

Abstract
Background
The animal gastrointestinal tract contains a complex community of microbes, whose composition ultimately reflects the co-evolution of microorganisms with their animal host and the diet adopted by the host. Although the importance of gut microbiota of humans has been well demonstrated, there is a paucity of research regarding non-human primates (NHPs), especially herbivorous NHPs.
Results
In this study, an analysis of 97,942 pyrosequencing reads generated from Rhinopithecus bieti fecal DNA extracts was performed to help better understanding of the microbial diversity and functional capacity of the R. bieti gut microbiome. The taxonomic analysis of the metagenomic reads indicated that R. bieti fecal microbiomes were dominated by Firmicutes, Bacteroidetes, Proteobacteria and Actinobacteria phyla. The comparative analysis of taxonomic classification revealed that the metagenome of R. bieti was characterized by an overrepresentation of bacteria of phylum Fibrobacteres and Spirochaetes as compared with other animals. Primary functional categories were associated mainly with protein, carbohydrates, amino acids, DNA and RNA metabolism, cofactors, cell wall and capsule and membrane transport. Comparing glycoside hydrolase profiles of R. bieti with those of other animal revealed that the R. bieti microbiome was most closely related to cow rumen.
Conclusions
Metagenomic and functional analysis demonstrated that R. bieti possesses a broad diversity of bacteria and numerous glycoside hydrolases responsible for lignocellulosic biomass degradation which might reflect the adaptations associated with a diet rich in fibrous matter. These results would contribute to the limited body of NHPs metagenome studies and provide a unique genetic resource of plant cell wall degrading microbial enzymes. However, future studies on the metagenome sequencing of R. bieti regarding the effects of age, genetics, diet and environment on the composition and activity of the metagenomes are required.
Electronic supplementary material
The online version of this article (doi:10.1186/s12864-015-1378-7) contains supplementary material, which is available to authorized users.
Keywords: Gastrointestinal microbiota, Rhinopithecus bieti, Metagenomics, Lignocellulose degradation, Pyrosequencing
Background
The Yunnan snub-nosed monkey (Rhinopithecus bieti) is an endangered colobine endemic to high-altitude forests ranging from 3000 to 4400 meters in southwestern China and southeastern Tibet [1]. Overall, these colobines can be classified as herbivores, ingesting flowers, fruits, leaves, and seeds to varying degrees [2]. The R. bieti possesses specialized S-shaped and partitioned stomachs where microbial fermentation of cellulose takes place [3,4]. This adaptation enables them to eat food containing high levels of structural polysaccharides, i.e., cellulose and related compounds. Given the above features, this species has received significant attention from researchers and serves as an important model organism for studying the evolution of the primate diet. However, researches on R. bieti have mostly focused on aspects of taxonomy, ecology, anatomy and conservation genetics.
The gastrointestinal tract of animals harbors a complex microbial community, and the composition of this community ultimately reflects the co-evolution of microorganisms with their animal host and the diet adopted by the host [5]. It’s well known that herbivores lack the enzymatic capacity needed to degrade plant polysaccharides, particularly cellulose, and instead rely on community of microorganisms that have this capacity. Therefore, R. bieti are expected to have a well-adapted gut microbiota. Indeed, the limited studies published to date suggest the microbiomes of R. bieti possess a large number of bacteria that may be involved in degradation of cellulose [6]; however, very little is currently known about the genetic potential and structure-function relationships intrinsic to these microbiomes.
Recently, next-generation sequencing technologies have been used to characterize the microbial diversity and functional capacity of a range of microbial communities in the gastrointestinal tracts of humans [7-10] as well as in several animal species [11-25]. The most important advantages of this cloning-independent approach are the avoidance of cloning bias and the bias introduced by PCR amplification. To the best of our knowledge, this study was the first to apply a random sample pyrosequencing approach to analyze the metagenome of R. bieti, an herbivore whose habitat and diet are very different compared to the herbivores studied so far.
Results and discussion
The analysis of the reads yielded a high percentage of species identification in complex metagenomes and even higher in less complex samples. Long sequence reads from 454 GS FLX Titanium pyrosequencing provided the high specificity needed to compare the sequenced reads with the DNA or protein databases and allowed the unambiguous assignment of closely related species. The initial pyrosequencing runs yielded 97,942 reads containing 37,482,416 bases of sequence, with an average read length of 382 bp. Prior to further processing, the raw read data were subjected to the Metagenome Rapid Annotation using Subsystem Technology (MG-RAST) v.3.0 online server quality control pipeline [26] to remove duplicate and low quality reads (Additional file 1). The filtering step removed 9.6% of reads in the sample. The unique sequence reads that passed the quality control (QC) filtering step were then subjected to further analysis focusing on biodiversity and functional annotation. All reads were deposited in the National Center for Biotechnology Information (NCBI) and can be accessed in the Short Read Archive (SRA) under the accession number SRX493843.
Phylogenetic analysis of R. bieti fecal bacteria, eukaryota, archaea, and viruses
The overview of the phylogenetic computations provided 97.81% bacteria, 1.27% eukaryota, 0.73% archaea, and 0.17% viruses. In the R. bieti intestinal metagenome, Firmicutes was the most predominant phylum (39.36%), followed by Bacteroidetes (27.6%), Proteobacteria (19.41%), Actinobacteria (3.61%) and Spirochaetes (2.01%) (Figure 1). Compared with the previous 16S rRNA gene-based data [6], Firmicutes were the dominant bacteria phylum. In addition, higher percentages of Bacteroidetes and lower percentages of Spirochaetes in the R. bieti intestinal metagenome were observed. This discrepancy may have been caused by the biases associated with the primers, PCR reaction conditions, or selection of clones [27].
Figure 1.
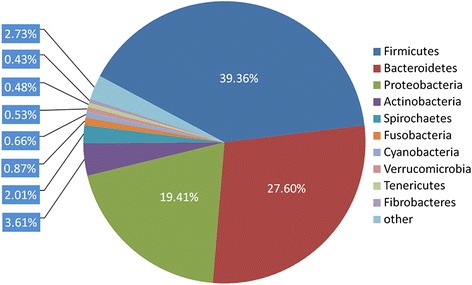
Bacterial phylum profiles of the R. bieti microbiome. The percentage of the R. bieti fecal metagenomic sequences assigned to M5NR database is shown. Through the “Organism Abundance” tool in MG-RAST, the R. bieti fecal sequencing runs were determined from the M5NR database with the BLASTx algorithm. The e-value cutoff for the metagenomic sequence matches to the M5NR database was 1 × 10−5, with a minimum alignment length of 30 bp.
Within the Firmicutes group, Clostridiales were most predominant, among which Clostridium was overrepresented (Additional file 2), which is consistent with previous 16S rRNA gene-based data [6]. The presence of a large proportion of Clostridia was likely to be important for lignocellulosic biomass degradation [28]. Several cellulolytic microbes such as C. phytofermentans, Ruminococcus albus, C. thermocellum, C. cellulolyticum, R. flavefaciens and C. cellulovorans were abundant in the R. bieti metagenome (Additional file 2). Among clostridial genomes sequenced to date, C. phytofermentans has the highest number of genes encoding enzymes for the modification and breakdown of complex carbohydrates. It contains genes for 161 carbohydrate-active enzymes (CAZy), which include 108 glycoside hydrolases (GH) spreading across 39 families [29]. C. thermocellum is an anaerobic thermophilic bacterium that exhibits one of the highest rates of cellulose utilization among described microorganisms [28]. R. flavefaciens is a specialist cellulolytic bacterial species characterized in the rumen, other herbivorous animals and humans. Currently it is the only rumen bacterium known to produce a defined cellulosome [30] which is usually associated with improved cellulolytic efficiency [31]. These cellulolytic Clostridia, which are ubiquitous in cellulosic anaerobic environments, represent a major paradigm for efficient biological degradation of cellulosic biomass [32].
Bacteroidetes were the second predominant phylum in the R. bieti gastrointestinal tract with Bacteroidales as the primary contributor to the Bacteroidetes populations, followed by Flavobacteria, Sphingobacteriales and Cytophagales. The major genus in the Bacteroidetes phylum was Bacteroides. Bacteroides are commonly found in the human intestine where they have a symbiotic host-bacterial relationship with humans. They assist in breaking down food and producing valuable nutrients and energy that the body needs. B. vulgatus, B. fragilis and Flavobacterium johnsoniae comprise about 1.1%, 1.1%, and 1.08% of the reads analyzed respectively (Additional file 2); therefore, it is considered the predominant species in the R. bieti metagenome. B. fragilis is a ubiquitous Gram-negative anaerobic bacterium that inhabits the lower gastrointestinal tract of most mammals [5]. Recent findings have revealed that this organism possesses the ability to direct the cellular and physical maturation of the host immune system and protect its host from experimental colitis [33-35]. B. vulgatus is among the most commonly isolated microbes from the human gastrointestinal tract, and it has been found to constitute part of the core gut microbiota in healthy humans [10,36]. According to the CAZy classification scheme, B. vulgatus is the only sequenced gut Bacteroidetes with a gene encoding a xylanase [37]. F. johnsoniae digests many polysaccharides and proteins, but it is best known for its ability to rapidly digest insoluble chitin [38]. F. johnsoniae and other members of the Bacteroidetes phylum are thought to play important roles in the turnover of this compound in many environments [39]. Perhaps the major metabolic function of these dominant intestinal bacteria is the fermentation of nondigestible carbohydrates including large polysaccharides (i.e., pectins and cellulose), which are key sources of energy in the R. bieti colon.
Similarly, Burkholderiales were the primary contributors to the Proteobacteria populations, followed by Enterobacteriales and Pseudomonadales. The major genus in the Proteobacteria phylum was Pseudomonas. P. fluorescens was the predominant species among the Pseudomonas in the R. bieti metagenome. P. fluorescens is a common Gram-negative bacterium that can be found in the low section of the human digestive tract [40].
A distinctive feature of the R. bieti metagenome is the abundance of phylum Fibrobacteres and Spirochaetes, and this abundance is unexpected and far greater than in other animals (Figure 2). Fibrobacter succinogenes was the only species existed in the R. bieti gut (Additional file 2) that is recognised as a major bacterial degrader of lignocellulosic material in the herbivore gut [41]. It was originally thought that members of the genus Fibrobacter were restricted to the mammalian intestinal tract, but the occurrence and distribution of members of the Fibrobacteres phylum has recently been extended to include termite intestinal contents where cellulose is again the primary carbon source for the host organisms [42]. Spirochaetes form a distinct monophyletic phylum of bacteria, and contain four genera that contain important pathogenic species, these being Treponema, Borrelia, Leptospira and Brachyspira, which were all exist in the R. bieti metagenome (Additional file 2). Morphologically diverse Spirochaetes are consistently present in the hindgut of all termites [43], and are found out as ectosymbionts attached to the surface of cellulose-digesting protists [44].
Figure 2.
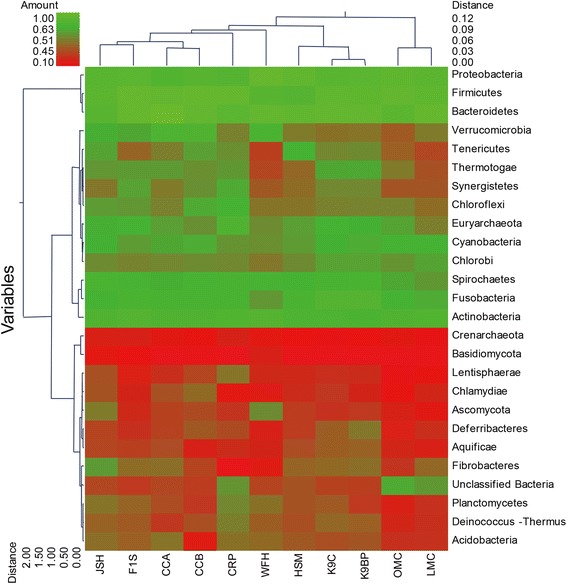
Phylogenetic clustering of R. bieti , pygmy loris, human, mouse, canine, cow, and chicken gastrointestinal metagenomes. A double hierarchical dendrogram was established through weight-pair group clustering methods based on the non-scaling Manhattan distance. The dendrogram shows the phylogenetic distribution of the microorganisms among the eleven metagenomes from the seven different hosts, including R. bieti (JSH), pygmy loris (WFH), human (HSM and F1S), mouse (LMC and OMC), dog (K9C and K9BP), cow (CRP), and chicken (CCA and CCB). The linkages of the dendrogram do not show the phylogenetic relationship of the bacterial phylum and are based on the relative abundance of taxonomic profiles. The heat map depicts the relative percentage of each phylum of microorganism (variables clustering on the y axis) in each sample (x axis clustering). The heat map color represents the relative percentage of the microbial descriptions in each sample, with the legend indicated at the upper left corner. Branch length indicates the Manhattan distances of the samples along the x axis (scale at the upper right corner) and of the microbial phyla along the y axis (scale at the lower left corner).
Eukaryota were a minor constituent (1.27%) in the R. bieti metagenome. Species of Blastocystis which have been reported as the most commonly occurring micro-eukaryote in human feces [45,46] were also represented in small quantities (<0.01%) in the R. bieti metagenome. In addition, the presence of Blastocystis has been linked to a number of gut-related diseases. Some of these diseases could be the outcome of the predation of beneficial bacteria by Blastocystis in light of the similar observations in ruminant cattle and their communalistic protozoa [47].
Fungi have very low abundance sequences (0.26%), with Ascomycota being the primary contributor (Additional file 3). Compared with humans, more diverse fungi species belonging to 21 different genera exist in the R. bieti metagenome. The most abundant fungi genera in the R. bieti metagenome were Aspergillus (0.06%), Gibberella (0.03%), Magnaporthe (0.02%) and Neurospora (0.02%) (Additional file 3). Fungi in the intestinal ecosystem of NHPs have not yet been studied extensively. Comparative analyses indicate that both fungal populations in the R. bieti and pygmy loris metagenomes [25] were predominated by Aspergillus. However, the most abundant fungi species within Aspergillus of the R. bieti metagenome, A. fumigatus, has not been detected in pygmy loris. A. fumigatus is one of the most common human pathogen. The whole genome sequence of A. fumigatus shows that up to 18 different genes encoding endoglucanases have been annotated, which indicates that this species own a good capacity in lignocellulose hydrolysis [48]. Moreover, two fungi species (Neurospora crassa and Gibberella zeae) have been identified predominantly both in the R. bieti (Additional file 3) and pygmy loris metagenome [25], which were also identified in feline [19], canine (K9C and K9BP) [16], and mouse metagenomes (OMC) [49].
Unlike other NHPs, R. bieti are herbivorous mammals that have a specialized gut in which plant materials degraded by microbial processes apparently similar to those that occur in the rumen. In rumen ecosystems, fungi interact with other microbes to take part in decomposing cellulose. Thus, it seems likely that anaerobic fungi may play significant role in fiber degradation in the R. bieti. Future studies are required by next-generation sequencing to gain further insight into the fungal diversity in the R. bieti gastrointestinal tract.
Archaea are a minor component of the R. bieti metagenome, comprising 0.73% of the total sequencing reads. Archaea consist of two phyla, Euryarchaeota and Crenarchaeota, which diverged into night classes and eleven orders (Additional file 4). Among the groups of archaea, methanogenic archaea is the most predominant and diverse group. Methanogenic archaea is also widespread in chicken, dogs, felines, mice, ruminants, NHPs and humans [12,16,19,25,49-51]. In the R. bieti fecal metagenome, Methanocorpusculum labreanum is the major component of archaea, having a percentage of 0.05% in all the analytic sequences (Additional file 4), which is consistent with pygmy loris [25]. The majority of the Archaea in the rumen are methanogens which provide thermodynamically favorable conditions for ruminal microbial fibre degradation [52]. Although methanogenic archaea make up only a small part of the R. bieti microbial population, they may play an important role in microbial fermentation like in the rumen system. Archaea are considered commensals; however, they contribute to pathogeny in humans because of mutual interactions with other microorganisms [53]. For instance, methanogens consume hydrogen and create an environment that enhances the growth of polysaccharide fermenting bacteria, leading to higher energy utilization. Higher numbers of methanogenic archaea have been observed in obese humans [54]. However, the prevalence and medical importance of archaea in R. bieti need to be determined.
Only 0.17% of the total reads have viral origin, with only the order Caudovirales being identified. Three families were observed (Myoviridae, Podoviridae and Siphoviridae) within the Caudovirales order, and all sequences were classified as bacteriophages (Additional file 5). Bacteriophages influence food digestion by regulating microbial communities in the human gastrointestinal tract through lytic and lysogenic replication [55]. Bacteriophages also contribute to human health by controlling invading pathogens [56]. Recent metagenomic analyses of the DNA viruses from human feces have revealed that the majority of DNA viruses in human feces are novel, and most of the recognizable sequences also belong to bacteriophages [57]. The close phylogenetic relationship between humans and NHPs, coupled with the exponential expansion of human populations and human activities within the primate habitats, has resulted in the exceptionally high possibility of pathogen exchange [58]. Therefore, studies on the viral community of NHPs and the potential for cross-transmission between humans and NHPs are needed. Given the type of methodology (shotgun DNA pyrosequencing approach) that we utilized, our study could only determine the dsDNA virus. Future studies need to provide a richer understanding of both RNA and dsDNA viruses to complete human knowledge of the viral intestinal ecosystem.
Studies on cow [59], cats [60], mice [61], rats [62] as well as humans [63] and NHPs [64] revealed a correlation between host diet and microbial community composition. However, the characterization of the dietary-induced changes in NHPs microbiomes through high-throughput sequencing technologies has not been performed thus far. Hence, more attention should be given to it in future experiments.
Metagenomics-based metabolic profiles
Protein and carbohydrate metabolism are the most abundant functional categories, representing 9.87% and 9.59% of the R. bieti fecal metagenomes respectively (Figure 3). Genes associated with amino acids and derivatives, DNA metabolism, RNA metabolism, cofactors (vitamins, prosthetic groups, pigments), cell wall and capsule and membrane transport are also very abundant in the R. bieti metagenomes. Approximately 16.85% of the annotated reads from the R. bieti fecal metagenomes were categorized within the clustering-based subsystems, most of which have unknown or putative functions.
Figure 3.
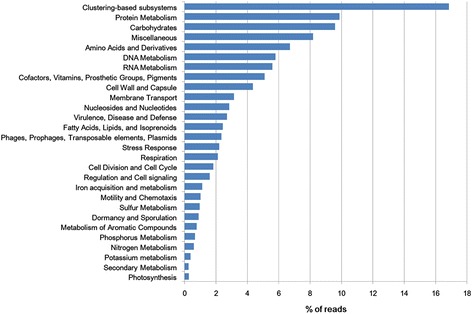
Functional composition of the R. bieti microbiome. The percentage of the R. bieti fecal metagenomic sequences assigned to the general SEED subsystems is shown. Through the “Functional Abundance” tool in MG-RAST, the R. bieti fecal sequencing runs were determined from the SEED database with the BLASTx algorithm. The e-value cutoff for the metagenomic sequence matches to the SEED subsystem database was 1 × 10−5 with a minimum alignment length of 30 bp.
Compared with NHPs pygmy loris gastrointestinal microbiomes [25], protein metabolism was more enriched in the gut microbiomes of R. bieti. On further analysis, it seemed that R. bieti had enriched protein biosynthesis in which universal GTPases were the most abundant. Because R. bieti eat foods rich in fibre, it is likely R. bieti have more available substrate for bacterial fermentation. This increase in protein biosynthesis may simply be the result of higher metabolic activity and/or growth of microbial populations.
Diversity of fibrolytic enzymes in the R. bieti metagenome
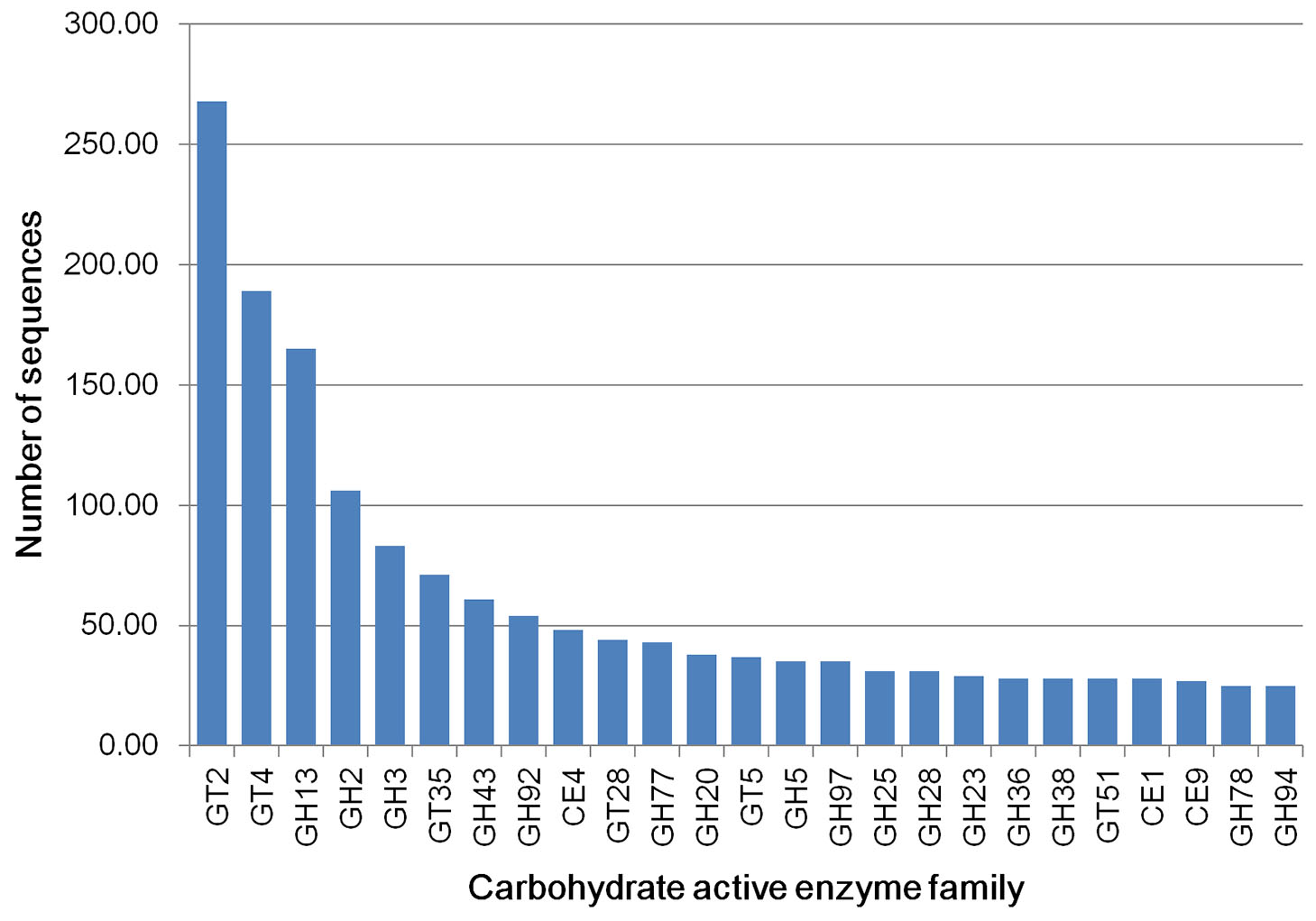
To obtain a more in-depth view of the carbohydrate enzymes present in R. bieti fecal microbiome, we subjected our samples to the CAZy database (http://www.cazy.org), as described by Cantarel et al. [29]. The comparison of the 88,514 metagenome reads post-QC processing based on the CAZy database provided 2,315 hits at an e-value restriction of 1 × 10−6. Candidate sequences that belong to the glycosyl transferase (GT) families GT2 (268) are the most abundant, followed by members of the GT4 (189) and glycoside hydrolase (GH) families GH13 (165) (Additional files 6 and 7).
In the R. bieti fecal metagenomes there is a wide diversity of GH catalytic modules with >1,300 sequences belonging to 79 GH families. GHs are a prominent group of enzymes that hydrolyze the glycosidic bond among the carbohydrate molecules. The most frequently occurring GH families in the R. bieti metagenome were GH13 (165; 12.61% of the total GH matches), 2 (106; 8.1%), 3 (83; 6.35%) and 43 (61; 4.66%) (Additional files 6 and 7). Family GH13 is the major glycoside hydrolase family acting on substrates containing α-glucoside linkages. The majority of the enzymes acting on starch, glycogen, and related oligo- and polysaccharides, are found within family GH13, which represents the largest family of glycoside hydrolases. In adition to α-amylases, it contains pullulanase, cyclomaltodextrin glucanotransferase (CGTase), cyclomaltodextrinase, neopullulanase, α-glucosidase, etc. (www.cazy.org). Some members of family GH13 bearing a variable number of supplemental N- or C-terminal extensions such as starch-binding modules (carbohydrate-binding modules (CBM) 26, CBM41 and CBM34 in CAZy) [65] were all annotated in the R. bieti metagenome (Additional file 7).
Families GH2 and GH3, which contain a large range of glycosidases cleaving nonreducing carbohydrates in oligosaccharides and the side chains of hemicelluloses and pectins, were abundant less than GH13. GH2 components are β-D-galactosidases, β-glucuronidases, β-D-mannosidases, and exo-β-glucosaminidases. The most common activities of GH3 include β-D-glucosidases, α-L-arabinofuranosidases, β-D-xylopyranosidases, and N-acetyl-β-D-glucosaminidases [66]. GH43 shows β-xylosidase, β-1,3-xylosidase, α-L-arabinofuranosidase, arabinanase, xylanase, and galactan 1,3-β-galactosidase activity (www.cazy.org). Glycosyl transferases are ubiquitous enzymes that catalyze the attachment of sugars to a glycone [67]. Candidate genes that belong to the glycosyl transferase families GT2 (268; 36.12% of the total GT matches) and GT4 (189; 25.47%) are the most abundant (Additional file 6).
Comparative analysis of GH families was done between metagenomes of the R. bieti, human, Tammar, termite hindgut, and cow rumen. Clustering analysis of the GH family distribution implied that the R. bieti metagenome was most closely related to cow rumen (Additional file 8). The GH5 were the most abundant cellulases in all metagenomes, which occurred at the highest frequency in the termite metagenome. Similar to the termite [11] and rumen [15] metagenomes, R. bieti was more evenly balanced with GH5 and GH9 cellulases. However, GH9 cellulases occurred at a lower frequency in human [10] metagenomes and were not even detected in the macropod [14] metagenomic datasets. GH28 hemicellulases were prevalent in the R. bieti microbiome; however, they occurred at a lower frequency in other animal gut microbiomes. Debranching enzymes profile in the R. bieti microbiome was similar to those reported for wallaby foregut microbiome [14]. The R. bieti microbiome possessed a large number of reads matching GH family specific for oligosaccharides, in which the most abundant oligosacchride-degrading enzymes were GH2, GH3, and GH43. Although both R. bieti and humans belong to primates, the GH profiles targeting plant structural polysaccharides in the two metagenomes are not more similar than other animals. This could be the result of the diet differences. Overall, the distribution of GH family enzymes in the micobiomes of R. bieti generally reflected its adaptation to special food types.
Comparative metagenomic analysis
Despite the extensive variation among individuals, the gut microbiota of members of the same species are often more similar to one another compared with those of other species. Thus, it is important to provide a comparison between the gastrointestinal microbiomes of primates and those of other animals. The results of this study were compared with data sets from different animals and even humans in the MG-RAST database. Paired data from other studies were chosen, such as lean (LMC) and obese (OMC) mouse cecal metagenomes [49], two chicken cecal metagenomes (CCA, CCB) [12], two canine intestinal metagenomes (K9C, K9BP) [16], and two human fecal metagenomes (F1S, HSM). F1S was a healthy human fecal metagenome [51], whereas HSM was defined as human feces from a malnourished subject. In addition,a cow rumen and a NHPs pygmy loris [25] fecal metagenomes were also utilized for comparison. The comprehensive overview of the data sets is shown in Additional file 9.
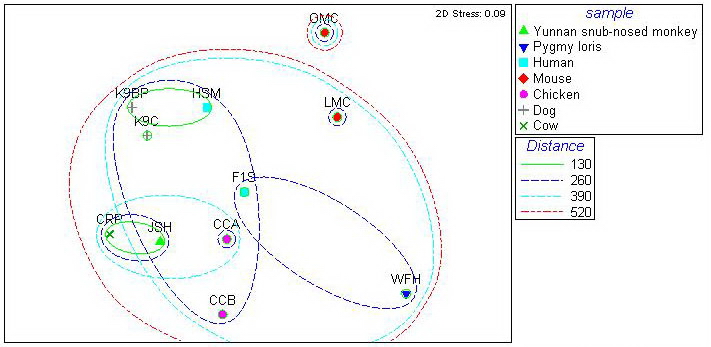
Clustering the metagenomes was carried out with unscaled Manhattan variance distances and presented through a double hierarchical dendrogram. The clustering-based comparisons were demonstrated at the phylogenetic level (Figure 2) and the metabolic level (Figure 4). In the phylogenetic comparison, the R. bieti samples clustered with the human fecal metagenomes (F1S), two chicken cecal metagenomes (CCA and CCB), a cow rumen metagenome and a pygmy loris fecal metagenome and separated from those of the two mouse metagenomes (OMC and LMC). Non-metric multidimensional scaling (MDS) plot illustrated that the distances among the R. bieti and cow rumen as well as among HSM, K9C and K9BP were the nearest (Additional file 10). This may be due to similar bacterial diversity influenced by similar diet rich in fibre within R. bieti and cow. In all the samples, the Actinobacteria, Bacteroidetes, Firmicutes and Proteobacteria were the most abundant (Figure 2). The heat map also demonstrates that the R. bieti metagenome was most distinguished by the greater prevalence of Fibrobacteres, an important phylum of cellulose-degrading bacteria, compared with other animals.
Figure 4.
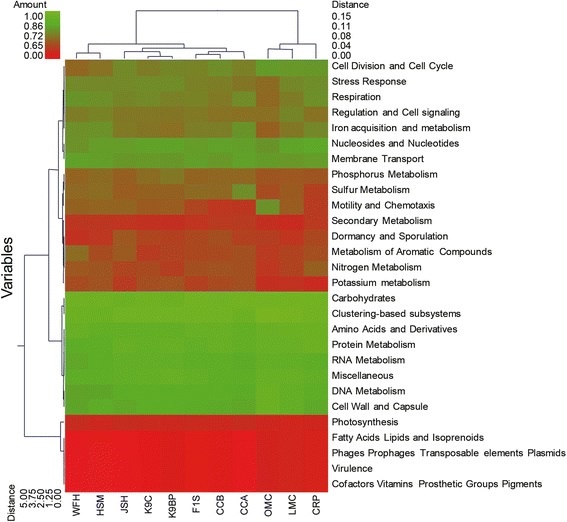
Metabolic clustering of R. bieti , pygmy loris, human, mouse, canine, cow, and chicken gastrointestinal metagenomes. A double hierarchical dendrogram was established through a weight-pair group clustering method based on the non-scaling Manhattan distance. The dendrogram shows the distribution of the functional categories among the eleven metagenomes from the seven different hosts, including R. bieti (JSH), pygmy loris (WFH), humans (HSM and F1S), murine (LMC and OMC), canine (K9C and K9BP), cow (CRP), and chicken (CCA and CCB). The linkages of the dendrogram are based on the relative abundance of metabolic profiles. The heat map depicts the relative percentage of each category of function (variables clustering on the y axis) in each sample (x axis clustering). The heat map color represents the relative percentage of functional categories in each sample, with the legend indicated at the upper left corner. Branch length indicates the Manhattan distances of the samples along the x axis (scale at the upper right corner) and of the microbial classes along the y axis (scale at the lower left corner).
Metabolism-based hierarchical clustering demonstrates that the R. bieti, dog, chicken, human and pygmy loris samples clustered together and separated from those of the mice and cow (Figure 4). Non-metric MDS plot illustrated that all metagenomics data have more than 80% similarity (Additional file 11). As expected, all the gut metagenomes were dominated by carbohydrate metabolism subsystems with amino acids, protein, and cell wall and capsule; the DNA and RNA subsystems were represented in relatively high abundance as well (Figure 4).
Conclusions
We presented for the first-time the application of the shotgun metagenomic pyrosequencing approach to study the fecal microbiome of the R. bieti. The overall goal of this study was to characterize the species composition and the functional capacity of the R. bieti fecal microbiome. Taxonomic analysis of the metagenomic reads showed similarities among the gut microbiomes of the R. bieti, humans and other animals. Four phyla dominated the microbiomes, namely, Actinobacteria, Bacteroidetes, Firmicutes and Proteobacteria. However, the relative proportion of the phyla was different. At the genus-level taxonomic resolution, Bacteroides species were the most abundant, most of which were represented by B. vulgates and B. fragilis. The organisms belonging to the same genus also represent one of the most abundant microbial taxa in the human intestinal microbiota [7,68]. The R. bieti faecal samples contained more bacteria belonging to the phylum Fibrobacteres, all of which were represented by the lignocellulose-degrading bacterium F. succinogenes. The high amount of F. succinogenes present in the R. bieti feces indicates a high turnover of lignocellulose in this NHPs. Archaea, fungi, and viruses are minor constituents of the R. bieti fecal microbiome. All archaea are members of Crenarchaeota and Euryarchaeota, with methanogens being the most abundant and diverse. Two fungi phylotypes were present in the R. bieti fecal microbiome, namely, Ascomycota and Basidiomycota, with Ascomycota being the primary contributor. Only about 0.17% of sequences were of viral origin, and all sequences were classified as bacteriophages. Overall, the microbial populations of the R. bieti gut system appeared to consist of taxa with known capacities for degradation and utilizing foods high in fibrous matter.
The comparative metagenomic analyses identified unique and/or overabundant taxonomic and functional elements in the R. bieti distal gut microbiomes. Relatively abundant bacteria of phylum Fibrobacteres and Spirochaetes were found in the R. bieti metagenome compared with all the other gut metagenomes. Primary functional categories were similar to those of other gut microbiomes and were associated mainly with protein, carbohydrates, amino acids, DNA and RNA metabolism, cofactors (vitamins, prosthetic groups, pigments), cell wall and capsule and membrane transport. Comparing GH profiles of R. bieti with those of herbivores, we found that the R. bieti microbiome possessed a large number of GH family specific for oligosaccharides, similar to that for the cow rumen. These findings may reflect the evolutionary adaptations for the highly specialized herbivory of R. bieti.
These results contribute to the limited body of primate metagenome studies and provide a unique genetic resource of plant cell wall degrading microbial enzymes. More studies involving the deeper sequencing of metagenomes are required to fully characterize the gastrointestinal microbiome of the R. bieti and other NHPs in healthy and diseased states, of varying ages or genetic backgrounds, and in the wild or in captivity.
Methods
Fecal sample collection
Fresh fecal samples from eight R. bieti were collected at a single time point from the Baimaxueshan National Nature Reserve of Weixi, Yunnan Province, China, with the permission of the authorities of Baimaxueshan National Nature Reserve. We tracked the R. bieti until they defecated; the fecal samples were immediately collected aseptically. The fresh fecal samples were transported to the laboratory on dry ice within 24 hours of collection, and then stored at −80°C until DNA extraction. We brought no toxic substance that would have adverse effects on the biotic community to minimize disturbance in the animal habitats. The research complied with the protocols established by the China Wildlife Conservation Association and adhered to the American Society of Primatologists (ASP) Principles for the Ethical Treatment of Non-Human Primates as well as the legal requirements of China.
DNA extraction and shotgun pyrosequencing
Genomic DNA were extracted from the fecal samples with the QIAamp DNA stool mini kit (Qiagen, Valencia, CA, USA) following the protocol provided by the supplier (0.25 g of each fecal sample). The quality and quantity of the DNA were determined with a nanodrop (ND-1000) spectrophotometer (Nanodrop Technologies, Wilmington, DE, USA) through agarose gel electrophoresis. DNA samples were stored frozen (−20°C) until use.
A total of 500 ng of pooled DNA was subjected to library preparation and shotgun pyrosequencing using the Roche 454 GS FLX Titanium System (Roche, Basel, Switzerland) as it was not feasible to distinguish which fecal sample corresponded to which individual. The obtained reads were uploaded to MG-RAST [26] under the name JSH_Metagenome and were assigned the Metagenome ID: 4452795.3. The MG-RAST v.3.0 online server quality control pipeline was utilized to remove reads of short length and poor quality before annotation and the analysis of metagenomic data [26]. The pipeline parameters were kept at default settings.
Bioinformatics and statistical analysis
Comparative metagenomic analysis was performed with MG-RAST pipelines. The metagenomic runs from the R. bieti data were compared with the current publicly available gut metagenomes in the databases. In the MG-RAST metagenomic annotation pipeline, the R. bieti fecal metagenomic datasets were compared with ten public sets of data from animals, including chicken cecum A (CCA 4440283), chicken cecum B (CCB 4440284), two dog metagenome data sets (K9C 4444164 and K9BP 4444165), lean mouse cecum (LMC 4440463), obese mouse cecum (OMC 4440464), cow rumen (CRP 4441682), human stool metagenome (HSM 4444130), human F1-S feces metagenome (F1S 4440939) and pygmy loris fecal metagenome (WFH 4476304). The organisms in MG-RAST were classified through the M5NR protein database (http://tools.metagenomics.anl.gov/m5nr/). The functional annotation and classification relied on the SEED subsystem ([69]; http://www.theseed.org/wiki/ Home_of_the_SEED) databases. The maximum e-value of 1e-5, minimum percent identity of 60, and minimum alignment length of 30 were applied as the parameter settings in the analysis. The taxonomic and functional profiles were normalized to determine the differences in the sequencing coverage by calculating the percent distribution prior to downstream statistical analysis. Clustering was performed using Ward’s minimum variance with unscaled Manhattan distances [70]. Heat maps were drawn by hierarchal clustering performed with NCSS 2007 (Kaysville, Utah). Non-metric MDS analysis based on Bray-Curtis’ similarity and Euclidean distance were performed using PRIMER 6 statistical software (PRIMER-E Ltd., Plymouth Marine Laboratory, Plymouth, U.K.).
Annotations based on the carbohydrate-active enzymes database ([29]; http://www.cazy.org) were provided for all the reads that passed the MG-RAST QC filter. Sequences are subject to Blast analysis against a library composed of modules derived from CAZy at an e-value restriction of 1 × 10−6. The cluster analysis of metagenomes on the basis of GH profile was carried out using the PAST v.2.17b data analysis package [71].
Acknowledgements
This work was supported by the National Natural Science Foundation of China (31360268 and 31160229). We thank Dr. Bernard Henrissat for his assistance with parts of the data analysis based on CAZy. We also thank Dr Hein Min Tun for his assistance with the non-metric MDS analysis.
Abbreviations
- NHPs
Non-human primates
- MG-RAST
Metagenome Rapid Annotation using Subsystem Technology
- QC
Quality control
- NCBI
National Center for Biotechnology Information
- SRA
Short Read Archive
- CAZy
Carbohydrate-Active enzymes
- GH
Glycoside hydrolase
- GT
Glycosyl transferase
- CBM
Carbohydrate-binding modules
- MDS
Multidimensional scaling
- ASP
American Society of Primatologists
Additional files
Information regarding the sequence datas.
Phylogenetic classification of bacteria in the R. bieti metagenome.
Phylogenetic classification of fungi in the R. bieti metagenome.
Phylogenetic classification of archaea in the R. bieti metagenome.
Phylogenetic classification of viruses in the R. bieti metagenome.
{kind=link}
Number of occurrences for the 25 most dominant carbohydrate active enzyme families. In total, these families contain 1557 sequences, representing 67.3% of the putative carbohydrate active enzyme family.
Presence of carbohydrate active enzyme families in the R. bieti metagenome.
GH profiles targeting plant structural polysaccharides in the R. bieti and other metagenomes.
Overview of the MG-RAST metagenomes chosen for comparison.
{kind=link}
Non-metric multidimensional scaling analysis based on relative abundance of taxonomic profiles among R. bieti , pygmy loris, human, mouse, canine, cow, and chicken gastrointestinal metagenomes. Samples of the same host species are indicated by the same symbol. Superimposed circles represent clusters of samples at different distance values (Euclidean distance).
{kind=link}
Non-metric multidimensional scaling analysis based on relative abundance of metabolic profiles among R. bieti , pygmy loris, human, mouse, canine, cow, and chicken gastrointestinal metagenomes. Samples of the same host species are indicated by the same symbol. Superimposed circles represent clusters of samples at different similarity values (Bray-Curtis similarity).
Footnotes
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
BX participated with the study design, bioinformatic analyses and manuscript preparation. WX contributed with bioinformatic and statistical analyses. JL contributed reagents and materials for the study. LD and CX carried out sample collection and sample processing. XT and YY participated with statistical analyses. YM contributed analysis tools for the study. JZ participated with bioinformatic analyses. JD and QW carried out sample processing. ZH collaborated in the design and coordination and helped to draft the manuscript. All authors read and approved the final manuscript.
Contributor Information
Bo Xu, Email: xubo128028@163.com.
Weijiang Xu, Email: sn-xwj@163.com.
Junjun Li, Email: 1979ljsx@163.com.
Liming Dai, Email: 690058128@qq.com.
Caiyun Xiong, Email: xiongcaiyun928@126.com.
Xianghua Tang, Email: txh13888381045@163.com.
Yunjuan Yang, Email: fcyangyunjuan@163.com.
Yuelin Mu, Email: muyuelin@sohu.com.
Junpei Zhou, Email: zhoujunpei_yn@126.com.
Junmei Ding, Email: djm3417@163.com.
Qian Wu, Email: qian99223@163.com.
Zunxi Huang, Email: huangzunxi@163.com.
References
- 1.Long YC, Kirkpatrick CR, Zhong T, Xiao L. Report on distribution, population and ecology of the Yunnan snub-nosed monkey (Rhinopithecus bieti) Primates. 1994;35:241–250. doi: 10.1007/BF02382060. [DOI] [Google Scholar]
- 2.Bennett EL, Davies AG. The ecology of Asian colobines. In: Davies AG, Oates JF, editors. Colobine monkeys: Their ecology, behaviour and evolution. Cambridge, UK: Cambridge University Press; 1994. pp. 129–171. [Google Scholar]
- 3.Peng YZ, Zhang YP, Ye ZZ, Liu RL. Study on the stomachs in three species of snub-nosed monkeys. Zoonoses Res. 1983;4:167–175. [Google Scholar]
- 4.Chen JJ, Lu T, Liu JS, Huang ZR. Observations on the stomach of Rhinopithecus Roxellanae. Acta Theriologica Sinica. 1995;15:176–180. [Google Scholar]
- 5.Ley RE, Hamady M, Lozupone C, Turnbaugh PJ, Ramey RR, Bircher JS, et al. Evolution of mammals and their gut microbes. Science. 2008;320:1647–1651. doi: 10.1126/science.1155725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wu C, Yang F, Gao R, Huang Z, Xu B, Dong Y, et al. Study of fecal bacterial diversity in Yunnan snub-nosed monkey (Rhinopithecus bieti) using phylogenetic analysis of cloned 16S rRNA gene sequences. Afr J Biotechnol. 2010;9:6278–6289. [Google Scholar]
- 7.Eckburg PB, Bik EM, Bernstein CN, Purdom E, Dethlefsen L, Sargent M, et al. Diversity of the human intestinal flora. Science. 2005;308:1635–1638. doi: 10.1126/science.1110591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gill SR, Pop M, Deboy RT, Eckburg PB, Turnbaugh PJ, Samuel BS, et al. Metagenomic analysis of the human distal gut microbiome. Science. 2006;312:1355–1359. doi: 10.1126/science.1124234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Turnbaugh PJ, Hamady M, Yatsunenko T, Cantarel BL, Duncan A, Ley RE, et al. A core gut microbiome in obese and lean twins. Nature. 2009;457:480–484. doi: 10.1038/nature07540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Qin J, Li R, Raes J, Arumugam M, Burgdorf KS, Manichanh C, et al. A human gut microbial gene catalogue established by metagenomic sequencing. Nature. 2010;464:59–65. doi: 10.1038/nature08821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Warnecke F, Luginbühl P, Ivanova N, Ghassemian M, Richardson TH, Stege JT, et al. Metagenomic and functional analysis of hindgut microbiota of a wood-feeding higher termite. Nature. 2007;450:560–565. doi: 10.1038/nature06269. [DOI] [PubMed] [Google Scholar]
- 12.Qu A, Brulc JM, Wilson MK, Law BF, Theoret JR, Joens LA, et al. Comparative metagenomics reveals host specific metavirulomes and horizontal gene transfer elements in the chicken cecum microbiome. PLoS One. 2008;3:e2945. doi: 10.1371/journal.pone.0002945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Brulc JM, Antonopoulos DA, Miller ME, Wilson MK, Yannarell AC, Dinsdale EA, et al. Gene-centric metagenomics of the fiber-adherent bovine rumen microbiome reveals forage specific glycoside hydrolases. Proc Natl Acad Sci U S A. 2009;106:1948–1953. doi: 10.1073/pnas.0806191105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Pope PB, Denman SE, Jones M, Tringe SG, Barry K, Malfatti SA, et al. Adaptation to herbivory by the Tammar wallaby includes bacterial and glycoside hydrolase profiles different from other herbivores. Proc Natl Acad Sci U S A. 2010;107:14793–14798. doi: 10.1073/pnas.1005297107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hess M, Sczyrba A, Egan R, Kim TW, Chokhawala H, Schroth G, et al. Metagenomic discovery of biomass-degrading genes and genomes from cow rumen. Science. 2011;331:463–467. doi: 10.1126/science.1200387. [DOI] [PubMed] [Google Scholar]
- 16.Swanson KS, Dowd SE, Suchodolski JS, Middelbos IS, Vester BM, Barry KA, et al. Phylogenetic and gene-centric metagenomics of the canine intestinal microbiome reveals similarities with humans and mice. ISME J. 2011;5:639–649. doi: 10.1038/ismej.2010.162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lamendella R, Domingo JW, Ghosh S, Martinson J, Oerther DB. Comparative fecal metagenomics unveils unique functional capacity of the swine gut. BMC Microbiol. 2011;11:103. doi: 10.1186/1471-2180-11-103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dai X, Zhu Y, Luo Y, Song L, Liu D, Liu L, et al. Metagenomic insights into the fibrolytic microbiome in yak rumen. PLoS One. 2012;7:e40430. doi: 10.1371/journal.pone.0040430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tun HM, Brar MS, Khin N, Jun L, Hui RK, Dowd SE, et al. Gene-centric metagenomics analysis of feline intestinal microbiome using 454 junior pyrosequencing. J Microbiol Methods. 2012;88:369–376. doi: 10.1016/j.mimet.2012.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Singh KM, Ahir VB, Tripathi AK, Ramani UV, Sajnani M, Koringa PG, et al. Metagenomic analysis of Surti buffalo (Bubalus bubalis) rumen: a preliminary study. Mol Biol Rep. 2012;39:4841–4848. doi: 10.1007/s11033-011-1278-0. [DOI] [PubMed] [Google Scholar]
- 21.Li RW, Connor EE, Li C, Baldwin Vi RL, Sparks ME. Characterization of the rumen microbiota of pre-ruminant calves using metagenomic tools. Environ Microbiol. 2012;14:129–139. doi: 10.1111/j.1462-2920.2011.02543.x. [DOI] [PubMed] [Google Scholar]
- 22.Pope PB, Mackenzie AK, Gregor I, Smith W, Sundset MA, McHardy AC, et al. Metagenomics of the Svalbard reindeer rumen microbiome reveals abundance of polysaccharide utilization loci. PLoS One. 2012;7:e38571. doi: 10.1371/journal.pone.0038571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lu HP, Wang YB, Huang SW, Lin CY, Wu M, Hsieh CH, et al. Metagenomic analysis reveals a functional signature for biomass degradation by cecal microbiota in the leaf-eating flying squirrel (Petaurista alborufus lena) BMC Genomics. 2012;13:466. doi: 10.1186/1471-2164-13-466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bhatt VD, Dande SS, Patil NV, Joshi CG. Molecular analysis of the bacterial microbiome in the forestomach fluid from the dromedary camel (Camelus dromedarius) Mol Biol Rep. 2013;40:3363–3371. doi: 10.1007/s11033-012-2411-4. [DOI] [PubMed] [Google Scholar]
- 25.Xu B, Xu W, Yang F, Li J, Yang Y, Tang X, et al. Metagenomic analysis of the pygmy loris fecal microbiome reveals unique functional capacity related to metabolism of aromatic compounds. PLoS One. 2013;8:e56565. doi: 10.1371/journal.pone.0056565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Meyer F, Paarmann D, D’Souza M, Olson R, Glass EM, Kubal M, et al. The metagenomics RAST server - a public resource for the automatic phylogenetic and functional analysis of metagenomes. BMC Bioinformatics. 2008;9:386. doi: 10.1186/1471-2105-9-386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chandler DP, Fredrickson JK, Brockman FJ. Effect of PCR template concentration on the composition and distribution of total community 16S rDNA clone libraries. Mol Ecol. 1997;6:475–482. doi: 10.1046/j.1365-294X.1997.00205.x. [DOI] [PubMed] [Google Scholar]
- 28.Lynd LR, Weimer PJ, van Zyl WH, Pretorius IS. Microbial Cellulose Utilization: Fundamentals and Biotechnology. Microbiol Mol Biol Rev. 2002;66:506–577. doi: 10.1128/MMBR.66.3.506-577.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cantarel BL, Coutinho PM, Rancurel C, Bernard T, Lombard V, Henrissat B. The Carbohydrate-Active EnZymes database (CAZy): an expert resource for Glycogenomics. Nucleic Acids Res. 2009;37:D233–D238. doi: 10.1093/nar/gkn663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Jindou S, Borovok I, Rincon MT, Flint HJ, Antonopoulos DA, Berg ME, et al. Conservation and divergence in cellulosome architecture between two strains of Ruminococcus flavefaciens. J Bacteriol. 2006;188:7971–7976. doi: 10.1128/JB.00973-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bayer EA, Belaich JP, Shoham Y, Lamed R. The Cellulosomes: Multienzyme machines for degradation of plant cell wall polysaccharides. Annu Rev Microbiol. 2004;58:521–554. doi: 10.1146/annurev.micro.57.030502.091022. [DOI] [PubMed] [Google Scholar]
- 32.Demain AL, Newcomb M, Wu JHD. Cellulase, clostridia, and ethanol. Microbiol Mol Biol Rev. 2005;69:124–154. doi: 10.1128/MMBR.69.1.124-154.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mazmanian SK, Liu CH, Tzianabos AO, Kasper DL. An immunomodulatory molecule of symbiotic bacteria directs maturation of the host immune system. Cell. 2005;122:107–118. doi: 10.1016/j.cell.2005.05.007. [DOI] [PubMed] [Google Scholar]
- 34.Mazmanian SK, Round JL, Kasper DL. A microbial symbiosis factor prevents intestinal inflammatory disease. Nature. 2008;453:620–625. doi: 10.1038/nature07008. [DOI] [PubMed] [Google Scholar]
- 35.Round JL, Mazmanian SK. Inducible Foxp3+ regulatory T-cell development by a commensal bacterium of the intestinal microbiota. Proc Natl Acad Sci U S A. 2010;107:12204–12209. doi: 10.1073/pnas.0909122107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Tap J, Mondot S, Levenez F, Pelletier E, Caron C, Furet JP, et al. Towards the human intestinal microbiota phylogenetic core. Environ Microbiol. 2009;11:2574–2584. doi: 10.1111/j.1462-2920.2009.01982.x. [DOI] [PubMed] [Google Scholar]
- 37.Xu J, Mahowald MA, Ley RE, Lozupone CA, Hamady M, Martens EC, et al. Evolution of symbiotic bacteria in the distal human intestine. PLoS Biol. 2007;5:e156. doi: 10.1371/journal.pbio.0050156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Stanier RY. Studies on non-fruiting myxobacteria. I. Cytophaga johnsonae, n. sp., a chitin-decomposing myxobacterium. J Bacteriol. 1947;53:297–315. [PMC free article] [PubMed] [Google Scholar]
- 39.Kirchman DL. The ecology of Cytophaga-Flavobacteria in aquatic environments. FEMS Microbiol Ecol. 2002;39:91–100. doi: 10.1111/j.1574-6941.2002.tb00910.x. [DOI] [PubMed] [Google Scholar]
- 40.Wei B, Huang T, Dalwadi H, Sutton CL, Bruckner D, Braun J. Pseudomonas fluorescens encodes the Crohn’s disease-associated I2 sequence and T-cell superantigen. Infect Immun. 2002;70:6567–6575. doi: 10.1128/IAI.70.12.6567-6575.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Montgomery L, Flesher B, Stahl D. Transfer of Bacteroides succinogenes (Hungate) to Fibrobacter gen-nov as Fibrobacter succinogenes comb nov and description of Fibrobacter intestinalis sp-nov. Int J Syst Bacteriol. 1988;38:430–435. doi: 10.1099/00207713-38-4-430. [DOI] [Google Scholar]
- 42.Hongoh Y, Deevong P, Hattori S, Inoue T, Noda S, Noparatnaraporn N, et al. Phylogenetic diversity, localization, and cell morphologies of members of the candidate phylum TG3 and a subphylum in the phylum Fibrobacteres, recently discovered bacterial groups dominant in termite guts. Appl Environ Microbiol. 2006;72:6780–6788. doi: 10.1128/AEM.00891-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Breznak JA, Brune A. Role of microrganisms in the digestion of lignocellulose by termites. Annu Rev Entomol. 1994;39:453–487. doi: 10.1146/annurev.en.39.010194.002321. [DOI] [Google Scholar]
- 44.Wenzel M, Radek R, Brugerolle G, Konig H. Identification of the ectosymbiotic bacteria of Mixotricha paradoxa involved in movement symbiosis. Eur J Protistol. 2003;39:11–23. doi: 10.1078/0932-4739-00893. [DOI] [Google Scholar]
- 45.Scanlan PD, Marchesi JR. Micro-eukaryotic diversity of the human distal gut microbiota: qualitative assessment using culture-dependent and –independent analysis of faeces. ISME J. 2008;2:1183–1193. doi: 10.1038/ismej.2008.76. [DOI] [PubMed] [Google Scholar]
- 46.Pandey PK, Siddharth J, Verma P, Bavdekar A, Patole MS, Shouche YS. Molecular typing of fecal eukaryotic microbiota of human infants and their respective mothers. J Biosci. 2012;37:221–226. doi: 10.1007/s12038-012-9197-3. [DOI] [PubMed] [Google Scholar]
- 47.Hobson PN, Wallace RJ. Microbial ecology and active ties in the rumen. Crit Rev Microbiol. 1982;9:165–225. doi: 10.3109/10408418209104490. [DOI] [PubMed] [Google Scholar]
- 48.Nierman WC, Pain A, Anderson MJ, Wortman JR, Kim HS, Arroyo J, et al. Genomic sequence of the pathogenic and allergenic filamentous fungus Aspergillus fumigatus. Nature. 2005;438:1151–1156. doi: 10.1038/nature04332. [DOI] [PubMed] [Google Scholar]
- 49.Turnbaugh PJ, Ley RE, Mahowald MA, Magrini V, Mardis ER, Gordon JI. An obesity-associated gut microbiome with increased capacity for energy harvest. Nature. 2006;444:1027–1031. doi: 10.1038/nature05414. [DOI] [PubMed] [Google Scholar]
- 50.Skillman LC, Evans PN, Strompl C, Joblin KN. 16S rDNA directed PCR primers and detection of methanogens in the bovine rumen. Lett Appl Microbiol. 2006;42:222–228. doi: 10.1111/j.1472-765X.2005.01833.x. [DOI] [PubMed] [Google Scholar]
- 51.Kurokawa K, Itoh T, Kuwahara T, Oshima K, Toh H, Toyoda A, et al. Comparative metagenomics revealed commonly enriched gene sets in human gut microbiomes. DNA Res. 2007;14:169–181. doi: 10.1093/dnares/dsm018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hook SE, Wright AD, McBride BW. Methanogens: methane producers of the rumen and mitigation strategies. Archaea. 2010;30:945785. doi: 10.1155/2010/945785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Conway de Macario E, Macario AJ. Methanogenic archaea in health and disease: a novel paradigm of microbial pathogenesis. Int J Med Microbiol. 2009;299:99–108. doi: 10.1016/j.ijmm.2008.06.011. [DOI] [PubMed] [Google Scholar]
- 54.Zhang H, DiBaise JK, Zuccolo A, Kudrna D, Braidotti M, Yu Y, et al. Human gut microbiota in obesity and after gastric bypass. Proc Natl Acad Sci U S A. 2009;106:2365–2370. doi: 10.1073/pnas.0812600106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Weinbauer MG. Ecology of prokaryotic viruses. FEMS Microbiol Rev. 2004;28:127–181. doi: 10.1016/j.femsre.2003.08.001. [DOI] [PubMed] [Google Scholar]
- 56.Gorski A, Weber-Dabrowska B. The potential role of endogenous bacteriophages in controlling invading pathogens. Cell Mol Life Sci. 2005;62:511–519. doi: 10.1007/s00018-004-4403-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Breitbart M, Hewson I, Felts B, Mahaffy JM, Nulton J, Salamon P, et al. Metagenomic analyses of an uncultured viral community from human feces. J Bacteriol. 2003;185:6220–6223. doi: 10.1128/JB.185.20.6220-6223.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Davies TJ, Pedersen AB. Phylogeny and geography predict pathogen community similarity in wild primates and humans. Proc Biol Sci. 2008;275:1695–1701. doi: 10.1098/rspb.2008.0284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Tajima K, Aminov RI, Nagamine T, Matsui H, Nakamura M, Benno Y. Diet-dependent shifts in the bacterial population of the rumen revealed with real-time PCR. Appl Environ Microbiol. 2001;67:2766–2774. doi: 10.1128/AEM.67.6.2766-2774.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Lubbs DC, Vester BM, Fastinger ND, Swanson KS. Dietary protein concentration affects intestinal microbiota of adult cats: a study using DGGE and qPCR to evaluate differences in microbial populations in the feline gastrointestinal tract. J Anim Physiol Anim Nutr (Berl) 2009;93:113–121. doi: 10.1111/j.1439-0396.2007.00788.x. [DOI] [PubMed] [Google Scholar]
- 61.Turnbaugh PJ, Backhed F, Fulton L, Gordon JI. Diet-induced obesity is linked to marked but reversible alterations in the mouse distal gut microbiome. Cell Host Microbe. 2008;3:213–223. doi: 10.1016/j.chom.2008.02.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Abnous K, Brooks SP, Kwan J, Matias F, Green-Johnson J, Selinger LB, et al. Diets enriched in oat bran or wheat bran temporally and differentially alter the composition of the fecal community of rats. J Nutr. 2009;139:2024–2031. doi: 10.3945/jn.109.109470. [DOI] [PubMed] [Google Scholar]
- 63.Li F, Hullar MAJ, Schwarz Y, Lampe JW. Human gut bacterial communities are altered by addition of cruciferous vegetables to a controlled fruit- and vegetable-free diet. J Nutr. 2009;139:1685–1691. doi: 10.3945/jn.109.108191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Kisidayová S, Váradyová Z, Pristas P, Piknová M, Nigutová K, Petrzelková KJ, et al. Effects of high- and low-fiber diets on fecal fermentation and fecal microbial populations of captive chimpanzees. Am J Primatol. 2009;71:548–557. doi: 10.1002/ajp.20687. [DOI] [PubMed] [Google Scholar]
- 65.Machovic M, Janecek S. Starch-binding domains in the post-genome era. Cell Mol Life Sci. 2006;63:2710–2724. doi: 10.1007/s00018-006-6246-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Harvey AJ, Hrmova M, De Gori R, Varghese JN, Fincher GB. Comparative modeling of the three-dimensional structures of family 3 glycoside hydrolases. Proteins. 2000;41:257–269. doi: 10.1002/1097-0134(20001101)41:2<257::AID-PROT100>3.0.CO;2-C. [DOI] [PubMed] [Google Scholar]
- 67.Lairson LL, Henrissat B, Davies GJ, Withers SG. Glycosyltransferases: structures, functions, and mechanisms. Annu Rev Biochem. 2008;77:521–555. doi: 10.1146/annurev.biochem.76.061005.092322. [DOI] [PubMed] [Google Scholar]
- 68.Ley RE, Peterson DA, Gordon JI. Ecological and evolutionary forces shaping microbial diversity in the human intestine. Cell. 2006;124:837–848. doi: 10.1016/j.cell.2006.02.017. [DOI] [PubMed] [Google Scholar]
- 69.Overbeek R, Begley T, Butler RM, Choudhuri JV, Chuang HY, Cohoon M, et al. The subsystems approach to genome annotation and its use in the project to annotate 1000 genomes. Nucleic Acids Res. 2005;33:5691–5702. doi: 10.1093/nar/gki866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Kaufman L, Rousseeuw PJ. Finding Groups in Data: An Introduction to Cluster Analysis. New York: Wiley; 1990. [Google Scholar]
- 71.Hammer Ø, Harper DAT, Ryan PD. PAST: paleontological statistics software package for education and data analysis. Palaeontol Electron. 2001;4:1–9. [Google Scholar]