

Abstract
In mammalian cells, cargo-laden secretory vesicles leave the endoplasmic reticulum (ER) en route to ER-Golgi intermediate compartments (ERGIC) in a manner dependent on the COPII coat complex. We report here that COPII-coated transport carriers traverse a submicron, TFG (Trk-fused gene)-enriched zone at the ER/ERGIC interface. The architecture of TFG complexes as determined by three-dimensional electron microscopy reveals the formation of flexible, octameric cup-like structures, which are able to self-associate to generate larger polymers in vitro. In cells, loss of TFG function dramatically slows protein export from the ER and results in the accumulation of COPII-coated carriers throughout the cytoplasm. Additionally, the tight association between ER and ERGIC membranes is lost in the absence of TFG. We propose that TFG functions at the ER/ERGIC interface to locally concentrate COPII-coated transport carriers and link exit sites on the ER to ERGIC membranes. Our findings provide a new mechanism by which COPII-coated carriers are retained near their site of formation to facilitate rapid fusion with neighboring ERGIC membranes upon uncoating, thereby promoting interorganellar cargo transport.
Keywords: COPII vesicle transport, intrinsic disorder, secretion, single particle electron microscopy
Introduction
Biosynthetic secretory cargoes leave the endoplasmic reticulum (ER) in membrane-bound transport carriers, which assemble via the action of the coat protein complex II (COPII) machinery (Zanetti et al, 2011; Brandizzi & Barlowe, 2013; Lord et al, 2013; Venditti et al, 2014). The COPII coat consists of two distinct layers, both of which exhibit a high degree of flexibility to accommodate a wide range of membrane curvatures. The inner layer is composed of heterodimers of Sec23 and Sec24 that associate directly with the small GTPase Sar1, which harbors an amino-terminal amphipathic helix that inserts into lipid bilayers when bound to GTP (Matsuoka et al, 1998; Lee et al, 2005; Zanetti et al, 2013). The outer layer consists of heterotetramers of Sec13 and Sec31, which form lattices that surround the inner coat and provide further structural integrity (Barlowe et al, 1994; Fath et al, 2007; Stagg et al, 2008). Each layer is capable of enclosing both small vesicles (∽50–100 nm) and elongated tubules (∽300–400 nm), thereby enabling the transport of small and large cargoes (Zanetti et al, 2013).
The process of coat assembly has been reconstituted with a minimal set of components in vitro, fostered by the use of poorly hydrolyzable GTP analogs or mutant forms of Sar1 that are constitutively active (Matsuoka et al, 1998; Antonny et al, 2001; Schindler & Schekman, 2006; Zanetti et al, 2013). In vivo, COPII coat formation is tightly regulated. Somewhat paradoxically, the coat encodes a GTPase-activating protein (GAP), which promotes Sar1 dissociation from membranes. Specifically, the inner coat component Sec23 drives GTP hydrolysis on Sar1, in a manner that is stimulated by the outer coat subunit Sec31 (Antonny et al, 2001; Bi et al, 2007). However, the conserved scaffolding protein Sec16, which accumulates on ribosome-free regions of the ER that produce COPII vesicles, inhibits Sec23 GAP activity (Watson et al, 2006; Bhattacharyya & Glick, 2007; Kung et al, 2012; Yorimitsu & Sato, 2012). Additionally, Sec16 promotes GTP loading onto Sar1 by binding and potentially directing the localization of its guanine nucleotide exchange factor (GEF) Sec12 (Witte et al, 2011; Montegna et al, 2012). Thus, by regulating the actions of its GEF and GAP, Sec16 locally promotes Sar1 activity on specialized ER subdomains.
The COPII coat disassembly process is initiated rapidly following the scission of transport carriers from the ER, to enable downstream fusion events (Barlowe et al, 1994; Sato & Nakano, 2005; Lord et al, 2011). In addition to GTP hydrolysis on Sar1, phosphorylation of coat components (Sec23 and Sec31) has been proposed to drive uncoating (Lord et al, 2011; Koreishi et al, 2013). However, the timing of coat disassembly remains unclear, as does the precise sequence of events that underlie the redistribution of COPII subunits from membranes. In most metazoan systems, the majority of COPII transport carriers fuse with ER-Golgi intermediate compartments (ERGIC) that are juxtaposed to budding sites on ER membranes, prior to microtubule-dependent cargo trafficking to the Golgi apparatus (Martinez-Menarguez et al, 1999; Breuza et al, 2004; Appenzeller-Herzog & Hauri, 2006). Electron tomographic reconstructions of the ∽200–500 nm ER/ERGIC interface in numerous cell types highlight the presence of free COPII-associated transport carriers, which must traverse a region of cytoplasm prior to uncoating and fusing with other ER-derived vesicles or pre-existing ERGIC clusters (Zeuschner et al, 2006; Hughes et al, 2009; Witte et al, 2011). Microtubules are dispensable for this short-range movement (Presley et al, 1997), but it remains unclear whether there exist alternative mechanisms that limit vesicle diffusion away from the ER/ERGIC interface. Moreover, how the positioning of ERGIC membranes relative to the ER is maintained, as well as its functional significance, remains unknown.
We previously identified Trk-fused gene (TFG) as an additional conserved regulator of COPII-mediated transport that accumulates at the ER/Golgi interface in a variety of Caenorhabditis elegans cell types (Witte et al, 2011). Its depletion impairs the rate of cargo secretion from the ER, both in C. elegans and human tissue culture cells, suggesting an important role for TFG in coordinating transport in the early secretory pathway. Here, we find that human TFG functions at the ER/ERGIC interface in mammalian cells. We further define the structure of TFG complexes using three-dimensional electron microscopy and demonstrate that TFG can homo-polymerize to generate large polymers in vitro. Additionally, based on siRNA-mediated depletion studies in human cells, we show that TFG controls the distribution of COPII transport carriers subsequent to their scission from the ER, and its inhibition uncouples sites of COPII budding from ERGIC membranes, which leads to the dramatic reduction in the rate of cargo secretion. Together, our findings demonstrate that TFG regulates COPII-mediated transport by locally concentrating COPII-coated vesicular carriers and thereby enhances their ability to undergo homotypic and/or heterotypic fusion events at or near juxtaposed ERGIC membranes.
Results
TFG oligomerizes to form octameric cup-like structures
Based on sequence analysis, TFG isoforms contain at least three conserved regions, including an amino-terminal PB1 (Phox and Bem1p) domain, a coiled-coil motif, and a carboxyl-terminal proline/glutamine (PQ)-rich region (Supplementary Fig S1A). Our prior work showed that TFG expressed in both C. elegans and human cells are capable of oligomerization, in vitro and in vivo (Witte et al, 2011). Together, the PB1 domain and the coiled-coil motif form stable octamers in solution (Witte et al, 2011; Beetz et al, 2013). In addition, based on hydrodynamic studies, the PQ-rich region of C. elegans TFG is capable of homodimerization (Witte et al, 2011). To investigate the morphological nature of TFG oligomers, we performed negative staining EM analysis using recombinant proteins. For these studies, we took advantage of isoforms expressed in both human cells and C. elegans, enabling us to investigate the evolutionary conservation of TFG structure. Strikingly, both full-length proteins formed ∽11 nm ring-like structures, with a ∽4 nm central pore (Supplementary Fig S1B and C). By creating a series of truncations, we determined that the carboxyl-terminal half of TFG (including a majority of the PQ-rich region) was dispensable for ring formation in both cases (Supplementary Fig S1B and C). Consistent with subunit stoichiometry determined previously for TFG complexes, two-dimensional class averaging of particles formed by the TFG amino-terminus (C. elegans TFG1-195 and human TFG1-193) highlighted eight interconnected densities within the ring (Fig1A and B). Together, these data suggest that the PB1 domain and coiled-coil motif are sufficient to form octameric ring-like particles in solution.
Figure 1.
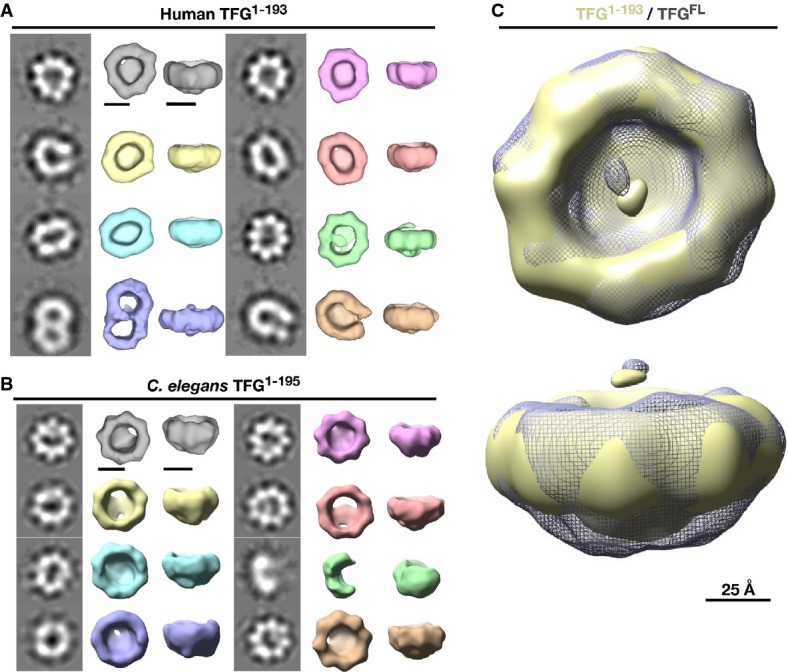
- Montage of class averages and 3D volumes of human TFG (amino acids 1-193; EMDataBank accession code EMD-6076). Two sets of 2D class averages and 3D volumes are shown with the left columns showing the class averages generated by reference-free alignment and classification and the right columns showing 3D RCT volumes corresponding to the class average to the left. Scale bars, 50 Å.
- Montage of class averages and 3D volumes of C. elegans TFG (amino acids 1-195; EMDataBank accession code EMD-6075), generated as described in (A). Scale bars, 50 Å.
- Superposition of 3D volumes of full-length human TFG and truncated TFG (amino acids 1-193). The truncated form is depicted in yellow, while the full-length form is depicted as gray mesh. The top view (top) exhibits limited differences in the structures, while the side view (bottom) shows extra density in the full-length TFG isoform. Scale bar, 25 Å.
Given their conformational similarity and high degree of purity, we focused on the amino-terminal fragments of TFG to conduct three-dimensional EM. Particles exhibited a strongly preferred orientation along the eightfold axis, enabling us to use the single particle technique known as random conical tilt (Radermacher, 1988). Reference-free single particle alignment and classification revealed that TFG assembles into octameric cup-like structures (Fig1A and B, Supplementary Tables S1 and S2). The class averages observed were all highly asymmetric and exhibited substantial flexibility, suggesting that TFG can adopt multiple conformations. Nonetheless, the resemblance observed between human and C. elegans TFG was striking, especially considering that the proteins exhibit < 23% identity (∽43% similarity) throughout their amino-termini (Supplementary Fig S2A).
To determine the relative positions of the amino- and carboxyl-terminal halves of TFG, we compared one of the structures (TFG1-193) to that of full-length human TFG and observed little change in the conformation of the TFG ring. However, the density observed at the base of the cup was noticeably diminished when the PQ-rich region was truncated (Fig1C). These data are again consistent with the idea that the PB1 domain and coiled-coil motif form the octameric ring, while the carboxyl-terminal PQ-rich region of each TFG monomer is dispensable for ring assembly and plays an alternative role in TFG complex formation and/or function.
To rule out artifacts that can arise from the use of stain, we also examined TFG complexes using cryogenic EM (cryo-EM). Consistent with our observations using negative staining, TFG formed ∽11 nm rings in vitreous ice (Supplementary Fig S2B). Taken together, our findings demonstrate that TFG subunits homo-polymerize to form small cup-like structures, which facilitate the export of COPII vesicles from the ER.
The PQ-rich region can promote further polymerization of octameric TFG complexes
Although the majority of TFG particles formed individual octameric cup-like structures, at least one highly represented population of human TFG1-193 assembled into complexes that resembled conjoined rings (Fig1A, bottom left). These findings raised the possibility that TFG octamers may co-assemble into larger polymers, which function during COPII vesicle transport. To better mimic biological processes that likely control TFG assembly in cells, we adjusted the ionic environment of solutions containing recombinant C. elegans TFG (100 mM NaCl and 50 mM HEPES, pH 7.6), a procedure used previously to enable the assembly of COPII cages in vitro (Stagg et al, 2008). Following the addition of specific salts in the Hofmeister series (potassium acetate and ammonium sulfate; Zhang & Cremer, 2006), TFG (1 μM) rapidly formed particles ∽200–300 nm in size, as judged by dynamic light scattering (Fig2A, Supplementary Fig S2C). Importantly, their inability to assemble in the presence of other salts (sodium chloride and magnesium chloride) and their highly limited size distribution argues against the possibility that these particles are aggregates (Supplementary Fig S2C).
Figure 2.
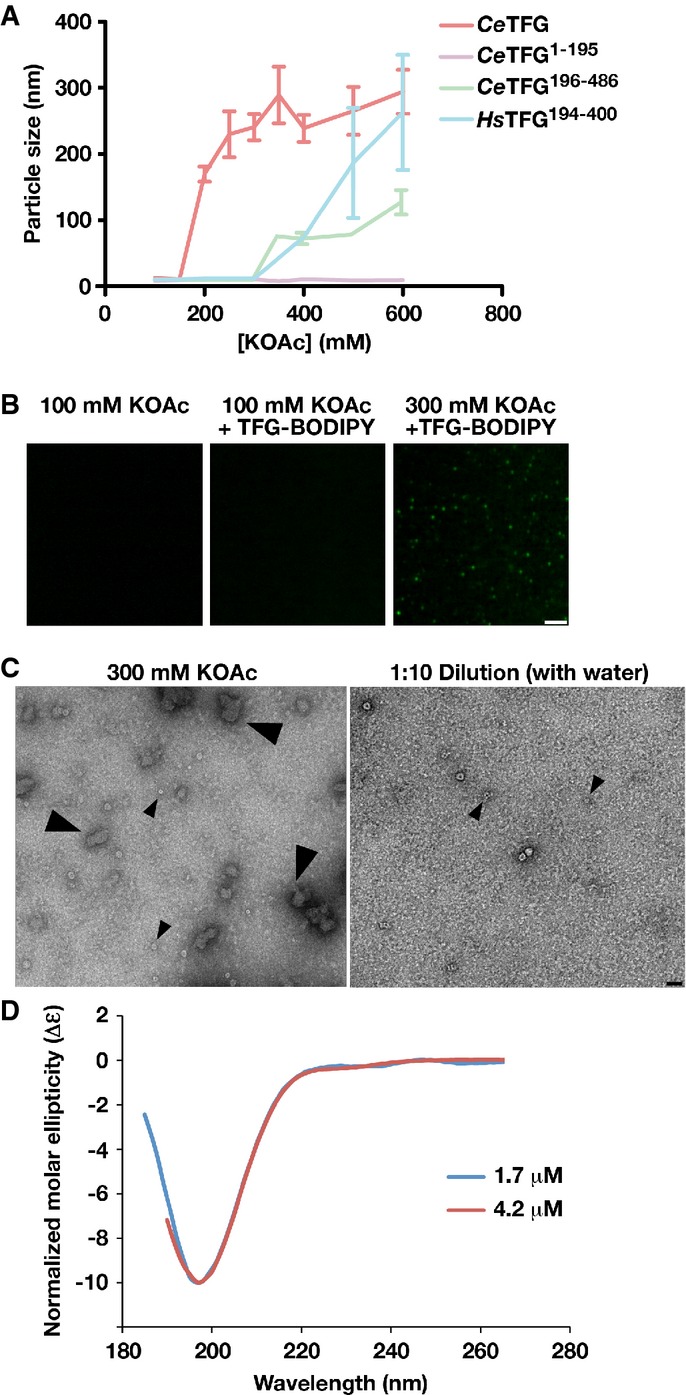
- TFG particle size was determined using dynamic light scattering under increasing potassium acetate concentrations. Error bars represent mean ± SEM; n = 10 replicates.
- Confocal images of recombinant, BODIPY-labeled TFG in the presence of varying potassium acetate concentrations. Scale bar, 2 μm.
- Recombinant TFG in the presence of varying potassium acetate concentrations was imaged by negative stain-EM. Large arrowheads highlight TFG polymers (only found in the presence of elevated potassium acetate). Small arrowheads highlight individual 11 nm cup-like TFG octamers. Scale bar, 30 nm.
- Circular dichroism spectroscopy was used to characterize the carboxyl-terminus of C. elegans TFG (amino acids 195-486). Samples were analyzed at different concentrations, and the data were normalized relative to one another. CD spectra were collected at 25°C in 25 mM sodium phosphate (pH 7.2) using a 1 mm path length quartz cell. The spectra are characteristic of an intrinsically disordered protein.
To further study the dynamics of TFG assembly, we labeled recombinant TFG with a fluorescent dye (BODIPY-FL) and examined particle formation using confocal microscopy. Upon addition of potassium acetate, visible TFG particles were immediately observed to assemble (Fig2B and Supplementary Movie S1). Analysis by negative staining EM confirmed the presence of amorphous 200–300 nm particles (Fig2C). However, upon dilution of the salt with water, the particles disassembled rapidly, again arguing against the idea that they are aggregates (Fig2C and Supplementary Movie S1).
To determine the region of TFG responsible for polymerization, we conducted salt shifts with recombinant forms of both the amino- and carboxyl-termini. In contrast to full-length TFG, the amino-terminus (1 μM) purified in the same buffer was refractory to assembling into larger structures upon addition of any salt (Fig2A). However, the carboxyl-terminal half of TFG (1 μM) behaved similarly to full-length TFG, although slightly higher concentrations of salt were necessary to promote particle formation (Fig2A). Together, these data suggest that TFG octamers can undergo further polymerization in a manner dependent on the PQ-rich domain to generate a structurally heterogeneous polymer.
Based on amino acid content, several prediction algorithms (DISOPRED2, IUPRED, DisEMBL, PredictProtein, etc.) suggest that the carboxyl-terminus of TFG, including its PQ-rich domain, is intrinsically unstructured (Supplementary Fig S3A and B; Campen et al, 2008). To confirm these in silico findings, we performed circular dichroism spectroscopy to characterize the secondary structure of the C. elegans TFG carboxyl-terminal region. Our data revealed a spectrum characteristic of a random coil architecture (Fig2D). Since exposed, unstructured domains are more sensitive to protease digestion (Johnson et al, 2012), we subjected full-length C. elegans TFG (4 μM) to limited proteolysis with the goal of obtaining further insight into the accessibility of its carboxyl-terminus in both cup-shaped TFG octamers and the larger 200–300 nm TFG polymers. In the absence of potassium acetate, we observed proteolytic cleavage of TFG after only 30 min of protease treatment, which proceeded toward completion within hours. In contrast, following salt-induced polymerization, TFG became more protease resistant, exhibiting only mild proteolysis during the time course of the experiment (Supplementary Fig S3C). Collectively, these findings suggest that the TFG carboxyl-terminus is intrinsically disordered, but can facilitate the formation of large TFG polymers from individual octameric cup-like structures.
TFG accumulates at the interface between ER and ERGIC membranes
We found previously that an ectopically expressed, fluorescently labeled version of TFG accumulates at or near sites of COPII vesicle formation (Witte et al, 2011). However, the resolution limit of the confocal microscope, combined with the analysis of a tagged gene product, prevents us from making a definitive conclusion regarding the distribution of endogenous TFG in human cells. To unambiguously determine the site of TFG action, we took advantage of affinity-purified antibodies directed against native TFG, combined with super-resolution structured illumination microscopy (SR-SIM), which provided spatial resolution to approximately 120 nm. For our studies, we used human telomerase reverse transcriptase (hTERT)-immortalized RPE-1 cells, which in some cases stably expressed low levels of mApple-Sec16B, a marker for sites of COPII vesicle formation on the ER (Supplementary Fig S4A). Importantly, we demonstrated that ectopically expressed mApple-Sec16B co-localizes precisely with endogenous Sec16A using SR-SIM, validating our use of this marker (Supplementary Fig S4B). Co-labeling mApple-Sec16B expressing cells with polyclonal α-TFG antibodies and monoclonal antibodies directed against endogenous ERGIC-53, which accumulates on ER-Golgi intermediate compartments (ERGIC), demonstrated that TFG is juxtaposed to both ER and ERGIC membranes (Fig3A). These data were further confirmed in wild-type cells, which did not express mApple-Sec16B (Supplementary Fig S4C–E). Additionally, in contrast to Sec16A, mApple-Sec16B and TFG, ERGIC-53 exhibited a substantially higher density throughout the peri-nuclear region of cells, consistent with its ability to traffic between ERGIC and Golgi membranes (Supplementary Fig S4E–G). Together, these data suggest that TFG accumulates specifically at the interface between ER and ERGIC membranes and, unlike ERGIC-53, does not localize to the Golgi apparatus (Supplementary Fig S4H).
Figure 3.
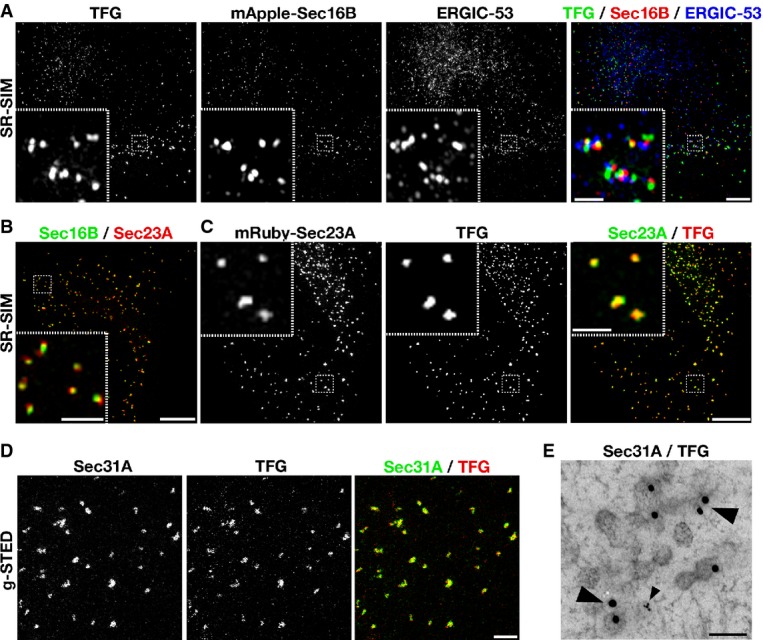
- The distributions of endogenous TFG and ERGIC-53 were examined in hTERT-immortalized RPE-1 cells stably expressing low levels of mApple-Sec16B using SR-SIM (more than 15 cells analyzed on 3 separate occasions). Scale bar, 5 μm. Inset scale bar, 1 μm.
- Cells stably expressing low levels of GFP-Sec16B and mRuby-Sec23A (Sec23A) were fixed and imaged using SR-SIM (n = 12). Scale bar, 5 μm. Inset scale bar, 1 μm.
- The distribution of endogenous TFG was examined in cells stably expressing low levels of mRuby-Sec23A (Sec23A) using SR-SIM (n = 10). Scale bar, 5 μm. Inset scale bar, 1 μm.
- The distributions of endogenous TFG and Sec31A were examined using g-STED (n = 8). Scale bar, 1 μm.
- The distributions of endogenous TFG and Sec31A were examined using immunogold EM. Large arrowheads highlight 15 nm gold particles bound to α-TFG antibodies. The small arrowhead highlights 5 nm gold particles bound to α-Sec31A antibodies. At least 15 different cells were examined, and representative images are shown. Scale bar, 100 nm.
The localization of TFG relative to Sec16 was markedly similar to that exhibited by inner (Sec23A) and outer (Sec31A) components of the COPII coat complex (Fig3B). We therefore explored the possibility that TFG and COPII-coated transport carriers occupy an identical space in cells. Using multiple super-resolution imaging techniques, including SR-SIM and gated stimulated emission depletion (g-STED) microscopy, which provided spatial resolution to ∽50 nm, we found that endogenous TFG co-localized tightly with both inner and outer COPII coat subunits (Fig3C and D). Immunogold-electron microscopy (EM) was further used to demonstrate that TFG accumulates throughout areas that are enriched with COPII-coated transport carriers (Fig3E). Collectively, these data conclusively demonstrate that TFG functions specifically at the interface between ER and ERGIC membranes.
The PQ-rich region is required for TFG localization at the ER/ERGIC interface
Our previous work showed that the TFG amino-terminus (TFG1-193), including the PB1 domain, the coiled-coil motif, and approximately one quarter of the PQ-rich region, is sufficient for its localization in wild-type cells (Witte et al, 2011). However, since the TFG amino-terminus co-assembles with the full-length isoform (Supplementary Fig S5A), the localization of the truncated mutant may be driven by the presence of endogenous TFG and thereby obscure prior conclusions. To address this issue, we expressed truncated forms of TFG in cells depleted of the endogenous protein. For these studies, we employed a siRNA that targets the 3′UTR of chromosomally expressed TFG specifically, which is absent in all transgenic constructs used. We investigated two truncation mutants, lacking either the final 50 (TFG1-350) or 100 (TFG1-300) amino acids of human TFG, and expressed each as a GFP fusion protein or untagged (to further verify that the GFP epitope did not interfere with localization). Untagged transgenes were expressed in tandem with soluble GFP, to enable the identification of transfected cells and to determine the relative level of transgene expression. Consistent with our previous work (Witte et al, 2011), both mutant isoforms targeted to sites of COPII vesicle accumulation in wild-type cells, similar to the full-length protein (Supplementary Fig S5B and C). In contrast, following depletion of endogenous TFG, we found that neither mutant protein was capable of assembly at the ER/ERGIC interface. At low expression levels, both truncated mutants exhibited a mostly diffuse, cytoplasmic distribution (Fig4A), although in some cases, TFG1-350 appeared to coalesce into puncta, which largely did not overlap with COPII staining. At higher levels of TFG1-350 expression (based on an elevated intensity of soluble GFP), we observed the formation of numerous submicron foci throughout the cytoplasm (Supplementary Fig S5D). In contrast to full-length TFG, however, the majority of these punctate structures failed to co-localize with Sec31, indicating that their formation was dysregulated. Regardless of TFG1-300 expression level, we were unable to detect it above cytoplasmic staining at sites of COPII vesicle formation (Supplementary Fig S5E). Taken together, these data strongly suggest that the TFG intrinsically disordered carboxyl-terminus plays a critical role in assembly of TFG complexes at the ER/ERGIC interface.
Figure 4.

- A Cells depleted of endogenous TFG were transfected with a variety of TFG transgenes (including full-length, untagged TFG, a truncation mutant encoding amino acids 1-300 of untagged TFG, and a truncation mutant encoding amino acids 1-350 of untagged TFG) and stained using α-TFG and α-Sec31A antibodies. Scale bar, 5 μm. Inset scale bar, 1 μm. Images shown are representative of at least 15 individual cells analyzed for each condition.
- B A recombinant polyhistidine- and SUMO-tagged TFG fragment (Sumo-TFGc, amino acids 194-400; total molecular mass of 42 kDa) was expressed and purified from Escherichia coli extracts using nickel affinity resin. Coomassie-stained gels of the peak elution fractions after separation of the recombinant protein on a S200 gel filtration column (top) or a 10–30% glycerol gradient (bottom) are shown. To determine the native molecular weight of the protein, the following equation was used: M = 6πηNas/(1 − υρ), where M is the native molecular weight, η is the viscosity of the medium, N is Avogadro's number, a is the Stokes radius, s is the sedimentation value, υ is the partial specific volume, and ρ is the density of the medium (Siegel & Monty, 1966). These data suggest that this fragment of TFG is capable of forming dimers in solution.
- C, D Control cells (C) or cells depleted of endogenous TFG (D) were transfected with a construct expressing TFG with a carboxyl-terminal GFP tag and stained using α-Sec31A antibodies. Scale bar, 5 μm. Images shown are representative of at least 20 individual cells analyzed for each condition.
One potential mechanism by which the carboxyl-terminal domains of TFG subunits could facilitate polymerization is through their self-association. In this manner, individual octameric rings could be tethered to one another and generate a meshwork. To investigate this possibility, we purified a recombinant form of the TFG PQ-rich region and subjected it to hydrodynamic analysis. Based on size exclusion chromatography and sedimentation through a glycerol gradient, we found that it was capable of forming dimers in vitro (Fig4B), consistent with our prior work using C. elegans TFG (Witte et al, 2011) and the idea that the carboxyl-terminus can link TFG octamers to one another. Based on these data, we reasoned that a large epitope tag appended onto the carboxyl-terminus of full-length TFG could impair its ability to polymerize in cells, as it would create a steric block to self-association. To test this possibility, we expressed a carboxyl-terminal GFP-tagged form of TFG in cells. Although the fusion protein was able to accumulate at sites of COPII vesicle formation in the presence of wild-type TFG, we found it was incapable of doing so when endogenous TFG was depleted (Fig4C and D). Instead, the fusion protein accumulated mostly in the cytoplasm and in large foci that were devoid of COPII subunits. These data are consistent with our findings that TFG assembles at the ER/ERGIC interface in a manner that requires its unobstructed PQ-rich region.
Depletion of TFG uncouples sites of COPII vesicle formation from ERGIC membranes
We previously demonstrated that depletion of TFG impairs the rate of cargo secretion from the ER in C. elegans and human cells (Witte et al, 2011). To gain mechanistic insight into this effect, we investigated the consequences of TFG depletion on organization of the early secretory pathway. We took advantage of multiple distinct siRNA sequences targeting TFG, all of which yielded highly similar results. We noted that extended periods of depletion (∽72 h after siRNA treatment) resulted in an inhibition of cellular proliferation, underscored by a lack of cells entering mitosis, the appearance of highly elongated cells, and an increase in apoptosis, as indicated by elevated caspase-3 activation (Supplementary Fig S6A and B). These data are consistent with previous findings, which demonstrated that inhibition of COPII vesicle secretion results in cell death as a consequence of elevated ER stress (Xu et al, 2005). After 72 h of siRNA treatment, fewer than 15% of cells remained attached to the growth substrate (Supplementary Fig S6C). In contrast, following 60 h of siRNA treatment, when levels of TFG were nearly undetectable at sites of COPII vesicle formation (Fig4A), the majority of cells remained attached and did not show signs of caspase-3 activation (Supplementary Fig S6B and C). As shorter periods of depletion did not effectively eliminate the presence of TFG in cells, we chose the 60-h timepoint of siRNA treatment for all subsequent studies. Importantly, we only examined morphologically normal cells, eliminating the possibility that perturbations observed were a consequence of apoptosis initiation.
Strikingly, we found that reduction of TFG levels resulted in an ∽threefold increase in the number of COPII-positive structures throughout the cytoplasm (Figs4A and 5A and B; Supplementary Movie S2). In contrast, the number of endogenous Sec16A-labeled sites on the ER did not increase significantly at this timepoint of siRNA treatment (Fig5A and C). Consistent with these data, we identified numerous COPII-labeled puncta, which lacked juxtaposed Sec16A, suggesting that COPII-coated structures were no longer retained at their original site of formation (Fig5A). Although the scattered COPII puncta remained mostly at or near ER membranes in TFG-depleted cells (Supplementary Fig S6D), they could represent vesicles, soluble pools of COPII coat proteins, or even aggregated empty cages. To help distinguish between these possibilities, we conducted a series of photobleaching experiments using cells stably expressing GFP-tagged Sec23A. We reasoned that the kinetics of fluorescence recovery after photobleaching would differ between bona fide transport carriers and alternative structures harboring COPII components. Our analysis indicated that GFP-Sec23A fluorescence recovered to a similar extent in the presence or absence of TFG, with half-times of 3.8 ± 0.4 s in control cells versus 4.2 ± 0.5 s in TFG-depleted cells, a statistically insignificant difference (Supplementary Fig S6E and Supplementary Movie S3). These data argue that the additional COPII-labeled structures are likely transport intermediates.
Figure 5.
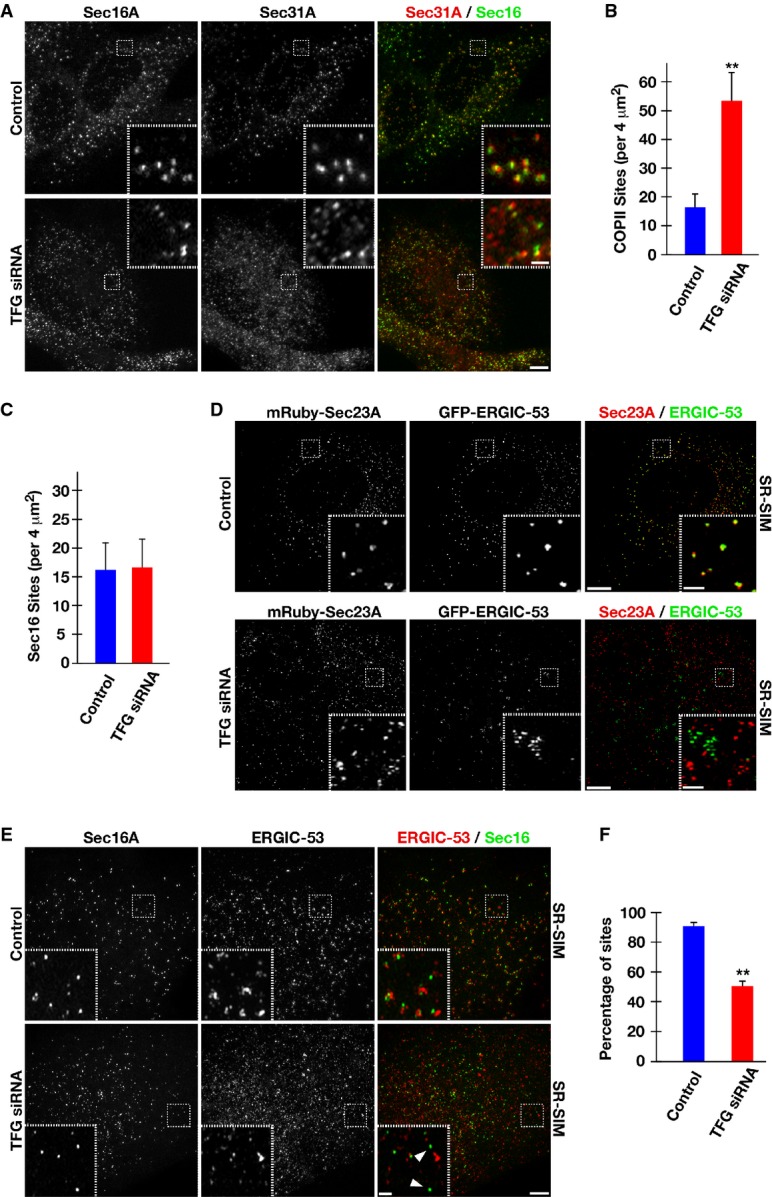
- Human RPE-1 cells were mock-transfected (control) or transfected with a TFG siRNA for 60 h. Cells were fixed and stained using α-Sec16A and α-Sec31 antibodies and imaged using confocal microscopy. Images shown are projections of 3D data sets (4 μm in z). Both individual and merged images with Sec16A in green and Sec31A in red are shown (at least 15 cells for each condition). Scale bar, 5 μm. Higher magnification views of the indicated regions (boxed) are also shown in the lower or upper right portion of each image. Inset scale bar, 1 μm.
- Quantification of the number of Sec31A-labeled structures in a 4 μm2 region. For both conditions (control or TFG siRNA-treated), at least 50 distinct regions away from the peri-nuclear Golgi were examined. Error bars represent mean ± SEM; at least 15 different cells per condition. **P < 0.01 compared with control, calculated using a paired t-test.
- Quantification of the number of Sec16A-labeled structures in a 4 μm2 region. For both conditions (control or TFG siRNA-treated), at least 50 distinct regions away from the peri-nuclear Golgi were examined. Error bars represent mean ± SEM; at least 15 different cells per condition. No statistically significant difference was observed, following analysis using a paired t-test.
- Cells stably expressing low levels of GFP-ERGIC-53 and mRuby-Sec23A were fixed and imaged using SR-SIM (top), or depleted of endogenous TFG prior to fixation and SR-SIM imaging (bottom). Images shown are representative of at least 15 individual cells analyzed for each condition. Scale bar, 5 μm. Inset scale bar, 1 μm.
- Human RPE-1 cells were mock-transfected (control) or transfected with a TFG siRNA for 60 h. Cells were fixed and stained using α-Sec16A and α-ERGIC-53 antibodies and imaged using SR-SIM. Images shown are projections of 3D data sets (4 μm in z). Both individual and merged images with Sec16A in green and ERGIC-53 in red are shown (at least 15 cells for each condition). Scale bar, 5 μm. Higher magnification views of the indicated regions (boxed) are also shown in the lower or upper right portion of each image. Arrowheads highlight Sec16A-labeled structures that do not exhibit juxtaposed ERGIC-53 staining. Inset scale bar, 1 μm.
- Based on structured illumination microscopy, quantification of the number of Sec16A-labeled structures juxtaposed (within 500 nm) to ERGIC-53-labeled structures is shown. For both conditions (control or TFG siRNA-treated), at least 1,000 unique Sec16A-labeled structures were examined. Error bars represent mean ± SEM; at least 15 different cells per condition. **P < 0.01 compared with control, calculated using a paired t-test.
In addition to altering the number of COPII-coated transport carriers, the tight apposition between ectopically expressed ERGIC-53 (fused to GFP) and COPII-labeled membranes (mRuby-Sec23A) was also dramatically diminished in TFG-depleted cells (Fig5D), as was the juxtaposed positioning of endogenous Sec16A and ERGIC-53 (Fig5E and F). These data suggest that numerous COPII-coated transport carriers were no longer restricted in their distribution, potentially as a result of disruption of the ER/ERGIC interface. Together, these data suggest that TFG plays an important role in locally retaining COPII vesicles and maintaining the ER/ERGIC interface. Consistent with this idea, immunogold EM experiments suggested that COPII-coated transport carriers were no longer clustered in the absence of TFG, in contrast to our observations in control cells (Supplementary Fig S6F).
Surprisingly, the disruption in early secretory pathway organization caused by TFG depletion failed to have a dramatic impact on steady state Golgi morphology (Fig6A). To determine a role for TFG in de novo Golgi formation, we treated cells with brefeldin A (BFA), a fungal metabolite that reversibly induces the Golgi to fuse with the ER (Lippincott-Schwartz et al, 1989), following TFG depletion and examined the rate at which the Golgi reassembled after washout. In contrast to control cells, TFG-depleted cells exhibited a substantial delay in regenerating a morphologically normal Golgi (Fig6A–C). Specifically, following TFG depletion, Golgi matrix proteins failed to coalesce near the nuclear periphery 30 min after BFA removal, and integral membrane proteins such as alpha-mannosidase II (ManII) remained trapped in the ER (Fig6A–C).
Figure 6.
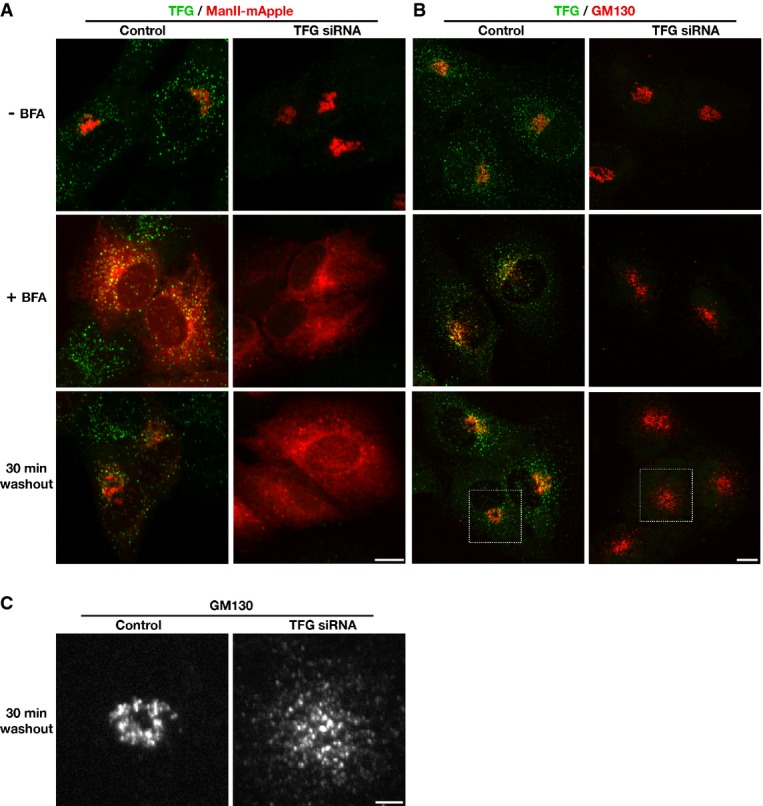
- Human RPE-1 cells stably expressing low levels of mannosidase II-mApple (ManII-mApple) were mock-transfected (control) or transfected with a TFG siRNA for 60 h, then fixed and stained using TFG antibodies and imaged using confocal microscopy. Alternatively, cells were either treated with DMSO (top row) or brefeldin A (BFA) for 1 h (middle row), followed by fixation, or washed into fresh media following DMSO or BFA treatment for 30 min (bottom row), prior to fixation. Images shown are projections of 3D data sets (4 μm in z). Merged images with TFG in green and ManII in red are shown. Scale bar, 5 μm. Images shown are representative of at least 10 individual cells analyzed for each condition.
- Human RPE-1 cells were mock-transfected (control) or transfected with a TFG siRNA for 60 h, then fixed and stained using TFG and GM130 antibodies and imaged using confocal microscopy. Alternatively, cells were either treated with DMSO (top row) or brefeldin A (BFA) for 1 h (middle row), followed by fixation, or washed into fresh media following DMSO or BFA treatment for 30 min (bottom row), prior to fixation. Images shown are projections of 3D data sets (4 μm in z). Merged images with TFG in green and GM130 in red are shown. Scale bar, 5 μm. Images shown are representative of at least 10 individual cells analyzed for each condition.
- Higher magnification views of the indicated regions in (B, boxed) are shown (GM130 distribution only). Scale bar, 2 μm.
Although Golgi reformation was slowed in TFG-depleted cells, it was not halted. Two hours after BFA removal, many cells exhibited a loosely organized Golgi apparatus in the absence of TFG (Fig7A). Additionally, following 1 h after BFA washout, several punctate structures containing ManII were observed throughout the cytoplasm. By co-staining cells with antibodies against Sec16 and COPII subunits, we found that many of these puncta corresponded to sites of COPII carrier formation at the ER (Fig7B). However, in many cases, ManII co-localized with COPII at sites that were devoid of juxtaposed Sec16 (Fig7C), indicating again that several cargo containing, COPII-coated transport carriers were no longer associated with their sites of biogenesis. These data further support a model in which TFG functions to retain cargo-containing COPII vesicles at the ER/ERGIC interface. In the absence of TFG, COPII carriers fail to remain clustered normally, which likely impairs the rate at which they fuse with ERGIC membranes to deliver cargo.
Figure 7.
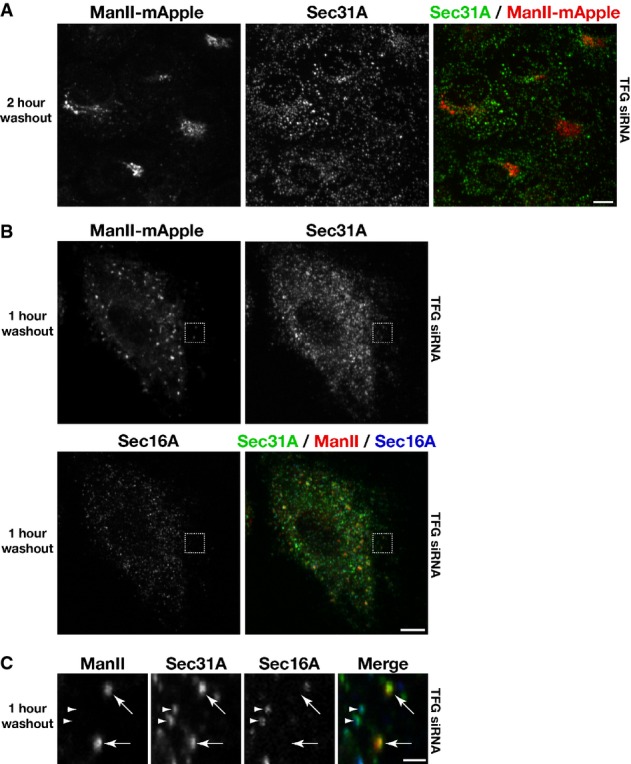
- Human RPE-1 cells stably expressing low levels of mannosidase II-mApple were transfected with a TFG siRNA for 60 h. Cells were subsequently treated with brefeldin A (BFA) for 1 h, followed by a wash into fresh media and further incubation for 2 h in the absence of BFA, prior to fixation. Images shown are projections of 3D data sets (4 μm in z). Merged images with Sec31A in green and ManII in red are shown (representative of at least 15 cells analyzed). Scale bar, 5 μm.
- Human RPE-1 cells stably expressing low levels of mannosidase II-mApple were transfected with a TFG siRNA for 60 h. Cells were subsequently treated with BFA for 1 h, followed by a wash into fresh media and further incubation for 1 h in the absence of BFA, prior to fixation and staining using α-Sec16A and α-Sec31A antibodies. Images shown are projections of 3D data sets (4 μm in z). Merged images with Sec31A (green), ManII (red), and Sec16A (blue) are shown (representative of at least 15 cells analyzed). Scale bar, 5 μm.
- Higher magnification views of the indicated regions in (B, boxed) are shown. Arrows highlight COPII-positive transport carriers that contain the cargo ManII, which are not juxtaposed to Sec16A-labeled sites on the ER. Additionally, arrowheads point out distinct foci in which COPII continues to associate with Sec16A-labeled sites, indicating that COPII vesicle formation continues in the absence of TFG. Scale bar, 1 μm.
Overexpression of TFG results in the titration of COPII subunits into enlarged foci
Since loss of TFG function results in the dispersal of COPII transport carriers, we also sought to determine the impact of excess TFG in cells. We first overexpressed the protein with an amino-terminal GFP tag and found that it formed enlarged foci relative to endogenous TFG (Supplementary Fig S7A). Strikingly, while the foci exhibited GFP fluorescence throughout their interior, TFG antibodies only stained the periphery of the structures, indicating a lack of antibody accessibility within them (Supplementary Fig S7A). We also overexpressed untagged TFG and found that the size of the structures formed was substantially larger as compared to those generated after GFP-TFG overexpression (Supplementary Fig S7B). These data raised concerns regarding the functionality of amino-terminally tagged TFG. To address this issue, we depleted endogenous TFG in cells stably expressing low levels of GFP-TFG (that did not form large foci) and measured the number of COPII-labeled structures that accumulated in cells. While depleted cells ectopically expressing untagged TFG harbored a similar number of COPII structures per unit area as compared to wild-type cells, those expressing GFP-TFG contained more than twice as many (Supplementary Fig S7C). Additionally, GFP-TFG expression did not rescue cells from apoptosis following extended treatment with siRNA targeting the 3′UTR of endogenous TFG. Together, these data indicate that an epitope tag on the TFG amino-terminus impairs its function. Consistent with this conclusion, we found that even a small 9 amino acid hemagglutinin-tag appended onto the amino-terminus of recombinant TFG perturbs its ring-like morphology in vitro (Supplementary Fig S7D).
Based on these findings, we focused on the use of untagged TFG for our overexpression studies. Strikingly, when we counterstained the enlarged foci generated in response to TFG overexpression using reagents against Sec23A or Sec31A, we found that they were highly enriched for COPII subunits (Supplementary Figs S7B and S8A). These data are consistent with the idea that TFG acts to locally concentrate COPII transport carriers. To determine whether the enlarged TFG foci contain membranous vesicles, we conducted a series of immunogold EM experiments. To do so, we first had to generate a clonal cell line that universally overexpressed untagged TFG, which we accomplished using a doxycycline-inducible system (Supplementary Fig S7E and F). Examination of the TFG-labeled foci by EM revealed elevated electron density as compared to the cytoplasm, which impaired our ability to easily visualize membranes. However, in several cases, we observed vesicular structures surrounding the foci, and when their electron density was minimal, we could observe 60–80 nm vesicles within the structures that were induced by TFG overexpression (Supplementary Fig S7G). These data are consistent with the idea that the TFG-induced foci trap COPII transport carriers.
We also found that ER membranes labeled by Sec61β wrapped around the enlarged TFG structures (Supplementary Fig S8B) and fluorescently tagged Sec16 clustered around their periphery (Supplementary Fig S8C). In a similar fashion, markers of the ERGIC (the COPI coat component β-COP and ERGIC-53) were also enriched at the foci, supporting the idea that TFG promotes the juxtaposed organization of ER and ERGIC membranes (Supplementary Fig S9A–C). Effects on Golgi morphology subsequent to TFG overexpression were more modest, resulting in only mild distention, but failing to substantially redistribute GM130, a marker of the cis-Golgi, onto the aberrant structures (Supplementary Fig S9D and E). Unfortunately, we were unable to determine the impact of TFG overexpression on the kinetics of secretory protein transport, as the overexpression of commonly used cargoes (e.g. VSVG-GFP) resulted in additional ER stress and caused cells to undergo apoptosis before measurements could be accurately made.
The enlarged foci generated by the overexpression of TFG are highly reminiscent of those formed upon overexpression of another PB1 domain-containing protein, SQSTM1 (Bjorkoy et al, 2005). To determine whether the effect on COPII carrier sequestration is specific to TFG foci, we overexpressed SQSTM1 in cells and counterstained using Sec31A antibodies. Unlike TFG-labeled structures, SQSTM1 foci failed to titrate COPII subunits (Supplementary Fig S9F). These data suggest that TFG is uniquely capable of recruiting and sequestering COPII carriers among PB1 domain-containing proteins. Another component of the early secretory pathway that is believed to function similarly to concentrate COPII transport carriers is the multimeric TRAPP tethering complex (Yu et al, 2006). To determine whether inhibition of TRAPP complex function phenocopies loss of TFG, we depleted a core TRAPP complex subunit (TRAPPC3/human Bet3) and determined its impact on COPII distribution in cells. Consistent with previous work (Yu et al, 2006), we found that TRAPPC3 depletion altered Golgi architecture and resulted in a dispersal of COPII transport carriers away from the perinuclear region (Supplementary Fig S9G and H). However, we failed to observe disengagement of Sec16A from COPII-labeled structures, suggesting that the TRAPP tethering complex acts at a distinct step of the early secretory pathway as compared to TFG. Overall, our findings define a new component of the ER/ERGIC interface that is present in metazoan cells to concentrate budded COPII transport carriers and link exit sites on the ER to ERGIC membranes.
Discussion
Although a stepwise series of reactions to facilitate COPII vesicle transport has been postulated (Cai et al, 2007; Lord et al, 2011), a mechanism to concentrate vesicles within the ER/ERGIC interface has gone unappreciated. In the absence of cytoskeletal elements, which have not been observed in reconstructions of the early secretory pathway (Zeuschner et al, 2006; Witte et al, 2011), an alternative pathway may exist to promote the clustering of COPII vesicles prior to their uncoating and fusion. Likewise, the juxtaposed organization of COPII budding sites on the ER relative to ERGIC membranes (Bannykh et al, 1996; Hughes et al, 2009) must be maintained to enable efficient secretory efflux. Our data support a model in which TFG functions in both capacities by potentially forming a meshwork at the ER/ERGIC interface, which transiently retains COPII-coated vesicles to provide sufficient time for uncoating and fusion with neighboring target compartments (Fig8).
Figure 8.
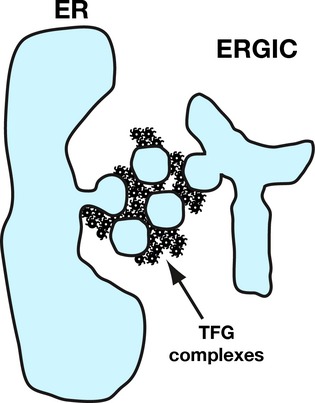
Model for TFG function at the ER/ERGIC interface
A model depicting the assembly of TFG at the ER/ERGIC interface, which facilitates the transient, local retention of COPII vesicles prior to vesicle uncoating and maintains the juxtaposed organization of ER and ERGIC membranes.
Regulation of COPII-mediated secretion
Although a minimal set of COPII coat proteins are sufficient to deform lipid bilayers and generate membrane vesicles and tubules in vitro, the formation and trafficking of COPII transport carriers in cells is tightly regulated by additional factors. Efficient coat polymerization requires strict control over Sar1 GTPase activity, which is mediated at least in part by Sec16 at ER membranes. Additionally, post-translational modifications on coat components have been suggested to further modulate coat assembly and stability (Aridor & Balch, 2000; Dudognon et al, 2004; Lord et al, 2011; Sharpe et al, 2011; Jin et al, 2012). In particular, phosphorylation of inner and outer coat subunits by casein kinases (CK1δ and CK2) inhibits their membrane association, leading to coat disassembly (Lord et al, 2011; Koreishi et al, 2013). Both kinases are found throughout cells, but enriched at ERGIC and Golgi membranes (Faust et al, 2001; Milne et al, 2001). These data suggest that the uncoating of COPII transport carriers, which is a prerequisite for fusion and cargo delivery, occurs at or near target compartments. By functioning at the interface between ER and ERGIC membranes, TFG polymers appear to regulate COPII transport in a unique manner, retaining vesicles locally and thereby promoting COPII subunit phosphorylation by casein kinases and coat disassembly. Consistent with this idea, overexpression of TFG generates enlarged polymers within cells that titrate COPII transport carriers. Moreover, depletion of TFG leads to an increased number of COPII-coated carriers, many of which are no longer juxtaposed to their sites of formation, as is observed in control cells. The random distribution of vesicles, coupled with a delay in uncoating, results in a diminished rate of cargo secretion. As a consequence, elevated ER stress caused by the secretory defect ultimately leads to apoptosis in cells lacking TFG (Xu et al, 2005).
Distinct functions for the amino- and carboxyl-termini of TFG
We previously showed that the amino-terminus of TFG was sufficient for its accumulation at sites of COPII vesicle formation in cells, in part through an interaction with Sec16 (Witte et al, 2011). However, our new findings indicate that the localization of this fragment additionally relies on the presence of endogenous TFG. Upon depleting the native protein, we found that both the amino-terminus and the carboxyl-terminal PQ-rich region of TFG play critical roles in its assembly at the ER/ERGIC interface. Based on our single particle EM analysis, the PB1 domain and coiled-coil motif co-assemble to form octameric cup-like structures. By contrast, the PQ-rich region of TFG is highly unstructured, harboring a high percentage of disorder-promoting residues (> 70%), including proline, glutamine, serine, alanine, and glycine (Ohashi et al, 2002; Campen et al, 2008). Thus, it is not surprising that we observed only modest differences in electron density when comparing full-length TFG to its amino-terminus, since much of the intrinsically disordered region is likely averaged out due to its inherent flexibility. Together with our finding that the PQ-rich region can self-associate, our data support a structural model for TFG octamers, in which a ring-like platform composed of PB1 domains and coiled-coil motifs functions as a hub to nucleate a set of intrinsically disordered domains, which can self-associate to form a polymer. In many ways, this architecture is analogous to the FG repeat nucleoporins, which play a key role in nuclear pore function by facilitating the selective transport of importin-bound cargo complexes into the nucleus (Frey & Gorlich, 2007; Hulsmann et al, 2012). When purified at high concentration, the Nup153 FG domain forms a hydrogel, which exhibits unique permeability to importin β, a nuclear transport receptor, while excluding other proteins of similar size (Frey et al, 2006; Frey & Gorlich, 2007). Similarly, TFG can form polymers in vitro, 200–300 nm in diameter, which may selectively retain transport carriers at the ER/ERGIC interface. In addition, it is feasible that the PB1 and coiled-coil domains of TFG also participate in the polymerization of TFG, perhaps by directly linking individual TFG cup-like structures, independently of the intrinsically disordered carboxyl-terminal region.
The importance of early secretory pathway organization
Many eukaryotes, including the budding yeast Saccharomyces cerevisiae, appear to lack an intermediate compartment between ER and Golgi membranes. Instead, cargoes are carried directly to the Golgi from sites of COPII vesicle formation on the ER. Recent studies using high-speed imaging highlight a transient association between the ER and cis-Golgi, enabling COPII vesicles to uncoat and rapidly deliver biosynthetic cargo to their target compartment (Kurokawa et al, 2014). In contrast, metazoans have evolved a network of vesicular-tubular clusters, commonly referred to as ERGIC membranes, which likely act as additional cargo sorting stations prior to microtubule-dependent Golgi transport (Appenzeller-Herzog & Hauri, 2006). Unlike the momentary association between the ER and Golgi in yeast, ER and ERGIC membranes are juxtaposed constitutively during interphase in metazoans. The development of such a secretory system necessitates new components to maintain its organization. Our data indicate that TFG functions in this capacity, linking sites of COPII vesicle formation on the ER to ERGIC membranes.
The precise function of the ERGIC remains unclear, although studies indicate that the organelle possesses a unique identity that is distinct from sites of COPII vesicle formation on the ER (Martinez-Menarguez et al, 1999; Appenzeller-Herzog & Hauri, 2006). Our data are consistent with this model and further suggest that the majority of COPII vesicles undergo uncoating and fusion at or near these membranes in mammalian cells. Until now, regulatory factors that specifically maintain the ER/ERGIC interface have not been clearly defined. A number of tethering factors, including p115 and the mammalian TRAPP complexes, have been implicated in the regulation of COPII vesicle fusion at the ERGIC. However, their roles in the early secretory pathway may be more general. Localization studies indicate that p115 accumulates at the Golgi apparatus, in addition to its distribution at ERGIC membranes (Nelson et al, 1998). Additionally, p115 depletion dramatically affects Golgi architecture, while its impact on cargo secretion is more variable, suggesting a dual role in general membrane tethering and selective cargo sorting (Grabski et al, 2012). In contrast to the relatively well-defined localization of p115, the distribution of TRAPPC3, a core subunit of all potential mammalian TRAPP complexes, is largely cytosolic, although a pool of this protein also accumulates at the ER/ERGIC interface (Yu et al, 2006). Similar to p115, depletion of TRAPPC3 causes Golgi fragmentation. However, it is unclear whether this effect is mediated by the loss of TRAPPC3 at the ER/ERGIC interface, or its role in COPI trafficking within the Golgi apparatus (Yu et al, 2006; Barrowman et al, 2010). Unlike p115 and TRAPPC3, TFG exhibits a unique distribution that is strictly limited to the ER/ERGIC interface, and its depletion dramatically affects the juxtaposed organization of these membranes without substantially affecting Golgi architecture at steady state. Nevertheless, export of proteins from the ER is substantially delayed in TFG-depleted cells, indicating for the first time that the juxtaposed organization of ER and ERGIC membranes is critical for efficient secretory flow.
Collectively, our findings support a new model in which TFG polymers form novel organelle contact sites that are critical for coupling efficient COPII vesicle budding from the ER with their uncoating and fusion at ERGIC membranes.
Materials and Methods
Antibody production, protein dye labeling and circular dichroism
Antibodies directed against human TFG were raised in rabbits by immunization with a GST fusion to a fragment of TFG (amino acids 1-193) produced in Escherichia coli. The antibodies were affinity-purified from serum by binding to columns of the same antigen following removal of the GST tag. For dye labeling of recombinant TFG, a single cysteine was added to the carboxyl-terminus of the protein and conjugated to a thiol reactive form of BODIPY-FL.
For circular dichroism studies, purified proteins at defined concentrations were analyzed using a 0.1 cm path length quartz cell. Protein was dialyzed overnight into 25 mM sodium phosphate (pH 7.2), and spectra were collected using a Model 202SF Circular Dichroism Spectrophotometer at 25°C.
Immunofluorescence, antibodies, fluorescent fusion proteins and fluorescence recovery after photobleaching
All images shown are representative of experiments conducted at least three times independently and include the analysis of at least 15 fields of cells for each condition, unless otherwise noted. Confocal images were acquired on a swept field confocal microscope (Nikon Ti-E) equipped with a Roper CoolSnap HQ2 CCD camera using a Nikon 60X, 1.4NA Planapo oil objective lens. Acquisition parameters were controlled by Nikon Elements software, and image analysis was conducted using Metamorph software. SR-SIM images were acquired on an OMX V4 Blaze Fast Structured Illumination system equipped with three custom high-speed sCMOS cameras using an API certified U-PLANAPO 60X, 1.42 NA objective lens. Acquisition parameters were controlled by API DeltaVision OMX Master acquisition software, and image analysis was conducted using the same software. Gated STED images were acquired on a Leica TCS STED system equipped with a DFC 360FX CCD camera using a HCX Planapo 100X, 1.4NA oil STED objective lens. Acquisition parameters were controlled by the Leica Application Suite platform (LAS AF), and image analysis was conducted using the same software. Alexa Fluor (488, 568 and 647) secondary antibodies were used for SR-SIM and confocal microscopy, and coverslips were mounted using Vectashield. For g-STED, the following dyes were used: Abberior STAR 440SX and DyLight 488, and coverslips were mounted using Prolong AntiFade.
RPE-1 cells were grown in DME/F-12 supplemented with 10% FBS, penicillin/streptomycin and l-glutamine (Invitrogen) and maintained at 37°C and 5% CO2. BSC-40 cells were grown similarly, except that DME media was used. Both types of cells were grown on glass coverslips in tissue culture dishes maintained at 37°C and 5% CO2, prior to fixation using paraformaldehyde or cold methanol. Immunofluorescence of fixed cells was performed as described previously (Audhya et al, 2007). Briefly, 40–60 Z sections at 0.2 μm steps were acquired (depending on sample thickness). To calculate the number of Sec16-, Sec31A- and ERGIC-53-labeled structures within a specified area (located distally from the peri-nuclear Golgi), the total number of structures (from a maximum intensity projection generated from no more then 1.4 μm in z) was measured after thresholding. The lower limit of intensity thresholds was defined by staining with secondary antibodies alone. The fluorescence intensity of each Sec16- or Sec31A-labeled structure was always confined to a maximum of seven Z planes (spaced 0.2 μm apart), which were used to generate maximum intensity projections. In some cases, a labeled structure from one Z section interfered with the analysis of another in a different Z section. These areas were not analyzed further. To quantify the juxtapositioning of Sec16- and ERGIC-53-labeled structures, a 1 μm diameter region around each Sec16-labeled structure was analyzed for the presence of ERGIC-53.
Staining using commercially available antibodies directed against Sec16A (Bethyl laboratories, A300-648A), and β-COP (Sigma, G6160) was preceded by fixation using cold methanol, as these antibodies were incompatible with paraformaldehyde fixation. Staining using antibodies directed against ERGIC-53 (Enzo Life Sciences, ALX-804-602), Sec31A (BD Biosciences, 612351), ERp57 (Proteintech, 15967-1-AP), GM130 (BD Biosciences, 610822) and TFG antibodies was typically preceded by fixation using 4% paraformaldehyde at 37°C, except in cases where cells were counterstained using Sec16A or β-COP antibodies. Constructs used to express fluorescently labeled proteins, including mRuby-Sec23A, mApple-Sec16B, GFP-TFG, GFP-SQSTM1, α-mannosidase II-mApple and mCherry-Sec61β, have been described previously (Pecot & Malhotra, 2004; Voeltz et al, 2006; Ichimura et al, 2008; Hughes & Stephens, 2010; Witte et al, 2011).
To analyze de novo Golgi assembly, cells were treated with the fungal metabolite brefeldin A (BFA, 10 μg/ml) for 1 h at 37°C and 5% CO2. Cells were subsequently washed with PBS three times to remove the drug and incubated with fresh, prewarmed media to allow export from the ER to proceed again. NucView (Biotium) staining was conducted as suggested by the manufacturer.
Photobleaching was carried out as described previously (Witte et al, 2011). The signal at the first post-bleach time was subtracted from all post-bleach measurements, and the percentage of fluorescence recovered at each time was calculated by dividing by the difference between the pre-bleach and first post-bleach measurements. Kaleidagraph software was used to fit the data and calculate the maximal fractional recovery and the half-time for recovery.
Transfections, electroporation, siRNAs, TALENs, and other plasmids
Plasmid transfections were conducted using FuGENE HD (Promega) as recommended by the manufacturer. TFG depletion studies were carried out using siRNAs (3′UTR TFG: 5′-CCAAAAGACUCCAGUACUA-3′ and exon 1 TFG: 5′-ACUUCUGAGUAAUGAUGAA-3′) that were transfected by using Lipofectamine RNAiMAX (Invitrogen). Cells were analyzed less than 72 h after transfection of siRNAs, and depletions were confirmed by immunoblotting cell extracts and/or staining using TFG antibodies. To overexpress untagged TFG, a cDNA encoding wild-type TFG was cloned into the bicistronic pSIN4 construct (Yu et al, 2009), which co-expresses GFP (using an internal ribosome entry site element). Cells were fixed and stained within 24 h after transfection (due to toxicity of TFG overexpression that results in apoptosis 36 h post-transfection), and TFG overexpressing cells were identified based on the presence of high levels of soluble GFP expression. To conduct studies in which endogenous TFG was depleted and replaced by a wild-type or truncated forms of TFG, cells were first transfected with a pSIN4-based plasmid harboring a specific TFG cDNA. After 24 h, cells were transfected with a siRNA targeting the endogenous 3′UTR of TFG, which is absent in the ectopically expressed transgene, and incubated for 60 h (at which point, less than 10% of endogenous TFG remains) prior to fixation and staining. Only cells expressing GFP were analyzed further.
To generate clonal doxycycline-inducible cell lines to overexpress TFG, we used a transcription activator-like effector nuclease (TALEN)-mediated targeting system, which incorporated the transgene at the AAVS1 locus (Qian et al, 2014). The puromycin-resistant donor cassette encoded TFG followed by an internal ribosome binding site and a gene encoding mApple. The cassette was electroporated into human RPE-1 cells together with the two AAVS1-specific TALEN plasmids at a ratio of 8:1:1, and cells were treated with puromycin (1 μg/ml) for approximately 3 weeks. Individual clones were picked and tested for doxycycline-induced mApple expression. For overexpression studies, cells were treated with doxycycline (1 μg/ml) for 48 h prior to analysis.
Three-dimensional electron microscopy
Recombinant samples were prepared for electron microscopy (EM) and subsequent RCT-particle processing as described previously (Leschziner & Nogales, 2006). All proteins were gel-filtered into 50 mM HEPES (pH 7.6) and 100 mM NaCl and analyzed by negative staining at the following concentrations: 0.01 mg/ml for C. elegans TFG1-195, 0.008 mg/ml for full-length C. elegans TFG, 0.009 mg/ml for human TFG1-193 and 0.005 mg/ml for full-length human TFG. Particles were picked using template-based matching followed by manual tilt pair editing with TiltPicker (Voss et al, 2009), resulting in 33,720 particle pairs for C. elegans TFG1-195, 18,000 pairs for full-length C. elegans TFG, 18,102 pairs for human TFG1-193 and 12,000 pairs for full-length human TFG. Samples were prepared for visualization by electron microscopy by placing 4 μl of a given sample on carbon-coated grids (Electron Microscopy Sciences) that had been plasma-cleaned for 15 s using a Gatan SOLARIS model 950 advanced plasma cleaner. Samples were then washed with H2O and stained with 2% uranyl formate. The samples were imaged using an FEI Titan Krios transmission electron microscope operated at 120 keV that was equipped with a Gatan Ultrascan 4000 × 400 CCD camera and the Leginon software (Suloway et al, 2005) for automatic data acquisition. Tilt pair images were collected at 59,000× magnification, 1 μm defocus and 15 e−/Å2 dose for the four different TFG constructs: full-length human TFG (2,248 pairs), truncated human TFG (amino acids 1-193; 1,181 pairs), full-length C. elegans TFG-1 (1,886 pairs) and truncated C. elegans TFG-1 (amino acids 1-195; 1,831 pairs). The tilt angles were 0° and 55° for all samples except truncated C. elegans TFG (amino acids 1-195), which was 0° and 45°.
For RCT-particle analysis, images were processed using Appion (Lander et al, 2009). Particles were picked using template-based matching followed by manual tilt pair editing with TiltPicker (Voss et al, 2009). For all datasets, XMIPP maximum likelihood alignment (Scheres et al, 2008) was used to align and classify the untilted particles. Forty class averages were generated, and averages with clearly discernable densities were selected for RCT reconstruction. 3D volumes were reconstructed using SPIDER (Frank et al, 1996) with Eulers determined from the tilt angle and the XMIPP alignment.
For cryo-EM, TFG particles were preserved in vitreous ice. Four microliters of the sample was placed on a C-flat holey carbon grid that had been plasma-cleaned for 8 s. Samples were blotted for 2.5 s and vitrified by using an FEI Vitrobot (FEI Company). Images were collected using a Titan Krios (FEI) transmission electron microscope operated at 120 keV at a dose of 15 e−/Å2 and a magnification of 59,000×.
Protein expression and purification and salt shifts
Recombinant protein expression was performed using BL21 (T1R, Sigma B2685) E. coli, and purifications were conducted using glutathione agarose beads (for GST fusions) or nickel affinity resin (for His-SUMO-tagged proteins). For samples applied to a S200 gel filtration column, the Stokes radius of each protein or protein complex was calculated from its elution volume based on the elution profiles of characterized standards. Glycerol gradients (10–30%) were poured using a Gradient Master and fractionated from the top by hand. Sedimentation values were calculated by comparing the position of the peak with that of characterized standards run on a separate gradient in parallel. For gel filtration chromatography, 1 ml samples were loaded onto a S200 column, and 1 ml fractions were subsequently collected. In all cases, 1/100 of the fractions were separated by SDS-PAGE and stained with Coomassie. For glycerol gradients, a 4 ml gradient (10–30% glycerol) was generated using a Gradient Master, and 100 μl fractions were collected by hand from the top of the gradient. In all cases, 1/10 of the fractions were separated by SDS-PAGE and stained with Coomassie. To determine the native molecular weight of proteins, the following equation was used: M = 6πηNas/(1 − υρ), where M is the native molecular weight, η is the viscosity of the medium, N is Avogadro's number, a is the Stokes radius, s is the sedimentation value, υ is the partial specific volume, and ρ is the density of the medium (Siegel & Monty, 1966). Immunoblotting of extracts were performed as described previously (Audhya et al, 2005), using antibodies directed against Sec23A (a gift from Dr. Randy Schekman), caspase-3 (Cell Signaling Technology, 9662) and α-tubulin (Sigma, T9026). Tunicamycin treatment (10 μg/ml) for 72 h was conducted as a positive control for examining apoptosis. To determine whether recombinant, truncated human TFG (amino acids 1-193, His-SUMO-tagged) could interact with full-length TFG (GST-tagged), transgenes were co-expressed in bacteria, and proteins were purified using glutathione agarose resin. Co-expression of truncated TFG with GST alone was performed as a control.
To study TFG polymerization in vitro, purified TFG isoforms (both wild-type and truncation mutants) were supplemented with a variety of salts at the concentrations indicated and analyzed using dynamic light scattering (DynaPro NanoStar; Wyatt Technology). At least 10 measurements were made at each concentration of salt, and the experiments were repeated at least three times independently. For partial proteolysis experiments, chymotrypsin was used at a 1:1,000 molar ratio with the protein under examination.
Immunogold labeling
Immunogold EM was conducted as described previously (Svitkina, 2009), with the exception that cells were subjected to high-pressure freezing and freeze substitution to maintain organelle morphology. Unless otherwise indicated, cells grown on sapphire disks were high-pressure frozen in a Baltec HPM 010 unit (Technotrade), freeze-substituted in 0.2% uranyl acetate (Electron Microscopy Sciences) and 0.2% glutaraldehyde (Electron Microscopy Sciences) in acetone at −90°C for 72 h, and warmed to −60°C for 24 h. After several acetone rinses, these samples were infiltrated with Lowicryl HM20 (Electron Microscopy Sciences) for 72 h and polymerized at −50°C under UV light for 48 h. Sections were mounted on formvar-coated nickel grids and blocked for 20 min with a 10% (w/v) solution of nonfat milk in Tris-buffered saline (TBS) containing 0.1% Tween-20. The sections were incubated sequentially with anti-TFG antibodies (100 μg/ml in TBS-Tween-20) for 1 h, secondary anti-rabbit IgG conjugated to 15-nm gold particles (1:10 in TBS-Tween-20), anti-Sec31A antibodies for 1 h (25 μg/ml in TBS-Tween 20), and secondary anti-mouse IgG conjugated to either 5-nm or 10-nm gold particles (1:10 in TBS-Tween-20). After each antibody incubation, grids were rinsed in TBS containing 0.5% Tween-20. Controls omitted the primary antibodies. Sections were stained with 2% uranyl acetate 70% methanol for 1 min.
Statistical analysis
Statistical significance was evaluated by performing a two-tailed Student's t-test.
Acknowledgments
This work was supported in part by grants from the NIH (GM088151 to AA and GM086892 to SMS), NSF (MCB1157824 to MSO), and the American Cancer Society (123268-RSG-12-139-01-CSM to AA). Circular dichroism data were obtained at the UW-Madison Biophysics Instrumentation Facility, which was established with support from the UW-Madison and grants BIR-9512577 (NSF) and S10 RR13790 (NIH). We thank Dr. Ed Chapman (UW-Madison) for providing us access to a dynamic light scattering instrument and Dr. David Stephens (University of Bristol, UK) and members of the Audhya lab for suggestions and critically reading this manuscript.
Author contributions
AJ, NB, MH, MSO, SMS, and AA conceived and designed experiments. AJ, NB, MH, ALS, JGP, LW, MSO, SMS, and AA performed experiments and analyzed data. MSO, SMS, and AA contributed reagents/materials/analysis tools. AA wrote the paper.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting Information
Supplementary Information
Supplementary Movie S1
Supplementary Movie S2
Supplementary Movie S3
Review Process File
References
- Antonny B, Madden D, Hamamoto S, Orci L, Schekman R. Dynamics of the COPII coat with GTP and stable analogues. Nat Cell Biol. 2001;3:531–537. doi: 10.1038/35078500. [DOI] [PubMed] [Google Scholar]
- Appenzeller-Herzog C, Hauri HP. The ER-Golgi intermediate compartment (ERGIC): in search of its identity and function. J Cell Sci. 2006;119:2173–2183. doi: 10.1242/jcs.03019. [DOI] [PubMed] [Google Scholar]
- Aridor M, Balch WE. Kinase signaling initiates coat complex II (COPII) recruitment and export from the mammalian endoplasmic reticulum. J Biol Chem. 2000;275:35673–35676. doi: 10.1074/jbc.C000449200. [DOI] [PubMed] [Google Scholar]
- Audhya A, Hyndman F, McLeod IX, Maddox AS, Yates JR, Desai A, Oegema K. A complex containing the Sm protein CAR-1 and the RNA helicase CGH-1 is required for embryonic cytokinesis in Caenorhabditis elegans. J Cell Biol. 2005;171:267–279. doi: 10.1083/jcb.200506124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Audhya A, McLeod IX, Yates JR, Oegema K. MVB-12, a fourth subunit of metazoan ESCRT-I, functions in receptor downregulation. PLoS One. 2007;2:e956. doi: 10.1371/journal.pone.0000956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bannykh SI, Rowe T, Balch WE. The organization of endoplasmic reticulum export complexes. J Cell Biol. 1996;135:19–35. doi: 10.1083/jcb.135.1.19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barlowe C, Orci L, Yeung T, Hosobuchi M, Hamamoto S, Salama N, Rexach MF, Ravazzola M, Amherdt M, Schekman R. COPII: a membrane coat formed by Sec proteins that drive vesicle budding from the endoplasmic reticulum. Cell. 1994;77:895–907. doi: 10.1016/0092-8674(94)90138-4. [DOI] [PubMed] [Google Scholar]
- Barrowman J, Bhandari D, Reinisch K, Ferro-Novick S. TRAPP complexes in membrane traffic: convergence through a common Rab. Nat Rev Mol Cell Biol. 2010;11:759–763. doi: 10.1038/nrm2999. [DOI] [PubMed] [Google Scholar]
- Beetz C, Johnson A, Schuh AL, Thakur S, Varga RE, Fothergill T, Hertel N, Bomba-Warczak E, Thiele H, Nurnberg G, Altmuller J, Saxena R, Chapman ER, Dent EW, Nurnberg P, Audhya A. Inhibition of TFG function causes hereditary axon degeneration by impairing endoplasmic reticulum structure. Proc Natl Acad Sci USA. 2013;110:5091–5096. doi: 10.1073/pnas.1217197110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhattacharyya D, Glick BS. Two mammalian Sec16 homologues have nonredundant functions in endoplasmic reticulum (ER) export and transitional ER organization. Mol Biol Cell. 2007;18:839–849. doi: 10.1091/mbc.E06-08-0707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bi X, Mancias JD, Goldberg J. Insights into COPII coat nucleation from the structure of Sec23.Sar1 complexed with the active fragment of Sec31. Dev Cell. 2007;13:635–645. doi: 10.1016/j.devcel.2007.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bjorkoy G, Lamark T, Brech A, Outzen H, Perander M, Overvatn A, Stenmark H, Johansen T. p62/SQSTM1 forms protein aggregates degraded by autophagy and has a protective effect on huntingtin-induced cell death. J Cell Biol. 2005;171:603–614. doi: 10.1083/jcb.200507002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brandizzi F, Barlowe C. Organization of the ER-Golgi interface for membrane traffic control. Nat Rev Mol Cell Biol. 2013;14:382–392. doi: 10.1038/nrm3588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breuza L, Halbeisen R, Jeno P, Otte S, Barlowe C, Hong W, Hauri HP. Proteomics of endoplasmic reticulum-Golgi intermediate compartment (ERGIC) membranes from brefeldin A-treated HepG2 cells identifies ERGIC-32, a new cycling protein that interacts with human Erv46. J Biol Chem. 2004;279:47242–47253. doi: 10.1074/jbc.M406644200. [DOI] [PubMed] [Google Scholar]
- Cai H, Yu S, Menon S, Cai Y, Lazarova D, Fu C, Reinisch K, Hay JC, Ferro-Novick S. TRAPPI tethers COPII vesicles by binding the coat subunit Sec23. Nature. 2007;445:941–944. doi: 10.1038/nature05527. [DOI] [PubMed] [Google Scholar]
- Campen A, Williams RM, Brown CJ, Meng J, Uversky VN, Dunker AK. TOP-IDP-scale: a new amino acid scale measuring propensity for intrinsic disorder. Protein Pept Lett. 2008;15:956–963. doi: 10.2174/092986608785849164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dudognon P, Maeder-Garavaglia C, Carpentier JL, Paccaud JP. Regulation of a COPII component by cytosolic O-glycosylation during mitosis. FEBS Lett. 2004;561:44–50. doi: 10.1016/S0014-5793(04)00109-7. [DOI] [PubMed] [Google Scholar]
- Fath S, Mancias JD, Bi X, Goldberg J. Structure and organization of coat proteins in the COPII cage. Cell. 2007;129:1325–1336. doi: 10.1016/j.cell.2007.05.036. [DOI] [PubMed] [Google Scholar]
- Faust M, Jung M, Gunther J, Zimmermann R, Montenarh M. Localization of individual subunits of protein kinase CK2 to the endoplasmic reticulum and to the Golgi apparatus. Mol Cell Biochem. 2001;227:73–80. [PubMed] [Google Scholar]
- Frank J, Radermacher M, Penczek P, Zhu J, Li Y, Ladjadj M, Leith A. SPIDER and WEB: processing and visualization of images in 3D electron microscopy and related fields. J Struct Biol. 1996;116:190–199. doi: 10.1006/jsbi.1996.0030. [DOI] [PubMed] [Google Scholar]
- Frey S, Richter RP, Gorlich D. FG-rich repeats of nuclear pore proteins form a three-dimensional meshwork with hydrogel-like properties. Science. 2006;314:815–817. doi: 10.1126/science.1132516. [DOI] [PubMed] [Google Scholar]
- Frey S, Gorlich D. A saturated FG-repeat hydrogel can reproduce the permeability properties of nuclear pore complexes. Cell. 2007;130:512–523. doi: 10.1016/j.cell.2007.06.024. [DOI] [PubMed] [Google Scholar]
- Grabski R, Balklava Z, Wyrozumska P, Szul T, Brandon E, Alvarez C, Holloway ZG, Sztul E. Identification of a functional domain within the p115 tethering factor that is required for Golgi ribbon assembly and membrane trafficking. J Cell Sci. 2012;125:1896–1909. doi: 10.1242/jcs.090571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughes H, Budnik A, Schmit K, Palmer KJ, Mantell J, Noakes C, Johnson A, Carter DA, Verkade P, Watson P, Stephens DJ. Organisation of human ER-exit sites: requirements for the localization of Sec16 to transitional ER. J Cell Sci. 2009;122:2924–2934. doi: 10.1242/jcs.044032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughes H, Stephens DJ. Sec16A defines the site for vesicle budding from the endoplasmic reticulum on exit from mitosis. J Cell Sci. 2010;123:4032–4038. doi: 10.1242/jcs.076000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hulsmann BB, Labokha AA, Gorlich D. The permeability of reconstituted nuclear pores provides direct evidence for the selective phase model. Cell. 2012;150:738–751. doi: 10.1016/j.cell.2012.07.019. [DOI] [PubMed] [Google Scholar]
- Ichimura Y, Kominami E, Tanaka K, Komatsu M. Selective turnover of p62/A170/SQSTM1 by autophagy. Autophagy. 2008;4:1063–1066. doi: 10.4161/auto.6826. [DOI] [PubMed] [Google Scholar]
- Jin L, Pahuja KB, Wickliffe KE, Gorur A, Baumgartel C, Schekman R, Rape M. Ubiquitin-dependent regulation of COPII coat size and function. Nature. 2012;482:495–500. doi: 10.1038/nature10822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson DE, Xue B, Sickmeier MD, Meng J, Cortese MS, Oldfield CJ, Le Gall T, Dunker AK, Uversky VN. High-throughput characterization of intrinsic disorder in proteins from the protein structure initiative. J Struct Biol. 2012;180:201–215. doi: 10.1016/j.jsb.2012.05.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koreishi M, Yu S, Oda M, Honjo Y, Satoh A. CK2 phosphorylates Sec31 and regulates ER-To-Golgi trafficking. PLoS One. 2013;8:e54382. doi: 10.1371/journal.pone.0054382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kung LF, Pagant S, Futai E, D'Arcangelo JG, Buchanan R, Dittmar JC, Reid RJ, Rothstein R, Hamamoto S, Snapp EL, Schekman R, Miller EA. Sec24p and Sec16p cooperate to regulate the GTP cycle of the COPII coat. EMBO J. 2012;31:1014–1027. doi: 10.1038/emboj.2011.444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurokawa K, Okamoto M, Nakano A. Contact of cis-Golgi with ER exit sites executes cargo capture and delivery from the ER. Nat Commun. 2014;5:3653. doi: 10.1038/ncomms4653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lander GC, Stagg SM, Voss NR, Cheng A, Fellmann D, Pulokas J, Yoshioka C, Irving C, Mulder A, Lau PW, Lyumkis D, Potter CS, Carragher B. Appion: an integrated, database-driven pipeline to facilitate EM image processing. J Struct Biol. 2009;166:95–102. doi: 10.1016/j.jsb.2009.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee MC, Orci L, Hamamoto S, Futai E, Ravazzola M, Schekman R. Sar1p N-terminal helix initiates membrane curvature and completes the fission of a COPII vesicle. Cell. 2005;122:605–617. doi: 10.1016/j.cell.2005.07.025. [DOI] [PubMed] [Google Scholar]
- Leschziner AE, Nogales E. The orthogonal tilt reconstruction method: an approach to generating single-class volumes with no missing cone for ab initio reconstruction of asymmetric particles. J Struct Biol. 2006;153:284–299. doi: 10.1016/j.jsb.2005.10.012. [DOI] [PubMed] [Google Scholar]
- Lippincott-Schwartz J, Yuan LC, Bonifacino JS, Klausner RD. Rapid redistribution of Golgi proteins into the ER in cells treated with brefeldin A: evidence for membrane cycling from Golgi to ER. Cell. 1989;56:801–813. doi: 10.1016/0092-8674(89)90685-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lord C, Bhandari D, Menon S, Ghassemian M, Nycz D, Hay J, Ghosh P, Ferro-Novick S. Sequential interactions with Sec23 control the direction of vesicle traffic. Nature. 2011;473:181–186. doi: 10.1038/nature09969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lord C, Ferro-Novick S, Miller EA. The highly conserved COPII coat complex sorts cargo from the endoplasmic reticulum and targets it to the golgi. Cold Spring Harb Perspect Biol. 2013;5:pii: a013367. doi: 10.1101/cshperspect.a013367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez-Menarguez JA, Geuze HJ, Slot JW, Klumperman J. Vesicular tubular clusters between the ER and Golgi mediate concentration of soluble secretory proteins by exclusion from COPI-coated vesicles. Cell. 1999;98:81–90. doi: 10.1016/S0092-8674(00)80608-X. [DOI] [PubMed] [Google Scholar]
- Matsuoka K, Orci L, Amberdt M, Bednarek SY, Mamamoto S, Schekman R, Yeung T. COPII-coated vesicle formation reconstituted with purified coat proteins and chemically defined liposomes. Cell. 1998;93:263–275. doi: 10.1016/s0092-8674(00)81577-9. [DOI] [PubMed] [Google Scholar]
- Milne DM, Looby P, Meek DW. Catalytic activity of protein kinase CK1 delta (casein kinase 1delta) is essential for its normal subcellular localization. Exp Cell Res. 2001;263:43–54. doi: 10.1006/excr.2000.5100. [DOI] [PubMed] [Google Scholar]
- Montegna EA, Bhave M, Liu Y, Bhattacharyya D, Glick BS. Sec12 binds to Sec16 at transitional ER sites. PLoS One. 2012;7:e31156. doi: 10.1371/journal.pone.0031156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson DS, Alvarez C, Gao YS, Garcia-Mata R, Fialkowski E, Sztul E. The membrane transport factor TAP/p115 cycles between the Golgi and earlier secretory compartments and contains distinct domains required for its localization and function. J Cell Biol. 1998;143:319–331. doi: 10.1083/jcb.143.2.319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohashi T, Hale CA, de Boer PA, Erickson HP. Structural evidence that the P/Q domain of ZipA is an unstructured, flexible tether between the membrane and the C-terminal FtsZ-binding domain. J Bacteriol. 2002;184:4313–4215. doi: 10.1128/JB.184.15.4313-4315.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pecot MY, Malhotra V. Golgi membranes remain segregated from the endoplasmic reticulum during mitosis in mammalian cells. Cell. 2004;116:99–107. doi: 10.1016/s0092-8674(03)01068-7. [DOI] [PubMed] [Google Scholar]
- Presley JF, Cole NB, Schroer TA, Hirschberg K, Zaal KJ, Lippincott-Schwartz J. ER-to-Golgi transport visualized in living cells. Nature. 1997;389:81–85. doi: 10.1038/38001. [DOI] [PubMed] [Google Scholar]
- Qian K, Huang CL, Chen H, Blackbourn LW, Chen Y, Cao J, Yao L, Sauvey C, Du Z, Zhang SC. A simple and efficient system for regulating gene expression in human pluripotent stem cells and derivatives. Stem Cells. 2014;32:1230–1238. doi: 10.1002/stem.1653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Radermacher M. Three-dimensional reconstruction of single particles from random and nonrandom tilt series. J Electron Microsc Tech. 1988;9:359–394. doi: 10.1002/jemt.1060090405. [DOI] [PubMed] [Google Scholar]
- Sato K, Nakano A. Dissection of COPII subunit-cargo assembly and disassembly kinetics during Sar1p-GTP hydrolysis. Nat Struct Mol Biol. 2005;12:167–174. doi: 10.1038/nsmb893. [DOI] [PubMed] [Google Scholar]
- Scheres SH, Nunez-Ramirez R, Sorzano CO, Carazo JM, Marabini R. Image processing for electron microscopy single-particle analysis using XMIPP. Nat Protoc. 2008;3:977–990. doi: 10.1038/nprot.2008.62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schindler AJ, Schekman R. In vitro reconstitution of ER-stress induced ATF6 transport in COPII vesicles. Proc Natl Acad Sci USA. 2006;106:17775–17780. doi: 10.1073/pnas.0910342106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharpe LJ, Luu W, Brown AJ. Akt phosphorylates Sec24: new clues into the regulation of ER-to-Golgi trafficking. Traffic. 2011;12:19–27. doi: 10.1111/j.1600-0854.2010.01133.x. [DOI] [PubMed] [Google Scholar]
- Siegel LM, Monty KJ. Determination of molecular weights and frictional ratios of proteins in impure systems by use of gel filtration and density gradient centrifugation. Application to crude preparations of sulfite and hydroxylamine reductases. Biochim Biophys Acta. 1966;112:346–362. doi: 10.1016/0926-6585(66)90333-5. [DOI] [PubMed] [Google Scholar]
- Stagg SM, LaPointe P, Razvi A, Gurkan C, Potter CS, Carragher B, Balch WE. Structural basis for cargo regulation of COPII coat assembly. Cell. 2008;134:474–484. doi: 10.1016/j.cell.2008.06.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suloway C, Pulokas J, Fellmann D, Cheng A, Guerra F, Quispe J, Stagg S, Potter CS, Carragher B. Automated molecular microscopy: the new Leginon system. J Struct Biol. 2005;151:41–60. doi: 10.1016/j.jsb.2005.03.010. [DOI] [PubMed] [Google Scholar]
- Svitkina T. Imaging cytoskeleton components by electron microscopy. Methods Mol Biol. 2009;586:187–206. doi: 10.1007/978-1-60761-376-3_10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venditti R, Wilson C, De Matteis MA. Exiting the ER: what we know and what we don't. Trends Cell Biol. 2014;24:9–18. doi: 10.1016/j.tcb.2013.08.005. [DOI] [PubMed] [Google Scholar]
- Voeltz GK, Prinz WA, Shibata Y, Rist JM, Rapoport TA. A class of membrane proteins shaping the tubular endoplasmic reticulum. Cell. 2006;124:573–586. doi: 10.1016/j.cell.2005.11.047. [DOI] [PubMed] [Google Scholar]
- Voss NR, Yoshioka CK, Radermacher M, Potter CS, Carragher B. DoG Picker and TiltPicker: software tools to facilitate particle selection in single particle electron microscopy. J Struct Biol. 2009;166:205–213. doi: 10.1016/j.jsb.2009.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watson P, Townley AK, Koka P, Palmer KJ, Stephens DJ. Sec16 defines endoplasmic reticulum exit sites and is required for secretory cargo export in mammalian cells. Traffic. 2006;7:1678–1687. doi: 10.1111/j.1600-0854.2006.00493.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Witte K, Schuh AL, Hegermann J, Sarkeshik A, Mayers JR, Schwarze K, Yates JR, Eimer S, Audhya A. TFG-1 function in protein secretion and oncogenesis. Nat Cell Biol. 2011;13:550–558. doi: 10.1038/ncb2225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu C, Bailly-Maitre B, Reed JC. Endoplasmic reticulum stress: cell life and death decisions. J Clin Invest. 2005;115:2656–2664. doi: 10.1172/JCI26373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yorimitsu T, Sato K. Insights into structural and regulatory roles of Sec16 in COPII vesicle formation at ER exit sites. Mol Biol Cell. 2012;23:2930–2942. doi: 10.1091/mbc.E12-05-0356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu S, Satoh A, Pypaert M, Mullen K, Hay JC, Ferro-Novick S. mBet3p is required for homotypic COPII vesicle tethering in mammalian cells. J Cell Biol. 2006;174:359–368. doi: 10.1083/jcb.200603044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu J, Hu K, Smuga-Otto K, Tian S, Stewart R, Slukvin II, Thomson JA. Human induced pluripotent stem cells free of vector and transgene sequences. Science. 2009;324:797–801. doi: 10.1126/science.1172482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zanetti G, Pahuja KB, Studer S, Shim S, Schekman R. COPII and the regulation of protein sorting in mammals. Nat Cell Biol. 2011;14:20–28. doi: 10.1038/ncb2390. [DOI] [PubMed] [Google Scholar]
- Zanetti G, Prinz S, Daum S, Meister A, Schekman R, Bacia K, Briggs JA. The structure of the COPII transport-vesicle coat assembled on membranes. Elife. 2013;2:e00951. doi: 10.7554/eLife.00951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeuschner D, Geerts WJ, van Donselaar E, Humbel BM, Slot JW, Koster AJ, Klumperman J. Immuno-electron tomography of ER exit sites reveals the existence of COPII-coated transport carrier. Nat Cell Biol. 2006;8:377–383. doi: 10.1038/ncb1371. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Cremer PS. Interactions between macromolecules and ions: the Hofmeister series. Curr Opin Chem Biol. 2006;10:658–663. doi: 10.1016/j.cbpa.2006.09.020. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Information
Supplementary Movie S1
Supplementary Movie S2
Supplementary Movie S3
Review Process File