

Abstract
The establishment of reproducible mouse models of acute lymphoblastic leukemia (ALL) is necessary to provide in vivo therapeutic models that recapitulate human ALL, and for amplification of limiting amounts of primary tumor material. A frequently used model is the primary xenograft model that utilizes immunocompromised mice and involves injection of primary patient tumor specimens into mice, and subsequent serial passaging of the tumors by retransplants of cells harvested from the mouse bone marrow and spleen. The tumors generated can then be used for genomic profiling, ex vivo compound testing, mechanistic studies and retransplantation. This unit describes detailed procedures for the establishment and maintenance of primary ALL xenograft panels for potential use in basic research or translational studies.
Keywords: Xenograft, Acute lymphoblastic leukemia, Mouse model
INTRODUCTION
Acute lymphoblastic leukemia (ALL) is believed to originate from various genetic lesions in hematopoietic progenitor cells that are committed to differentiate into T-cells or B-cells. ALL affects both children and adults, with a peak incidence at age 2–5 years and approximately 3000 new pediatric cases diagnosed per year in the US. Proliferation of leukemic cells (called “blasts”) results in bone marrow failure, leading to symptoms such as fever, infection, bone pain, bruising and bleeding, and lethargy from anemia. The blasts may also migrate from the bone marrow and infiltrate other organs such as the central nervous system (CNS). ALL comprises multiple subtypes that are characterized by recurring submicroscopic gains and losses of DNA, and inter- and intra-chromosomal rearrangements. These rearrangements commonly lead to dysregulation of expression of oncogenes and hematopoietic growth factors, or result in gene fusions and the expression of chimeric proteins(Mullighan 2013).
Several genetically engineered mouse models (GEMM) of ALL have been described, including a BCR-ABL1 positive, Arf null model (Williams, Roussel et al. 2006) that has been used for in vivo studies of various drug treatments (e.g. (Boulos, Mulder et al. 2011)), and models of acute lymphoblastic and myeloid leukemia induced by expression of MLL fusion oncoproteins (Bernt, Zhu et al. 2011). While useful, these models only study leukemias arising from mouse hematopoietic cells, and do thus not represent a true human leukemia. Moreover, faithful GEMM models of many subtypes of ALL are lacking. In part, this is because most ALL subtypes are polygenic disease with genetic alterations targeting multiple key cellular pathways, and until recently knowledge of the full repertoire of genomic alterations in ALL required to build these models has been unknown. Pre-clinical models involving human ALL cells are desirable to provide a more accurate response to in vivo therapies.
Recently, there has been increasing interest in the utilization of immunocompromised mice with variable severity of immune deficiency to establish mouse models of human ALL. Several studies have carefully assessed the efficiency of engraftment of human normal and malignant hematopoietic cells in various mouse strains (Morisot, Wayne et al. 2010, Notta, Mullighan et al. 2011). Although some tumors can engraft in mice with less severely compromised immune systems (e.g. NOD.Cg-Prkdcscid, or NOD-SCID mice), albeit with a longer latency, the strain with the highest average frequency of successful engraftment was the NOD.Cg-Prkdcscid Il2rgtm1Wjl/SzJ (NSG) mouse (Shultz, Lyons et al. 2005). The genetic background of the NSG mouse results in a lack of mature T cells, B cells and functional NK cells, and a deficiency in cytokine signalling, that together facilitate an enhanced engraftment of human cells in this strain. A version of the NSG mouse has been generated that expresses human hematopoietic cytokines (NOD.Cg-Prkdcscid Il2rgtm1Wjl Tg(CMV-IL3,CSF2,KITLG)1Eav/MloySzJ (NSG-SGM3))(Wunderlich, Chou et al. 2010). The lack of immune cells combined with the presence of human interleukin-3 (IL-3), granulocyte/macrophage-stimulating factor (GM-CSF), and stem cell factor (kit ligand) in the NSG-SGM3 mouse further support engraftment and expansion, particularly of human myeloid cells
Previous studies have shown that the gender of the mice may influence the success of engraftment for human normal hematopoietic stem cells, with female mice showing a superior engraftment rate (Notta, Doulatov et al. 2010). However, certain ALL subtypes show equal engraftment in both females and males, while others do not engraft at all in males (C.G. Mullighan et. al., unpublished data). The reason for these differences is currently unknown, but it is possible that the different sex hormones influence the niche where the cells engraft. It should also be noted that there may be differences in engraftment efficiencies when comparing normal versus malignant hematopoietic cells, and also when comparing lymphoid versus myeloid tumors. Care does thus have to be taken in deciding if all available animals should be used or if the males should not be used for transplantation, and a smaller pilot experiment might be preferable whenever working with a new leukemia subtype. A suggestion is to use females for primary grafts, and males or females for later passages.
This Unit describes the procedures employed for establishing and maintaining primary ALL xenografts for the purpose of using them for basic and/or translational studies. Details regarding housing of the animals, various considerations in the preparations of the cells, monitoring of engraftment and harvesting of the tumor cells are discussed. These models are highly valuable for the evaluation of novel therapeutic approaches of various ALL subtypes prior to moving compounds into the clinic.
NOTE; When compounds are being evaluated in vivo, it is imperative that some preliminary pharmacokinetic (PK) data are generated to ensure that the compound is known to be present at the time of testing and in what amount. Increasingly, in vivo studies rely more on the measured plasma level of a compound, rather than the dose administered, to construct accurate dose-response curves. Considerations for compound studies are for instance the route of compound administration and if females or males are used for xenografting, since these factors may significantly alter the compound PK, which in turn can compromise the outcome of the experiment. PK data can either be found published if previous studies have been performed with the compound in question and the same experimental paradigm was used (e.g. (Maude, Tasian et al. 2012) for JAK1/2 and mTOR inhibitors), or a de novo PK study needs to be performed prior to initiating a therapeutic study using an established ALL xenograft model.
Xenograft model of human Ph-like leukemic cells engrafted in NSG mice have been successfully used to demonstrate activity of the ABL1 inhibitor dasatinib in tumors harboring ABL1, ABL2, CSF1R and PDGFRB rearrangements, and the JAK2 inhibitor ruxolitinib in tumors harboring JAK2 and erythropoietin receptor rearrangements that lead to activation of JAK-STAT signaling (Roberts, Morin et al. 2012, Roberts, Li et al. 2014). Starting when the level of engraftment of human ALL cells exceeded 5% of peripheral blood leukocytes, mice were randomized to receive either ruxolitinib (30 mg/kg/day sc or vehicle) via implanted mini-osmotic pumps. After four weeks of ruxolitinib treatment, a marked decrease in leukemic burden based on engraftment levels in peripheral blood and spleen was observed compared to vehicle treated controls.
The same study also evaluated the use of the tyrosine kinase inhibitor,dasatinib in the case of an ALL tumor harboring a NUP214-ABL1 fusion that was transplanted into NSG mice (Roberts, Morin et al. 2012). Dasatinib-treated mice (20 mg/kg, 5 days a week po) responded to dasatinib up to 8 weeks of treatment while the tumor burden in vehicle treated mice constantly increased. Together these studies suggest that the use of ruxolitinib and dasatinib should be transferred into the clinic for treatment of ALL cases harboring the respective JAK2 or ABL1 fusion.
BASIC PROTOCOL 1: Transplantation of primary leukemia cells into immunocompromised mice
The step-by-step procedure for generation of ALL xenograft mice is outlined below. The specific sections are as follows:
Preparations prior to thawing the tumor cells
Processing of the patient tumor sample in preparation for injection
Injection of the tumor sample into the mouse
Monitoring of xenograft mice and harvest of leukemic cells
NOTE: All experiments using live animals must be reviewed and approved by an Institutional Animal Care and Use Committee (IACUC) prior to initiation, and must follow officially approved procedures for care and use of laboratory animals.
NOTE: The use of human tissue must be approved by an Institutional Review Board (IRB) together with patient and/or guardian consent/assent.
NOTE: If not used immediately after collection from a patient, the tissue to be transplanted should be cryopreserved using liquid nitrogen, preferably at −180°C, until use. The time between thawing the cells and transplanting them into the recipient mice should be minimized to avoid unnecessary cell death.
NOTE: Considering the lack of immune competence of the mice, all steps need to be carried out with great care to avoid potential contamination. All solutions and equipment coming into contact with living cells must thus be sterile, and aseptic technique should be used accordingly.
NOTE: NOD.Cg-Prkdcscid Il2rgtm1Wjl/SzJ mice, as well as other immunocompromised mouse strains, are available from The Jackson Laboratory. If mice are bred “in house”, care must be taken to reduce the risk of colonization with pathogenic bacteria – for example breeding in isolators, housing in ventilated racks, and minimization of contact with non-immunocompromised strains. Dedicated housing and staff are highly desirable. Consultation with local animal facility and veterinary staff is encouraged prior to procuring, breeding or manipulation of immunocompromised mouse strains. Detailed guidelines regarding husbandry can also be found at The Jackson Laboratories web site (http://jaxmice.jax.org/support/husbandry/index.html).
NOTE: The mice should preferably be housed in vented racks. In addition, the drinking water should be acidified using hydrochloric acid to achieve a pH range of 2.5–3.0 to minimize the risk of bacterial growth, especially Pseudomonas spp. that is a major health issue for immunocompromised mice. Alternative approaches include continual or periodic exposure to trimethoprim-sulphamethoxazole or quinolones according to local practices. These additions may, however, affect the experimental outcomes for instance during compound treatment studies, and appropriate comparisons need to be performed in pilot experiments.
Materials
NOD.Cg-Prkdcscid Il2rgtm1Wjl/SzJ (NSG) mice at an age of 8–12 weeks (The Jackson Laboratory)
Cesium irradiator or equivalent.
Primary patient sample(s) or tumor cell line(s)
Medium for cell thawing: Iscove’s Modified Dulbecco’s Medium (IMDM; Gibco, cat. no. 12440)
Sterile fetal bovine serum (FBS; e.g. Sigma-Aldrich, cat. no. F0926)
Penicillin-Streptomycin-Glutamine (Gibco, cat. no. 10378-016)
Optional: DNAse I from bovine pancreas (Sigma-Aldrich, cat. no. D4263)
Optional: Hanks’ Balanced Salt Solution (Gibco, cat. no. 14185-052)
Optional: Anticoagulant citrate-dextrose solution A (Sigma-Aldrich, cat. no. C3821)
Optional: HEPES (Sigma-Aldrich, cat. no. H4034)
Phosphate Buffered Saline (PBS; Lonza, cat. no. 17-516F)
3% Acetic Acid with Methylene Blue (StemCell Technologies, cat. no. 07060)
15 ml conical centrifuge tubes (Becton Dickinson, cat. no. 352097)
Hemacytometer
Microscope
5 ml round bottom tube with filter cap (Corning, cat. no. 352235)
Biosafety cabinet
70% Ethanol
All-purpose sponges (e.g. Kendall Healthcare, cat. no. 9022)
1 ml syringe (Becton Dickinson, cat. no. 309659)
27 gauge 1/2 inch needle (Becton Dickinson, cat. no. 305109)
25 gauge 5/8 inch needle (Becton Dickinson, cat. no. 305122)
Mouse restrainer (See Fig. 1 for an example; an inner diameter of 25 mm is usually appropriate for NSG mice at the age of 8–12 weeks)
Heating lamp
FITC conjugated human CD45 antibodies (Becton Dickinson, cat. no. 340664)
PE conjugated human CD19 antibodies (Becton Dickinson, cat. no. 349209)
APC conjugated human CD7 antibodies (Life Technologies, cat. no. MHCD0705)
PE-Cy7 conjugated mouse CD45 antibodies (Becton Dickinson, cat. no. 552848)
4′,6-diamidino-2-phenylindole (DAPI; e.g. Gibco, cat. No. D1306)
Fluorescence activated cell sorter
Fig. 1.

Pictured is a custom designed mouse restrainer constructed at the Biomedical Engineering department at St. Jude Children’s Research Hospital. Restrainers at various sizes are also available for purchasing from a number of different companies, e.g., Plas-Labs, Inc. and Braintree Scientific, Inc.
Preparations prior to thawing the tumor cells
-
1
Uniquely identify the mice to be used for transplants (e.g. by ear tagging, toe clipping or chip implants) according to approved local practices.
-
2
Irradiate the mice at a maximum of 24 hours prior to transplant. A sublethal dose at 250 Gy is administrated to facilitate efficient engraftment.
It should be noted that not all laboratories irradiate their mice prior to transplantation to avoid unnecessary stress for the mice, minimize the risk for infection or due to lack of access to an irradiator. Preferably, a small side-by-side comparison should be performed when a new leukemia subtype is being investigated to evaluate whether irradiation improves engraftment success and is thus desirable or can be avoided
Processing of the patient ALL tumor sample in preparation for injection
Primary patient ALL specimens may, after appropriate IRB and institutional approvals, be obtained through clinical tissue banks that collect tumor specimens. The specimens may be either fresh bone marrow aspirates or peripheral blood samples, or frozen viable cells stored in RPMI supplemented with 10% fetal calf serum and 10% DMSO, or similar. Fresh bone marrow and peripheral blood samples need to be purified by density gradient centrifugation prior to use, (Support Protocol 1), followed by sample preparation from step 5 in Basic Protocol 1. Cryopreserved specimens or established ALL cell lines (See Table 1 for examples) should be prepared as detailed starting at step 3, below.
Table 1.
Tabulated are cell lines that successfully have engrafted in NSG mice (Mullighan et al, unpublished data).
| Cell line | ALL subtype | Cells transplanted per mouse | Engraftment rate | Latency until detectable engraftment | Order number |
|---|---|---|---|---|---|
| SUP-B15 | BCR-ABL1 ALL | 1 million | 100% (2/2) | 1 week | DSMZ: ACC 389 |
| OP-1 | BCR-ABL1 ALL | 1 million | 100% (2/2) | 1 week | Not available for purchasing |
| NALM-16 | Hypodiploid ALL | 2 million | 100% (6/6) | 1 week | DSMZ: ACC 680 |
The cell line of choice all depends on the subtype of ALL of interest, for example according to underlying chromosomal alterations or mutations.
-
3
Keep cryo preserved cells on dry ice after they have been removed from liquid nitrogen until it is time to thaw them. Thaw the cryo preserved cells by placing them in a 37°C water bath until ice has melted.
-
4
Immediately after thawing, transfer the thawed cells to a 15 ml conical centrifuge tube. Using pre-warmed medium (37°C; IMDM supplemented with 20% FBS and 1x Penicillin-Streptomycin-Glutamine), wash the cells by slowly adding 12 ml medium (approximately 2–3 seconds per ml). Gently invert the tube a couple of times to mix.
A problem with cell clumping may occur when thawing the cells due to non-viable granulocytes especially for specimens with a lower frequency of tumor cells and more normal cells present (e.g. less than 70% tumor cells), as stated from the clinical evaluation of the specimen, Addition of 200 U/ml DNase I to the thawing media may help to avoid this problem. Another option is the replacement of IMDM/FBS/Penicillin-Streptomycin-Glutamine by Hanks’ Balanced Salt Solution supplemented with 5% FBS, 10mM HEPES and 5% anticoagulant citrate-dextrose solution A. The choice between these two options is not evidence based, but depends on user preference and availability. Enrichment of human CD19 positive cells by magnetic bead selection of fluorescence activated cell sorting may be used to improve purity (Support Protocol 3). -
5
Pellet the cells by centrifugation for 3 min at 450 x g at room temperature. Aspirate the supernatant and gently flick the tube to loosen cell pellet. Resuspend the cells in room temperature PBS, to minimize negative responses from the mice due to non-murine serum proteins. Use approximately 1ml PBS/estimated 5–10 million cells, based on statement for the specimen vial and pellet size.
At this stage it is optional to perform an enrichment of human CD19-positive cells to deplete any putative normal T-cells present in the primary tumor sample that may engraft and result in graft-versus-host disease. For this, Dynabeads® CD19 Pan B may be used, followed by treatment with DETACHaBEAD® CD19. See Support Protocol 2 for details. Alternatively, cells may be purified by fluorescence activated cell sorting (FACS) prior to transplantation, according to Support Protocol 3. -
6
Gently pipet up and down a few times to generate a homogenous suspension. Determine tumor cell concentration using 3% Acetic Acid with Methylene Blue, which lyses any red blood cells present, and a hemacytometer. Mix 10 μl cell suspension with the cell counting solution to reach an approximate estimated cell concentration of 0.5–1 million cells/ml to facilitate cell counting. Incubate for a few seconds to allow for red blood cell lysis prior to transferring 5–10 μl to the hemacytometer and count the cells. For the most accurate concentration determination, make sure not to overfill the chamber.
-
7
Preferably, 0.5–3 x 106 cells should be transplanted/mouse into a minimum of 3 mice/tumor, but a lower cell amount can be injected if material is sparse or if detailed analyses of leukemia initiating cell frequencies are to be performed (e.g. (Notta, Mullighan et al. 2011)). Adjust the tumor cell suspension as appropriate using PBS, to approximate a concentration with the target number of cells per mouse in 200μl.
When preparing the tumor cell suspension, take into account that extra volume will be needed, e.g., due to syringe dead space. If possible, prepare inoculum for 15% – 20% more injections than needed.Filter the cell suspension through the filter cap of a 5ml round bottom tube.
Injection of the tumor sample into the mouse
Perform the transplants in a Biosafety cabinet
-
8
Keep the tubes with the filtered cells at room temperature, and minimize the time from preparation of the cells until time for transplant to avoid unnecessary death of the frequently fragile primary tumor cells.
-
9
Place cage with mouse under a heating lamp for a few minutes prior to transplant to dilate the tail vein.
-
10
Flick tube to resuspend the cells prior to filling the 1 ml syringe. The same syringe can be prefilled and used for all mice to be transplanted with the same tumor. Do not attach the needle (27 g 1/2 inch) until after filling the syringe to avoid rupturing the cells.
-
11
Place one mouse at the time in a restrainer (Fig. 1), and disinfect the lateral tail vein area with 70% ethanol. Transplant 200 μl inoculum by tail vein injection. After removing needle, apply pressure using an all-purpose sponge to stop the bleeding prior to placing the mouse in a clean cage. Place cage in a vented rack.
Monitoring of xenograft mice and harvest of leukemic cells
-
12
Monitor the transplanted mice daily for signs of disease based on both development of leukemia and potential infections. These signs include poor grooming, hunching back, lethargy, labored breathing, and as indicators of CNS infiltrations, circling, doming of the head (indicative of hydrocephalus) and hind limb paralysis. Development of these signs requires euthanasia.
-
13
The level of engraftment may be monitored by the enumeration of human leukocytes in mouse peripheral blood, or bioluminescent imaging in the case of suitably marked cells (Terziyska, Castro Alves et al. 2012). At 4–5 weeks post-transplant, the presence of human leukemic cells in mouse peripheral blood may be investigated for the first time. Inside a Biosafety cabinet, draw blood either retro-orbitally or from the tail vein, depending on the technique approved by the institution.
-
14
Stain the blood cells with antibodies targeting human CD45 (FITC conjugated), human CD19 (PE-conjugated), human CD7 (APC-conjugated) and mouse CD45 (PE-Cy7- conjugated). Analyze the stained blood cells by fluorescence activated cell sorting for presence of engraftment of human leukemic cells (Fig. 2). See Support Protocol 4 for procedure.
The antibody targeting human CD7 should be included by analysis also of B-cell leukemia to verify that any potential engraftment is not based on normal human T-cells present in the tumor specimen. Further, normal human CD7 positive T-cells can often be distinguished from CD7 positive leukemic cells based on the frequently higher CD45 expression in normal T-cells versus leukemic cells.After the first peripheral blood analysis, repeat this analysis every 2–4 weeks until mouse is sacrificed. If no engraftment is seen, at least a 4 week gap is preferred until the next occasion of blood sampling and FACS analysis of the blood to avoid unnecessarily stressing the mice.. -
15
When the engraftment rate exceeds 50% in peripheral blood and/or by signs of disease, the mouse should be sacrificed in accordance with institution regulations. Animals are subjected to full necropsy to identify potential sites of organ infiltration by leukemia, and leukemic cells from hind limb long bone marrow, spleen, and optionally, other involved organs (step 16).
-
16
Dissect out the hind limbs and spleen under sterile conditions. Remove excess muscle tissue from the hind limbs using either scissors or a razor blade, and carefully remove the fibula from the tibia. Store dissected limbs in buffer (e.g. RPMI with 10% fetal calf serum) prior to flushing bone marrow from long bones. Cut open both ends of the femora and tibiae and wash out the bone marrow cells using a 25 gauge 5/8 inch needle, a 10 ml syringe and a total of 10 ml pre-warmed medium (37°C; IMDM supplemented with 20% FBS and 1x Penicillin-Streptomycin-Glutamine) into a 70μm cell strainer and filter the cell solution through the cell strainer. Mash the spleen using the plunger from a 1 ml syringe in a 70μm cell strainer and rinse the filter using 10–15 ml pre-warmed medium. Determine tumor cell concentrations by mixing 10 μl cell suspension with 90 μl 3% Acetic Acid with Methylene Blue and count the cells using a hemacytometer.
-
17
Harvested cells can be either directly transplanted back into new recipient mice, utilized for in vitro experimental purposes, or cryopreserved for later use.
Fig. 2.

Representative FACS data from the analysis of a bone marrow specimen harvested from an ALL xenograft mouse (xenograft mouse studied in (Holmfeldt, Wei et al. 2013). The engraftment of human leukemic cells in this mouse was at 51% in peripheral blood at the time of harvest.
The primary endpoint measured in the xenografted mice is engraftment e.g., the presence of human ALL cells in peripheral blood drawn from the transplanted mouse. The commencement and frequency of monitoring for engraftment is empiric and depends in part on cell proliferation rate. As this is usually not known for initial engraftment, a commonly used monitoring schedule is two-weekly measurement from 4 weeks after transplantation. Any sustained measurable level of human CD45+ cells in peripheral blood reflects engraftment. Typically, a threshold of 5% of human CD45+ cells (as a proportion of circulating mouse leukocytes) is used to guide commencement of drug treatment in preclinical studies. For initial expansion of human leukemic cells, one may follow mice until high levels of engraftment are observed (human CD45+ cells >50% circulating leukocytes) or the onset of signs of leukemia (above).
Circulating human CD45+ cells do not directly correlate with total leukemic burden, and may only emerge in peripheral blood after substantial engraftment has been achieved. Disease burden may also be measured by bioluminescent imaging for cells marked with lentiviral vectors expressing luciferase. In this approach, mice are anesthetized and luciferin administered by intraperitoneal injection. Leukemic cells marked with luciferase will metabolize luciferin and emitted light detected and quantitated (by the authors, using The Xenogen IVIS 100 and 200 imaging systems). Leukemic burden may be sensitively and reproducibly measured, and engraftment detected prior to measurable circulating CD45+ cells. (Terziyska, Castro Alves et al. 2012). Examples of B-ALL cell lines marked with luciferin and established as xenografts are shown in Table 1 and Figure 5
Fig. 5.
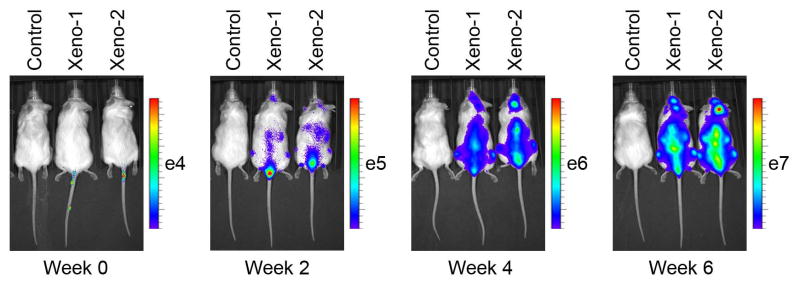
In vivo imaging of luciferase-tagged NALM-16 (Kohno, Minowada et al. 1980) cells using D-luciferin as substrate (Mullighan et al, unpublished data).
Secondary endpoints thus depend on the type of study and the assays available to measure engraftment. In preclinical studies evaluating activity of anti-leukemic drugs, end-points may be time to clinical manifestations of leukemia requiring euthanasia; serial measurement of leukemic burden (human CD45+ cell percentages or leukemic burden by bioluminescent imaging), or disease burden at time of euthanasia, which may be measured as spleen weight, tumor cell amount harvested from bone marrow or organ infiltration.
SUPPORT PROTOCOL 1: Density gradient centrifugation
To enrich for mononuclear cells, including the ALL tumor cells, from liquid samples such as peripheral blood or bone marrow, a density gradient centrifugation can be performed. This step should be carried out as soon as possible after collection of the specimen from the patient by clinical personnel, to avoid unnecessary cell death.
Materials
Cell suspension (e.g. bone marrow aspirate or peripheral blood)
Biosafety cabinet
50 ml conical test tubes (Fisher Scientific, cat. no. 2098,)
Hanks’ Balanced Salt Solution (Gibco, cat. no. 14185-052)
Hepes (Sigma Aldrich, cat. no. H4034)
Heparin (Sigma-Aldrich, cat. no. H3149)
15 ml conical centrifuge tubes (Becton Dickinson, cat. no. 352097)
Accu-Prep, Lymphocytes (Accurate Chemical and Scientific Corp, cat. no. AN5565)
Transfer pipets, sterile (Fisher Scientific, cat. no. 13-711-20)
Density gradient centrifugation method
Perform all steps possible in a laminar flow biosafety cabinet stipulated for work with biohazardous material.
Transfer the cell suspension from marrow/spleen samples to a 50 ml conical tube (maximum 20 ml/tube), and fill the tube with Hanks’-Hepes-Heparin (HHH; Hanks’ Balanced salt solution supplemented with 10 mM Hepes, pH 7.2 and 100 U/ml heparin). Mix by gently inverting tube 2–3 times, followed by centrifugation for 5 minutes at 1000 x g at room temperature
Aspirate the supernatant and resuspend the pellet in HHH according to the following formula: If the pellet size is 2.5 ml or smaller, add HHH to 10 ml. If the pellet size is 2.6 to 5 ml, add HHH to 20 ml.
Pipet 5 ml of room temperature, density gradient separation medium (Accu-Prep, Lymphocytes) into 15 ml tubes. Carefully layer 10 ml cell solution from step 2 onto the Accu-Prep. Do not allow mixing of the cell solution and Accu-Prep. If needed due to volume of cell solution, use multiple tubes.
Centrifuge the tubes at 500 x g for 20 minutes at room temperature, followed by immediately removing the mononuclear layer with a transfer pipet, carefully avoiding the density-gradient layer (See Fig. 3 for layer information). Place the interface into clean 15 ml tube and fill the 15 ml tube with HHH.
Centrifuge the tube at 1000 x g for 5 minutes at room temperature and aspirate the supernatant.
-
Continue with step 5 in Basic Protocol 1.
Avoid prolonged exposure to the density gradient medium, since contact with the medium may result in decreased cell viability.
Fig. 3.
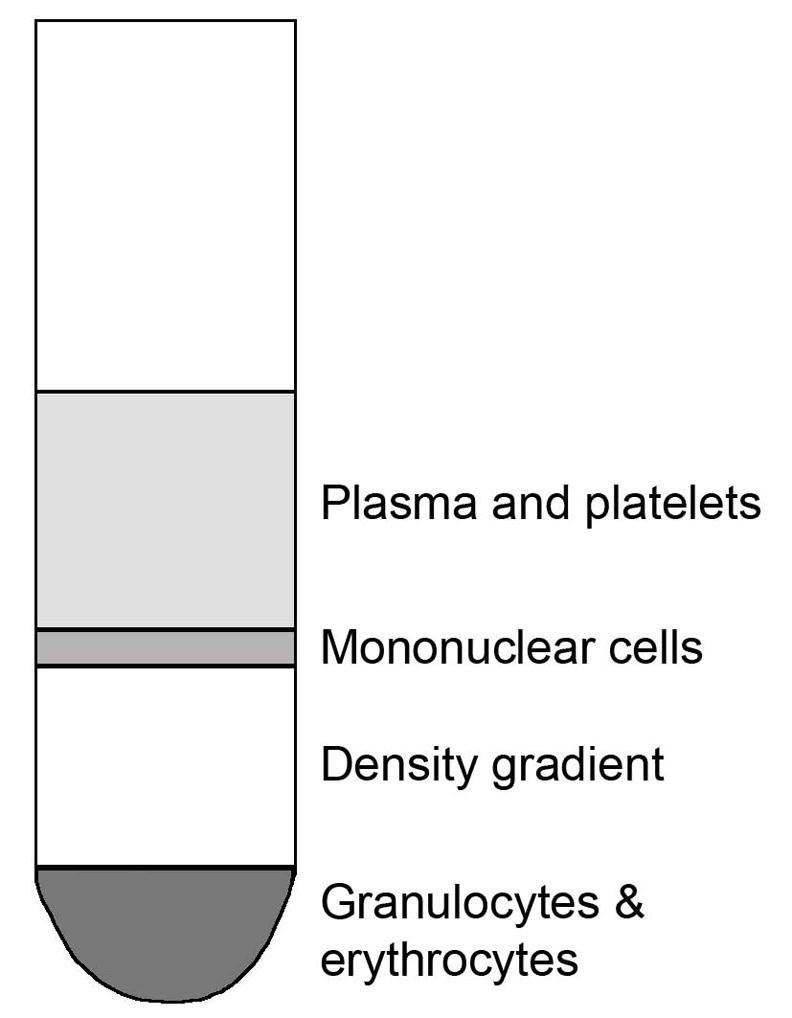
Fractions from density gradient centrifugation. The layer sizes may differ between different samples depending on the ratios between ALL cells and normal cells present in the specimen.
SUPPORT PROTOCOL 2: Enrichment of human CD19-positive cells
To avoid engraftment and expansion of normal human T-cells present in the primary tumor sample, enrichment for human CD19-positive cells may be carried out during the preparation of a B-cell leukemia for mouse transplants. This step would be carried out directly after washing and counting the thawed cells, and is performed according to the manufacturer’s instructions with slight modifications. However, the recommended ratio between number of cells and bead volume may need to be adjusted to avoid depletion of the beads when mixed with a tumor specimen that frequently harbors more than 70% CD19 positive cells.
Materials
Dynabeads® CD19 Pan B (Life Technologies, cat. no. 11143D)
DETACHaBEAD® CD19 (Life Technologies, cat. no. 12506D)
Ca2+ and Mg2+ free PBS (Lonza, cat. no. 17-516F)
Bovine Serum Albumin (BSA; e.g. Sigma-Aldrich, cat. no. A9418)
Ethylenediaminetetraacetic acid (EDTA; e.g. Sigma-Aldrich, cat. no. E5134)
15 ml conical centrifuge tubes (Becton Dickinson, cat. no. 352097)
MACSmix Tube Rotator (Miltenyi Biotec, cat. no. 130-090-753)
Magnet (e.g. DynaMag™-15 Magnet, Life Technologies, cat. no. 12301D)
Iscove’s Modified Dulbecco’s Medium (IMDM; Gibco, cat. no. 12440)
Sterile fetal bovine serum (FBS; e.g. Sigma-Aldrich, cat. no. F0926)
Penicillin-Streptomycin-Glutamine (Gibco, cat. no. 10378-016)
Trypan Blue Solution, 0.4% (Gibco, cat. no. 15250-061)
Hemacytometer
Microscope
Pre-washing of beads
-
1
Resuspend stock vial of Dynabeads® CD19 Pan B by vortexing for approximately 30 seconds.
-
2
Transfer needed amount to a 15 ml tube, add same volume of isolation buffer (see recipe below) (or at least 1 ml) and mix.
-
3
Place the tube in the magnet for 1 minute and remove and discard supernatant while the tube is still sitting in the magnet.
-
4
Add initial volume of Isolation buffer and remove tube from magnet
Treatment with Dynabeads® CD19 Pan B
-
5
Harvest cells from the tube removed from the magnetin a 15 ml conical tube and resuspend in Isolation buffer (see recipe below) to a concentration of 2.5 x 107 cells/ml or less.
-
6
Add 25 μl pre-washed and resuspended beads per ml cell solution.
This volume is based on the manufacturer’s recommendation, but for a tumor sample with a high blast content, 6 times or more than the recommended bead volume may be necessary to avoid depletion of the beads. The cells may be further diluted in Isolation buffer and a larger amount of beads may be used. -
7
Incubate for 20 minutes at 2–8°C with tilting and rotation, for instance using a MACSmix Tube Rotator.
-
8
Place in magnet. After 2 minutes, remove and discard supernatant while tube is still in magnet.
-
9
Remove tube from magnet and wash by adding 1 ml Isolation buffer per 25 μl beads.
-
10
Flick tube and place in magnet. After 2 minutes, remove and discard supernatant while tube is still in magnet.
-
11
Repeat wash steps (5–6) at least once.
-
12
Resuspend cells in 250 μl IMDM supplemented with 20% FBS and 1x Penicillin-Streptomycin-Glutamine per 25ul beads. Directly continue with DETACHaBEAD® CD19 treatment.
Treatment with DETACHaBEAD® CD19
-
13
Add 10 μl DETACHaBEAD® CD19 per 250 μl cell fraction.
-
14
Incubate for 45 minutes at room temperature with gentle mixing by hand swirling.
-
15
Pipet up and down a few times prior to placing tube in magnet.
-
16
Transfer supernatant containing released cells to a fresh 15 ml tube.
-
17
Wash the beads in 500 μl IMDM/FBS/Penicillin-Streptomycin-Glutamine and collect the supernatant in the same tube as in step 4.
-
18
To remove any remaining beads, place the tube containing the supernatant from steps 4–5 back in the magnet. After 2 minutes, transfer the supernatant to a new 15 ml tube.
-
19
Wash released cells by resuspending them in a total of 10 ml IMDM/FBS/Penicillin-Streptomycin-Glutamine.
-
20
Centrifuge for 6 minutes at 400 x g at room temperature to remove DETACHaBEAD® CD19.
-
21
Resuspend cells in approximately 1 ml room temperate PBS/estimated 5–10 million cells, and count the cells using Trypan Blue Solution and a hemacytometer (RBCs will be washed away at this stage). Mix 10 μl cell suspension with the cell counting solution to reach an approximate estimated cell concentration of 0.5–1 million cells/ml to facilitate cell counting. Transfer 5–10 μl cell solution/Trypan Blue mix to the hemacytometer and count the cells. For the most accurate concentration determination, make sure not to overfill the chamber.
Cell counting at this stage should preferably be done manually and not using an automated cell counter, since the presence of any contaminating beads can to lead to false viability rates using automated cell counters, and thus incorrect cell concentrations.
Continue with step 5 in the “Processing of the patient tumor sample in preparation for injection” section
SUPPORT PROTOCOL 3: Purification of primary ALL cells by fluorescence activated cell sorting (FACS)
Primary ALL specimens obtained from a tissue bank frequently are contaminated with normal hematopoietic cells, including normal T-cells that may cause graft-versus-host disease in the xenografted mice. To obtain a pure tumor population, the specimen can be subjected to flow sorting based on the immunophenotype of the tumor cells.
Materials
Iscove’s Modified Dulbecco’s Medium (IMDM; Gibco, cat. no. 12440)
Sterile fetal bovine serum (FBS; e.g. Sigma-Aldrich, cat. no. F0926)
Penicillin-Streptomycin-Glutamine (Gibco, cat. no. 10378-016)
5 ml round bottom tube with filter cap (Corning, cat. no. 352235)
Normal Rabbit Serum (Life Technologies, cat. no. 10510)
FITC conjugated human CD45 antibodies (Becton Dickinson, cat. no. 340664)
PE conjugated human CD19 antibodies (Becton Dickinson, cat. no. 349209)
APC conjugated human CD7 antibodies (Life Technologies, cat. no. MHCD0705)
4′,6-diamidino-2-phenylindole (DAPI; e.g. Gibco, cat. No. D1306)
Fluorescence activated cell sorter
Primary patient sample(s) or tumor cell line(s)
Staining of cells
Thaw and wash the cells (see Basic Protocol 1, step 3–4 for details).
Pellet the cells by centrifugation for 3 min at 450 x g at room temperature. Aspirate the supernatant and gently flick the tube to loosen cell pellet.
Resuspend pellet in 1 ml Staining medium (IMDM supplemented with 20% FBS and 1x Penicillin-Streptomycin-Glutamine). Transfer 10 μl cell suspension into four different 5 ml round bottom tubes, to be used for single color controls.
Add 50 μl Normal rabbit serum to the sort sample and 10 μl to the controls. Flick the tubes, and add 50 μl each of human specific CD45-FITC, CD19-PE and CD7-APC antibodies to the sort sample and either without antibody or 10 μl of a single antibody as appropriate to the respective control tubes (one tube without antibody, one with human CD45-FITC antibodies, one with human CD19-PE and one with human CD7-APC antibodies). Flick tubes and incubate in the dark for 10 minutes at room temperature. These amounts of antibody are based on the authors experience but may require empiric titration for different antibodies, vendors and lots to ensure accurate separation of marker positive and negative populations.
Add 10 ml of Staining medium to the sort sample and 3–4 ml to the control tubes. Invert tubes 2–3 times and centrifuge for 3 min at 450 x g at room temperature.
Decant supernatants and resuspend cell pellet for sorting in 1 ml Sorting medium supplemented with DAPI, and the control samples in 400 μl Sorting medium. Only add DAPI to the negative control lacking an antibody.
Filter the cell suspensions through the filter cap of clean 5 ml round bottom tubes. Keep tubes on ice in the dark until used for flow sorting. Minimize the time from staining until flow sorting to avoid cell death and internalization of the antibodies. AU are there guidelines to these time constraints?
-
Sort the stained samples according to the manual of the Flow sorter used, and place the gates for instance as exemplified in Fig. 4.
Note that the gating strategies may vary for different tumors, and should be guided by diagnostic immunophenotyping data. Diagnostic immunophenotypic data will provide guidance on which markers are positive, and at what intensity of staining. In the absence of such information, we typically stain tumor samples with antibodies directed against CD45 (pan leukocyte marker, typically bright in mature cells and of intermediate intensity in leukemic cells), CD19 (B-cell marker), CD7 (T cell marker) and a granulocyte marker (CD13 or CD33). The choice of fluorochromes conjugated to each antibody will depend on availability and compatibility with cell sorters. We find this panel of antibodies enables purification of leukemic and normal hematopoietic cells from the majority of samples.
Fig. 4.

Representative FACS data from the sorting of a primary ALL tumor to obtain a pure tumor cell population (B-ALL tumor) and populations of normal B-cells and T-cells. Data are from sorting a hypodiploid ALL specimen investigated in (Holmfeldt, Wei et al. 2013).
SUPPORT PROTOCOL 4: FACS analysis of peripheral blood, bone marrow and spleen cells from xenografted mice
By staining the specimens collected from xenografted mice with appropriate antibodies followed by flow cytometry analysis, the level of engraftment of human hematopoietic cells can be established. For access to appropriate single color controls in the advent of unknown engraftment of human cells, the protocol describes the use of fluorochrome conjugated beads instead of antibody labeling of cells harvested from the xenografted mouse.
Materials
Lysis machine, e.g. BD Lyse Wash Assistant (Becton Dickinson, cat. no. 337146)
Phosphate Buffered Saline (PBS; Lonza, cat. no. 17-516F)
Sterile fetal bovine serum (FBS; e.g. Sigma-Aldrich, cat. no. F0926)
5 ml round bottom tube with filter cap (Corning, cat. no. 352235)
CD16/CD32 (Becton Dickinson, cat. no. 553142)
FITC conjugated human CD45 antibodies (Becton Dickinson, cat. no. 340664)
PE conjugated human CD19 antibodies (Becton Dickinson, cat. no. 349209)
APC conjugated human CD7 antibodies (Life Technologies, cat. no. MHCD0705)
PE-Cy7 conjugated mouse CD45 antibodies (Becton Dickinson, cat. no. 552848)
4′,6-diamidino-2-phenylindole (DAPI; e.g. Gibco, cat. No. D1306)
Fluorescence activated cell sorter
BD Calibrite Beads (Unlabeled, FITC and PE beads; Becton Dickinson, cat. no. 349502)
BD Calibrite APC Beads (Becton Dickinson, cat. no. 340487)
Red blood cell lysis and staining of cells
Peripheral blood is obtained by retroorbital bleeding, tail vein venipuncture or cheek bleeding according to experience and local approval. Bone marrow is obtained by flushing of mouse hind limb long bones, or crushing with a mortar and pestle and rinsing/filtering. Typically 1 million cells are used per antibody staining cocktail, but less may be used if samples are limiting. Antibody titration should be performed prior to establishing staining procedures to ensure reproducible separation of cell populations.
Lyse RBCs in the cell suspension using for instance a BD Lyse Wash Assistant.
After lysis, dispense 0.5 million bone marrow or spleen cells can these be treated in the same manner as a peripheral blood sample - are other cell types from marrow and spleen a complicating factor?, alternatively peripheral blood, in a 5 ml round bottom tube for analysis, and 0.1 million cells in a tube for a single color control. Pellet by centrifugation for 3 minutes at 450 x g at room temperature and decant supernatant.
Add 25 μl staining solution (PBS supplemented with 1.5% FBS, 1:200 diluted CD16/CD32 (blocking unspecific binding), human specific antibodies (CD45-FITC, CD19-PE, and CD7-APC; all diluted 1:25) and mouse specific CD45-PE-Cy7 antibodies (diluted 1:400) to the analysis sample. Add 25 μl staining solution and mouse specific CD45-PE-Cy7 antibodies (diluted 1:400) to the control sample. Vortex briefly and incubate in the dark at room temperature for 15 minutes.
Add 1 ml PBS supplemented with 1.5% FBS, mix and centrifuge for 3 minutes at 450 x g at room temperature and decant the supernatant. Place tubes on ice and add 300 μl PBS supplemented with 1.5% FBS and DAPI. Filter the cell suspension through the filter cap of a clean 5 ml round bottom tube. Keep in the dark on ice until analysis.
Analyze the stained samples according to Flow cytometer manual used. Use unlabeled, FITC, PE and APC conjugated beads and the mouse specific CD45-PE-Cy7 control as single color controls, and place the gates as exemplified in Fig. 2. Data are analyzed to determine the proportion of leukocytes that are of mouse or human origin (ie mouse CD45+ or human CD45+). If there is evidence of human leukemic cell engraftment, the proportion of cells that are of B (CD19), T (e.g. CD7) or myeloid (e.g. CD13 or CD33) cell lineage are enumerated.
REAGENTS AND SOLUTIONS
Isolation buffer for enrichment of human CD19 positive cells
Supplement Ca2+ and Mg2+ free PBS with 0.1% BSA and 2mM EDTA.
Adjust pH to 7.4.
Store buffer at +2–8°C.
COMMENTARY
Background Information
The survival rates for pediatric patients with ALL has improved dramatically over the past few decades (Pui, Campana et al. 2009), but to be able to reach a 100% cure rate and decrease problems with side effects from the chemotherapy treatment, new treatment alternatives are necessary. For a more accurate analysis of the effect of various test substances and drugs on tumor cells, preferably human tumors should be used instead of mouse counterparts. Primary tumor material is frequently limiting, and the establishment of xenograft mice by transplantation of human ALL cells into immunocompromised mice cannot only be of use for the expansion of tumor cells, but also to serve as in vivo therapeutic models.
Albeit being a more accurate model for in vivo drug studies compared with pure mouse models, the xenograft model still has its disadvantages. One potential problem is the lack of an immune response in the NSG mouse that in the human usually has a role in the clearance of a tumor. Further, the niche differs in the mouse bone marrow compared to in the human bone marrow, which might lead to activation of different signaling pathways in the mouse compared to in the human. Finally, compound bioavailability is likely to differ in a human being compared to in a mouse. These potential issuess should be taken into consideration in the evaluation of the data obtained from in vivo studies.
As a means to follow the engraftment and localization of xenografted cells, the cells may be tagged with a luciferase marker prior to transplant. Two examples are the lentiviral vectors pLenti CMV PuroLuc (Campeau, Ruhl et al. 2009) and pHIV-Luc-ZsGreen (Addgene plasmid 39196), which both allow for in vivo imaging. The fluorescent marker ZsGreen in the latter also allows for fluorescence activated cell sorting prior to transplantation, while the former is dependent on Puromycin selection to gain a pure population of transduced cells. By an intraperitoneal injection of an appropriate substrate (e.g. D-Luciferin) followed by in vivo imaging using for instance the Xenogen IVIS-200 Optical In Vivo Imaging System, monitoring of the transplanted cells can be performed (Fig. 5).
Human ALL xenograft mice have been used for pre-clinical studies (e.g. (Roberts, Morin et al. 2012)). Depending on the characteristics of each compound, different routes of administration can be used, e.g. po for the tyrosine kinase inhibitor dasatinib, via drinking water for dexamethasone, or osmotic pumps (Alzet pumps) for the JAK inhibitor ruxolitinib (Roberts et al. 2012).
Critical Parameters and Troubleshooting
The most critical parameter influencing this protocol is the quality of the primary tumor specimen. It is of utmost importance that the human leukemic tissue is carefully processed by a Ficoll density gradient and stored at a sufficiently low temperature to avoid cell death. Further, the cells should be washed immediately after thawing to remove the dimethyl sulfoxide from the medium that otherwise might affect cell viability.
Great care has to be taken when handling the immunocompromised mice to avoid infection. Never open a cage outside a Biosafety cabinet. For irradiation of the mice, the animals should preferably be transferred to a sealed, autoclaved container that allows access to air and fits inside the irradiator.
Clonal evolution may occur through serial transplants, and/or different subclones present in the primary tumor may emerge in different mice transplanted with the same tumor (Notta, Mullighan et al. 2011). It may thereby be beneficial to investigate the presence or absence of key lesions identified in the primary tumor in the tumors harvested from various mice before using the xenograft cells for additional analyses. As an example, Affymetrix SNP 6.0 microarray analysis can be performed to compare the presence of DNA copy alterations and loss-of-heterozygosity in the primary tumor with the tumors emerging in the xenograft mice (for instance as performed by (Notta, Mullighan et al. 2011, Holmfeldt, Wei et al. 2013)). Similarly, if a leukemia sample is engrafted with a key chromosomal fusion, it is prudent to confirm the presence of this in the xenograft sample, for example by RT-PCR. With the advent of relatively cheap sequencing approaches, exome or whole genome sequencing may be considered for particularly valuable tumors.
The growth characteristics for different tumors may vary (Fig. 6), and gaining experience with these tumors through serial transplants can improve the efficiency of their use for instance for in vivo studies. Once a leukemia sample has been expanded in intial passages in NSG mice, and/or if luciferase marking is performed, it is suggested that multiple aliquots of each tumor are cryopreserved, and genomic analyses (tailored to the key underlying leukemogenic lesions) performed prior to reengrafting the tumor for (e.g.) preclinical drug studies.
Fig. 6.
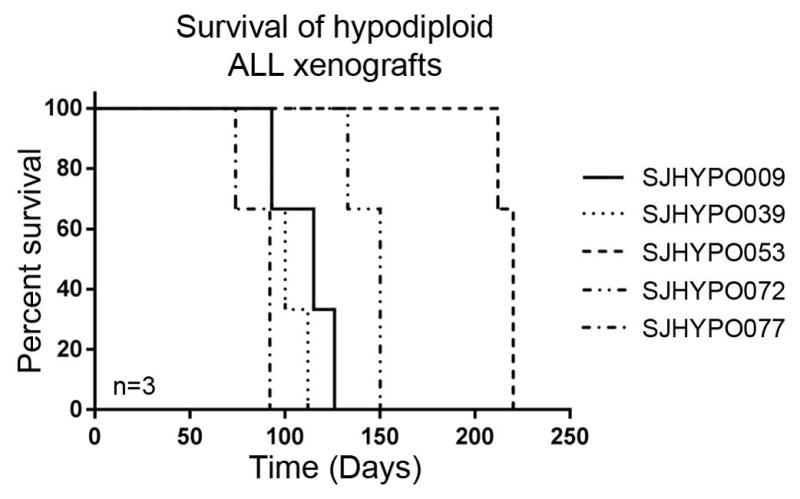
Kaplan-Meier graph showing engraftment rates for a number of representative hypodiploid ALL tumors transplanted into NSG mice (n=3 per tumor; xenograft mice used in (Holmfeldt, Wei et al. 2013)).
The technique described above involves iv adminstration, which is a straightforward method. The tail vein is easily recognized on the pale NSG mice, especially at age 8 weeks or older, and no sedation is necessary. One alternative is intrafemoral injections, which are more invasive and require sedation of the mice. This technique might be preferred if homing of the transplanted cells to the bone marrow is an issue, or if detailed studies of regional tumor dissemination are desired. Further, a lower amount of cells might need to be transplanted using intrafemoral injections, since the cells end up directly in the bone marrow, while it is unlikely that all cells will migrate to the bone marrow niche by iv injection.
Anticipated Results
The result of this procedure should be the development of a human leukemia in mouse. When harvesting bone marrow cells from a mouse with 50% or more human leukemic cells in peripheral blood, these usually consist of greater than 90% human leukemic cells, and 50–100 x 106 leukemic cells can frequently be obtained. Most tumors that develop in the mouse over the course of a few months give rise to an enlarged spleen, which can be up to 10–15 times larger than a normal spleen. Enlarged spleens usually consist of more than 80% human leukemic cells, and several hundred million leukemic cells can be obtained from a single spleen.
Time Considerations
The entire transplant procedure can be carried out within one working day. The time needed for efficient high-rate engraftment depends entirely on the specific tumor being transplanted, with time frames ranging from 5–6 weeks up to several months. For some tumors, no engraftment is achieved even in the most immunocompromised NSG mouse strain. A possible explanation for this might be that a certain microenvironment is needed for the tumor to engraft that does not exist in the mouse.
Acknowledgments
This work was supported by The Henry Schueler 41&9 Foundation in conjunction with Partnership for Cures, the St. Baldrick’s Foundation, US National Cancer Institute (NCI) grant RC4CA156329, US National Institutes of Health (NIH) grants CA21765 and U01 GM92666, the American Association for Cancer Research (AACR) Gertrude B. Elion Cancer Research Award and the American Lebanese and Syrian Associated Charities (ALSAC) of St. Jude Children’s Research Hospital. L.H. is an American Society of Hematology Scholar. C.G.M. is a Pew Scholar in the Biomedical Sciences and a St. Baldrick’s Scholar.
Contributor Information
Linda Holmfeldt, Email: linda.holmfeldt@igp.uu.se, Pathology, MS342, St. Jude Children’s Research Hospital, 262 Danny Thomas Place, Memphis, Tennessee 38105, Phone: 901-595-5463, Fax: 901-595-5947.
Charles G. Mullighan, Email: charles.mullighan@stjude.org, Pathology, MS342, St. Jude Children’s Research Hospital, 262 Danny Thomas Place, Memphis, Tennessee 38105, Phone: 901-595-3387, Fax: 901-595-5947.
LITERATURE CITED
- Bernt KM, Zhu N, Sinha AU, Vempati S, Faber J, Krivtsov AV, Feng Z, Punt N, Daigle A, Bullinger L, Pollock RM, Richon VM, Kung AL, Armstrong SA. MLL-rearranged leukemia is dependent on aberrant H3K79 methylation by DOT1L. Cancer Cell. 2011;20(1):66–78. doi: 10.1016/j.ccr.2011.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boulos N, Mulder HL, Calabrese CR, Morrison JB, Rehg JE, Relling MV, Sherr CJ, Williams RT. Chemotherapeutic agents circumvent emergence of dasatinib-resistant BCR-ABL kinase mutations in a precise mouse model of Philadelphia chromosome-positive acute lymphoblastic leukemia. Blood. 2011;117(13):3585–3595. doi: 10.1182/blood-2010-08-301267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campeau E, Ruhl VE, Rodier F, Smith CL, Rahmberg BL, Fuss JO, Campisi J, Yaswen P, Cooper PK, Kaufman PD. A versatile viral system for expression and depletion of proteins in mammalian cells. PLoS One. 2009;4(8):e6529. doi: 10.1371/journal.pone.0006529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holmfeldt L, Wei L, Diaz-Flores E, Walsh M, Zhang J, Ding L, Payne-Turner D, Churchman M, Andersson A, Chen SC, McCastlain K, Becksfort J, Ma J, Wu G, Patel SN, Heatley SL, Phillips LA, Song G, Easton J, Parker M, Chen X, Rusch M, Boggs K, Vadodaria B, Hedlund E, Drenberg C, Baker S, Pei D, Cheng C, Huether R, Lu C, Fulton RS, Fulton LL, Tabib Y, Dooling DJ, Ochoa K, Minden M, Lewis ID, To LB, Marlton P, Roberts AW, Raca G, Stock W, Neale G, Drexler HG, Dickins RA, Ellison DW, Shurtleff SA, Pui CH, Ribeiro RC, Devidas M, Carroll AJ, Heerema NA, Wood B, Borowitz MJ, Gastier-Foster JM, Raimondi SC, Mardis ER, Wilson RK, Downing JR, Hunger SP, Loh ML, Mullighan CG. The genomic landscape of hypodiploid acute lymphoblastic leukemia. Nat Genet. 2013;45(3):242–252. doi: 10.1038/ng.2532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohno S, Minowada J, Sandberg AA. Chromosome evolution of near-haploid clones in an established human acute lymphoblastic leukemia cell line (NALM-16) Journal of the National Cancer Institute. 1980;64(3):485–493. [PubMed] [Google Scholar]
- Maude SL, Tasian SK, Vincent T, Hall JW, Sheen C, Roberts KG, Seif AE, Barrett DM, Chen IM, Collins JR, Mullighan CG, Hunger SP, Harvey RC, Willman CL, Fridman JS, Loh ML, Grupp SA, Teachey DT. Targeting JAK1/2 and mTOR in murine xenograft models of Ph-like acute lymphoblastic leukemia. Blood. 2012;120(17):3510–3518. doi: 10.1182/blood-2012-03-415448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morisot S, Wayne AS, Bohana-Kashtan O, Kaplan IM, Gocke CD, Hildreth R, Stetler-Stevenson M, Walker RL, Davis S, Meltzer PS, Wheelan SJ, Brown P, Jones RJ, Shultz LD, Civin CI. High frequencies of leukemia stem cells in poor-outcome childhood precursor-B acute lymphoblastic leukemias. Leukemia : official journal of the Leukemia Society of America, Leukemia Research Fund, UK. 2010;24(11):1859–1866. doi: 10.1038/leu.2010.184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mullighan CG. Genomic characterization of childhood acute lymphoblastic leukemia. Semin Hematol. 2013;50(4):314–324. doi: 10.1053/j.seminhematol.2013.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Notta F, Doulatov S, Dick JE. Engraftment of human hematopoietic stem cells is more efficient in female NOD/SCID/IL-2Rgc-null recipients. Blood. 2010;115(18):3704–3707. doi: 10.1182/blood-2009-10-249326. [DOI] [PubMed] [Google Scholar]
- Notta F, Mullighan CG, Wang JC, Poeppl A, Doulatov S, Phillips LA, Ma J, Minden MD, Downing JR, Dick JE. Evolution of human BCR-ABL1 lymphoblastic leukaemia-initiating cells. Nature. 2011;469(7330):362–367. doi: 10.1038/nature09733. [DOI] [PubMed] [Google Scholar]
- Pui CH, Campana D, Pei D, Bowman WP, Sandlund JT, Kaste SC, Ribeiro RC, Rubnitz JE, Raimondi SC, Onciu M, Coustan-Smith E, Kun LE, Jeha S, Cheng C, Howard SC, Simmons V, Bayles A, Metzger ML, Boyett JM, Leung W, Handgretinger R, Downing JR, Evans WE, Relling MV. Treating childhood acute lymphoblastic leukemia without cranial irradiation. N Engl J Med. 2009;360(26):2730–2741. doi: 10.1056/NEJMoa0900386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts KG, Li Y, Payne-Turner D, Harvey RC, Yang YL, Pei D, McCastlain K, Ding L, Lu C, Song G, Ma J, Becksfort J, Rusch M, Chen SC, Easton J, Cheng J, Boggs K, Santiago-Morales N, Iacobucci I, Fulton RS, Wen J, Valentine M, Cheng C, Paugh SW, Devidas M, Chen IM, Reshmi S, Smith A, Hedlund E, Gupta P, Nagahawatte P, Wu G, Chen X, Yergeau D, Vadodaria B, Mulder H, Winick NJ, Larsen EC, Carroll WL, Heerema NA, Carroll AJ, Grayson G, Tasian SK, Moore AS, Keller F, Frei-Jones M, Whitlock JA, Raetz EA, White DL, Hughes TP, Guidry Auvil JM, Smith MA, Marcucci G, Bloomfield CD, Mrozek K, Kohlschmidt J, Stock W, Kornblau SM, Konopleva M, Paietta E, Pui CH, Jeha S, Relling MV, Evans WE, Gerhard DS, Gastier-Foster JM, Mardis E, Wilson RK, Loh ML, Downing JR, Hunger SP, Willman CL, Zhang J, Mullighan CG. Targetable kinase-activating lesions in Ph-like acute lymphoblastic leukemia. N Engl J Med. 2014;371(11):1005–1015. doi: 10.1056/NEJMoa1403088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts KG, Morin RD, Zhang J, Hirst M, Zhao Y, Su X, Chen SC, Payne-Turner D, Churchman ML, Harvey RC, Chen X, Kasap C, Yan C, Becksfort J, Finney RP, Teachey DT, Maude SL, Tse K, Moore R, Jones S, Mungall K, Birol I, Edmonson MN, Hu Y, Buetow KE, Chen IM, Carroll WL, Wei L, Ma J, Kleppe M, Levine RL, Garcia-Manero G, Larsen E, Shah NP, Devidas M, Reaman G, Smith M, Paugh SW, Evans WE, Grupp SA, Jeha S, Pui CH, Gerhard DS, Downing JR, Willman CL, Loh M, Hunger SP, Marra MA, Mullighan CG. Genetic alterations activating kinase and cytokine receptor signaling in high-risk acute lymphoblastic leukemia. Cancer Cell. 2012;22(2):153–166. doi: 10.1016/j.ccr.2012.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shultz LD, Lyons BL, Burzenski LM, Gott B, Chen X, Chaleff S, Kotb M, Gillies SD, King M, Mangada J, Greiner DL, Handgretinger R. Human lymphoid and myeloid cell development in NOD/LtSz-scid IL2R gamma null mice engrafted with mobilized human hemopoietic stem cells. J Immunol. 2005;174(10):6477–6489. doi: 10.4049/jimmunol.174.10.6477. [DOI] [PubMed] [Google Scholar]
- Terziyska N, Castro Alves C, Groiss V, Schneider K, Farkasova K, Ogris M, Wagner E, Ehrhardt H, Brentjens RJ, zur Stadt U, Horstmann M, Quintanilla-Martinez L, Jeremias I. In vivo imaging enables high resolution preclinical trials on patients’ leukemia cells growing in mice. PLoS One. 2012;7(12):e52798. doi: 10.1371/journal.pone.0052798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams RT, Roussel MF, Sherr CJ. Arf gene loss enhances oncogenicity and limits imatinib response in mouse models of Bcr-Abl-induced acute lymphoblastic leukemia. Proc Natl Acad Sci U S A. 2006;103(17):6688–6693. doi: 10.1073/pnas.0602030103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wunderlich M, Chou FS, Link KA, Mizukawa B, Perry RL, Carroll M, Mulloy JC. AML xenograft efficiency is significantly improved in NOD/SCID-IL2RG mice constitutively expressing human SCF, GM-CSF and IL-3. Leukemia. 2010;24(10):1785–1788. doi: 10.1038/leu.2010.158. [DOI] [PMC free article] [PubMed] [Google Scholar]