

Abstract
Lysine-specific demethylase 1 (LSD1) has been reported to repress and activate transcription by mediating histone H3K4me1/2 and H3K9me1/2 demethylation, respectively. The molecular mechanism that underlies this dual substrate specificity has remained unknown. Here we report that an isoform of LSD1, LSD1+8a, does not have the intrinsic capability to demethylate H3K4me2. Instead, LSD1+8a mediates H3K9me2 demethylation in collaboration with supervillin (SVIL), a new LSD1+8a interacting protein. LSD1+8a knockdown increases H3K9me2, but not H3K4me2, levels at its target promoters and compromises neuronal differentiation. Importantly, SVIL co-localizes to LSD1+8a-bound promoters, and its knockdown mimics the impact of LSD1+8a loss, supporting SVIL as a cofactor for LSD1+8a in neuronal cells. These findings provide insight into mechanisms by which LSD1 mediates H3K9me demethylation and highlight alternative splicing as a means by which LSD1 acquires selective substrate specificities (H3K9 versus H3K4) to differentially control specific gene expression programs in neurons.
Introduction
Histone modifications collectively contribute to chromatin template-based nuclear events including gene expression (Jenuwein and Allis, 2001; Shi et al., 2004; Strahl and Allis, 2000). Histone methylation, long considered to be permanent, has been shown to be reversible by the discovery of the first histone demethylase LSD1 (also known as KDM1A) (Shi et al., 2004). LSD1 was initially identified as a component of an HDAC-containing, transcriptional repressor complex (Ballas et al., 2001; Shi et al., 2005). Consistent with its proposed repressive role, LSD1 was subsequently characterized as a histone demethylase dedicated to removing mono- and di-methylation of histone H3 at lysine 4 (H3K4me1/2) (Shi et al., 2004), modifications that are associated with active promoters and either latent or active enhancers (Forneris et al., 2005; Rudolph et al., 2007; Shi et al., 2004; Whyte et al., 2012). LSD1 also associates with nuclear hormone receptors (e.g., androgen receptor [AR] and estrogen receptor [ER]) (Garcia-Bassets et al., 2007; Metzger et al, 2005; Nair et al., 2010). Interestingly, during nuclear-receptor-mediated gene expression, LSD1 appears to function as a co-activator and mediate demethylation of the repressive H3K9me2 mark (Garcia-Bassets et al., 2007; Metzger et al., 2005; Nair et al., 2010; Perillo et al., 2008). However, at the mechanistic level, it has remained unclear how LSD1 can mediate both H3K4me and H3K9me demethylation, a scenario that is not compatible with the LSD1-histone peptide co-crystal structure (Forneris et al., 2006, 2007; Hou and Yu, 2010; Yang et al., 2006).
The LSD1 gene contains 19 exons that are highly conserved among vertebrates. Through RNA alternative splicing, two additional exons, exon E2a and exon E8a, can be included in the mature mRNA, generating four possible LSD1 isoforms, namely the conventional LSD1, LSD1 plus exon E2a (LSD1+2a), exon E8a (LSD1+8a), or both (LSD1+2a+8a). While the inclusion of exon E2a can occur in all tissues, LSD1 transcripts containing the 12-nt-long exon E8a (LSD1+8a) are essentially restricted to the nervous system (Zibetti et al., 2010). In mouse neurons, the LSD1+8a isoforms (LSD1+8a and LSD1+2a+8a) represent a substantial proportion of the total pool of LSD1 transcripts (30%–40%). During the perinatal period, LSD1+8a is the predominantly expressed form of LSD1 (Rusconi et al., 2014; Zibetti et al., 2010). Multiple lines of evidence suggest an important role for LSD1 in neural differentiation. For instance, knockdown or overexpression of LSD1+8a isoforms in mouse cortical neurons, respectively, inhibits or enhances neurite morphogenesis, demonstrating their importance in the execution of the neuronal program (Toffolo et al., 2014; Zibetti et al., 2010). Furthermore, inhibition of LSD1 activity or knockdown of its expression leads to a dramatically reduced proliferation of mouse neural stem cells (NSCs) (Sun et al., 2010, 2011) and a decrease in the number of neurons in zebrafish due to an excessive apoptosis (Jie et al., 2009). Finally, the LSD1/CoREST complex is essential for the development of cortical neurons by controlling their radial migration (Fuentes et al., 2012). Although the role of LSD1 in neuronal differentiation was originally thought to be tied to its repressive functions on several genes essential for the neuronal phenotype (Andrés et al., 1999; Ballas et al., 2001; Hakimi et al., 2002), LSD1 has also been proposed to function as an activator of transcription (Metzger et al., 2005; Wang et al., 2007), leading us to speculate that the LSD1+8a isoform may be responsible for gene activation and possibly H3K9me2 demethylation.
Here we report that unlike the conventional LSD1, LSD1+8a protein complex purified from neuronal cells exhibits robust H3K9me2, but not H3K4me1/2, demethylase activity. Consistently, LSD1+8a functions as a transcriptional co-activator in vivo, and its knockdown is correlated with a rise in the H3K9me2 but not H3K4me2 levels at its direct target genes. We further identify the supervillin protein (SVIL) as a specific LSD1+8a interacting partner. We demonstrate that SVIL protein localizes to LSD1+8a target promoters and is important for LSD1+8a-mediated H3K9me2 demethylation in vitro and in vivo. We also demonstrate that both LSD1+8a and SVIL are essential for neuronal maturation, possibly by working together to activate target gene expression through H3K9me2 demethylation. Taken together, our findings identify a molecular mechanism by which LSD1 mediates H3K9me demethylation via a specific isoform in collaboration with other factors including SVIL. Our findings also highlight RNA alternative splicing as an important mechanism through which LSD1 can acquire histone substrate specificity and therefore differentially controlling specific gene expression programs during neuronal differentiation.
Results
LSD1+8a Is Essential for Neuronal Maturation
The expression of the LSD1+8a isoforms (LSD1+8a and LSD1+2a+8a) is regulated during mouse brain development (Zibetti et al., 2010). We asked whether LSD1+8a expression was also dynamically regulated during human neuronal differentiation using SH-SY5Y cells, which are human neuronal cells derived from neuroblastoma. Sequential exposure of these cells to retinoic acid (RA) for 4 days (day D0 to D4) and then to brain-derived neurotrophic factor (BDNF) in a serum-free medium for 5 days (day D4/B0 to B5) yields a homogeneous population of differentiated neuronal cells that possess many features of primary neurons (Figure S1A) (Encinas et al., 2000; Le et al., 2009). While exposure of SH-SY5Y cells to RA slightly reduced the level of both types of LSD1 transcripts (LSD1-8a and LSD1+8a), BDNF treatment increased the expression level of LSD1+8a isoforms more than that of LSD1-8a isoforms between day B0 and B5 (3.85 ± 0.3 versus 1.47 ± 0.15) (Figure 1A). At day B3 of differentiation, when an extensive neurite outgrowth became apparent, LSD1+8a transcripts represented almost 40% (38.3% ± 5.1%) of the total LSD1 mRNA level (Figure S1B, right panel). The level of the LSD1+8a transcripts is strongly correlated with that of the LSD1+8a proteins during neuronal maturation (Figure 1B). Consistent with the previous study conducted in mouse (Zibetti et al., 2010), these results suggest a potentially important function for the LSD1+8a isoforms in human neuronal differentiation.
Figure 1. LSD1+8a Is Essential for Neuronal Maturation of SH-SY5Y Cells.
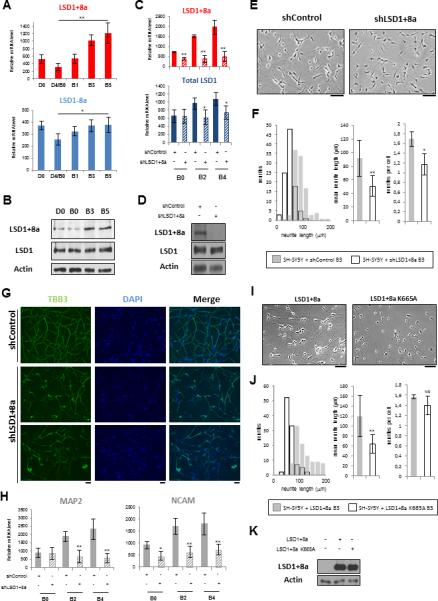
(A and B) SH-SY5Y cells were induced to differentiation.
(A) mRNA levels of LSD1-8a and LSD1+8a isoforms were measured by RT-qPCR and normalized to that of GAPDH. Results for mRNA data are shown as mean ± SEM from at least five independent experiments. (**) p 9 10−4; (*) p 9 0.002.
(B) Protein levels were analyzed with a LSD1+8a-specific antibody and a LSD1 antibody that recognizes all LSD1 isoforms.
(C–H) Stable SH-SY5Y cells infected with control or LSD1+8a shRNA were induced to differentiation.
(C) LSD1+8a and total LSD1 isoforms mRNA levels were measured by RT-qPCR and normalized to that of GAPDH. Results for mRNA data are shown as mean ± SEM from at least three independent experiments. (**) p 9 10−4; (*) p 9 0.01.
(D) Protein levels of SH-SY5Y cells at day B3 of differentiation were analyzed with a LSD1+8a-specific antibody and a LSD1 antibody that recognizes all LSD1 isoforms.
(E–G) The cell phenotypes were examined (E) with phase-contrast microscopy at day B3 (scale bar, 100 μm) and evaluated (F) by quantification of neurite length and number; results are indicated as mean ± SEM, (**) p 9 10−5 (*) p 9 0.01, and (G) by immunostaining for the neuron marker BIII tubulin (TBB3) at day B5 (scale bar, 100 μm).
(H) mRNA levels of NCAM and MAP2 were measured by RT-qPCR and normalized to that of GAPDH and are shown as mean ± SEM from at least three independent experiments. (**) p 9 0.003 (*) p 9 0.01.
(I and J) SH-SY5Y cells stably expressing LSD1+8a or the catalytic mutant LSD1+8a K665A were induced to differentiation. The cell phenotypes were examined (I) with phase-contrast microscopy at day B3 (scale bar, 100 μm) and evaluated (J) by quantification of neurite length and number; results are indicated as mean ± SEM (**) p 9 10−5; NS, non-specific.
(K) Protein levels were analyzed with a LSD1+8a specific antibody.
See also Figure S1.
To address this possibility further, we first established SH-SY5Y cell lines stably expressing either control short hairpin RNA (shRNA) or a shRNA against the neuro-specific exon E8a and performed differentiation assays. The increase of LSD1+8a expression during neuronal differentiation was abrogated by the LSD1+8a shRNA, compared with the control shRNA (Figures 1C and 1D). The LSD1+8a shRNA appeared to be specific to the LSD1+8a isoforms, as it did not affect the level of LSD1-8a isoforms (Figures 1D, S1C, and S1D). In response to differentiation cues, SH-SY5Y cells stop proliferating (day D0 to D4) to become a stable population that shows extensive neurite outgrowth (day B0 to B5). We observed no significant differences in cell growth by day D4/B0 of differentiation between cells infected either with control or LSD1+8a shRNA (Figure S1E). However, between days B0 and B5, inhibition of expression of the LSD1+8a isoforms strongly reduced the neurite outgrowth (92 ± 27 μm versus 51 ± 15 μm at day B3) (Figures 1E and 1F), consistent with the previous findings that LSD1+8a is involved in neurite morphogenesis (Zibetti et al., 2010). We then performed immunofluorescent stainings at day B5, using antibodies against the mature neuron marker BIII tubulin (TBB3). As shown in Figure 1G, the LSD1+8a knockdown cells displayed strong abnormalities in the establishment of the neurite network. Furthermore, inhibition of the LSD1+8a isoforms significantly reduced the expression of NCAM and MAP2, two additional markers for mature neurons (Figure 1H).
To determine whether dynamic expression of LSD1+8a isoforms during human neuronal differentiation is a general phenomenon, we also examined LSD1+8a expression in the human iNGN cell line, which is a human induced pluripotent stem cell line with an integrated copy of doxycycline-inducible neurogenin 1 and neurogenin 2 genes (Busskamp et al., 2014). Induction of neurogenin 1 and 2 expression by doxycycline yields a homogeneous population of differentiated neuronal cells in 4 days, with a significantly more elevated LSD1+8a expression than that of the conventional LSD1, at both mRNA and protein levels (Figures S1F and S1G). Moreover, inhibition of expression of LSD1+8a isoforms also strongly reduced neurite outgrowth (99 ± 37 μm versus 49 ± 13 μm at day B3) (Figures S1H–S1K), consistent with our findings obtained with the SH-SY5Y cell line.
We next asked whether LSD1+8a enzymatic activity was important for neuronal differentiation. Inhibition of LSD1 demethylase activity by a LSD1 inhibitor (S2101 inhibitor; Millipore) strongly reduced neurite outgrowth (98 ± 32 μm versus 58 ± 18 μm at day B3) (Figures S1L and S1M). We next evaluated neurite morphogenesis of SH-SY5Y cell lines stably expressing either LSD1+8a or its catalytic mutant, LSD1+8a K665A (Figure 1K). We focused on the specific LSD1+8a isoform because this splice variant is predominantly expressed in SH-SY5Y neuronal cells compared to that of LSD1+2a+8a (data not shown). Consistent with the previous findings (Toffolo et al., 2014; Zibetti et al., 2010), overexpression of the LSD1+8a isoform led to increased neurite outgrowth (120 ± 42 μm for SH-SY5Y cells overexpressing LSD1+8a versus 92 ± 27 μm for SH-SY5Y cells at day B3 of differentiation) (Figures 1F and 1J). Interestingly, overexpression of the LSD1+8a catalytic mutant strongly reduced neurite morphogenesis at day B3 (120 ± 42 μm versus 64 ± 19 μm) (Figures 1I and 1J), suggesting that the catalytic mutant may act in a dominant negative manner. This finding further suggests that the enzymatic activity of LSD1+8a is necessary for neuronal development. Taken together, these findings suggest that the neuro-specific LSD1+8a isoform is essential for differentiation of SH-SY5Y cells and that its enzymatic activity is required for this biological process.
LSD1+8a Mainly Functions as an Activator of Its Target Genes
To address the underlying molecular mechanisms of how LSD1+8a regulates neuronal differentiation, we set out to identify direct target genes of LSD1+8a by chromatin immunoprecipitation sequencing (ChIP-seq), using a general LSD1 antibody that recognizes both the conventional and the neuro-specific isoforms (LSD1-8a and LSD1+8a isoforms, respectively) and a LSD1+8a antibody that specifically recognizes the LSD1+8a isoforms. ChIP experiments were performed on SH-SY5Y cells at day B3 of differentiation, when the level of LSD1+8a expression is high (Figures 1A and 1B) and the neurite morphogenesis is in progress (Figure 1E). We found all LSD1 isoforms significantly enriched at the promoters of their target genes at around TSS (13.6% versus 2.9%; p < 10−4) (Figures 2A–2C). LSD1+8a isoforms also appeared to bind to the promoter regions of its targets (46.6% versus 2.9%; p < 10−4) and, to a less extent, exons (5.7% versus 1.9; p < 10−4) (Figure 2A).
Figure 2. LSD1+8a Mainly Binds the Promoters of Genes Activated during Neuronal Differentiation.
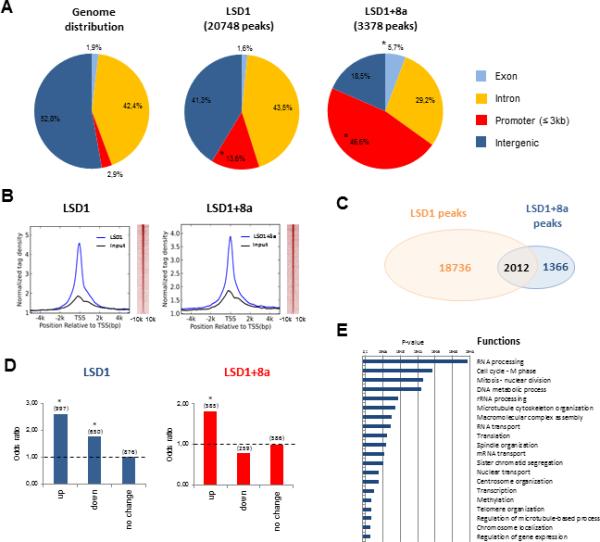
(A) Genomic distribution of LSD1 (20,748) and LSD1+8a (3,378) binding sites in SH-SY5Y cells at day B3 of differentiation. (*) p 9 10−4 assessed using one-sided binomial test.
(B) Normalized LSD1 and LSD1+8a tag density distribution at all genes’ promoters, in a relative distance of 5,000 bp from the TSS (left panel). Heatmap analysis of LSD1 and LSD1+8a distribution at promoter regions, in a relative distance of 10 kb from the TSS (right panel).
(C) Venn diagram showing the overlap between LSD1 and LSD1+8a peaks in the SH-SY5Y cells at day B3 of differentiation. p 9 10−4 assessed using hypergeometric test.
(D) LSD1 and LSD1+8a binding site enrichment for genes upregulated (up), downregulated (down) or with stable expression (no change) during neuronal differentiation of SH-SY5Y cells. Odds ratios were calculated by comparing the expression levels of LSD1 and LSD1+8a targets from day B0 to B3 of differentiation. (*) p 9 10−4.
(E) Gene ontology analysis of LSD1+8a target genes upregulated during neuronal differentiation.
See also Figure S2.
To identify the gene expression network regulated by LSD1+8a, we next performed microarray analysis and found an upregulation and downregulation of 247 and 1,354 genes, respectively, upon LSD1+8a inhibition in the differentiated SH-SY5Y cells (day B3). The preponderance of downregulated genes (1,354) suggests that LSD1+8a could function as a co-activator during neuronal differentiation. These downregulated genes were enriched for mitosis or microtubule cytoskeleton organization pathways, which are important for neurogenesis (Figure S2A). However, only 14.2% of these genes are directly bound by LSD1+8a (Figure S2B), suggesting that inhibition of LSD1+8a both directly and indirectly affects the expression of genes important for the neuronal differentiation. To better determine the precise functions of LSD1+8a during neurogenesis, we next made use of the published expression microarray data of SH-SY5Y cells undergoing neuronal differentiation (Nishida et al., 2008). We found that between days B0 and B3 of differentiation, target genes of the LSD1 isoforms, LSD1-8a and LSD1+8a, were either upregulated or downregulated. We noticed a slight binding preference of LSD1 (LSD1-8a and LSD+8a) for the activated genes than to the repressed genes (2.60 versus 1.75) (Figure 2D, left panel), while the LSD1+8a isoforms exhibited a stronger preference for genes, which were upregulated during neuronal maturation (Figure 2D, right panel). These activated genes were functionally enriched for pathways critical for neurogenesis (Figure 2E), and the promoters of LSD1+8a-bound activated genes were significantly enriched for nuclear receptor-binding motifs (Figure S2C). Importantly, knockdown of LSD1+8a reduced the transcription of the upregulated target genes, but not those target genes whose transcription remains unchanged or decreased during neuronal differentiation (Figures 3 and S2D). Collectively, our data suggest that LSD1+8a mainly functions as a transcriptional co-activator during neuronal differentiation.
Figure 3. Inhibition of LSD1+8a Isoforms Decreases the Expression of Its Target Genes Upregulated during Neuronal Maturation.
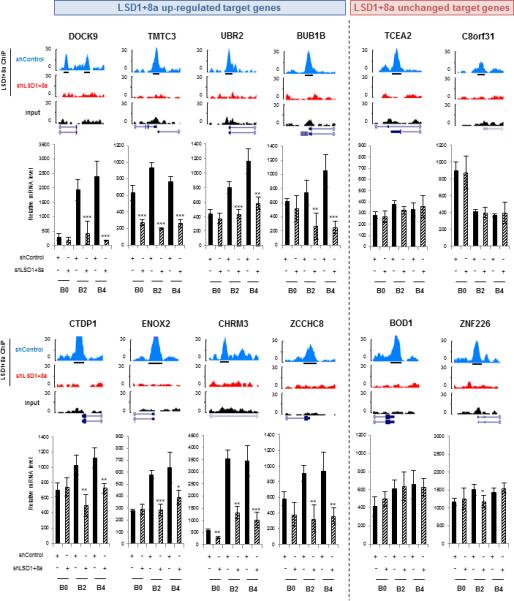
Stable SH-SY5Y cells infected with control or LSD1+8a shRNA were induced to differentiation. Two groups of LSD1+8a target genes were studied: target genes upregulated (left panel) and target genes with a stable or downregulated expression (right panel) between days B0 and B3 of differentiation. For each target, ChIP-seq signal profiles at day B3 of differentiation are shown (top part). LSD1+8a ChIP-seq profiles from both SH-SY5Y cells infected with control or LSD1+8a shRNA are shown. Peak calling track is displayed (black boxes); mRNA levels of target genes were measured by RT-qPCR and normalized to that of GAPDH (bottom part). Results for mRNA data are shown as mean ± SEM from at least three independent experiments. (***) p 9 0.001, (**) p 9 0.01, and (*) p 9 0.025.
See also Figure S2.
LSD1+8a Mediates H3K9me2 Demethylation In Vitro and In Vivo
To further investigate the co-activator function of LSD1+8a, we performed additional ChIP-seq experiments on SHSH5Y cells at day B0 of differentiation, when LSD1+8a expression is low and its inhibition largely does not affect the transcription of its target genes (Figures 1A, 1B, and 3). While LSD1 binding mainly occurred at promoters at day B0 of differentiation (Figures S2E and S2F), we were unable to detect any specific binding of the LSD1+8a isoform at this stage. The conventional LSD1 is largely bound to LSD1+8a target promoters before differentiation (day B0) (Figure S2G), suggesting that a switch of LSD1 isoforms may occur during neuronal differentiation to promote gene activation. This finding further suggests that LSD1-8a and LSD1+8a isoforms could differentially affect the epigenetic landscape and therefore expression of their respective target genes.
To explore the molecular mechanisms by which LSD1+8a promotes transcription, we next performed demethylation assays on peptides and histones using recombinant LSD1 and LSD1+8a purified from insect cells (Figures 4A and 4B). While conventional LSD1 demethylated H3K4me2 as expected, LSD1+8a was unable to remove methyl groups from either H3K4me2 or H3K9me2 peptides and from histones purified from HeLa cells. Similar results were obtained using recombinant LSD1 proteins purified from bacteria (Figure S3A).
Figure 4. LSD1+8a Demethylates H3K9me2 In Vitro and In Vivo.
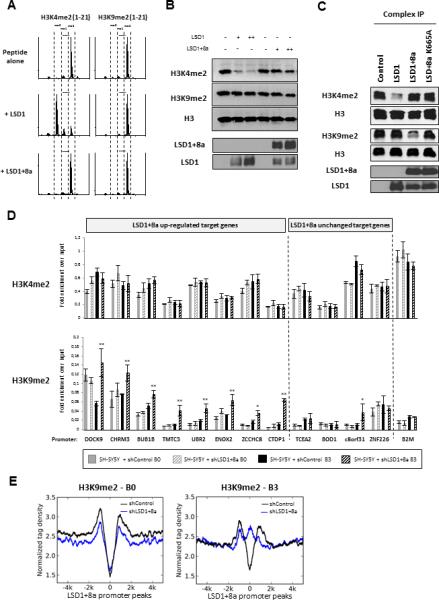
(A) Histone demethylase activity of LSD1 and LSD1+8a proteins purified from sf9 insect cells, on H3K4me2 and H3K9me2 peptides (residues 1 to 21 of histone H3 tail). Activity was examined by MALDI-TOF mass spectrometry.
(B) Histone demethylase assay on calf histones with LSD1 and LSD1+8a proteins purified from sf9 insect cells. Histone lysates were analyzed by western blotting as indicated.
(C) Histone demethylase assay on calf histones with LSD1 complexes purified from SH-SY5Y cells stably expressing Flag-HA-tagged LSD1, LSD1+8a, or the catalytic mutant LSD1+8a K665A. Protein complexes were purified on FLAG tag and analyzed by western blotting as indicated.
(D) ChIP-qPCR analysis of H3K4me2 and H3K9me2 levels at the promoters of LSD1+8a target genes. Stable SHSY5Y cells infected with control or LSD1+8a shRNA were induced to differentiation. Experiments were performed before and during the neuronal maturation (day B0 and day B3, respectively). β2 microglobulin (B2M) promoter was used as a control. Results are shown as the relative fold enrichment over the input (mean ± SEM) of at least three independent experiments. (**) p 9 0.006; (*) p 9 0.03.
(E) Normalized H3K9me2 tag density at LSD1+8a target promoters, in a relative distance of 5,000 bp from the TSS. Tag densities are shown for SH-SY5Y cells infected with control or LSD1+8a shRNA, at day B0 and B3 of differentiation (left and right panels, respectively).
See also Figure S3 and Table S1.
What might be the reason for the apparent inactivity of LSD1+8a in vitro? The recombinant LSD1+8a isoform appears to be stable (Figure S3B) and probably not misfolded, judging from the gel filtration chromatography elution profiles (Figure S3C). Given that LSD1+8a function requires its catalytic activity, the failure to demethylate H3K4me or H3K9me suggests that additional cofactors and/or post-translational modifications might be required for its enzymatic activity. To address this possibility, we performed in vitro demethylation assays using TAP-tagged LSD1 and LSD1+8a complexes purified from SH-SY5Y cells. We found that the TAP-LSD1 complex demethylated H3K4me2 in vitro as expected. Importantly, the TAP-LSD1+8a complex, but not the LSD1+8a catalytic mutant complex, removed methyl groups on H3K9 (but not H3K4) (Figure 4C), indicating that the demethylation was mediated by LSD1+8a itself and not an associated histone demethylase. The specificity of histone H3K9me demethylation mediated by LSD1+8a was further confirmed by mass spectrometry analysis (Table S1). As an important control, the conventional LSD1 complex purified from SH-SY5Y cells showed no activity toward H3K9me2 (Figure 4C). Taken together, these findings indicate that the H3K9me2 demethylase activity exhibited by the LSD1+8a complex is intrinsic to the LSD1+8a isoform.
To determine whether LSD1+8a regulates its target genes by demethylating H3K9me in vivo, we performed ChIP-qPCR experiments using SH-SY5Y cells expressing either control or LSD1+8a shRNA, before and during the neuronal maturation (day B0 and day B3, respectively). We found that inhibition of LSD1+8a isoform increased the H3K9me2 level at its target genes that are upregulated during neuronal development (Figure 4D and Figure S3D). No significant changes were observed for the H3K4me2 level at the same target promoters. Similar results were obtained using the iNGN cells (Figure S3E). We next investigated the impact of LSD1+8a inhibition on H3K9me2 at the genome-wide level by performing ChIP-seq experiments on SH-SY5Y cells expressing either control or LSD1+8a shRNA, at day B0 and B3 of differentiation. In agreement with previous reports (Chandra et al., 2012), we found a lower abundance of H3K9me2 at around the TSS compared to the gene body, both before and during differentiation (Figures S3F and S3G). Notably, inhibition of LSD1+8a specifically increased the H3K9me2 level at its upregulated target genes at day B3 but not at day B0 of differentiation (Figure 4E), suggesting that LSD1+8a regulates local changes of H3K9me2 at its target promoters rather than a global change in epigenetic repression during neuronal differentiation. Collectively, our data suggest that LSD1+8a functions as an H3K9me demethylase to promote transcription of its target genes during neuronal differentiation.
LSD1+8a Interacts with the SVIL Protein
As discussed above, the fact that only the LSD1+8a complex but not the recombinant LSD1+8a protein mediated H3K9 demethylation suggests that LSD1+8a may require additional cofactors for H3K9me demethylation. To investigate, TAP-LSD1+8a complex was purified at different times of differentiation and analyzed by mass spectrometry. Apart from the known interacting proteins including CoREST and HDACs, we identified the Supervillin (SVIL) protein as a potentially new LSD1+8a-associated partner (Table 1tbl1). We confirmed the interaction of SVIL with the LSD1+8a isoform by western blot analysis, by endogenous co-immunoprecipitation, and by purification of the TAP-SVIL complex in SH-SY5Y cells (Figures 5A and 5B and Table S2, respectively).
Table 1.
LSD1+8a complexes purification.
| D0 | B0 | B1 | |
|---|---|---|---|
| KDM1A | 307 | 364 | 285 |
| ZMYM2 | 37 | 42 | 36 |
| GSE1 | 46 | 44 | 26 |
| HDAC2 | 42 | 40 | 35 |
| RCOR2 | 24 | 16 | 10 |
| HSP90A | 13 | 7 | 9 |
| HSP90B | 13 | 6 | 7 |
| RCOR3 | 34 | 33 | 28 |
| RCOR1 | 32 | 24 | 27 |
| HDAC1 | 20 | 20 | 17 |
| CH60 | 7 | 2 | 1 |
| ZNF516 | 10 | 11 | 10 |
| PHF21A | 13 | 9 | 16 |
| HMG20B | 14 | 8 | 12 |
| CTBP1 | 9 | 12 | 9 |
| SVIL | 1 | 2 | 15 |
Total peptide numbers of proteins found by MS/MS analysis of the Flag-LSD1+8a complexes purified from SH-SY5Y cells at different times of neuronal differentiation i.e. day D0, B0 and B1. A summary of the components previously identified in LSD1 complex is shown in addition to the new LSD1+8a partner, the supervillin protein (SVIL).
Figure 5. SVIL Is Essential for Neuronal Maturation.
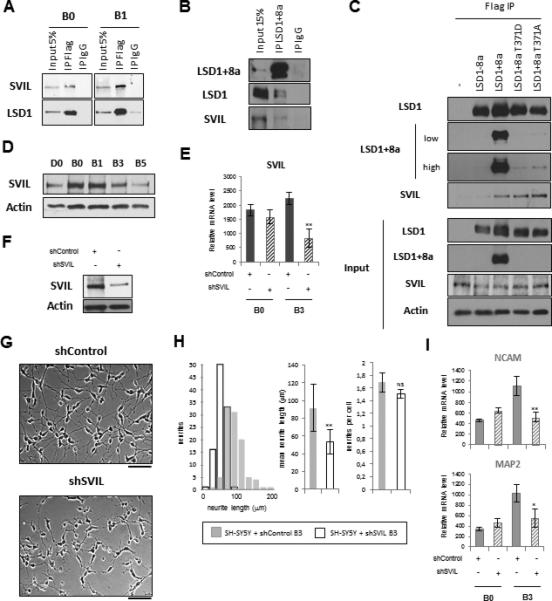
(A) SH-SY5Y cells stably expressing Flag/HA-tagged LSD1+8a were induced to differentiation. LSD1+8a was immunoprecipitated at day B0 and B1 using a Flag antibody, followed by western blotting as indicated.
(B) SH-SY5Y cells were induced to undergo differentiation, and cellular extracts of cells at day B3 of differentiation were immunoprecipitated with a LSD1+8a-specific antibody, followed by western blotting as indicated.
(C) Cellular extracts of SH-SY5Y cells stably expressing Flag/HA-tagged LSD1-8a, LSD1+8a, LSD1+8a T371D phospho-mimetic mutant, and LSD1+8a T371A phospho-defective mutant were immunoprecipitated with a Flag antibody, followed by western blotting as indicated. In the top panel, LSD1+8a western blot is shown with two exposures, one low and one high. The LSD1+8a specific antibody was generated against the exon 8a and therefore does not recognize the LSD1+8a mutants.
(D) SH-SY5Y cells were induced to undergo differentiation. Protein levels were analyzed with a SVIL antibody.
(E–I) SH-SY5Y cells were induced to differentiation and infected at day D2 with control or SVIL shRNA.
(E) SVIL mRNA level was measured by RT-qPCR and normalized to that of GAPDH. Results for mRNA data are shown as mean ± SEM from at least three independent experiments. (**) p 9 0.001.
(F) Protein levels at day B3 of differentiation were analyzed with a SVIL antibody. Actin was used as a loading control.
(G and H) (G) The cell phenotypes were examined with phase-contrast microscopy at day B3 (scale bar, 100 μm) and evaluated (H) by quantification of neurite length and number; results are indicated as mean ± SEM (**) p 10−5 9; NS, non-specific.
(I) NCAM and MAP2 mRNA levels were measured by RT-qPCR and normalized to that of GAPDH. Results for mRNA data are shown as mean ± SEM from at least three independent experiments. (**) p 9 0.003, (*) p 9 0.01.
See also Figure S4 and Table S2.
SVIL belongs to the gelsolin gene family, which encodes a number of actin-binding proteins (ABPs). These proteins have been shown to control important actin-dependent functions in the cytoplasm, such as cell division, mobility, and morphology (Bhuwania et al., 2012; Fang et al., 2010; Smith et al., 2010). However, these ABPs also have specific nuclear functions such as nuclear transport, structural maintenance, and regulation of transcription (Ting et al., 2002; Wulfkuhle et al., 1999). In the nucleus, ABPs interact with nuclear receptors (e.g., AR and ER) through their steroid-receptor-binding domain (Figure S4A) (Archer et al., 2005). SVIL has been reported to be an AR-interacting protein that functions as a co-activator of AR-mediated transcription by an unknown molecular mechanism (Ting et al., 2002). To investigate whether SVIL functions as a cofactor for LSD1+8a during neuronal differentiation, we first confirmed that SVIL protein was present in the nucleus of SH-SY5Y cells (Figure S4B). We next asked whether SVIL specifically associated with LSD1+8a isoform. To address this question, we stably overexpressed Flag/HA-tagged LSD1-8a, LSD1+8a, LSD1+8a phospho-mimetic (T371D), and phospho-defective (T371A) mutants in SH-SY5Y cells and then performed Flag immunoprecipitations (Figure 5C). Importantly, SVIL protein specifically associated with LSD1+8a, but interestingly, single mutations (T371D or T371A) within exon 8a did not disturb this interaction (Figure 5C). We then attempted to identify the region within SVIL that is responsible for LSD1+8a binding and found the steroid-receptor-binding domain of SVIL was sufficient for its interaction with LSD1+8a (Figure S4C).
SVIL Is Essential for Neuronal Maturation
To further address the potential cofactor function of SVIL, we first analyzed SVIL expression during neuronal differentiation. In SH-SY5Y cells, SVIL expression is strongly elevated after exposure to RA (day D0 to B0) and then progressively decreased during neuronal maturation (Figure 5D). Differentiation of the iNGN cells also strongly induced SVIL expression, which remained stable during maturation (Figure S4D). We next investigated SVIL role during neuronal differentiation but were unable to establish a SVIL knockdown SH-SY5Y cell line (data not shown), consistent with a previous report showing that SVIL is essential for cell survival (Fang and Luna, 2013). To overcome this, we inhibited SVIL expression while SH-SY5Y cells were undergoing differentiation upon RA treatment, which drastically slows down SH-SY5Y proliferation (days D2/D3 of treatment). Under this condition, cell survival and differentiation were possible, and as shown in Figures 5E–5F, SVIL expression was reduced at mRNA and protein levels, respectively (Figures 5E and 5F). Importantly, inhibition of SVIL expression significantly reduced neurite outgrowth (92 ± 26 μm versus 53 ± 13 μm at day B3) (Figures 5G and 5H) and consistently also reduced the expression of NCAM and MAP2, similar to that of inhibition of LSD1+8a (Figure 5I). Comparable results were also obtained with the siRNA-mediated inhibition of SVIL expression during differentiation of the iNGN cells (Figures S4E–S4I). Taken together, these findings show that SVIL protein plays a role similar to that of LSD1+8a during neuronal differentiation and are consistent with our hypothesis that SVIL may function as a cofactor for LSD1+8a in neurons.
SVIL Controls LSD1+8a-Mediated H3K9 Demethylation
To investigate the molecular functions of SVIL, we determined the genomic location of SVIL in the SH-SY5Y cells at day B3 of differentiation by ChIP-seq. We found that SVIL bound promoters at and around the TSS (Figures S5A and S5B), and importantly, 20% of the SVIL peaks showed an overlap with the LSD1+8a peaks (Figure S5C). SVIL preferentially bound the promoters of genes that were upregulated rather than downregulated during neuronal differentiation (Figures S5D and S5E), similar to that of LSD1+8a (Figure 2D). These findings further support our hypothesis that SVIL functions as a cofactor for LSD1+8a.
As a cofactor for LSD1+8a, is SVIL sufficient to enable LSD1+8a-mediated H3K9 demethylation? To address this question, we performed in vitro demethylation assays on histones using recombinant SVIL and LSD1+8a purified from insect cells. The addition of recombinant SVIL purified from insect cells or Flag-tagged SVIL purified from SH-SY5Y cells failed to reconstitute LSD1+8a-mediated H3K9me2 demethylation in vitro (Figures 6A and S5F). Notably, inclusion of purified SVIL in this assay inhibited LSD1-mediated demethylation of H3K4me2 (Figure 6A). We next carried out in vitro demethylation assays using recombinant LSD1, LSD1+8a, and LSD1+2a+8a isoforms purified from insect cells, mixed in with nuclear extracts prepared from SH-SY5Y cells that either express or don't express Flag-tagged SVIL. We found that addition of the nuclear extract expressing Flag-SVIL, LSD1+8a, but not the conventional LSD1, facilitated LSD1+8a-mediated H3K9me2 demethylation. Under the same assay conditions, LSD1+2a+8a also mediated H3K9me2 demethylation, consistent with the idea that exon 8a plays an important role in LSD1-mediated H3K9me2 demethylation (Figure 6B). Importantly, nuclear extracts alone showed no detectable H3K9me2 demethylation activity (Figure 6B). Furthermore, TAP-tagged LSD1+8a complex purified from SH-SY5Y cells in which SVIL was knocked down showed a reduced H3K9me2 demethylase activity (Figure S5G).
Figure 6. SVIL Regulates LSD1+8a-Mediated H3K9me2 Demethylation In Vitro and In Vivo.
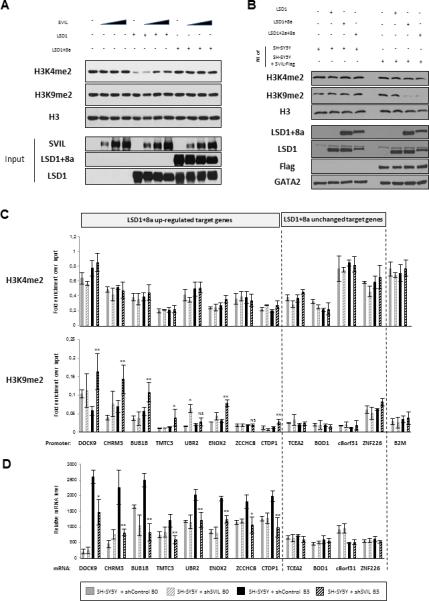
(A) Histone demethylase assay on calf histones with LSD1, LSD1+8a, and SVIL proteins purified from sf9 insect cells. Histone lysates were analyzed with H3K4me2, H3K9me2 and H3 antibodies.
(B) Histone demethylase assay on calf histones with LSD1, LSD1+8a, and LSD1+2a+8a proteins purified from sf9 insect cells and with nuclear extracts from SH-SY5Y cells overexpressing or not Flag-tagged SVIL. Histone lysates were analyzed by western blotting as indicated.
(C and D) SH-SY5Y cells were induced to differentiation and infected at day D2 with control or SVIL shRNA. Experiments were performed before and during the neuronal maturation (day B0 and day B3, respectively).
(C) ChIP-qPCR analysis of H3K4me2 and H3K9me2 levels at the promoters of LSD1+8a target genes. β2 microglobulin (B2M) promoter was used as a control. Results are shown as the relative fold enrichment over the input (mean ± SEM) of at least three independent experiments. (**) p 9 0.006; (*) p 9 0.05. NS, non-specific.
(D) mRNA levels of LSD1+8a target genes were measured by RT-qPCR and normalized to that of GAPDH. Results for mRNA data are shown as mean ± SEM from at least five independent experiments. (**) p 9 0.006; (*) p 9 0.01.
See also Figure S5.
We then asked whether SVIL regulates H3K9 methylation at LSD1+8a target genes in vivo. We performed ChIP-qPCR and found that inhibition of SVIL increased the H3K9me2 level at the LSD1+8a-upregulated target genes (Figure 6C). In contrast, no significant changes were observed for the H3K4me2 level at the same target promoters. These findings support the in vitro finding that SVIL plays a role in LSD1+8a-mediated H3K9 demethylation. We additionally found that, similar to knockdown of LSD1+8a, knockdown of SVIL also reduced the expression of the LSD1+8a target genes, which are upregulated during differentiation, but had no effects on LSD1+8a target genes showing no changes in their expression during normal differentiation (Figure 6D). Similar results were obtained using iNGN cells (Figures S5H and S5I), confirming that inhibition of SVIL resulted in an increase in the H3K9me2 level at LSD1+8a target genes and a decrease in their expression. Collectively, these findings show that SVIL regulates LSD1+8a-mediated H3K9 demethylation and neuronal differentiation.
Discussion
The early structure work on LSD1 provided a molecular basis for how LSD1 mediates demethylation of H3K4me1/2 but not H3K9me1/2 (Forneris et al., 2006, 2007; Hou and Yu, 2010; Yang et al., 2006). Therefore, it has remained an enigma how LSD1 mediates both H3K4me1/2 and H3K9me1/2 demethylation. The main finding of this study is that the specific LSD1+8a isoforms (containing an extra 4-aa-long exon E8a) mediate H3K9me demethylation (Figure 6B). Importantly, our data show that LSD1+8a alone is not sufficient to mediate H3K9me1/2 demethylation (Figures 4A and 4B); however, the LSD1+8a protein complex, but not the complex of the conventional LSD1, purified from SH-SY5Y cells, is capable of specifically demethylating H3K9me2 (Figure 4C), indicating that additional cofactors are required for the demethylase activity of LSD1+8a. We identified SVIL as a new LSD1+8a-interacting protein, which is necessary for LSD1+8a-mediated H3K9me2 demethylation both in vitro and in vivo. We speculate that the inclusion of this additional mini-exon may have created a docking site that facilitates LSD1+8a/SVIL interaction.
Interestingly, the fact that only the nuclear extract expressing SVIL, but not recombinant SVIL alone, reconstitutes LSD1+8a-mediated H3K9me2 demethylase activity in vitro suggests that additional cofactors and/or neuro-specific posttranslational modifications of SVIL may be required. One possible auxiliary factor could be Proline-, glutamic acid-, and leucine-rich protein-1 (PELP1), which acts as a co-activator of several nuclear receptors including ER and AR (Nair et al., 2007; Vadlamudi et al., 2001) and can promote LSD1-mediated H3K9me demethylation (Nair et al., 2010). However, whether PELP1 is involved in the LSD1+8a/SVIL interaction and LSD1+8a-mediated H3K9 demethylation remains to be determined. As SVIL interacts with nuclear receptors (Archer et al., 2005), another possibility is that H3K9 demethylase activity of LSD1+8a could be due to a cooperative and ligand enhancement effect between SVIL and nuclear receptors. Consistently, the promoters of LSD1+8a-bound activated but not repressed genes were significantly enriched for nuclear receptor-binding motifs (Figure S2C).
The inclusion of exon E8a not only allows LSD1 to gain H3K9me demethylation capability but also seems to compromise its ability to demethylate H3K4 in vitro. How does this additional exon affect LSD1's enzymatic activity toward H3K4me? The crystal structure of LSD1+8a revealed that exon E8a, which is located within the amine oxidase domain, did not disturb the overall conformation, which is very similar to that of the conventional LSD1 protein (Zibetti et al., 2010). A computational study showed that the entrance of the H3 pocket is gated by the “lysine triad” of residues K355, K357, and K359, which are helped by the residues D375, E376, and E379 (Baron and Vellore, 2012a, b). The inclusion of exon E8a at position 369 may disturb the triad gate motions and therefore affects LSD1 substrate specificity toward H3K4me.
The idea that specific isoforms of LSD1 play a role in mediating H3K9me2 demethylation is appealing given that structural studies do not support the possibility that the conventional LSD1 itself could function as an H3K9me2 demethylase (Forneris et al., 2006, 2007; Hou and Yu, 2010; Yang et al., 2006). It will be interesting to determine which LSD1 isoforms are operating in prostate cells where LSD1 was first reported to also be an H3K9me2 demethylase (Metzger et al., 2005), especially given that the neuronal-cell-enriched LSD1+8a isoform is also expressed in testis (Zibetti et al., 2010). Furthermore, LSD1+8a isoforms may also be expressed in other biological contexts and/or in response to developmental or environmental stimuli, where it may function to promote gene transcription by mediating H3K9me2 demethylation. Regardless, our findings revealed an isoform and cofactor-dependent role in regulating the substrate specificity of LSD1, thus suggesting that alternative splicing provides a mechanism by which LSD1 can acquire dual substrate specificities. Given the central role of LSD1 in regulating gene expression, modulation of LSD1 pre-mRNA splicing could offer a promising therapeutic option to reprogramming the cell phenotype in brain cancers and neurodegenerative diseases.
Experimental Procedures
Cell Culture and Viral Transduction
SH-SY5Y and HEK293T cells were maintained in DMEM containing 4,500 mg/l glucose, 10% heat-inactivated fetal bovine serum, 110 mg/l sodium pyruvate, 2 mM L-glutamine, and 1% penicillin-streptomycin. For differentiation, SH-SY5Y cells were seeded onto collagen-coated plates at an initial density of 104 cells/cm2. RA (Sigma) was added at a final concentration of 10 μM the next day after plating. After 4 days, the cells were washed three times with PBS and incubated with 50 ng/ml of BDNF (Millipore) in serum-free medium for 5 days. The iNGN cells were maintained on a Matrigel hESC-qualified matrix (BD Biosciences) in mTeSR media (StemCell Technologies). For passaging, cells were dissociated using TrypLE Express (GIBCO) and replated using mTeSR supplemented with 3 μg/ml of InSolution Y-27632 Rho-kinase inhibitor (Millipore). For differentiation, cells were induced with 1 μg/ml of doxycycline (Sigma).
Lentiviruses were produced by cotransfection of the lentiviral plasmid with helper vectors (pHDM-VSV-G, pHDM-tat1b, pHDM-HgPM2, and pRC-CMV-RaII) into HEK293T cells, and viral supernatants were collected after 60–72 hr. Cells infected with lentiviral shRNAs or expression vectors were selected after transduction with 1 μg/ml puromycin.
Immunoprecipitation, Tandem Affinity Purification, and Mass Spectrometry
For immunoprecipitation assays, cell extracts from 107 cells were incubated with 2 μg of antibodies—i.e., FLAG M2 (F1804; Sigma), GFP (N86/38) (75-132; NeuroMab). Complexes were pelleted using 50 μl of protein A/G PLUS-Agarose beads (sc-2003; Santa Cruz).
Tandem affinity purification (TAP) experiments were carried out by using at least 4.00 × 108 cells. Cell extracts were incubated with Flag M2 beads (Sigma) and immunocomplexes were eluted with Flag peptide. For mass spectrometry analyses, the elution mixture was trichloroacetic acid (TCA) precipitated, trypsin digested, loaded onto stage tips, and resuspended in sample loading buffer (5% formic acid/5% acetonitrile). Samples were analyzed by HPLC (Eksigent NanoLC; AB Sciex) into PicoTips (New Objective) coupled to a LTQ ion trap mass spectrometer (Thermo Fisher Scientific) operated in positive ion mode.
ChIP
Cells were fixed with 1% formaldehyde for 10 min at room temperature before termination with 0.125 M glycine. Cells were then lysed in ChIP buffer (0.6% SDS, 10 mM EDTA, and 50 mM Tris-HCl [pH 8.1]), and cross-linked chromatin was sonicated to obtain DNA fragments of 300–800 bp. Immunoprecipitations were performed following the Millipore protocol (http://www.emdmillipore.com). Antibodies used were as follows: LSD1 (ab17721; Abcam), Supervillin (S8695 ; Sigma), H3 (ab1791; Abcam), H3K9me2 (ab1220; Abcam), H3K4me2 (07-030; Millipore), and the homemade polyclonal antibody against LSD1+8a. DNA was recovered by phenol-chloroform extraction and ethanol precipitation. RT-qPCR analyses were performed on immunoprecipitated DNA using specific primers described in supplementary material. The results were calculated and presented as relative fold enrichment over the input.
ChIP-seq libraries were prepared using NEBNext DNA library preparation reagents (E6000) and the protocol and reagents concentrations described in the Illumina Multiplex ChIP-seq DNA sample Prep Kit. Libraries were indexed using a single indexed PCR primer. Libraries were quantified by Qubit (Invitrogen) and sequenced using a HiSeq 2000 (Illumina) to generate 50 bp single-end reads. The raw data are deposited at the Gene Expression Omnibus (GEO) under the subseries entry GSE58258. The significantly enriched peaks were determined by Model-Based Analysis of ChIP-Seq (MACS) package based on a p value 9 10−5 and a false discovery rate (FDR) cut-off 9 0.05.
Protein Purification and Demethylation Assay
LSD1, LSD1+8a, LSD1+2a+8a, and SVIL cDNA were cloned into a bactulovirus expression vector; pFastBacHT A (Invitrogen); and transfected into Sf9 cells by using the Bac-to-Bac HT Vector kit (Invitrogen). His-tagged proteins were produced according to the manufacturer's specifications, purified on Ni-NitriloTriacetic Acid (NTA)-agarose beads (QIAGEN), and eluted by imidazole (Sigma). Demethylation assays were carried out on calf histones or peptides at 37°C for 4 hr with the following buffer: 50 mM Tris (pH 8.5), 50 mM KCl, 5 mM MgCl, 0.5% BSA, and 5% glycerol. For a typical reaction, the volume of the reaction is 50 μl, in which either 30 μg of calf histones or 3 μg of modified histone peptides were used as substrates. Different amounts of LSD1 isoforms and SVIL protein ranging from 10–20 μg were used in the reaction. For histone assays, the reaction mixture was analyzed by western blotting using methyl-specific antibodies. For peptide assays, the reaction mixture was analyzed by matrix-assisted laser desorption/ionization (MALDI) mass spectroscopy as previously described (Shi et al., 2004). The samples were analyzed by a MALDI-TOF/TOF mass spectrometer at the Molecular Biology Core Facility of Dana Farber Cancer Institute.
Also see Supplemental Information.
Supplementary Material
Highlights.
LSD1+8a isoform does not have the intrinsic property to demethylate H3K4
LSD1+8a functions as a co-activator by demethylating the repressive H3K9me2 mark
LSD1+8a interacts with SVIL; LSD1+8a/SVIL-containing complex demethylates H3K9me2
SVIL regulates neuronal maturation by controlling LSD1+8a mediated H3K9 demethylation
Benoit Laurent et al. find that the neuro-enriched LSD1 isoform, LSD1+8a, mediates H3K9me2 demethylation in collaboration with the supervillin protein (SVIL), a new LSD1+8a partner. These findings highlight alternative splicing as a means by which LSD1 acquires selective substrate specificities (H3K9 versus H3K4) to control specific transcriptional programs in neurons.
Acknowledgments
We thank members of the Shi lab for helpful discussions. In particular, we wish to thank James Lee (Dana Farber Cancer Institute, Boston) for help with the mass spectrometer and Angeles Fernandez-Gonzalez (Boston Children's Hospital, Boston) for help with immunofluorescence microscopy. We thank Dr. Elizabeth J. Luna (University of Massachusetts Medical School, Worcester) for the pEGFP-SVIL constructs. B.L. was supported by fellowships from Association pour la Recherche sur le Cancer (ARC) and the Philippe Foundation. This work was supported by a grant from the NIH to Y.S. (R01 CA118487). Y.S. is an American Cancer Society Research Professor.
Footnotes
Supplemental Information
Supplemental Information includes five figures, two tables, and Supplemental Experimental Procedures and can be found with this article online at *bxs.
References
- Andrés ME, Burger C, Peral-Rubio MJ, Battaglioli E, Anderson ME, Grimes J, Dallman J, Ballas N, Mandel G. CoREST: a functional corepressor required for regulation of neural-specific gene expression. Proc. Natl. Acad. Sci. USA. 1999;96:9873–9878. doi: 10.1073/pnas.96.17.9873. PubMed. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Archer SK, Claudianos C, Campbell HD. Evolution of the gelsolin family of actin-binding proteins as novel transcriptional coactivators. Bioessays. 2005;27:388–396. doi: 10.1002/bies.20200. PubMed. [DOI] [PubMed] [Google Scholar]
- Ballas N, Battaglioli E, Atouf F, Andres ME, Chenoweth J, Anderson ME, Burger C, Moniwa M, Davie JR, Bowers WJ, et al. Regulation of neuronal traits by a novel transcriptional complex. Neuron. 2001;31:353–365. doi: 10.1016/s0896-6273(01)00371-3. PubMed. [DOI] [PubMed] [Google Scholar]
- Baron R, Vellore NA. LSD1/CoREST is an allosteric nanoscale clamp regulated by H3-histone-tail molecular recognition. Proc. Natl. Acad. Sci. USA. 2012a;109:12509–12514. doi: 10.1073/pnas.1207892109. PubMed. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baron R, Vellore NA. LSD1/CoREST reversible opening-closing dynamics: discovery of a nanoscale clamp for chromatin and protein binding. Biochemistry. 2012b;51:3151–3153. doi: 10.1021/bi300068r. PubMed. [DOI] [PubMed] [Google Scholar]
- Bhuwania R, Cornfine S, Fang Z, Krüger M, Luna EJ, Linder S. Supervillin couples myosin-dependent contractility to podosomes and enables their turnover. J. Cell Sci. 2012;125:2300–2314. doi: 10.1242/jcs.100032. PubMed. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Busskamp V, Lewis NE, Guye P, Ng AH, Shipman SL, Byrne SM, Sanjana NE, Murn J, Li Y, Li S, et al. Rapid neurogenesis through transcriptional activation in human stem cells. Mol. Syst. Biol. 2014;10:760. doi: 10.15252/msb.20145508. PubMed. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chandra T, Kirschner K, Thuret JY, Pope BD, Ryba T, Newman S, Ahmed K, Samarajiwa SA, Salama R, Carroll T, et al. Independence of repressive histone marks and chromatin compaction during senescent heterochromatic layer formation. Mol. Cell. 2012;47:203–214. doi: 10.1016/j.molcel.2012.06.010. PubMed. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Encinas M, Iglesias M, Liu Y, Wang H, Muhaisen A, Ceña V, Gallego C, Comella JX. Sequential treatment of SH-SY5Y cells with retinoic acid and brain-derived neurotrophic factor gives rise to fully differentiated, neurotrophic factor-dependent, human neuron-like cells. J. Neurochem. 2000;75:991–1003. doi: 10.1046/j.1471-4159.2000.0750991.x. PubMed. [DOI] [PubMed] [Google Scholar]
- Fang Z, Luna EJ. Supervillin-mediated suppression of p53 protein enhances cell survival. J. Biol. Chem. 2013;288:7918–7929. doi: 10.1074/jbc.M112.416842. PubMed. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang Z, Takizawa N, Wilson KA, Smith TC, Delprato A, Davidson MW, Lambright DG, Luna EJ. The membrane-associated protein, supervillin, accelerates F-actin-dependent rapid integrin recycling and cell motility. Traffic. 2010;11:782–799. doi: 10.1111/j.1600-0854.2010.01062.x. PubMed. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forneris F, Binda C, Vanoni MA, Mattevi A, Battaglioli E. Histone demethylation catalysed by LSD1 is a flavin-dependent oxidative process. FEBS Lett. 2005;579:2203–2207. doi: 10.1016/j.febslet.2005.03.015. PubMed. [DOI] [PubMed] [Google Scholar]
- Forneris F, Binda C, Dall'Aglio A, Fraaije MW, Battaglioli E, Mattevi A. A highly specific mechanism of histone H3-K4 recognition by histone demethylase LSD1. J. Biol. Chem. 2006;281:35289–35295. doi: 10.1074/jbc.M607411200. PubMed. [DOI] [PubMed] [Google Scholar]
- Forneris F, Binda C, Adamo A, Battaglioli E, Mattevi A. Structural basis of LSD1-CoREST selectivity in histone H3 recognition. J. Biol. Chem. 2007;282:20070–20074. doi: 10.1074/jbc.C700100200. PubMed. [DOI] [PubMed] [Google Scholar]
- Fuentes P, Cánovas J, Berndt FA, Noctor SC, Kukuljan M. CoREST/LSD1 control the development of pyramidal cortical neurons. Cereb. Cortex. 2012;22:1431–1441. doi: 10.1093/cercor/bhr218. PubMed. [DOI] [PubMed] [Google Scholar]
- Garcia-Bassets I, Kwon YS, Telese F, Prefontaine GG, Hutt KR, Cheng CS, Ju BG, Ohgi KA, Wang J, Escoubet-Lozach L, et al. Histone methylation-dependent mechanisms impose ligand dependency for gene activation by nuclear receptors. Cell. 2007;128:505–518. doi: 10.1016/j.cell.2006.12.038. PubMed. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hakimi MA, Bochar DA, Chenoweth J, Lane WS, Mandel G, Shiekhattar R. A core-BRAF35 complex containing histone deacetylase mediates repression of neuronal-specific genes. Proc. Natl. Acad. Sci. USA. 2002;99:7420–7425. doi: 10.1073/pnas.112008599. PubMed. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hou H, Yu H. Structural insights into histone lysine demethylation. Curr. Opin. Struct. Biol. 2010;20:739–748. doi: 10.1016/j.sbi.2010.09.006. PubMed. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jenuwein T, Allis CD. Translating the histone code. Science. 2001;293:1074–1080. doi: 10.1126/science.1063127. PubMed. [DOI] [PubMed] [Google Scholar]
- Jie Z, Li T, Jia-Yun H, Qiu J, Ping-Yao Z, Houyan S. Trans-2-phenylcyclopropylamine induces nerve cells apoptosis in zebrafish mediated by depression of LSD1 activity. Brain Res. Bull. 2009;80:79–84. doi: 10.1016/j.brainresbull.2009.04.013. PubMed. [DOI] [PubMed] [Google Scholar]
- Le MT, Xie H, Zhou B, Chia PH, Rizk P, Um M, Udolph G, Yang H, Lim B, Lodish HF. MicroRNA-125b promotes neuronal differentiation in human cells by repressing multiple targets. Mol. Cell. Biol. 2009;29:5290–5305. doi: 10.1128/MCB.01694-08. PubMed. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Metzger E, Wissmann M, Yin N, Müller JM, Schneider R, Peters AH, Günther T, Buettner R, Schüle R. LSD1 demethylates repressive histone marks to promote androgen-receptor-dependent transcription. Nature. 2005;437:436–439. doi: 10.1038/nature04020. PubMed. [DOI] [PubMed] [Google Scholar]
- Nair SS, Guo Z, Mueller JM, Koochekpour S, Qiu Y, Tekmal RR, Schüle R, Kung HJ, Kumar R, Vadlamudi RK. Proline-, glutamic acid-, and leucine-rich protein-1/modulator of nongenomic activity of estrogen receptor enhances androgen receptor functions through LIM-only coactivator, four-and-a-half LIM-only protein 2. Mol. Endocrinol. 2007;21:613–624. doi: 10.1210/me.2006-0269. PubMed. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nair SS, Nair BC, Cortez V, Chakravarty D, Metzger E, Schüle R, Brann DW, Tekmal RR, Vadlamudi RK. PELP1 is a reader of histone H3 methylation that facilitates oestrogen receptor-alpha target gene activation by regulating lysine demethylase 1 specificity. EMBO Rep. 2010;11:438–444. doi: 10.1038/embor.2010.62. PubMed. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishida Y, Adati N, Ozawa R, Maeda A, Sakaki Y, Takeda T. Identification and classification of genes regulated by phosphatidylinositol 3-kinase- and TRKB-mediated signalling pathways during neuronal differentiation in two subtypes of the human neuroblastoma cell line SH-SY5Y. BMC Res. Notes. 2008;1:95. doi: 10.1186/1756-0500-1-95. PubMed. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perillo B, Ombra MN, Bertoni A, Cuozzo C, Sacchetti S, Sasso A, Chiariotti L, Malorni A, Abbondanza C, Avvedimento EV. DNA oxidation as triggered by H3K9me2 demethylation drives estrogen-induced gene expression. Science. 2008;319:202–206. doi: 10.1126/science.1147674. PubMed. [DOI] [PubMed] [Google Scholar]
- Rudolph T, Yonezawa M, Lein S, Heidrich K, Kubicek S, Schäfer C, Phalke S, Walther M, Schmidt A, Jenuwein T, Reuter G. Heterochromatin formation in Drosophila is initiated through active removal of H3K4 methylation by the LSD1 homolog SU(VAR)3-3. Mol. Cell. 2007;26:103–115. doi: 10.1016/j.molcel.2007.02.025. PubMed. [DOI] [PubMed] [Google Scholar]
- Rusconi F, Paganini L, Braida D, Ponzoni L, Toffolo E, Maroli A, Landsberger N, Bedogni F, Turco E, Pattini L, et al. LSD1 Neurospecific Alternative Splicing Controls Neuronal Excitability in Mouse Models of Epilepsy. Cereb Cortex. 2014 doi: 10.1093/cercor/bhu070. Published online April 15, 2014. 10.1093/cercor/bhu070. PubMed. [DOI] [PubMed] [Google Scholar]
- Shi Y, Lan F, Matson C, Mulligan P, Whetstine JR, Cole PA, Casero RA, Shi Y. Histone demethylation mediated by the nuclear amine oxidase homolog LSD1. Cell. 2004;119:941–953. doi: 10.1016/j.cell.2004.12.012. PubMed. [DOI] [PubMed] [Google Scholar]
- Shi YJ, Matson C, Lan F, Iwase S, Baba T, Shi Y. Regulation of LSD1 histone demethylase activity by its associated factors. Mol. Cell. 2005;19:857–864. doi: 10.1016/j.molcel.2005.08.027. PubMed. [DOI] [PubMed] [Google Scholar]
- Smith TC, Fang Z, Luna EJ. Novel interactors and a role for supervillin in early cytokinesis. Cytoskeleton (Hoboken) 2010;67:346–364. doi: 10.1002/cm.20449. PubMed. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strahl BD, Allis CD. The language of covalent histone modifications. Nature. 2000;403:41–45. doi: 10.1038/47412. PubMed. [DOI] [PubMed] [Google Scholar]
- Sun G, Alzayady K, Stewart R, Ye P, Yang S, Li W, Shi Y. Histone demethylase LSD1 regulates neural stem cell proliferation. Mol. Cell. Biol. 2010;30:1997–2005. doi: 10.1128/MCB.01116-09. PubMed. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun G, Ye P, Murai K, Lang MF, Li S, Zhang H, Li W, Fu C, Yin J, Wang A, et al. miR-137 forms a regulatory loop with nuclear receptor TLX and LSD1 in neural stem cells. Nat. Commun. 2011;2:529. doi: 10.1038/ncomms1532. PubMed. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ting HJ, Yeh S, Nishimura K, Chang C. Supervillin associates with androgen receptor and modulates its transcriptional activity. Proc. Natl. Acad. Sci. USA. 2002;99:661–666. doi: 10.1073/pnas.022469899. PubMed. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toffolo E, Rusconi F, Paganini L, Tortorici M, Pilotto S, Heise C, Verpelli C, Tedeschi G, Maffioli E, Sala C, et al. Phosphorylation of neuronal Lysine-Specific Demethylase 1LSD1/KDM1A impairs transcriptional repression by regulating interaction with CoREST and histone deacetylases HDAC1/2. J. Neurochem. 2014;128:603–616. doi: 10.1111/jnc.12457. PubMed. [DOI] [PubMed] [Google Scholar]
- Vadlamudi RK, Wang RA, Mazumdar A, Kim Y, Shin J, Sahin A, Kumar R. Molecular cloning and characterization of PELP1, a novel human coregulator of estrogen receptor alpha. J. Biol. Chem. 2001;276:38272–38279. doi: 10.1074/jbc.M103783200. PubMed. [DOI] [PubMed] [Google Scholar]
- Wang J, Scully K, Zhu X, Cai L, Zhang J, Prefontaine GG, Krones A, Ohgi KA, Zhu P, Garcia-Bassets I, et al. Opposing LSD1 complexes function in developmental gene activation and repression programmes. Nature. 2007;446:882–887. doi: 10.1038/nature05671. PubMed. [DOI] [PubMed] [Google Scholar]
- Whyte WA, Bilodeau S, Orlando DA, Hoke HA, Frampton GM, Foster CT, Cowley SM, Young RA. Enhancer decommissioning by LSD1 during embryonic stem cell differentiation. Nature. 2012;482:221–225. doi: 10.1038/nature10805. PubMed. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wulfkuhle JD, Donina IE, Stark NH, Pope RK, Pestonjamasp KN, Niswonger ML, Luna EJ. Domain analysis of supervillin, an F-actin bundling plasma membrane protein with functional nuclear localization signals. J. Cell Sci. 1999;112:2125–2136. doi: 10.1242/jcs.112.13.2125. PubMed. [DOI] [PubMed] [Google Scholar]
- Yang M, Gocke CB, Luo X, Borek D, Tomchick DR, Machius M, Otwinowski Z, Yu H. Structural basis for CoREST-dependent demethylation of nucleosomes by the human LSD1 histone demethylase. Mol. Cell. 2006;23:377–387. doi: 10.1016/j.molcel.2006.07.012. PubMed. [DOI] [PubMed] [Google Scholar]
- Zibetti C, Adamo A, Binda C, Forneris F, Toffolo E, Verpelli C, Ginelli E, Mattevi A, Sala C, Battaglioli E. Alternative splicing of the histone demethylase LSD1/KDM1 contributes to the modulation of neurite morphogenesis in the mammalian nervous system. J. Neurosci. 2010;30:2521–2532. doi: 10.1523/JNEUROSCI.5500-09.2010. PubMed. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.