initial report describing JUNQ and CytoQ in yeast (Kaganovich et al, 2008; Specht et al, 2011).
Figure 1.

- S. cerevisiae wt or hsp42Δ cells expressing mCherry-VHL (red) and GFP-luciferase-DM-NLS (green) were grown at 30°C and shifted to 37°C for 90 min. MG132 was added prior to temperature upshift. Changes in protein localizations were recorded. DNA was stained by DAPI (blue).
- S. cerevisiae wt cells or hsp42Δ cells expressing GFP-VHL (green) were treated as in (A). Nuclear membranes were visualized by Nsp1 immunofluorescence labeling (red).
- S. cerevisiae wt cells or hsp42Δ cells expressing mCherry-VHL (red) and GFP-Nup49 (green) were treated as in (A). Three-dimensional reconstructions of respective cells are given.
- Time-lapse microscopy of wt cells expressing mCherry-VHL (green) and GFP-Nup49 (red) upon temperature upshift from 30°C to 37°C. * indicates collision of peripheral foci leading to fusion, and an arrow indicates apparent encounters of peripheral foci and nuclear INQ, followed by foci separation.
Upon shift to 37°C mCherry-VHL formed one peripheral cytosolic and one apparently juxtanuclear focus in most cells, as reported earlier (Kaganovich et al, 2008; Specht et al, 2011). In contrast, upon temperature upshift, nuclear GFP-luciferase-DM-NLS formed a single inclusion adjacent to the DAPI staining. Strikingly, the juxtanuclear mCherry-VHL foci almost perfectly colocalized with aggregates of GFP-luciferase-DM-NLS. Colocalization between mCherry-VHL and GFP-luciferase-DM-NLS was observed in 92% of wt cells (n = 104), indicating codeposition of cytosolic and nuclear misfolded proteins (Fig1A). In hsp42Δ cells, coexpression of mCherry-VHL and GFP-luciferase-DM-NLS results in exclusively juxtanuclear deposit formation, confirming colocalization. The single mCherry-VHL juxtanuclear focus always colocalized (99% colocalization, n = 100) with the GFP-luciferase-DM-NLS aggregate (Fig1A).
The juxtanuclear deposit reported as JUNQ therefore represents a shared quality control compartment for aggregated misfolded proteins from both cytosol and nucleus. Transport of proteins from one compartment into the other must occur, depending on the precise localization of the deposit, nuclear, or cytosolic. We therefore expressed GFP-VHL in wt and hsp42Δ cells and stained the nuclear envelope for immunofluorescence against the nuclear pore protein Nsp1 (Fig1B). GFP-VHL foci generated upon stress application were always observed within the ring-like Nsp1 signal (97% of GFP-VHL foci were inside the nucleus of hsp42Δ cells, and 3% of foci were at the nuclear envelope, n = 100) providing evidence that JUNQ is located inside the nucleus. In an alternative approach, we coexpressed mCherry-VHL with GFP-Nup49 as label for the nuclear envelope in wt and hsp42Δ cells (Fig1C). Three-dimensional reconstruction of microscopic images confirmed the presence of intra-nuclear mCherry-VHL foci after stress application (Fig1C, panels on the right).
To generalize our findings, we used thermolabile GFP-Ubc9ts as an alternative JUNQ substrate (Kaganovich et al, 2008; Escusa-Toret et al, 2013). Localization of GFP-Ubc9ts foci occurred within the Nsp1 ring in wt and hsp42Δ cells supporting JUNQ localization within the nucleus (Supplementary Fig S1). We also determined the localization of stress-induced endogenous yeast aggregates in the absence of exogenous reporter proteins, through visualization of the aggregate-specific disaggregase Hsp104-GFP expressed from its authentic promoter. One of the several Hsp104-GFP foci in wt cells and the single Hsp104-GFP focus in hsp42Δ cells were present within the nuclear envelope marked by Nsp1 (Supplementary Fig S2). Accordingly, Hsp104-GFP foci adjacent to the DAPI signal did not colocalize with Hsp42 (Supplementary Fig S2). Together, these findings indicate the deposit previously identified as JUNQ is a nuclear deposition site devoid of Hsp42. We therefore redefine this deposit as INQ (intranuclear quality control compartment).
To exclude the possibility that the proteasome inhibition regime used in our experiments changes the aggregation pattern, we shifted the cells from 30°C to 38°C for 30 min in the absence of MG132. This slight increase in heat-shock temperature superseded the requirement for MG132 addition upon upshift to 37°C to induce protein aggregation in most cells. Foci numbers and localizations determined for either GFP-VHL or Hsp104-GFP in wt and hsp42Δ cells were very similar to those in MG132-treated cells. Only the persistence of protein aggregation was increased by MG132 (Supplementary Fig S3). Proteasome inhibition therefore does not alter the formation and sorting of protein aggregates.
In order to determine the relationship between formation of CytoQ and INQ aggregates, we performed time-lapse microscopy. We used wt cells coexpressing mCherry-VHL as misfolding protein and GFP-Nup49, which clearly distinguishes cytosolic from nuclear aggregates. Upon heat shock to 37°C, cytosolic and nuclear mCherry-VHL foci formed with similar kinetics (Fig1D). Collision and fusion of cytosolic mCherry-VHL foci reduced aggregate numbers with time, in agreement with the previously reported reduction in foci numbers upon prolonged stress (Kaganovich et al, 2008; Specht et al, 2011). While apparent encounters of cytosolic aggregates and nuclear INQ foci were occasionally observed, they did not fuse and separated shortly after apparent contact (Fig1D). The observation that INQ and CytoQ form independently from one another and do not converge is entirely congruent with the spatial separation across the nuclear membrane.
Immunoelectron microscopy provides higher resolution evidence for nuclear localization of INQ in wt cells expressing GFP-luciferase-DM-NLS and hsp42Δ cells expressing GFP-VHL (Fig2A and B, Supplementary Fig S4). In wt cells, stress treatment led to the appearance of electron-dense areas located in the cytosol and the nucleus (Fig2A, Supplementary Fig S4). Electron-dense structures were not observed in non-stressed cells, indicating that they represent protein aggregates (Supplementary Fig S4). Nuclear but not cytosolic electron-dense particles were labeled with protein A–gold (PAG) employing a GFP-specific antibody, demonstrating that they represent aggregates containing GFP-luciferase-DM-NLS (FigA and E, Supplementary Fig S4). Cytosolic electron-dense regions visible in the EM represent CytoQs of endogenous yeast proteins in the presence of Hsp42. Indeed, in hsp42Δ cells expressing GFP-VHL, stress-induced electron-dense areas were exclusively observed inside the nucleus after stress application. Electron-dense GFP-VHL aggregates were detected inside a continuous nuclear membrane in multiple serial sections (representing a segment of 0.35-μm thickness) (Supplementary Fig S5). Identity of nuclear GFP-VHL aggregates was confirmed by GFP-specific antibody and labeling with PAG (FigB and E, Supplementary Fig S4).
Figure 2.

- Cryo-sections of (A) S. cerevisiae wt cells expressing GFP-luciferase-DM-NLS, (B) hsp42Δ cells expressing GFP-VHL, and (C, D) wt or hsp42Δ cells expressing Hsp104-GFP. Sections were immunogold-labeled with GFP-specific antibodies. Gold particles are marked (black arrows). Electron-dense regions represent protein aggregates (A). Locations of cytosol (C), nuclear envelope (NE, orange arrows), and nucleus (N) are given. Scale bars, 200 nm.
- Average numbers of gold particles associated with nuclear and cytosolic protein aggregates or distributed throughout the cytosol or nucleus were determined.
To confirm that nuclear INQ forms independently of the misfolded reporters chosen or of proteasome inhibition, we visualized endogenous yeast aggregates in wt and hsp42Δ cells by EM immunostaining of Hsp104-GFP. In heat-shocked (38°C) wt cells expressing Hsp104-GFP, gold-labeled cytosolic and nuclear aggregates were visible (FigC and E). In hsp42Δ cells, Hsp104-GFP-decorated aggregates were only found in the nucleus (Fig2D and E, Supplementary Fig S4). The binding of Hsp104 to nuclear INQ aggregates suggested that disaggregase activity is involved in INQ dissolution. Indeed, inhibiting Hsp104 activity by the addition of low concentrations (3 mM) of guanidinium hydrochloride resulted in stabilization of INQ as revealed by time-lapse microscopy of hsp42Δ cells expressing mCherry-VHL (Supplementary Fig S6). Our findings are consistent with early reports, showing increased levels of nuclear aggregates upon heat shock in hsp104Δ mutant cells (Parsell et al, 1994).
Given the nuclear identity of INQ, we investigated whether this deposit maintains a defined localization within this compartment. We coexpressed mCherry-VHL in hsp42Δ cells with Spc42-GFP, a representative component of the spindle pole body (SPB) which is the yeast equivalent of the mammalian MTOC, or with Nop1-GFP, a marker for the nucleolus. We did not observe any colocalization of INQ, stained by mCherry-VHL, with the SPB. In most cells, these foci were instead located opposite to one another (Supplementary Fig S7A) and INQ was always found adjacent to the nucleolus (Supplementary Fig S7A and C). Very similar results were obtained when wt cells expressing GFP-luciferase-DM-NLS and Nop1-mCherry were analyzed (Supplementary Fig S7B and C). Close vicinity between nuclear aggregates and nucleolus was confirmed by 3D reconstructions and distance measurement of fluorescent foci (Supplementary Fig S7D and E).
INQ sorting does not require ubiquitination
Our finding that INQ is a shared deposit for aggregating proteins of both nucleus and cytosol raises the question of how the differentially localized substrates are targeted to the nuclear compartment. We considered ubiquitination as a sorting signal for misfolded proteins, in line with an earlier proposal made for sorting of cytosolic proteins to the JUNQ (Kaganovich et al, 2008). Ubiquitin immunostaining, however, failed to show specific colocalization of ubiquitin and INQ (Fig3A, Supplementary Fig S8A).
Figure 3.

- S. cerevisiae wt cells expressing GFP-VHL (green) were grown at 30°C and shifted to 37°C for 90 min in the presence of MG132. Ubiquitin (red) was stained by immunofluorescence using specific antibodies. DNA was stained by DAPI (blue). Changes in protein localizations were recorded. Scale bars, 2 μm.
- tGnd1-GFP is stabilized in ubr1Δ san1Δ cells. S. cerevisiae wt and ubr1Δ san1Δ mutant cells expressing tGnd1-GFP were grown at 30°C. Cycloheximide was added, and protein levels were determined at the indicated time points by Western blot using GFP-specific antibodies. Zwf1 levels are given as a loading control.
- Ubiquitination status of tGnd1-GFP expressed in S. cerevisiae wt and ubr1Δ san1Δ cells was determined by GFP pull-down and Western blot analysis using ubiquitin-specific antibodies. Levels of isolated tGnd1-GFP after GFP pull-down were determined by Western blot using GFP-specific antibodies and are given as a control.
- S. cerevisisae wt, ubr1Δ san1Δ, and ubr1Δ san1Δ btn2Δ cells expressing tGnd1-GFP (green) were grown at 30°C. DNA and nuclear envelopes were visualized by coexpressing Htb1-mCherry or Nic96-mCherry, respectively (red). The fraction of cells showing specific tGnd1-GFP foci numbers per cell was determined. INQ formation in cells containing tGnd1-GFP foci was determined (% cells with INQ) based on close vicinity of tGnd1-GFP foci to Htb1-mCherry or localization inside Nic96-mCherry signals, yielding comparable results. Scale bars, 2 μm.
In a second approach, we tested whether substrate ubiquitination is a prerequisite for INQ targeting. We took advantage of two unstable misfolded proteins for which the ubiquitinating E3 ligases are known: tGnd1-GFP, a permanently misfolded truncation variant of Gnd1, and ΔssCPY*, a cytosolic mutant variant of carboxypeptidase Y lacking the ER-targeting signal. Both proteins are targeted for degradation by joint action of the nuclear San1 and cytosolic Ubr1 E3 ligases (Eisele & Wolf, 2008; Heck et al, 2010; Prasad et al, 2010). In ubr1Δ san1Δ cells, degradation of tGnd1-GFP was almost entirely prevented and greatly diminished for ΔssCPY*-GFP (Fig3B, Supplementary Fig S8B). In agreement with these data, substrate ubiquitination was either abrogated (tGnd1-GFP) or reduced (ΔssCPY*-GFP) in the mutant cells (Fig3C, Supplementary Fig S8C). Stabilization of tGnd1-GFP and ΔssCPY*-GFP in ubr1Δ san1Δ cells caused protein aggregation. Nuclear accumulation of both substrates at the INQ was demonstrated by showing tGnd1-GFP and ΔssCPY*-GFP foci located inside the nuclear envelope stained by Nic96-mCherry or Nsp1, along with the detection of nuclear electron-dense structures in thin sections of ubr1Δ san1Δ cells expressing tGnd1-GFP (Fig3D, Supplementary Figs S8D and E and S9). INQ identity was further validated by demonstrating the absence of Hsp42 (Supplementary Fig S8E) and dependence on Btn2 for formation (Malinovska et al, 2012). Nuclear foci of tGnd1-GFP and ΔssCPY*-GFP did no longer form in ubr1Δ san1Δ btn2Δ cells, while the substrate amount remained unaffected (Fig3D, Supplementary Fig S8D and F). This finding excludes the possibility that nuclear aggregation of tGnd1-GFP and ΔssCPY*-GFP represents an uncontrolled chaotic process, and indicates aggregation is instead under control of spatial quality control. Taken together, these findings argue against an essential role of substrate ubiquitination for INQ sorting.
Targeting soluble cytosolic misfolded proteins to INQ involves nuclear import
To identify further factors affecting INQ formation, we performed a systematic genetic screen by mating hsp42Δ query cells, which have a single mCherry-VHL focus as marker for the INQ, with the deletion library of all nonessential yeast genes. We identified 62 double-mutant cells that revealed either no (n = 9) or multiple cytosolic (n = 53) mCherry-VHL foci. Importantly, the list of gene deletions affecting INQ formation included hsp42Δ nup42Δ cells, which lack the nonessential phenylalanine–glycine repeat nucleoporin Nup42 (Wente & Rout, 2010). hsp42Δ nup42Δ cells have multiple mCherry-VHL foci distributed throughout the cytosol at 30 min after stress treatment (Fig4). Only prolonged stress application for 90 min reduced the number of cytosolic mCherry-VHL foci and led to the appearance of either single foci or ring-like structures, both located within the nucleus (Fig4, Supplementary Fig S10E). The slow decrease in the number of cytosolic foci and the relocalization of aggregates into the nucleus was prevented by selective Hsp104 inhibition through the addition of low concentration (3 mM) of guanidinium hydrochloride (Jung et al, 2002) (Supplementary Fig S10A and B). These findings indicate that when misfolded cytosolic proteins are aggregated in the cytosol, they can still be transported into the nucleus via the nuclear pore for deposition at the INQ, provided that they become solubilized by the Hsp104 disaggregase. Importantly, INQ formation of nuclear-resident GFP-luciferase-DM-NLS was unaffected in nup42Δ cells (Supplementary Fig S10C). mCherry-VHL and Hsp104 levels were comparable in hsp42Δ nup42Δ and hsp42Δ cells, excluding differences in substrate or disaggregase levels as a basis for the observed altered aggregation phenotype (Supplementary Fig S10D).
Figure 4.
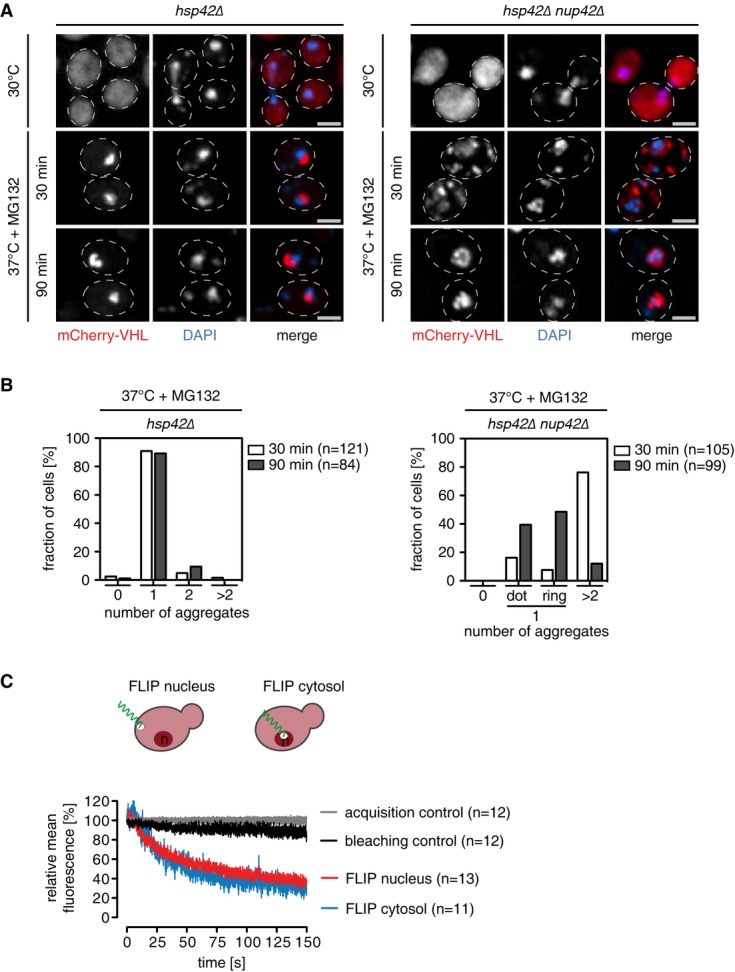
- S. cerevisiae hsp42Δ and hsp42Δ nup42Δ cells expressing mCherry-VHL (red) were grown at 30°C and shifted to 37°C in the presence of MG132. Changes in protein localization were monitored. Scale bars, 2 μm. The total number of mCherry-VHL foci per cell was determined.
- Dynamic exchange of a misfolded model protein between cytosol and nucleus. FLIP measurements of mCherry-VHL were performed in S. cerevisiae wild-type cells at 30°C. Nuclear or cytosolic areas were bleached, and loss of fluorescence intensities in non-bleached compartments was determined. Bleaching and acquisition controls are given. Error bars: SEM.
Together, these findings indicate that a compromised nuclear import of misfolded mCherry-VHL in nup42Δ cells leads to initial cytosolic aggregation and delayed INQ formation. Possibly, Nup42, which faces the cytosol, is involved at an initial phase of nuclear import for misfolded proteins, explaining the delay in INQ formation in nup42Δ cells.
FLIP analysis underline the trafficking of the mCherry-VHL reporter across nuclear pores (Fig4D). We continuously bleached either cytosol or nucleus and monitored the changes in reporter fluorescence in both compartments. Cytosolic mCherry-VHL fluorescence decreased upon bleaching the nucleus, indicating transport of the reporter away from the cytosol to the nuclear compartment. Notably, nuclear mCherry-VHL fluorescence also decreased upon bleaching the cytosol (Fig4D) indicating that cytosolic misfolded proteins enter the nucleus but are not retained in this compxartment. This dynamic shuttling of misfolded mCherry-VHL is consistent with its deposition at cytosolic and nuclear sites. Dynamic shuttling between cytosol and nucleus was also observed for tGnd1-GFP, which also forms cytosolic and nuclear aggregates (Supplementary Fig S11). The sizes of the chosen fluorescent reporters are beyond the size limit (45 kDa) allowing for passive diffusion in and out the nucleus, indicating active import and export processes (Supplementary Table S1).
The Hsp70 co-chaperones Sis1 and Sti1 control cytosolic protein aggregation
Identification of a nuclear protein quality control compartment for deposition of misfolded cytosolic proteins demands for cytosolic factors involved in nuclear import of misfolded proteins and INQ sorting. We tested for possible roles of the Hsp70 co-chaperones Sis1 and Sti1, as they were suggested previously to affect JUNQ formation (Kaganovich et al, 2008; Malinovska et al, 2012). Sis1 is an essential J-domain co-chaperone protein for Hsp70 recently implicated in nuclear import of misfolded proteins for San1-dependent degradation (Guerriero et al, 2013; Park et al, 2013). To explore whether Sis1 is also involved in targeting cytosolic proteins to INQ, we took advantage of tet-off sis1 cells, where SIS1 expression is controlled by a doxycycline repressible promoter. Sis1 was depleted in hsp42Δ cells, which under stress exclusively form INQ. We visualized INQ formation using the GFP-VHL reporter. Upon growth of the cells in the presence of doxycycline for 20 h, Sis1 was no longer detectable by Western blot analysis (Supplementary Fig S12A). Sis1 depletion did not affect cell viability or GFP-VHL levels compared to control cells, indicating that Sis1 remains at low residual levels (Supplementary Fig S12B). Sis1 depletion caused a strong increase in the number of cytosolic GFP-VHL foci, yet GFP-VHL aggregates still formed adjacent to the DAPI signal, indicating that INQ still forms (Fig5A). INQ identity was confirmed by demonstrating that a single GFP-VHL focus resided inside the nuclear Nsp1 ring (Supplementary Fig S12C). To generalize these observations, we depleted Sis1 levels as described above and tested misfolded substrates tGnd1-GFP and ΔssCPY*-GFP, which form INQ aggregates in ubr1Δ san1Δ double-knockout cells. Sis1 depletion strongly increased accumulation of cytosolic aggregates of both proteins (Supplementary Fig S12D). INQ formation was, however, not abolished, and misfolded reporters were still deposited in the nucleus as confirmed by Nsp1 and Hsp42 immunofluorescence microscopy (Supplementary Fig S12E). These findings suggest that while Sis1 appears to play a role in nuclear import of misfolded proteins for INQ formation, further unidentified factors are involved.
Figure 5.
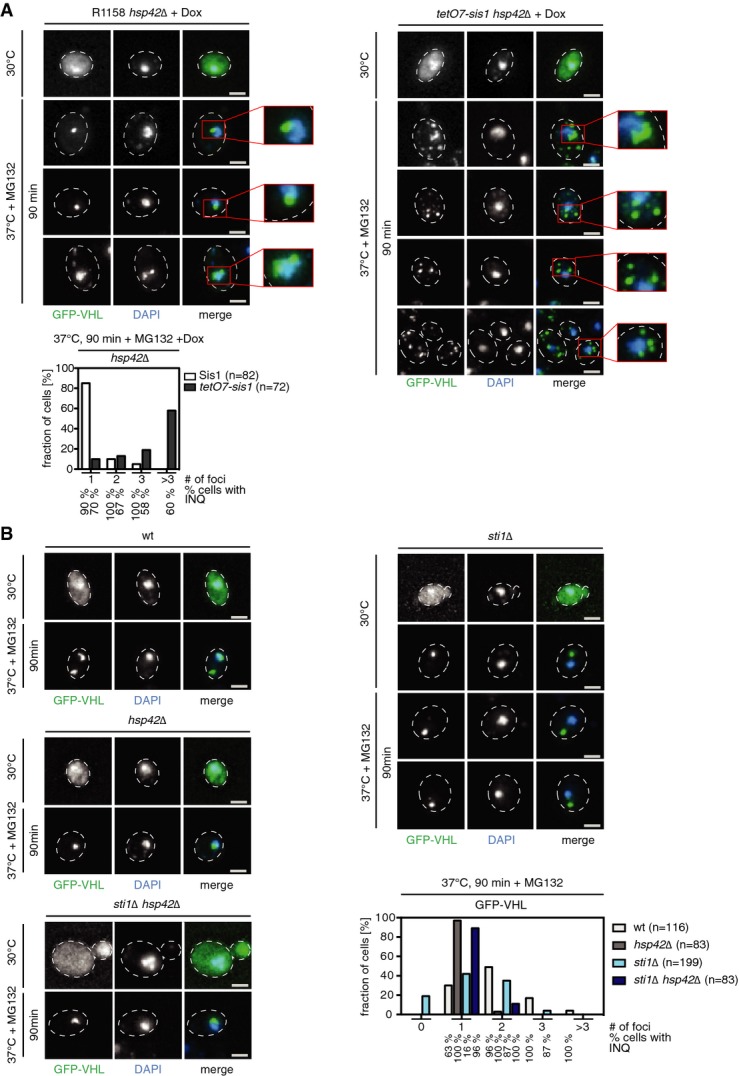
- S. cerevisiae hsp42Δ and hsp42Δ tet-off sis1 cells expressing GFP-VHL were grown for 20 h in the absence (−Dox) or presence (+Dox) of doxycycline at 30°C and shifted for 90 min to 37°C in the presence of MG132. Changes in protein localization were monitored. DNA was stained by DAPI (blue). The total number of GFP-VHL foci per cell and frequencies (%) of INQ formation were determined. Scale bars, 2 μm.
- S. cerevisiae wt, hsp42Δ, sti1Δ, and hsp42Δ sti1Δ cells expressing GFP-VHL were grown at 30°C and shifted to 37°C in the presence of MG132. Changes in protein localization were monitored. The total number of GFP-VHL foci per cell and frequencies (%) of INQ formation were determined. Scale bars, 2 μm.
The Hsp70/Hsp90 co-chaperone Sti1 has been implicated in JUNQ (INQ) targeting since mCherry-VHL forms exclusively peripheral cytosolic aggregates in sti1Δ cells (Kaganovich et al, 2008). We compared deposition of misfolded GFP-VHL upon stress application in sti1Δ and sti1Δ hsp42Δ mutant cells. In agreement with earlier findings in sti1Δ cells, GFP-VHL was most frequently deposited at CytoQs compared to wt, though INQ formation was not completely abolished (Fig5B). In contrast, in sti1Δ hsp42Δ knockouts, GFP-VHL was almost exclusively targeted to INQ, as in hsp42Δ cells (Fig5B). These data exclude an essential function for Sti1 in INQ targeting, but underline a crucial role in cellular protein quality control and hence suggest a more indirect role of Sti1 in INQ formation. The change in localization observed for aggregated VHL from cytosol to nucleus upon deleting hsp42 in sti1Δ cells (Fig5B) indicates that Hsp42 only indirectly affects the import of misfolded proteins, by sequestering these in cytosolic deposits. We conclude that substrate deposition is switched to either CytoQ or INQ dependent on the status of the protein quality control system and availability of Hsp42.
Btn2 promotes protein aggregation in the nucleus
In a separate approach, we set out to dissect the role of Btn2 in INQ formation and its relation to Hsp42-mediated aggregation in the cytosol. We initially considered the possibility that Btn2 plays a role in nuclear import of misfolded proteins since it has been reported to interact with Sis1 (Malinovska et al, 2012). We monitored the aggregation pattern of GFP-VHL in hsp42Δ and btn2Δ single- and double-knockout cells, which lack the essential cellular factors for CytoQ and/or INQ (Fig6A). Nuclear aggregates were identified by localization within the Nsp1 ring signal. GFP-VHL was exclusively deposited at INQ in hsp42Δ cells, but formed only CytoQ in btn2Δ mutants. In these, GFP-VHL foci appeared less intense, suggesting smaller aggregate size. Importantly, no GFP-VHL aggregates were visible in hsp42Δ btn2Δ double-mutant cells (Fig6A). Instead, the diffuse GFP-VHL fluorescence was enriched inside the nucleus. This suggests that nuclear aggregation, but not nuclear transport of misfolded GFP-VHL is compromised in the absence of Btn2. The lack of GFP-VHL aggregates was confirmed by determining the solubility of GFP-VHL before and after stress treatment. While GFP-VHL shifted from the soluble to the insoluble cell fraction after stress application in wt cells, it remained entirely soluble in hsp42Δ btn2Δ cells (Fig6A, Supplementary Fig S13A).
Figure 6.

- S. cerevisiae wt, hsp42Δ, btn2Δ, and hsp42Δ btn2Δ cells expressing GFP-VHL (green, A) or GFP-luciferase-DM-NLS (green, B) were grown at 30°C and shifted to 37°C for 90 min in the presence of MG132, and protein localizations were recorded. DNA was stained by DAPI (blue), and the nuclear envelope was stained by Nsp1 immunofluorescence (red). Solubilities of GFP-VHL and GFP-luciferase-DM-NLS were determined after stress application by Western blot using GFP-specific antibodies. Solubility of actin was determined as a control. T, total fraction; S, soluble fraction; P, pellet fraction. Scale bars, 2 μm.
- S. cerevisiae wt, hsp42Δ, btn2Δ, and hsp42Δ btn2Δ cells expressing mCherry-VHL were heat-shocked to 38°C. Upon return to 30°C, protein synthesis was stopped by cycloheximide (CHX) addition and degradation of mCherry-VHL was monitored by Western blot analysis. The stability of GFP-luciferase-DM-NLS expressed in S. cerevisiae wt and btn2Δ cells was determined accordingly. Ccs1 or Zwf1 levels are given as loading controls.
- S. cerevisiae hsp42Δ btn2Δ cells expressing either GFP or LuciDM-GFP and Htb1-mCherry were grown at 30°C and heat-shocked to 37°C for 30 min. Line intensity plots of luciferase-DM-GFP and Htb1-mCherry before and after heat shock are given. The ratio of nuclear and cytosolic luciferase-DM-GFP fluorescence intensity was determined at 30°C and 37°C (n > 25). Scale bars, 2 μm.
To substantiate the nuclear role of Btn2, we monitored the aggregation of GFP-luciferase-DM-NLS, which resides permanently in the nucleus. Stress treatment caused the formation of nuclear aggregates of GFP-luciferase-DM-NLS in wt and hsp42Δ cells, but GFP-luciferase-DM-NLS stayed soluble in btn2Δ cells with fluorescence remaining diffuse throughout the nucleus (Fig6B, Supplementary Fig S13B).
The observation that protein aggregation is prevented in hsp42Δ btn2Δ mutant cells allowed us to test the impact of misfolded protein deposition at either INQ or peripheral cytosolic sites on substrate degradation. Previous models suggest INQ as a major site of cellular proteolysis and predict INQ targets misfolded proteins for degradation (Kaganovich et al, 2008). We compared the stabilities of mCherry-VHL and GFP-luciferase-DM-NLS in yeast wt, hsp42Δ, btn2Δ, and hsp42Δ btn2Δ cells (Fig6C). Substrate sequestration was first induced by heat shock, and stability was determined during a recovery phase. We found no differences in degradation rates of mCherry-VHL and GFP-luciferase-DM-NLS comparing yeast wt and mutant cells (Fig6C), demonstrating INQ formation is not required for the degradation of these substrates. This suggests that triage decisions determining fate of misfolded proteins are not inherent to sequestration.
hsp42Δ btn2Δ cells also enabled us to analyze for preferential nuclear import of misfolded proteins, without the analysis being affected by protein aggregation. Here, we made use of hyperlabile, cytosolic luciferase-DM-GFP. Unfolding of luciferase was triggered by shifting cells from permissive (30°C) to non-permissive (37°C) temperature. Luciferase-DM-GFP stayed diffuse in hsp42Δ btn2Δ cells and was enriched in the nucleus at non-permissive temperatures, in contrast to cells expressing only GFP (Fig6D). This finding indicates preferential nuclear import of misfolded luciferase conformers and substantiates our previous findings.
Btn2 and Hsp42 are compartment-specific aggregases
Our findings suggest the existence of compartment-specific aggregases that trigger protein aggregation in the cytosol (Hsp42) and the nucleus (Btn2) during physiological stress conditions. Indeed, we observed strong depletion of Hsp42 from the nucleus when monitoring its localization by immunofluorescence microscopy at 30°C (Fig7A) and 39°C (Supplementary Fig S13C), which explains the missing colocalization of Hsp42 and INQ. Btn2 colocalized with peripheral cytosolic aggregates and was strongly enriched in INQ (Fig7B). This explains the partial impact of Btn2 on cytosolic aggregation and its essential role in nuclear aggregation.
Figure 7.

- Cellular localizations of Hsp42 and Btn2 were determined at the indicated temperature by immunofluorescence. DNA was stained by either Htb1-mCherry or DAPI. Scale bars, 2 μm. In case of Hsp42 immunofluorescence, a line intensity plot of a deconvoluted widefield image is given.
- S. cerevisiae btn2Δ hsp42Δ Hsp42-NLS cells expressing GFP-VHL (green) were grown at 30°C and heat-shocked to 37°C for 30 min in the presence of MG132. Changes in protein localizations were recorded. DNA was stained by DAPI (blue). The total number of GFP-VHL foci per cell and frequencies (%) of INQ formation were determined. Scale bars, 2 μm.
- S. cerevisiae btn2Δ hsp42Δ Hsp42-NLS cells expressing mCherry-VHL (red) were treated as described in (C). The nuclear membrane was visualized by Nsp1 immunofluorescence (green) and colocalization of mCherry-VHL foci with Hsp42 was probed by Hsp42 immunofluorescence (green). DNA was stained with DAPI (blue). Scale bars, 2 μm.
- Hsp42 and Btn2 levels were determined prior and post-heat shock at the indicated time points by Western blot. The numbers of molecules/cell were calculated using purified proteins as standard.
We asked to what extent these two proteins play equivalent roles or, alternatively, have unique features. We specifically tested whether nuclear targeting of Hsp42 can complement Btn2 function in nuclear protein aggregation in btn2Δ cells (Fig7C). We produced GFP-VHL or mCherry-VHL in btn2Δ cells expressing Hsp42-NLS from its authentic chromosomal locus as sole Hsp42 source. VHL deposited exclusively at the INQ after stress and colocalized with Hsp42-NLS, demonstrating that Hsp42 can trigger INQ formation if targeted to the nucleus (Fig7C and D). Hsp42 therefore complements Btn2 function if appropriately targeted. Notably, nuclear VHL foci already existed in 35% of HSP42-NLS cells prior to being stress treated. INQ formation at 30°C can be explained by the fact that Hsp42 is produced at higher basal levels at non-heat-shock conditions compared to Btn2 (see below; Malinovska et al, 2012).
Together, these findings support a model in which compartment-specific aggregases drive protein aggregation in the cytosol (Hsp42) and nucleus (Btn2), explaining the specific aggregation phenotypes of corresponding knockout cells. While Btn2 and Hsp42 share common functions, their expression profiles are remarkably different (Fig7E). Hsp42 is already abundant at 30°C (28,000 molecules/cell), whereas Btn2 is virtually undetectable. Heat shock induces massive accumulation of Btn2 to levels comparable to Hsp42 (each 46,000 molecules/cell) (Fig7E). However, in contrast to Hsp42, Btn2 is subjected to rapid degradation even upon continuous heat stress exposure and hence exhibits only a short burst of induction. Notably, protecting Btn2 from proteasomal degradation by the addition of MG132 led to INQ stabilization providing a rationale for Btn2 degradation (Supplementary Fig S14A and B).
Btn2 organizes DNA damage-induced nuclear inclusions
We explored whether other stress conditions known to induce formation of nuclear foci also involve Btn2 activity and thus relate to INQ. We focused on the DNA alkylating agent methyl methanesulfonate (MMS), which has recently been shown to induce non-canonical nuclear protein inclusions different to classical DNA repair foci (Tkach et al, 2012). Intriguingly, Btn2 began accumulating 60 min after MMS treatment. We therefore analyzed whether the formation of non-canonical DNA stress foci depends on Btn2 using the histone deacetylase Hos2-GFP, a known component of these foci, as marker (Tkach et al, 2012) (FigA and B). MMS treatment induced the formation of nuclear Hos2-GFP foci in wt but not btn2Δ mutants (Fig8B, Supplementary Fig S15A). These findings demonstrate that Btn2 organizes nuclear inclusions induced by both, protein damage and DNA replication stress. Misfolded GFP-VHL also formed nuclear foci upon MMS treatment in a Btn2-dependent manner, indicating that stress conditions causing Btn2 accumulation trigger protein aggregation (Fig8C, Supplementary Fig S15B). Together, these findings extend the role of Btn2 as a nuclear aggregase that controls protein aggregation responsive to diverse stress conditions.
Figure 8.

- S. cerevisiae wt cells were treated with MMS and Btn2 levels were determined at the indicated time points. Zwf1 levels are given as a loading control.
- S. cerevisiae wt and btn2Δ cells expressing Hos2-GFP (green, B) or GFP-VHL (green, C) were grown at 30°C and treated with MMS. Changes in protein localizations were recorded at the indicated time points. DNA was stained by DAPI (blue). The total number of GFP-VHL foci per cell and frequencies (%) of INQ formation were determined.
Discussion
This study determines the molecular organization of protein aggregation in yeast cells and the function of critical factors in controlling protein aggregation (Fig9). INQ represents a newly defined, general quality control compartment located in the nucleus. We demonstrate nuclear localization of INQ by using both fluorescence and electron microscopy to detect exogenous (VHL, Ubc9ts, tGnd1, ΔssCPY*) and endogenous misfolded proteins and the aggregate-specific chaperone Hsp104. Fluorescence microscopy criteria establishing nuclear localization of INQ include: (i) vicinity to DAPI-stained chromatin, (ii) localization within the fluorescently labeled nuclear envelope, and (iii) the absence of Hsp42. Previous identification of this deposit as a cytosolic, juxtanuclear site (JUNQ) (Kaganovich et al, 2008) is based solely on the close but non-overlapping DNA-staining DAPI signal, which on its own is inconclusive and likely led to misinterpretation.
Figure 9.

Compartment-specific aggregases control protein aggregation in S. cerevisiae
Misfolded proteins are deposited at cytosolic CytoQ and nuclear INQ compartments, while amyloidogenic proteins are sequestered at IPOD next to the vacuole. CytoQ formation depends on Hsp42. Misfolded, cytosolic proteins are imported through nuclear pore complexes by Sis1 and other, so far unknown sorting factors. Protein aggregation inside the nucleus is triggered by Btn2, which transiently accumulates upon stress. INQ is located adjacent to the nucleolus and harbors cytosolic and nuclear misfolded proteins. DNA replication stress causes formation of Btn2-dependent non-canonical DNA stress foci at the INQ and might signal to checkpoint controls.
Our data show INQ as a single focus of fluorescence adjacent to nucleolus and chromatin, opposite the spindle pole body. It is currently unclear whether a specific mechanism directs INQ location or whether occlusion restricts the space available for aggregate deposition elsewhere in the nucleus. The asymmetric inheritance observed for this body during cell division, however, (Spokoini et al, 2012) would be facilitated by defining INQ positioning within the nucleus. INQ proximity to the nucleolus invites further surmise, regarding provision for coupling ribosome biogenesis with the protein aggregation status of the cell.
The intranuclear status identified for INQ changes our understanding of how protein aggregation is organized in yeast cells and explains the previously puzzling specificity of Hsp42 and Btn2 in aggregate formation (Fig9). It also clarifies the previously murky relationship of INQ to other aggregates (peripheral aggregates/Q-bodies/stress foci), which are truly cytosolic. In the cytosol, peripheral aggregates/Q-bodies/stress foci collide and fuse, eventually forming a single deposit previously termed ‘IPOD’ (Kaganovich et al, 2008). However, as originally defined, IPOD includes amyloidogenic aggregates, the formation of which is, however, Hsp42-independent (Specht et al, 2011; Escusa-Toret et al, 2013). Moreover, IPODs do not colocalize with peripheral aggregates/Q-bodies (Specht et al, 2011; Escusa-Toret et al, 2013) and therefore represent stress-independent specific deposits for amyloidogenic substrates (Fig9). We propose clarifying nomenclature of amorphous deposits by referring to cytosolic, Hsp42-containing aggregates as ‘CytoQ’ (cytosolic quality control compartment). Cytosolic IPOD and CytoQ and nuclear INQ represent independent aggregate deposits, and separate cellular compartments, which do not interchange by fusion events.
Is nuclear sequestration of misfolded proteins conserved in mammalian cells? We found that stress-induced GFP-VHL inclusions in mammalian cells are located outside the nucleus and next to the MTOC (microtubule-organizing center) (Supplementary Fig S16). VHL inclusions also localize next to vimentin clusters, qualifying these inclusions as aggresomes (Kopito, 2000) (Supplementary Fig S16) in agreement with other recent findings (Weisberg et al, 2012; Ogrodnik et al, 2014). Evidently therefore, both yeast and mammalian cells actively sequester misfolded proteins, but employ different mechanisms and target aggregating protein to different cellular sites.
In yeast however, both cytosolic and nuclear substrates aggregate into INQ deposits (Fig9). This implies cytonuclear transport of substrate proteins. We show this transport occurs via the nuclear pore, involving nucleoporin Nup42. Transport is selective for misfolded proteins in soluble states, excluding proteins in aggregated state. The Hsp70 co-chaperone Sis1 plays a role in INQ formation, in agreement with earlier findings reporting on a crucial role of Sis1 in nuclear transport of unstable proteins destined for degradation by the UPS (Park et al, 2013). However, even efficient Sis1 depletion does not completely block misfolded protein import into the nucleus and deposition at the INQ, suggesting that additional factors may be involved in this process as well. Sis1 depletion may also indirectly affect INQ formation by reducing the protective capacity of cytosolic protein quality control systems causing increased cytosolic aggregation of GFP-VHL before it can be encountered by nuclear import factors.
Several observations indicate that substrate shuttling between cytosol and nucleus is actively biased toward transport into the nucleus. First, when CytoQ formation is compromised (in hsp42Δ mutants), the size of INQ increases. Second, when aggregate formation at CytoQ and INQ is simultaneously compromised (in btn2Δ hsp42Δ double mutants), soluble misfolded proteins accumulate in the nucleus. INQ is however not simply an overflow compartment for misfolded proteins of the cytosol. Live cell imaging shows that protein folding stress causes cytosolic mCherry-VHL to accumulate in INQ and CytoQ foci simultaneously and not sequentially, within minutes, in wt cells. Within normal limits of physiological stress, the nucleus therefore is still a major protein quality control compartment for the deposition of aggregation-prone cytosolic proteins. This represents a cellular strategy, which may serve to protect cytosolic functions, particularly protein synthesis and folding, against accumulation of damaged proteins. The involvement of the nucleus in protein quality control in yeast extends to protein degradation, since nuclear E3 ligase San1 is implicated in the ubiquitination of unstable proteins of the cytosol (Heck et al, 2010; Prasad et al, 2010; Guerriero et al, 2013) and the proteasome is long known to be enriched in the nucleus relative to the cytosol (Russell et al, 1999).
Our results challenge the current view of a ubiquitin-based sorting mechanism that targets misfolded proteins to JUNQ (now INQ) (Kaganovich et al, 2008). Two different misfolded proteins tested (tGnd1-GFP, ΔssCPY*-GFP), which are normally ubiquitinated and degraded in wt cells, in ubr1Δ san1Δ mutant cells are not ubiquitinated or degraded, but still efficiently targeted to INQ and CytoQ aggregates. Consistent with our findings, nuclear aggregates are also formed by nuclear E3 ligase San1 substrates in san1Δ cells (Fredrickson et al, 2011, 2013). INQ consistently shows only low-level staining for ubiquitin. Conversely, and again in contrast to the proposed ubiquitin sorting model, for a different protein (unstable VMA12-GFP-DegAB reporter), ubiquitination was required for Hsp42-dependent foci (CytoQ) formation upon Hsp70 depletion (Shiber et al, 2013). Ubiquitination therefore cannot be the general basis for sorting misfolded proteins to the INQ (JUNQ). Consistent with this, we show that deposition at INQ does not automatically target sequestered substrates for degradation. The fate of aggregated proteins is instead decided after Hsp70/Hsp104-driven protein disaggregation and evidently depends on substrate-intrinsic features. There is at present therefore no evidence for a unifying sorting principle for aggregating proteins other than the spatial features imposed by the distinct cellular localizations of CytoQ and INQ.
Our findings reveal a bipartite system for compartment-specific promotion of protein aggregation in the nucleus and the cytosol involving two distinct aggregases, Btn2 and Hsp42. We show that Hsp42 is virtually absent in the nucleus, vindicating earlier observations that Hsp42 does not colocalize with INQ and is not involved in INQ formation (Specht et al, 2011) (Fig9). So far, Btn2 has been thought to act in the cytosol in the formation of misfolded protein aggregates and amyloids (Kryndushkin et al, 2008; Malinovska et al, 2012). However, we find Btn2 is much less abundant in the cytosol compared to the nucleus. We further show INQ formation requires the activity of Btn2 inside the nucleus. Mammalian cells lack a direct homolog of Btn2, but human Hook2 displays limited sequence similarity to a C-terminal region of Btn2 (Supplementary Fig S17A). Hook2 overproduction promotes aggresome formation by CFTR (Szebenyi et al, 2007), suggesting that the related Btn2 segment is involved in promoting protein aggregation.
Why would two distinct aggregases be required to control cytosolic and nuclear aggregation? Hsp42 and Btn2 have similar activities but exhibit remarkably different expression profiles. Btn2 accumulates only in direct response to stress (Fig7E; Malinovska et al, 2012) and is rapidly degraded thereafter. Stabilizing Btn2 by proteasomal inhibition causes INQ stabilization (Supplementary Fig S14A and B). These observations imply tight control of aggregate deposition is required in the nuclear compartment. Btn2-triggered INQ formation may conceivably be linked to ribosome biogenesis, DNA damage repair, and cell cycle control and which would therefore require tight regulation. Supporting this model, we show that Btn2 is required for the formation of non-canonical DNA replication stress foci implicated in DNA damage repair and genome stability (Stirling et al, 2011; Tkach et al, 2012). Possibly, sequestration of factors such as Hos2, a histone deacetylase, might facilitate chromatin remodeling to enable binding of the chromatin-associated factor Cmr1 to damaged DNA (Choi et al, 2012; Tkach et al, 2012).
Both DNA and protein damaging conditions lead to Btn2-orchestrated INQ formation, suggesting potential INQ involvement in coordination with stress responses hitherto considered unrelated. INQ therefore is a newly defined, general quality control compartment found in the nucleus. Btn2-driven INQ formation may facilitate the orchestration and organization of downstream processes including DNA damage checkpoint control. Rapid degradation of Btn2 would ensure signaling was limited to periods of immediate stress. The additional role for INQ in coordination with stress responses and signaling processes related to nuclear physiology, however, awaits further investigation.
Materials and Methods
Yeast media, strains, plasmids, and antibodies
Yeast media preparation, growth, and transformations as well as recombinant DNA methods were performed as described previously (Abelson et al, 2003). All genes were cloned by PCR from yeast genomic DNA or a template plasmid and verified by sequencing. The genotypes of strains and plasmids used in this study are summarized in Supplementary Tables S2 and S3. Chromosomal tagging for the generation of fluorescent proteins was performed as described (Janke et al, 2004). Antibodies used are listed in Supplementary Table S4.
Growth conditions
To induce galactose promoter-driven expression of misfolded proteins, cells were treated as described elsewhere (Kaganovich et al, 2008). In short, cells were grown overnight in SD with 2% glucose (SD-Glu), diluted into SD with 2% galactose (SD-Gal), and grown for 16 h to reach mid-log-phase. Prior to heat-shock treatment, Gal promoter activity was shut off by the addition of 2% glucose. Cells were either subjected to acute heat stress or grown constantly at the indicated temperatures to test the fate of constantly misfolded proteins under steady-state conditions. Heat shock was performed by either shifting cells from 30°C to 37°C in the presence of 80 μM MG132 or to 38°C in the absence of MG132 for up to 90 min as indicated in a shaking waterbath (ThermoScientific MaxQ 7000).
To test the effect of Sis1 on protein aggregate formation, cells were grown overnight in SD-Glu or SD with 2% raffinose (SD-Raf) depending on the substrate, diluted to OD600 = 0.05 in SD-Glu or SD-Raf-Gal containing 10 μg/ml doxycycline and grown for 20 h. During growth, cells were diluted into fresh medium with doxycycline once prior to the experiment.
To test foci formation in the presence of methyl–methane-sulfonate (MMS), cells were grown in SD-Glu or SD-Gal to OD600 = 0.6–0.8 at 30°C. A total of 0.05% (v/v) MMS was added to the cultures and incubation continued at 30°C for up to 2 h.
For plating assays, yeast cultures were treated as indicated and plated on agar plates in fivefold dilution steps. Plates were subsequently incubated for 2–3 days at the indicated temperatures.
Image acquisition, processing, and data analysis
Cells were treated as indicated, harvested by centrifugation, fixed with 70% (v/v) ethanol for 5 min on ice, washed once with H2O, and resuspended in PBS. If indicated, DNA was stained with 50 ng/ml DAPI in PBS for 15 min at room temperature.
Optical sections of 0.2 μm were acquired to image the entire cell volume using a widefield system (xcellence IX81, Olympus) equipped with a Plan-Apochromat 100×/NA 1.45 oil immersion objective and an EMCCD camera (Hamamatsu). Acquired z-stacks were deconvolved with xcellence software using the Wiener filter.
All further processing of digital images was performed with ImageJ. If not indicated else, maximum projections are shown and contrast was adjusted allowing up to 0.2% pixels to be saturated.
Confocal images were acquired with a Plan-Apochromat 63×/NA 1.4 oil immersion objective on a Zeiss LSM 780 confocal microscope. If indicated, entire stacks were subjected to restoration by deconvolution using Huygens. Three-dimensional reconstructions of confocal images were carried out with ZenBlue software (Zeiss) following deconvolution using Huygens. Prior to surface rendering, a median filter with a kernel size of 3 was applied.
Distance measurements
Confocal optical sections of 0.17 μm were acquired of fixed cells by simultaneously imaging GFP and mCherry signals, using 488-nm and 568-nm laser light at a pinhole size of 0.92 AU (based on 488 nm) and separately acquiring the DAPI signal using 405 nm at a pinhole size of 0.91 AU. Image resolution was set to 512 × 512 pixels and a pixel size of 0.08 μm. Prior to distance determination, entire stacks were subjected to restoration by deconvolution using Huygens. All following steps were performed with ImageJ. To segment individual structures, deconvolved stacks of individual channels were converted to 16-bit images without scaling and the histogram stretched allowing 0.002% (spindle pole bodies (Spc42-GFP)) or 0.02% (INQ, mCherry-VHL) of all pixels to be saturated in order to avoid outlying pixel interference. Subsequently, Otsu thresholding was performed for both channels using ImageJ default settings. Segmentation and distance measurement (border to border and centroid to centroid) was performed with the RoiManager 3D V2.0 plugin (Ollion et al, 2013). Corresponding structures in one cell were manually selected based on their proximity to the DAPI signal. Only non-dividing cells with a single spindle pole body were used for quantification.
Nucleoli were detected using Nop1-GFP as a marker. In order to distinguish nucleoli from background nuclear signal, 0.35% pixels were allowed to be saturated after 16-bit conversion and thresholding was performed using the Minimum function with default settings. Segmentation and measurements were done as described before. Only non-dividing cells (not budding) were used for quantifications.
To determine the distance between INQ formed by GFP-luciferase-DM-NLS and the nucleolus (Nop1-mCherry), no histogram stretching was performed after 16-bit conversion for either channel and Max Entropy thresholding function with default settings was used for both. Segmentation and distance measurements were performed as described before using only non-dividing cells (not budding) for quantification.
Ratio quantification
A circular area (diameter = 0.5 μm) within nucleus or cytosol was used to quantify the fluorescence intensity using ImageJ. The nucleus-to-cytosol ratio is presented by boxplot overlayed with beeswarm plot using R.
Time-lapse microscopy
Aggregate formation was imaged on the microscope stage with a custom-made stage inlay (heat-shock stage) for the xcellence IX61 microscope linked to a water-cooled Peltier-type temperature control and feedback system and software (BelektroniG). Temperature was controlled and measured with a sensor located inside the sample in close proximity to the area imaged. A second temperature sensor allowed recording the temperature of the metal frame harboring the Peltier element and the sample inlay. Temperatures of both sensors were continuously recorded every 10 s by the control software.
For sample preparation, a round glass coverslip was sealed on the inlay of the heat-shock stage using laboratory grease and coated with concanavalin A (ConA). Cells were adhered on the ConA-coated coverslip for 15 min, unbound cells were removed by washing, and a final volume of 400 μl medium was added. The temperature sensor was fixed into the sample such that it touches the coverslip and the whole system was covered with a squared coverslip to prevent extensive evaporation. The heat-shock stage was then mounted onto the microscope, followed by initial temperature equilibration to 30°C. Temperature profiles were set using the BelektroniG Software module, including a 1-min 30°C pre-phase and heating or cooling at 3.5°C/min. Image acquisition was started simultaneously with the programmed temperature profile. Images were acquired using the xcellence IX61 microscope using a Plan-Apochromat 100×/NA 1.45 oil immersion objective including hardware autofocusing during the course of the experiment. 2 × 2 binning was used to increase signal intensities. Nine optical sections of 0.5 μm were acquired to image the entire cell volume. Stacks were deconvolved as described above. Simultaneously, to image acquisition, temperatures at both sensors were recorded.
Immunofluorescence microscopy
Immunostaining was performed as described elsewhere (Cherkasov et al, 2013). For immunostaining, cells were fixed with 4% p-formaldehyde/100 mM KPi (Sigma Aldrich) for 1 h prior to cell wall digestion with 500 μg/ml zymolase T-100 in wash buffer (1.2 M Sorbitol/100 mM KPi pH 6.5) supplemented with 20 mM β-mercaptoethanol for 30 min at 30°C. Spheroblasts were attached to poly-lysine-coated cover slides, permeabilized by washing three times with 1% Triton X-100/100 mM KPi pH 6.4 and blocked for 1 h with 1% (w/v) BSA in 100 mM KPi pH 6.4. All antibodies were diluted in blocking buffer. Primary antibody incubations were carried out either 2 h at room temperature or overnight at 4°C. Secondary antibody incubations were performed at room temperature for 2 h. Following secondary antibody incubation, spheroblasts were stained with 50 ng/ml DAPI in PBS and embedded in 55% glycerol. Antibodies and dilutions used in this study are listed in Supplementary Table S4.
FLIP analysis
Cells were grown to mid-log-phase at 30°C and adhered to glass-bottom microdishes (MatTec Corp) coated with ConA. Fluorescence loss in photobleaching was measured at room temperature using a Zeiss LSM 780 confocal microscope. Cells constitutively expressed mCherry-VHL and GFP-Nup49 or tGnd1-GFP and HTB-mCherry, respectively. Resolution of the image was set to 128 × 128 (0.176 μm/pixel). Equally sized ROIs were defined either in the cytosol or in the nucleus based on an initial two-color image. In total, 4 ROIs were bleached simultaneously per frame. The actual bleaching experiment was performed using the 561-nm laser (mCherry-VHL) or 488-nm laser (tGnd1-GFP) only. Images were acquired at 2.5% laser intensity with a pinhole diameter of 2 AU at maximum scanning speed. Selected ROIs were repeatedly bleached with 100% laser intensity at maximum scanning speed. After 5 initial frames of acquisition, 300 cycles of bleaching and acquisition were acquired.
Image analysis was performed with ZenBlue software, measuring mean fluorescence in the whole nuclear region or a small region in the cytosol and plotted after background subtraction and correcting for bleaching during acquisition if necessary.
Electron microscopy and immunoelectron microscopy
Yeast cells expressing the respective GFP fusion proteins were grown and subjected to heat shock in the presence or absence of MG132 as described above. Label-free control cells were grown in SD-Glu medium at 30°C. Cells were subsequently high-pressure-frozen, freeze-substituted, sectioned, labeled, and stained for electron microscopy as previously described (Giddings et al, 2001). Briefly, cells were collected onto a 0.45-μm polycarbonate filter (Millipore) using vacuum filtration and cryoimmobilized by high-pressure freezing using the EM PACT2 machine (Leica Microsystems, Vienna, Austria). Cells were freeze-substituted using the EM-AFS2 device (Leica Microsystems, Vienna, Austria) with 0.2% uranyl acetate, 1% water—dissolved in anhydrous acetone and stepwise infiltrated with Lowicryl HM20 (Polysciences, Inc., Warrington, PA, USA), started at low temperatures. For polymerization, the samples were exposed to UV light for 48 h at −45°C and were gradually warmed up to 20°C. Polymerized cells were serially sectioned (thickness: 60–70 nm) using a Reichert Ultracut S Microtome (Leica Instruments).
For immunoelectron microscopy, sectioned cells were incubated in blocking buffer (1.5% (w/v) BSA, 0,1% (w/v) fish skin gelatin in PBS), labeled with rabbit anti-GFP antibody (Clontech), diluted 1:5 in blocking buffer, and finally labeled with protein A–gold conjugates (10 nm, Utrecht University, Utrecht, the Netherlands). After post-staining with 2% uranyl acetate and lead citrate, sections were imaged at a CM120 electron microscope (Philips Electronics N.V., Eindhoven, the Netherlands), which was operated at 120 kV and equipped with a CCD camera (Keen View, Soft Imaging systems). A Digital Micrograph Software was used for visualization.
Identification of factors affecting INQ formation
Double deletion strains, lacking Hsp42 and any nonessential gene of interest expressing mCherry-VHL were created by crossing the query strain Moh14 (Y8205 hsp42Δ::hph p406 GPD mCherryVHL) with the nonessential gene knockout library (EUROSCARF) using the synthetic genetic array method (Tong et al, 2001). Mat a haploid progeny harboring both deletions and the substrate were selected for the screening procedure. High-throughput microscopy was performed in 384-well format. A total of 4 × 96 arrays of cells were grown in SD-Glu overnight, diluted to OD 0.1 the next day, and grown to mid-log-phase (OD600 = 0.5–0.7) prior to heat-shock treatment. All further treatments were performed in 96-well PCR plates. Cells were harvested by centrifugation, resuspended in SD-Glu + MG132, and shifted to 37°C for 30 min in a waterbath. Cells were subsequently harvested by centrifugation, fixed with 70% ethanol on ice, washed once with water, and stained with 50 ng/ml DAPI in PBS for 15 min at room temperature. Cells were finally resuspended in PBS and transferred to 384-well glass-bottom microscopy plates (MatTek corporation). After sedimenting cells by centrifugation, cells were overlaid with 87% glycerol to restrict movement during image acquisition.
Images were acquired using an Olympus IX81 microscope equipped with a PlanFl 100×/NA 0.95 air objective with hardware autofocus and an EM-CCD camera controlled by ScanR software (Olympus). Optical slices of 0.5 μm thickness were recorded to image the entire cell volume at 4 positions per well for both DAPI and mCherry signals.
Data analysis of acquired images was performed with a customized workflow built in KNIME 2.6.2 (Berthold et al, 2008) and its built-in ImageJ macro functionalities.
All images were initially converted to 8 bit, maximum projections of individual z-stacks built and background was subtracted. Subsequently, nuclei, aggregates, and cells were segmented. In order to segment spots (=aggregates) of mCherry-VHL, the find maxima within tolerance function with a noise level 40 was used. An area size threshold of 5–100 pixels was applied. Nuclei were segmented using the DAPI signal and applying a global threshold of 150. Identified nuclei were dilated with a neighbor count of 1 for 7 iterations. An area range of 60–900 pixels and a perimeter of 25–100 pixels were set as size thresholds.
Cells were identified by Voronoi segmentation in the mCherry images using the segmented nuclei as seeds and a background threshold of 30. Cells were selected based on an area size of 200–2,500 pixels, a perimeter of 80–300 pixels, and a convexity of 0.9–1.0.
The number of spots per cell was determined to identify genes that impact INQ formation. Hits were considered to have no aggregates with < 0.2 spots/cell and multiple aggregates with > 1.2 spots/cell. Potential hits were visually inspected.
Cycloheximide chase experiments
Yeast cultures were grown to OD600 = 0.6–1.0 and subjected to heat treatment as indicated. Translation was inhibited by the addition of 100 μg/ml cycloheximide, and degradation of substrates was monitored at different time points by SDS–PAGE and Western blot. To this end, total protein extracts were prepared in analogy to Knop et al (Knop et al, 1996). In short, 177 μl 1.85 M NaOH were added to 1 ml of yeast culture and incubated on ice for 10 min. Cells were then TCA-precipitated by the addition of 177 μl 55% TCA and further incubation on ice for 10 min. TCA pellets were resuspended in 100 μl HU buffer (8 M urea, 5% SDS, 200 mM Tris pH 6.8, 1 mM EDTA, 1.5% (w/v) DDT, 0.1% (w/v) bromphenolblue) per 1 OD600 unit and lysed by incubation at 65°C for 10 min.
Protein solubility assay
Protein aggregation was assessed as described earlier (Cherkasov et al, 2013). Cells were grown in SC medium to OD600 = 0.7 at 30°C and subjected to heat stress and proteasome inhibition as indicated. Cells were harvested by centrifugation, resuspended in 50 mM Tris/HCl and 500 mM NaCl pH 8.5 supplemented with protease inhibitors (1 mM phenylmethylsulfonfluoride (PMSF), 5 μg/ml leupeptin, 10 μg/ml pepstatin, 8 μg/ml aprotinin, Proteaseinhibitor Mix FY (Serva)), and flash-frozen in liquid nitrogen prior to pulverization by mixer milling (MM 400 (Retsch), 30 Hz, 2 min). Cell lysates were pre-cleared by centrifugation at 3,000 g for 20 min and total protein concentration of supernatants adjusted to equal levels before separating soluble and insoluble fractions by centrifugation at 16,000 g for 20 min. The soluble fraction was removed, and pellets were washed once with 50 mM Tris/HCl and 150 mM NaCl pH 8.5 supplemented with protease inhibitors, centrifuged at 16,000 g for 20 min, and resuspended in 50 mM Tris/HCl, 500 mM NaCl, 8 M urea, 2% SDS, and 2 mM DTT pH 8.5 supplemented with protease inhibitors. Distribution of substrate proteins to soluble or insoluble fractions was analyzed by SDS–PAGE and Western blotting with specific antisera.
Ubiquitination assay
Saccharomyces cerevisiae wt and ubr1Δ san1Δ strains expressing tGnd1-GFP or ΔssCPY*-GFP were grown at 30°C to OD600 = 1.0. Denatured cell lysates were prepared as described for CHX chase experiments except TCA precipitates were resuspended in 50 μl HU-IP buffer (8 M urea, 1% SDS, 200 mM Tris pH 6.8, 1 mM EDTA, 30 mM DDT) per OD600. A total of 50 μl of denatured lysate was diluted into 1 ml IP buffer (20 mM Tris pH 7.5, 150 mM NaCl, 1 mM EDTA, 0.1% Triton X-100) supplemented with 10 mM N-ethylmaleimide (NEM) and 1 mM PMSF. A total of 30 μl Protein A Dynabeads (Life Technologies) with pre-bound anti-YFP antibody was added and incubated while rotating for 1 h at room temperature. Beads were washed sequentially with IP buffer, IP buffer plus 500 mM NaCl, IP buffer, and eluted by boiling in HU buffer. The input, unbound, and eluted fractions were analyzed by Western blotting using YFP- and ubiquitin-specific antibodies.
Western blot and calculation of protein abundance
Protein mixtures were separated by SDS–PAGE according to standard procedures (Laemmli, 1970) and transferred to PVDF membrane by semi-dry transfer (TransBlot Turbo, Bio-Rad) or wet transfer. Protein bands were detected by specific antibodies (see Supplementary Table S3) and corresponding secondary antibodies coupled to alkaline phosphatase. Bands were visualized using ECF reagent according to the manufacturer's instructions (GE Healthcare). Fluorescence was detected with a Fuji LAS4000 system.
In vivo concentrations of Btn2 and Hsp42 were determined by quantitative Western blotting. Signals were detected with a LI-COR Odyssey IR imager, and band intensities were quantified by densitometry using ImageJ. Purified Hsp42 and Btn2 served as standard.
Mammalian cell culture
HeLa cells were cultured in DMEM supplemented with 10% (v/v) fetal bovine serum at 37°C and 5% CO2. Prior to transfection, cells were plated onto poly-L-lysine-coated coverslips and allowed to adhere for 1 day. Cells were transiently transfected with pCV17 (GFP-VHL). Twenty-fours hours after transfection, 20 μM MG132 dissolved in DMSO were added to the cells and incubated at 37°C, 5% CO2 for 8 h. Cells were subsequently fixed with 4% p-formaldehyde for 10 min at room temperature. For immunostaining of γ-tubulin, cells were fixed with ice-cold methanol (−20°C) for 5 min at −20°C. After fixation, cells were permeabilized with 0.1% (v/v) Triton X-100 in PBS and blocked at room temperature in blocking buffer (1% BSA in PBS). All antibodies were diluted in blocking buffer. First antibody incubation was carried out for 2 h at room temperature, and secondary antibodies were added for 30 min at room temperature. Finally, DNA was stained with 2 μg/ml Hoechst 33442 in PBS for 10 min and cells were embedded in ProLong®Gold (Life Technologies).
Acknowledgments
We are grateful to J. Buchner and M. Haslbeck for providing Hsp42-specific antibodies, O. Gruß for providing γ-tubulin antibodies, H. Hermann-Lerdon for providing vimentin antibodies, and D. Cyr for providing tet-off sis1 cells. We thank H. Lorenz for expert advice in microscopy and thank S. Druffel-Augustin for valuable technical support. S. Miller, C. Ho, and M. Khokhrina were supported by the Hartmut Hoffmann-Berling International Graduate School of Molecular and Cellular Biology (HBIGS). S. Miller was additionally supported by a fellowship of the Boehringer Ingelheim Fonds. This work was supported by the Deutsche Forschungsgemeinschaft (SFB1036) to ES, AM, and BB and AgeNet to BB.
Author contributions
SM, CH, JW, MK, AN, MM, KR, AM, and BB conceived and designed experiments. SM, CH, JW, MK, AN, MM, and KR performed experiments. SM, CH, JW, MK, AN, MM, KR, ML, ES, AM, and BB analyzed the data. LG, AM, and BB wrote the manuscript.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting Information
Supplementary Information
Review Process File
References
- Abelson JN, Guthrie C, Fink GR. Guide to Yeast Genetics and Molecular Biology. USA: Academic Press Inc; 2003. [Google Scholar]
- Berthold MR, Cebron N, Dill F, Gabriel TR, Kötter T, Meinl T, Ohl P, Sieb C, Thiel K, Wiswedel B. KNIME: The Konstanz Information Miner. In: Preisach C, Burkhardt H, Schmidt-Thieme L, Decker R, editors. Data Analysis, Machine Learning and Applications. Vol. 38. Berlin, Heidelberg: Springer; 2008. pp. 319–326. (eds), Vol. [Google Scholar]
- Chen B, Retzlaff M, Roos T, Frydman J. Cellular strategies of protein quality control. Cold Spring Harb Perspect Biol. 2011;3:a004374. doi: 10.1101/cshperspect.a004374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cherkasov V, Hofmann S, Druffel-Augustin S, Mogk A, Tyedmers J, Stoecklin G, Bukau B. Coordination of translational control and protein homeostasis during severe heat stress. Curr Biol. 2013;23:2452–2462. doi: 10.1016/j.cub.2013.09.058. [DOI] [PubMed] [Google Scholar]
- Choi DH, Kwon SH, Kim JH, Bae SH. Saccharomyces cerevisiae Cmr1 protein preferentially binds to UV-damaged DNA in vitro. J Microbiol. 2012;50:112–118. doi: 10.1007/s12275-012-1597-4. [DOI] [PubMed] [Google Scholar]
- Cohen E, Bieschke J, Perciavalle RM, Kelly JW, Dillin A. Opposing activities protect against age-onset proteotoxicity. Science. 2006;313:1604–1610. doi: 10.1126/science.1124646. [DOI] [PubMed] [Google Scholar]
- Douglas PM, Treusch S, Ren HY, Halfmann R, Duennwald ML, Lindquist S, Cyr DM. Chaperone-dependent amyloid assembly protects cells from prion toxicity. Proc Natl Acad Sci USA. 2008;105:7206–7211. doi: 10.1073/pnas.0802593105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eisele F, Wolf DH. Degradation of misfolded protein in the cytoplasm is mediated by the ubiquitin ligase Ubr1. FEBS Lett. 2008;582:4143–4146. doi: 10.1016/j.febslet.2008.11.015. [DOI] [PubMed] [Google Scholar]
- Escusa-Toret S, Vonk WI, Frydman J. Spatial sequestration of misfolded proteins by a dynamic chaperone pathway enhances cellular fitness during stress. Nat Cell Biol. 2013;15:1231–1234. doi: 10.1038/ncb2838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fredrickson EK, Gallagher PS, Clowes Candadai SV, Gardner RG. Substrate recognition in nuclear protein quality control degradation is governed by exposed hydrophobicity that correlates with aggregation and insolubility. J Biol Chem. 2013;288:6130–6139. doi: 10.1074/jbc.M112.406710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fredrickson EK, Rosenbaum JC, Locke MN, Milac TI, Gardner RG. Exposed hydrophobicity is a key determinant of nuclear quality control degradation. Mol Biol Cell. 2011;22:2384–2395. doi: 10.1091/mbc.E11-03-0256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Mata R, Bebok Z, Sorscher EJ, Sztul ES. Characterization and dynamics of aggresome formation by a cytosolic GFP-chimera. J Cell Biol. 1999;146:1239–1254. doi: 10.1083/jcb.146.6.1239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giddings TH, Jr, O'Toole ET, Morphew M, Mastronarde DN, McIntosh JR, Winey M. Using rapid freeze and freeze-substitution for the preparation of yeast cells for electron microscopy and three-dimensional analysis. Methods Cell Biol. 2001;67:27–42. doi: 10.1016/s0091-679x(01)67003-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guerriero CJ, Weiberth KF, Brodsky JL. Hsp70 targets a cytoplasmic quality control substrate to the San1p ubiquitin ligase. J Biol Chem. 2013;288:18506–18520. doi: 10.1074/jbc.M113.475905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta R, Kasturi P, Bracher A, Loew C, Zheng M, Villella A, Garza D, Hartl FU, Raychaudhuri S. Firefly luciferase mutants as sensors of proteome stress. Nat Methods. 2011;8:879–884. doi: 10.1038/nmeth.1697. [DOI] [PubMed] [Google Scholar]
- Hartl FU, Bracher A, Hayer-Hartl M. Molecular chaperones in protein folding and proteostasis. Nature. 2011;475:324–332. doi: 10.1038/nature10317. [DOI] [PubMed] [Google Scholar]
- Heck JW, Cheung SK, Hampton RY. Cytoplasmic protein quality control degradation mediated by parallel actions of the E3 ubiquitin ligases Ubr1 and San1. Proc Natl Acad Sci USA. 2010;107:1106–1111. doi: 10.1073/pnas.0910591107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janke C, Magiera MM, Rathfelder N, Taxis C, Reber S, Maekawa H, Moreno-Borchart A, Doenges G, Schwob E, Schiebel E, Knop M. A versatile toolbox for PCR-based tagging of yeast genes: new fluorescent proteins, more markers and promoter substitution cassettes. Yeast. 2004;21:947–962. doi: 10.1002/yea.1142. [DOI] [PubMed] [Google Scholar]
- Johnston JA, Ward CL, Kopito RR. Aggresomes: a cellular response to misfolded proteins. J Cell Biol. 1998;143:1883–1898. doi: 10.1083/jcb.143.7.1883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung G, Jones G, Masison DC. Amino acid residue 184 of yeast Hsp104 chaperone is critical for prion-curing by guanidine, prion propagation, and thermotolerance. Proc Natl Acad Sci USA. 2002;99:9936–9941. doi: 10.1073/pnas.152333299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaganovich D, Kopito R, Frydman J. Misfolded proteins partition between two distinct quality control compartments. Nature. 2008;454:1088–1095. doi: 10.1038/nature07195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawaguchi Y, Kovacs JJ, McLaurin A, Vance JM, Ito A, Yao TP. The deacetylase HDAC6 regulates aggresome formation and cell viability in response to misfolded protein stress. Cell. 2003;115:727–738. doi: 10.1016/s0092-8674(03)00939-5. [DOI] [PubMed] [Google Scholar]
- Knop M, Finger A, Braun T, Hellmuth K, Wolf DH. Der1, a novel protein specifically required for endoplasmic reticulum degradation in yeast. EMBO J. 1996;15:753–763. [PMC free article] [PubMed] [Google Scholar]
- Kopito RR. Aggresomes, inclusion bodies and protein aggregation. Trends Cell Biol. 2000;10:524–530. doi: 10.1016/s0962-8924(00)01852-3. [DOI] [PubMed] [Google Scholar]
- Kryndushkin DS, Shewmaker F, Wickner RB. Curing of the [URE3] prion by Btn2p, a Batten disease-related protein. EMBO J. 2008;27:2725–2735. doi: 10.1038/emboj.2008.198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laemmli UK. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature. 1970;227:680–685. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- Lelouard H, Gatti E, Cappello F, Gresser O, Camosseto V, Pierre P. Transient aggregation of ubiquitinated proteins during dendritic cell maturation. Nature. 2002;417:177–182. doi: 10.1038/417177a. [DOI] [PubMed] [Google Scholar]
- Malinovska L, Kroschwald S, Munder MC, Richter D, Alberti S. Molecular chaperones and stress-inducible protein-sorting factors coordinate the spatiotemporal distribution of protein aggregates. Mol Biol Cell. 2012;23:3041–3056. doi: 10.1091/mbc.E12-03-0194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McClellan AJ, Scott MD, Frydman J. Folding and quality control of the VHL tumor suppressor proceed through distinct chaperone pathways. Cell. 2005;121:739–748. doi: 10.1016/j.cell.2005.03.024. [DOI] [PubMed] [Google Scholar]
- Morimoto RI. The heat shock response: systems biology of proteotoxic stress in aging and disease. Cold Spring Harb Symp Quant Biol. 2011;76:91–99. doi: 10.1101/sqb.2012.76.010637. [DOI] [PubMed] [Google Scholar]
- Nystrom T, Liu B. The mystery of aging and rejuvenation-a budding topic. Curr Opin Microbiol. 2014;18C:61–67. doi: 10.1016/j.mib.2014.02.003. [DOI] [PubMed] [Google Scholar]
- Ogrodnik M, Salmonowicz H, Brown R, Turkowska J, Sredniawa W, Pattabiraman S, Amen T, Abraham AC, Eichler N, Lyakhovetsky R, Kaganovich D. Dynamic JUNQ inclusion bodies are asymmetrically inherited in mammalian cell lines through the asymmetric partitioning of vimentin. Proc Natl Acad Sci USA. 2014;111:8049–8054. doi: 10.1073/pnas.1324035111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ollion J, Cochennec J, Loll F, Escude C, Boudier T. TANGO: a generic tool for high-throughput 3D image analysis for studying nuclear organization. Bioinformatics. 2013;29:1840–1841. doi: 10.1093/bioinformatics/btt276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park SH, Kukushkin Y, Gupta R, Chen T, Konagai A, Hipp MS, Hayer-Hartl M, Hartl FU. PolyQ Proteins Interfere with Nuclear Degradation of Cytosolic Proteins by Sequestering the Sis1p Chaperone. Cell. 2013;154:134–145. doi: 10.1016/j.cell.2013.06.003. [DOI] [PubMed] [Google Scholar]
- Parsell DA, Kowal AS, Singer MA, Lindquist S. Protein disaggregation mediated by heat-shock protein Hsp104. Nature. 1994;372:475–478. doi: 10.1038/372475a0. [DOI] [PubMed] [Google Scholar]
- Prasad R, Kawaguchi S, Ng DT. A nucleus-based quality control mechanism for cytosolic proteins. Mol Biol Cell. 2010;21:2117–2127. doi: 10.1091/mbc.E10-02-0111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenbaum JC, Gardner RG. How a disordered ubiquitin ligase maintains order in nuclear protein homeostasis. Nucleus. 2011;2:264–270. doi: 10.4161/nucl.2.4.16118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rujano MA, Bosveld F, Salomons FA, Dijk F, van Waarde MA, van der Want JJ, de Vos RA, Brunt ER, Sibon OC, Kampinga HH. Polarised asymmetric inheritance of accumulated protein damage in higher eukaryotes. PLoS Biol. 2006;4:e417. doi: 10.1371/journal.pbio.0040417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russell SJ, Steger KA, Johnston SA. Subcellular localization, stoichiometry, and protein levels of 26 S proteasome subunits in yeast. J Biol Chem. 1999;274:21943–21952. doi: 10.1074/jbc.274.31.21943. [DOI] [PubMed] [Google Scholar]
- Shiber A, Breuer W, Brandeis M, Ravid T. Ubiquitin conjugation triggers misfolded protein sequestration into quality-control foci when Hsp70 chaperone levels are limiting. Mol Biol Cell. 2013;24:2076–2087. doi: 10.1091/mbc.E13-01-0010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sontag EM, Vonk WI, Frydman J. Sorting out the trash: the spatial nature of eukaryotic protein quality control. Curr Opin Cell Biol. 2014;26C:139–146. doi: 10.1016/j.ceb.2013.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Specht S, Miller SB, Mogk A, Bukau B. Hsp42 is required for sequestration of protein aggregates into deposition sites in Saccharomyces cerevisiae. J Cell Biol. 2011;195:617–629. doi: 10.1083/jcb.201106037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spokoini R, Moldavski O, Nahmias Y, England JL, Schuldiner M, Kaganovich D. Confinement to organelle-associated inclusion structures mediates asymmetric inheritance of aggregated protein in budding yeast. Cell Rep. 2012;2:738–747. doi: 10.1016/j.celrep.2012.08.024. [DOI] [PubMed] [Google Scholar]
- Stirling PC, Bloom MS, Solanki-Patil T, Smith S, Sipahimalani P, Li Z, Kofoed M, Ben-Aroya S, Myung K, Hieter P. The complete spectrum of yeast chromosome instability genes identifies candidate CIN cancer genes and functional roles for ASTRA complex components. PLoS Genet. 2011;7:e1002057. doi: 10.1371/journal.pgen.1002057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szebenyi G, Wigley WC, Hall B, Didier A, Yu M, Thomas P, Kramer H. Hook2 contributes to aggresome formation. BMC Cell Biol. 2007;8:19. doi: 10.1186/1471-2121-8-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szeto J, Kaniuk NA, Canadien V, Nisman R, Mizushima N, Yoshimori T, Bazett-Jones DP, Brumell JH. ALIS are stress-induced protein storage compartments for substrates of the proteasome and autophagy. Autophagy. 2006;2:189–199. doi: 10.4161/auto.2731. [DOI] [PubMed] [Google Scholar]
- Tkach JM, Yimit A, Lee AY, Riffle M, Costanzo M, Jaschob D, Hendry JA, Ou J, Moffat J, Boone C, Davis TN, Nislow C, Brown GW. Dissecting DNA damage response pathways by analysing protein localization and abundance changes during DNA replication stress. Nat Cell Biol. 2012;14:966–976. doi: 10.1038/ncb2549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tong AH, Evangelista M, Parsons AB, Xu H, Bader GD, Page N, Robinson M, Raghibizadeh S, Hogue CW, Bussey H, Andrews B, Tyers M, Boone C. Systematic genetic analysis with ordered arrays of yeast deletion mutants. Science. 2001;294:2364–2368. doi: 10.1126/science.1065810. [DOI] [PubMed] [Google Scholar]
- Tyedmers J, Mogk A, Bukau B. Cellular strategies for controlling protein aggregation. Nat Rev Mol Cell Biol. 2010;11:777–788. doi: 10.1038/nrm2993. [DOI] [PubMed] [Google Scholar]
- Weisberg SJ, Lyakhovetsky R, Werdiger AC, Gitler AD, Soen Y, Kaganovich D. Compartmentalization of superoxide dismutase 1 (SOD1G93A) aggregates determines their toxicity. Proc Natl Acad Sci USA. 2012;109:15811–15816. doi: 10.1073/pnas.1205829109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wente SR, Rout MP. The nuclear pore complex and nuclear transport. Cold Spring Harb Perspect Biol. 2010;2:a000562. doi: 10.1101/cshperspect.a000562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou C, Slaughter BD, Unruh JR, Eldakak A, Rubinstein B, Li R. Motility and segregation of Hsp104-associated protein aggregates in budding yeast. Cell. 2011;147:1186–1196. doi: 10.1016/j.cell.2011.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Information
Review Process File