

Abstract
The Pim protein kinases contribute to transformation by enhancing the activity of oncogenic Myc and Ras, which drives significant metabolic changes during tumorigenesis. In this report, we demonstrate that mouse embryo fibroblasts (MEFs) lacking all three isoforms of Pim protein kinases, triple knockout (TKO), cannot tolerate the expression of activated K-Ras (K-RasG12V) and undergo cell death. Transduction of K-RasG12V into these cells markedly increased the level of cellular reactive oxygen species (ROS). The addition of N-acetyl cysteine attenuates ROS production and reversed the cytotoxic effects of K-RasG12V in the TKO MEFs. The altered cellular redox state caused by the loss of Pim occurred as a result of lower levels of metabolic intermediates in the glycolytic and pentose phosphate pathways as well as abnormal mitochondrial oxidative phosphorylation. TKO MEFs exhibit reduced levels of superoxide dismutase (Sod), glutathione peroxidase 4 (Gpx4) and peroxiredoxin 3 (Prdx3) that render them susceptible to killing by K-RasG12V-mediated ROS production. In contrast, the transduction of c-Myc into TKO cells can overcome the lack of Pim protein kinases by regulating cellular metabolism and Sod2. In the absence of the Pim kinases, c-Myc transduction permitted K-RasG12V-induced cell growth by decreasing Ras-induced cellular ROS levels. These results demonstrate that the Pim protein kinases play an important role in regulating cellular redox, metabolism and K-Ras-stimulated cell growth.
Keywords: glycolysis, Ras, metabolism, oxidative phosphorylation, Pim kinase, reactive oxygen species
Introduction
The Pim family of serine/threonine kinases includes three isoforms that play a role in multiple cellular processes, including signal transduction, cell cycle progression, cell metabolism, and tumor growth. The Pim kinases were initially cloned as enzymes that enhance the ability of c-Myc to induce lymphomas1,2, and they are overexpressed in human myeloid leukemia, lymphoma, and myeloma3-5. In mice, Pim has been demonstrated to collaborate with Myc to induce lymphoma and prostate cancer1,6,7. We recently demonstrated that the knockout of the Pim kinases markedly slowed the growth of mouse embryo fibroblasts (MEFs) and increased the activity of AMP-kinase, while diminishing 5′cap-dependent translation8. Overexpression of either c-Myc or peroxisome proliferator-activated receptor gamma coactivator 1α (PGC-1α), two enzymes involved in energy metabolism and mitochondrial biogenesis, overcame the absence of Pim and markedly increased cell growth. These results suggest that Pim protein kinases play an important, but yet undefined, role in regulating cellular metabolism.
Oncogenic K-Ras also collaborates with Myc in tumorigenesis9-11, and it has been suggested to play an important role in modulating cellular metabolism12. Active K-Ras mutants promote glutamine entry into the tricarboxylic acid (TCA) cycle through glutaminase, and they uncouple glycolysis and the TCA cycle13,14. In pancreatic cancer, K-Ras regulates a distinct pathway of glutamine metabolism involving cytoplasmic aspartate transaminase. Recently, K-Ras was shown to redirect glucose flux into the pentose phosphate pathway (PPP), contributing to the biosynthesis of nucleotides and maintenance of cellular redox balance12. This generation of reactive oxygen species (ROS) is dependent on mitochondrial cytochrome oxidases, and it is necessary for Ras-induced anchorage-independent growth15,16. Moreover, K-Ras, enhances cell growth and transformation through transcriptional up-regulation and control of novel metabolic enzymes14.
Like Pim and Ras, the c-Myc protooncogene plays an important role in regulating metabolism, including glycolysis17 and the induction of mitochondrial glutaminase expression through a mechanism that involves the regulation of miRNAs18. By controlling anaplerotic input into the tricarboxylic cycle, Myc also modulates proline metabolism19. Myc regulates nicotinamide-phosphoribosyltransferase (Nampt), which controls NAD levels20 and the induction of the transcription factor Nrf221; thus, Myc modulates the intracellular redox cycle in cells.
In this report, we demonstrate that the Pim protein kinases control metabolic pathways that are necessary for the induction of cell growth by activated K-RasG12V. However, even in the absence of all three Pim kinase isoforms, c-Myc is able to regulate cellular metabolism and permit K-RasG12V-stimulated cell growth.
Results
K-RasG12V induces cell death in MEF cells lacking all three Pim kinases
To examine the role of Pim kinases in regulating cell growth, MEFs were generated from 14.5 day embryos lacking all three Pim protein kinase isoforms (triple knockout; TKO) and, in parallel, cells were derived from wild type (WT) mice (Supplementary Figure S1A). As we reported previously8, TKO cells grow more slowly than WT and Pim-1 and -3 double knockout cells (Supplementary Figure S1B). Re-expression of Pim kinases in TKO cells conferred the ability for these cells to proliferate (data not shown). To examine whether the absence of one or more Pim kinases regulated the ability of mutated, activated K-RasG12V to stimulate cell growth, WT and TKO MEF cells were infected with viruses carrying activated K-RasG12V or empty vector. K-RasG12V transduction increased the phosphorylation of ERK1/2 proteins in both WT and TKO MEFs to a similar extent, indicating that K-RasG12V was active in cells lacking all three Pim kinases (Supplementary Figure S1C). WT MEF cells expressing K-RasG12V grow similarly to cells expressing the control vector, whereas TKO MEFs expressing K-RasG12V die within four days post-infection (Figures 1a-c). Similarly, immortalized WT MEFs transduced with K-RasG12V form foci, while TKO MEFs failed to grow in the presence of this oncogene (Figures 1d). Expression of K-RasG12V resulted in a decrease in procaspase-9 and -3 expression, and subsequently induced a marked degradation of PARP-1(Poly [ADP-ribose] polymerase 1) in TKO but not in WT MEFs (Supplementary Figures S1D), suggesting the induction of an apoptotic mechanism. In contrast, MEFs isolated from mice containing single knockouts of individual Pim kinases (Supplementary Figure S1E) continued to grow when expressing K-RasG12V (Figure 1e). Ectopic expression of Pim-1, -2 or -3 in TKO MEFs (Supplementary Figure S1F) was sufficient to prevent K-RasG12V-mediated cell death (Figure 1f), as depicted in morphological changes, demonstrating that a single Pim isoform is sufficient to inhibit K-RasG12V-induced cell death.
Fig. 1. Knockout of Pim kinases switches oncogenic activity of K-Ras to cell death.

(a) Fluorescence microscopy of primary WT and TKO MEF cells expressing, FUCRW empty vector (EV) or K-RasG12V (K-Ras). At day 4 post-infection, expression of red fluorescent protein (RFP) and morphological changes were visualized. (b, c) Primary WT and TKO MEF cells were infected with empty vector (-) or K-Ras. After 7 days, cell viability was evaluated by the crystal violet assay. (d) A focus-forming assay was performed at 14 days post-infection using lentiviruses carrying EV or K-Ras in immortalized WT and TKO MEF cells. The numbers of cell foci were counted under inverted microscope. Mean value and standard deviation were obtained from triplicates. (e) Pim1, 2, or 3 single knockout MEF cells were infected with lentivirus expressing K-Ras (+) or empty vector (-). Cell viability was determined by a crystal violet assay on day 4. (f) Cell viability was examined 7 days after K-Ras expression in TKO MEF cells expressing EV, Pim1, 2, or 3. The experiment was done in triplicate.
Loss of Pim kinase exacerbates K-RasG12V-mediated ROS accumulation, leading to cell death
Because ectopic Ras transduction has been demonstrated to modulate cellular metabolic pathways and increase ROS production15, we evaluated the ability of Pim kinases to regulate cellular redox signaling. When WT and TKO MEFs were stained with H2-DCF (ROS), MitoSOX (superoxide) or C11-BODIPY (lipid peroxidation), flow cytometry analysis indicated that a basal level of multiple pro-oxidants was significantly higher in TKO MEFs compared to WT (Figure 2a; Supplementary Figures S2A-C). Similarly, a higher level of lipid peroxides in TKO MEFs was detected by thiobarbituric acid reactive substances (TBARS) assay compared to WT (Supplementary Figures S2D). Transduction of K-RasG12V into WT MEFs produced a low level of induction of cellular ROS, while K-RasG12V transduction in TKO MEF cells significantly elevated cellular ROS levels in multiple assays (Figures 2b and c; Supplementary Figures S2D). In MEFs isolated from mice lacking a single isoform of Pim (Pim-1, -2, or -3 knockout), K-RasG12V did not induce additional ROS accumulation (Supplementary Figure S2E), suggesting that the Pim kinases have overlapping activity, and inhibition of all three Pim kinases is necessary for K-RasG12V-mediated ROS accumulation. To test whether prevention of ROS accumulation blocks K-RasG12V-induced cell death, MEFs were treated with N-acetyl cysteine (NAC). NAC treatment promoted cell viability against K -RasG12V-induced cell death (Figures 2d and e). Induction of cell death by K-RasG12V in TKO MEFs was prevented by multiple ROS scavengers including polyethylene glycol (PEG)-superoxide dismutase (Sod) and PEG-catalase. In presence of diphenylene iodonium (DPI), a canonical NOX inhibitor, or N5-(Methylamidino)-L-ornithine acetate (L-NMMA), a nitric oxide synthase inhibitor, KRASG12V-mediated cell death was also suppressed (Supplementary Figure S2F and G). Iron chelators, including deferoxamine and ciclopriox olamine were toxic to these cells and could not be further evaluated (data not shown). To test whether inhibition of Pim was sufficient to induce ROS accumulation, WT MEF cells were treated with the small molecule pan-Pim kinase inhibitors, GNE-65222 or NVP-LGB32123,24. Pim inhibitor treatment induced an accumulation of ROS that was mildly cytotoxic, reducing cell viability (Supplementary Figures S2H and I). Pretreatment with NAC significantly improved the viability of WT MEFs after pan-Pim inhibitor treatment (Supplementary Figure S2J). Additionally, mutant K-Ras expressing Calu-1 and H358 non-small cell lung carcinoma cell (NSCLC) lines were found to be more sensitive to treatment with high doses of a Pim kinase inhibitor compared to wild-type K-Ras expressing H1299 cells (Supplementary Figure S2K and L). Ectopic expression of KRASG12V in the insensitive H1299 cells caused these cells to become sensitive to these compounds, suggesting that Pim kinases may play a role in KRASG12V cancer cell growth and survival.
Fig. 2. Lethal accumulation of reactive oxygen species (ROS) induced by K-RasG12V in absence of Pim kinases.

(a) WT and TKO MEF cells were exposed to 1 μM H2DCF-DA for 30 min and cells were evaluated by real-time confocal analysis for ROS production. (b) Fluorescence microscopy for detection of ROS accumulation in WT and TKO MEF cells at day 4 post-infection of empty vector (EV) or K-Ras lentiviruses. (c) Flow cytometric analysis of ROS in WT and TKO MEF cells expressing EV or K-Ras. (d, e) Cell viability was determined by crystal violet staining. After infection for 2 days, EV or K-Ras infected WT and TKO MEF cells were treated with 5 mM NAC for 48 h (triplicates).
Altered Ras-driven metabolism in TKO and WT MEFs
To understand the defect in TKO cells that sensitized these cells to increased ROS production by K-RasG12V, we compared cellular metabolism in TKO and WT MEFs using both global metabolic profiling and gene expression arrays. The results of LC/MS, GC/MS and UPLC/MS/MS indicate that loss of the three Pim kinases altered the intracellular concentrations of 178 metabolites (68 increased and 110 decreased relative to WT cells) with p < 0.05, as determined using Welch's t-tests. Among these small molecules, glycolytic intermediates, including glucose-6-phospate (G6P), fructose-6-phosphate (F6P), 2-phosphoglycerate (2-PG), and glucose-1-phospate (G1P), were strikingly decreased in TKO compared to WT MEFs (Figure 3a). This was not a general decrease in glycolytic metabolites, as the loss of Pim kinases did not affect the levels of intracellular glucose, 3-phosphoglycerate (3-PG), and pyruvate metabolites. Gene expression analysis was carried out on WT and TKO MEFs using Illumina bead arrays (∼45,200 transcripts). Over 1326 genes (690 downregulated and 636 upregulated genes with a fold change >2 and FDR adjusted p value <0.05) were identified as having significantly altered expression levels between these two cell lines. To characterize these gene expression changes, the results were compared with 522 publicly available gene sets (Molecular Signature Database, Broad Institute) using gene set enrichment analysis (GSEA). TKO MEFs were found to have significantly decreased gene expression signatures among 10 gene sets involved in regulating glycolysis, fatty acid oxidation, TCA cycle enzymes, mitochondrial respiration target genes, and genes involved in oxidative phosphorylation (OxPhos) (FDR q-value < 0.05) (Figure 3b; Supplementary Figure S3A and Table S1). Quantitative real time PCR (qPCR) validated these observations, demonstrating that glycolytic pathway rate-limiting enzymes, including glucose transporter 1 (Glut1/Slc2a1), hexokinase 1 (Hk1), Hk2, aldolase A (Aldoa), enolase 3 (Eno3), and pyruvate kinase muscle (Pfkm) transcripts, are lower in TKO MEF cells compared to WT MEF cells (Figure 3c). Similar to mRNA expression, western blotting demonstrated that the enzymes driving the formation of glycolytic metabolites (e.g., Hk2, phosphofructokinase-1 (Pfk-1), pyruvate dehydrogenase 4 (Pdk4), enolase, Pkm1/2 and lactate dehydrogenase A (Ldha) are reduced at the protein level, accounting for the lower levels of glycolytic metabolites seen in global profiling (Figure 3d). The observed changes in the levels of these glycolytic enzymes are not artifacts of in vitro culture of MEF cells, as similar results are seen in kidney tissues freshly isolated from TKO mice (Supplementary Figure S3B). The transduction of K-RasG12V into WT MEFs increases the protein expression of multiple enzymes in the glycolytic pathway (Figure 4a). However, transduction of K-RasG12V into TKO MEFs did not induce changes in the mRNA levels of metabolic enzymes, including Glut1, 1/2, Pfkm, and Ldha. Loss of Pim kinases blocked the increased transcription of these metabolic enzymes induced by activated Ras (Figure 4b). These results demonstrate that deletion of Pim kinases is sufficient to repress some, but not all, enzymes involved in glycolysis, and the addition of activated Ras does not overcome these defects.
Fig. 3. Pim kinases are required for regulation of glycolysis.

(a) Metabolomics analysis reveals that key glycolytic metabolites are repressed in TKO MEFs (mean +/- SD, n=5). The metabolites shown are glucose-1-phosphate (G6P), glucose-1-phosphate (G6P), fructose-6-phosphate (F6P), 2-phosphoglycerate (2PG). Pyruvate and 3-phosphoglycerate (3PG) levels were not different between WT and TKO MEFs. * denotes a statistical significance of p<0.05. (b) GSEA reveals the most significantly altered metabolic pathway in TKO MEFs. Selected metabolic gene sets are shown. NES, normalized enrichment score; q-values, FDR-adjusted p-value. The enrichment plot of the glucose metabolism gene set is found to be the most significantly associated with downregulation in TKO MEFs. The heatmap shows the expression profile of genes significantly downregulated in TKO MEFs. (c) Quantitative real-time PCR (qPCR) analysis of mRNA expression of glycolytic enzymes and PPP enzymes. Normalized fold expression of Slc2a1 (Glut1), Hk1 (hexokinase 1), Hk2, Eno3 (enolase 3), Aldoa (Aldolase A), Pfkm (phosphofructokinase muscle), Ldha (lactate dehydrogenase A) and Pgm1 (phosphoglucomutase 1) are shown (triplicates) and β-Actin was used for normalization. (d) Immunoblot analysis of key glycolytic enzymes in WT and TKO MEF cells. Protein expression of Hk2, Glut1, Pfk1, Pdk4 (pyruvate dehydrogenase kinase 4), enolase, Pkm1/2, Ldha and β-actin are shown.
Fig. 4. K-Ras requires Pim kinases for regulation of glycolysis.
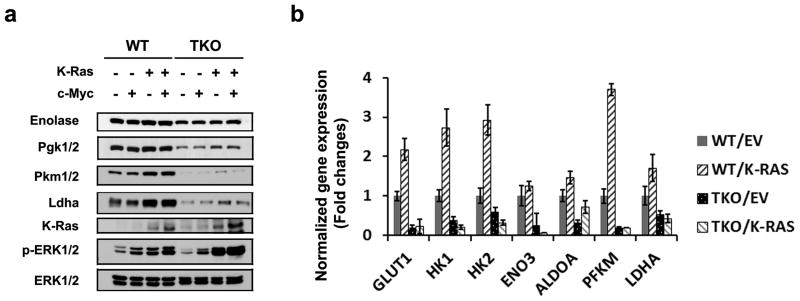
(a) Immunoblot analysis of glycolytic enzyme expression regulated by K-RasG12V. WT and TKO MEFs were infected with lentiviruses of EV, K-Ras or c-Myc. After transduction for 96 h, cells were lysed and subjected to immunoblottting. (b) K-RasG12V-mediated changes in gene expression were assayed using qPCR. Normalized expression of Glut1, Hk1, Hk2, Eno3, Aldoa, Pfkm, and Ldha mRNA to β-Actin mRNA is shown (Mean +/- SD, triplicates). WT and TKO MEF cells were infected with lentiviruses containing empty vector (EV) or K-RasG12V for 72 hours and then total RNA was isolated.
To assess whether loss of Pim kinases influences glucose flux into the pentose phosphate pathway (PPP), we compared the levels of metabolites in this pathway in TKO and WT MEFs. In TKO MEFs, both G6P and 6- phosphogluconate (6PG) are diminished, leading to a decrease in ribulose 5- phosphate (Ru5P) and sedoheptulose-7-phosphate (S7P) (Figure 5a). In contrast to the metabolites formed by enzymes that require NADP+ as a cofactor, the levels of ribose 5-phosphate (R5P) and ribose were similar in WT and TKO MEFs (not shown), suggesting that these metabolic effects are specific to Pim deletion. Similarly, the PPP enzymes, G6pd, Pgd, Pgsl, Rpi, Rpe, and Tkt transcripts, are lower in TKO MEFs (Figure 5b). The changes in cellular metabolism observed in MEFs were also observed in fresh primary tissues. In T cells derived from TKO mouse spleens, the reduced gene expression of PPP enzymes (G6pd, Pgd, Rpia, and Rpe) (Supplementary Figure S4A) is consistent with the lower proliferative rate observed in splenic T cells after TCR-mediated stimulation with anti-CD3 and anti-CD28 antibodies (Supplementary Figure S4B). Compared to splenocytes obtained from the WT mice, CFSE (carboxyfluorescein succinimidyl ester)-labeled TKO splenocytes showed a lag in proliferation. In WT MEFs, K-RasG12V transduction induced PPP enzymes (e.g., G6pd, Pgd, Rpia, and Rpe), but oncogenic Ras is markedly less effective in inducing these enzyme changes in TKO MEFs (Figure 5c). These results indicate that K-RasG12V mediates metabolic programming by controlling the mRNA transcription of key enzymes, and these mRNA changes require Pim kinases.
Fig. 5. Pim kinases are required for regulation of the pentose phosphate pathway (PPP).

(a) Key metabolites within the PPP were significantly altered in TKO MEFs compared to WT MEFs. Levels of G6P, 6-phosphoglucognate (6PG), sedoheptulose 7-phosphate (S7P), F6P and ribulose 5-phosphate (Ru5p) + xylulose 5-phosphate (Xu5P) are shown (n=5). (b) qPCR analysis of mRNA expression of PPP enzymes. Measurement of key enzymes in the PPP, including G6pd (glucose-6-phosphate dehydrogenase), Pgls (6-phosphoglucono-δ-lactone), Pgd (6-phosphogluconate dehydrogenase), Rpia (ribose-5-phosphate isomeras A), Rpe (ribose-5-phosphate epimerase), Tktl2 (transketolase 2), and Tald1 (transaldolase 1). (c) K-RasG12V-mediated changes in gene expression were assayed by qPCR. Normalized expression of G6pd, Pgd, Rpe, and Rpia to β-Actin mRNA is shown (triplicates). WT and TKO MEF cells were infected with lentiviruses of EV or K-RasG12V for 72 h and then total RNA was isolated.
Pim kinases are required for the regulation of mitochondrial OxPhos
Metabolic analysis demonstrated significant decreases in α-ketoglutarate (α-KG) levels, a key component of the TCA cycle, in TKO compared to WT MEFs. α-KG can be derived from the conversion of pyruvate to OAA by Pcx or by isocitrate dehydrogenase (Idh), which catalyzes the oxidative decarboxylation of isocitrate. The low levels of oxaloacetate (OAA) in TKO MEFs could be secondary to decreased pyruvate carboxylase (Pcx) mRNA expression. When comparing TKO and WT MEFs, qPCR analysis demonstrated significant down-regulation of Idh1, Idh2 and Pcx transcripts, suggesting a potential explanation for the low level of α-KG in TKO cells (Figure 6a). As demonstrated for the regulation of the glycolytic and PPP enzymes, K-RasG12V transduction into WT MEF cells markedly increased the expression of Pcx and Idh1 transcripts. However, Ras transduction only induced minor changes in the level of these enzymes in TKO MEFs, further demonstrating the importance of Pim kinases in the regulation of Ras-induced metabolism.
Fig. 6. Pim kinases are involved in regulation of the TCA cycle and mitochondrial oxidative phosphorylation.

(a) K-RasG12V-mediated changes in gene expression were assayed with a qPCR. Normalized expression of Pcx and Idh1 to β-Actin mRNA is shown (Mean +/- SD, triplicates). (b) OCR was evaluated in WT and TKO MEF cells. Oligomycin (2.5 μM), FCCP (2.5 μM), antimycin A (1 μM) and rotenone (1 μM) were sequentially added as indicated by an arrow (triplicates). (c) Immunoblot analysis of electron transport chain complex proteins. Two independently derived sets of WT and TKO MEF cells were subjected to subcellular fractionation to obtain mitochondria and cytosolic fractions. A cocktail of antibodies, CV-ATPA, CII-UQCR2, CII-SDHB and CI-NDUFB8, was used to probe the membranes of mitochondria of various MEF cells. β-actin protein was used as a loading control. (d) Mitochondrial DNA was quantified by qPCR by measuring the ratio of mitochondrially encoded Cox2 to an intron of the nuclear-encoded β-globin gene. The ratio of mitochondrial cytochrome b (Cyt b) to an intron of nuclear glucagon gene is shown.
To assess mitochondrial OxPhos function in WT versus TKO MEFs, oxygen consumption rates (OCR) were analyzed. Although the basal levels of OCR were not significantly different between WT and TKO MEFs, TKO MEFs exhibited markedly decreased responses to oligomycin, FCCP (Carbonyl cyanide 4-[trifluoromethoxy] phenylhydrazone), and the combination of antimycin B and rotenone (Figure 6b). The absence of Pim created potential abnormalities in both redox reactions and the production of ATP. The OxPhos activity of TKO MEFs, as measured by OCR, was partially restored by the re-expression of a single Pim isoform (Pim-1, Pim-2, or Pim-3) (Supplementary Figure S5A). In isolated mitochondria from TKO MEFs, western blotting revealed decreased levels of complex V-ATPA, complex III-UQCR2, and complex II-SDHB (Figure 6c). OxPhos capacity is determined in part by mitochondrial DNA content25, and the mitochondrial DNA content of cyclooxygenase 2 (Cox2) and Cytochrome b (Cyt b) was lower in TKO compared to WT MEFs (Figure 6d). In TKO MEFs, mRNAs encoding subunits of these complexes, such as Cox2, Cox5A, Cytb and Cytc, were all decreased (Supplementary Figure S5B)26. Additionally, gene expression array and qPCR analyses indicated that mitochondrial genes involved in oxidative phosphorylation (e.g., Ndufb9, Atp6ap2 and Suclg1) are significantly down regulated in TKO MEFs (Supplementary Figure S5C). Significant reductions in the level of TCA cycle intermediates in TKO MEFs and reduced ATP levels8 suggest that Pim plays an important role in for mitochondrial OxPhos activity.
NAD(P)+ levels are reduced by loss of Pim kinases
To better understand why K-Ras was ineffective in promoting glucose metabolism and mitochondrial redox balance in the absence of Pim kinases, NAD(P)+/NAD(P)H production was examined. NADP+, the phosphorylated from of NAD+, is an important metabolic coenzyme in the anabolic processes of highly proliferative cells27,28, and it enhances the detoxification of ROS by increasing the activity of glutathione peroxidase (Gpx) and peroxiredoxin29,30. Results from UPLC/MS/MS analysis indicated that intracellular levels of NADP+ but not NADPH were significantly lower in TKO compared to WT MEFs (Figure 7a; Supplementary Figure S6A). This difference was reflected in the ratio of NADP+/NADPH, which was reduced in TKO MEFs (Supplementary Figure S6B). Additionally, NAD+ was reduced in TKO compared to WT MEF cells, suggesting that nicotinamide metabolism is abnormal in TKO MEFs (Supplementary Figure S6A). Since the NADP+/NADPH ratio is critical for glutathione recycling, the level glutathione (GSH) and oxidized glutathione (GSSG) were examined. Compared to WT, TKO MEFs showed a slight reduction in GSH levels but a much lower level of GSSG, indicating that total the glutathione level was low in TKO MEFs (Figure 7b). K-Ras expression in TKO MEFs resulted in a significant reduction in GSH and an increase in GSSG levels, indicating that GSH was consumed for the detoxification of ROS induced by K-Ras. It is noteworthy to mention that the transcription and translation level of Gpx4 was low in TKO cells (Supplementary Figure S6C), which is in line with the recent report shown by others that Gpx4 depletion induced cell death in mutant Ras-expressing fibroblast cells31. It is thus not only the level of GSH that is important, but the level of enzymes which make use of this molecule. In this regards, Sod enzymes could play an important role in this process (as shown as Supplementary Figure S2F). Experiments demonstrate that cellular Sod mRNA and enzyme activities in TKO MEFs were significantly lower than WT and not increased when K-RasG12V was expressed (Supplementary Figure S6D and S6E). This loss of detoxifying activity could in part account for the marked increase in ROS observed in TKO MEFs after transduction with activated K-Ras.
Fig 7. NAD(P)+ coenzymes for metabolic redox reaction is modulated by loss of Pim kinases.

(a) NADP+, NADPH and total NADP (NADP+ plus NADPH) levels in WT and TKO MEF cells are shown (triplicates). NADP+ concentrations were calculated by the subtraction of NADPH from total NADP (nmol per mg protein). (b) The content of reduced glutathione (GSH), oxidized glutathione (GSSG) was normalized to cellular protein content. (Mean +/- SD, n=3). WT and TKO MEFs were transduced for 72 hours using lentiviruses carrying empty vector (EV), K-RasG12V (KRAS), c-Myc (MYC) or both. (c) Focus forming assay. WT and TKO MEFs were transduced with lentiviruses carrying empty vector (EV), K-RasG12V (KRAS), c-Myc (MYC) or both. After 10 days of growth, the number of foci was documented with crystal violet staining. Cell viability was evaluated by MTT assay (Mean +/- SD, n=4). (d) Quantitation of DCF labeling from flow analysis is shown with relative ROS levels (triplicates) with the control value 1 set in the RFP cells. (e) NADP+ and NAD+ production was measured in WT and TKO MEF cells expressing empty vector (EV) or c-Myc (MYC). (f) Immunoblotting assay for Sod2 protein expression in cells transduced with HA-Myc. (g) Model demonstrating the role of Pim, Ras, and Myc in regulating cellular metabolism.
Forced c-Myc expression rescues K-Ras-mediated cell death by lowering ROS accumulation
In a previous report8, we demonstrated that the level of c-Myc is considerably lower in TKO than WT MEFs. Ras transformation is enhanced by increased expression of Myc32, and these two oncogenes are known to function together to enhance Ras-mediated growth33. The GSEA corresponding to the loss of Pim in TKO cells is similar to the signature found in response to down regulation of the Myc pathway, suggesting that Pim may play a role in regulating genes controlled by Myc. Moreover, c-Myc is known to stimulate mitochondrial genes and mitochondrial biogenesis34. Having observed that c-Myc transduction restored mitochondrial DNA content in TKO MEFs to the level of WT MEFs (Supplementary Figure S6F), the role of c-Myc in these MEF cells was further examined. Since the level of c-Myc was low in TKO MEF cells, this cDNA was transduced into TKO MEF cells, with or without K-RasG12V, to study the effect of c-Myc expression on K-RasG12V killing. Vital staining of transfected TKO cells demonstrated that c-Myc re-expression rescued TKO MEF cells from the K-RasG12V-induced cell death (Figures 7c). Using dye labeling and FACS analysis in TKO MEFs, co-expression of c-Myc and K-RasG12V lowered ROS accumulation induced by K-RasG12V (Figures 7d; Supplementary Figure S6G), c-Myc controls the level of NAD through transcriptional regulation of Nampt enzymes20, and TKO MEFs expressing c-Myc demonstrated increased NAD+ and NADP+ levels equivalent to those of WT MEFs (Figure 7e; Supplementary Figure S6H). Additionally, the transduction of c-Myc into these cells enhanced Sod2 protein levels (Figure 7f). The improved NAD(P)+ cascade and increased Sod2 protein levels induced by c-Myc in TKO MEFs could account for the decreased ROS production observed with transduction of this protein. These results demonstrate that through transcriptional regulation of proteins involved in cellular metabolism, c-Myc is able to alter cell metabolism, block ROS generation by K-RasG12V, and enable the growth of cells expressing the K-RasG12V protein (Figure 7g), even in the absence of the Pim protein kinases.
Discussion
To decipher the molecular mechanism by which loss of Pim kinases increases ROS and sensitizes to Ras-induced cell death, we conducted global metabolic profiling and gene expression arrays. GSEA demonstrate significant down regulation of metabolic pathways in TKO cells, including glycolysis, the TCA cycle, and mitochondrial respiratory genes. However, not all metabolic pathways were regulated by Pim, as we observed no changes in key genes involved in regulation of hexobiosynthesis. Moreover, the loss of Pim kinases did not affect the levels of 3-PG or pyruvate metabolites, suggesting a possible reason for the lack of changes in the extracellular acidification. These suggest alternative glucose utilization could compensate for reduced glycolysis and PPP metabolism in Pim knockout cells. The GSEA analysis further revealed that genes that are important for mitochondrial β-oxidation to generate ATP including Pparggc1a, Acadvl, Acadm, Acads, Echs1, Hadhb, Adipor2 and Ctp1a were repressed in TKO cells, suggesting a possible rationale for how the deletion of Pim kinases leads to low levels of cellular ATP. These defects were shown to impact the ability of K-RasG12V to induce genes changes in these MEFs. For example, transduction of activated K-Ras into WT MEFs significantly elevated enzymes that enhance glycolysis, such as Pkm-1 and Pfk-1, and the activity of the pentose phosphate pathway. Similarly, K-RasG12V transduction into WT MEFs increased Pcx and Idh1 levels, which encode important enzymes that regulate the movement of metabolites through the TCA cycle. However, transduction of K-RasG12V into TKO MEFs was unable to induce the majority of these enzyme changes, suggesting that that the metabolic pathways normally elevated by Ras during the early stimulation of growth are not inducible in the absence of Pim kinases. Additionally, Ras transduction has been demonstrated to markedly increase OxPhos activity in MEFs containing the E1A protein35. Although baseline OCR rates in TKO are identical to WT, the ability of the uncoupler, FCCP, to modulate OCR was abolished, and oligomycin, which functions by blocking ATP synthesis by the F0/F1 ATPase and inhibits mitochondrial respiration, did not fully suppress OCR in TKO MEFs. Ectopic expression of H-RasV12 sensitizes cells to death after the addition of the complex 1 inhibitor, rotenone36. This result is consistent with the observation that increased oxidative stress was also reported in fibroblast cells carrying complex I deficiency37. In neurons38, not only complex I, but also complex III deficiency demonstrated severe oxidative damage. Additional experiments are needed to define the precise mechanism by which Pim kinases control redox balance. Our data suggest that Pim knockout results in changes to cytochrome complexes that mimic the effects of rotenone and function in a similar manner this agent in the presence of activated K-Ras. Taken together, these data suggest that the Pim protein kinases control the transcription of a broad array of genes that regulate a number key enzyme levels in specific metabolic pathways that influence the activity of the K-RasG12V protein.
To understand the biochemical basis for the increased ROS level in TKO cells and why mutant Ras expression increases the level of oxygen free radicals, we measured NADP+/NADPH, whose levels play an active role in regulating the activity of glutathione peroxidase and peroxiredoxins to detoxify ROS. UPLC/MS/MS analysis revealed that NAD+ metabolism was abnormal in TKO MEFs, and flux through the PPP was decreased, reducing the levels of NADP+ /NADPH. This result combined with abnormalities in the GSH/GSSG ratio in TKO cells suggests the ability of TKO cells to detoxify ROS is markedly compromised. Additionally, the levels of Sod 1, 2, and 3, Gpx4 and Prdx3 mRNAs and Sod2 and Gpx4 protein (Supplementary Figure S6C-E) were lower in TKO compared to WT MEFs, further suggesting that the ability of these cells to detoxify ROS produced through metabolic pathways was limited. Pim knockout cells can survive and divide, but, when stressed by activated K-Ras, they do not have sufficient detoxification capabilities to handle increased ROS production.
c-Myc expression restored NAD(P)+ levels in TKO MEFs, increased Sod2 protein to nearly wild type levels, and consequently, decreased ROS accumulation by K-RasG12V transduction. c-Myc transduction reduced the metabolic redox equivalents, preventing K-RasG12V-induced killing in TKO MEFs. Combined with the increase in NAD+ and Sod2, c-Myc plays an important role in the regulation of mitochondrial metabolism39, suggesting that changes in this enzyme level could play a part in the mechanism by which c-Myc regulates OxPhos activity in the TKO MEFs.
Taken together, our data reveal a critical role for Pim kinases in regulating cellular metabolism and define how deletion of these protein kinases modulates oxidative stress by regulating redox equivalents and ROS detoxification. These metabolic changes sensitized cells to K-RasG12V killing, but were reversed by (1) the action of NAC, (2) a single Pim kinase isoform, or (3) c-Myc transduction. These results have the potential to impact the development of small molecule Pim kinase family inhibitors that are now entering Phase I trials for the treatment of cancer patients.
Materials and Methods
Cell culture, lentivirus infection and plasmids
MEFs were generated from embryos (day 14.5) of WT and Pim1, 2, and 3 triple knockout40 of Friend virus B (FVB) mice. Cells were immortalized by culturing and reseeding them multiple times at the density of 3×105 cells in DMEM supplemented with 10% fetal bovine serum, 1% Glutamax, and 1 % Penicillin and Streptomycin (Life Technologies, Grand Island, NY). Lentiviruses were packaged in HEK293T cells using calcium phosphate transfection. Two days after transfection, medium was collected and concentrated by ultracentrifugation and then added to the target cells. Full-length cDNAs encoding mouse K-RasG12V, Pim-1, Pim-2, and Pim-3 were subcloned into the FUCRW vector41. HA-tagged c-Myc in pLEX vector (Open Biosystems) was used as previously reported8.
Cell viability and focus forming assays
To evaluate growth induced by K-RasG12V and c-Myc, MEF cells were plated in 1 ml of DMEM in a 6-well plate at the density of 500 cells and then infected with lentiviruses containing empty vector, K-RASG12V, or c-Myc. Infected cells were cultured at 37 °C for 10–14 days before scoring for foci formation. Foci were visualized with crystal violet staining. Cell viability was determined by the MTT assay42 or crystal violet staining43.
Measurements of ROS, glutathione, NAD, NADP and oxygen consumption rate
To determine oxidative stress, cells were washed once with PBS and incubated with PBS containing 2 μM CM-H2DCFDA (Life Technologies) for the determination of reactive oxygen species for 30 min at 37 °C. Live cells were imaged using an inverted fluorescence microscope. For quantitative comparison of cellular ROS between WT and TKO MEFs, DCF-labelled cells were analyzed by flow cytometry. Levels of NAD(H), NADP(H), and GSH were quantified using a NAD/NADH Quantification Kit Sigma-Aldrich, St. Louis, MO, NADP/NADPH Quantification Kit (Sigma-Aldrich), and GSH/GSSG-Glo assay kit (Promega, Madison, WI; V6611) according to the manufacturer's protocols. Oxygen consumption rates were measured using a XF-24 extracellular flux analyzer (SeaHorse Bioscience Inc., Chicopee, MA). To assess mitochondrial function, the XF Cell Mito Stress Test Kit (101706-100) was employed.
Quantitative real-time-PCR, gene expression array, and metabolic profiling analyses
Quantitative real-time-PCR (qPCR) was performed as reported previously42. Gene expression array analysis was performed by the Hollings Cancer Center Genomics Shared Resource at the Medical University of South Carolina using Illumina Bead-Array Chip (MouseWG-6 v2.0 Expression BeadChip, Illumina, San Diego, CA). Pathway enrichment analyses were performed using the HHMI/Keck Biotechnology Resource Laboratory Service (Dr. Xiting Yan, Yale University). In order to avoid false positives due to multiple testing in GSEA, the FDR approach is used to adjust the P-value. For the metabolic profiling analysis, immortalized WT and TKO MEF cells were collected from five separate replicates. Global metabolite analysis was performed by Metabolon, Inc (Durham, NC) using a combination of gas or ultra high performance liquid chromatography combined with mass spectrometry (GC/MS) as reported previously44,45.
Supplementary Material
Acknowledgments
This work was supported in part by the Genomics Shared Resource at Hollings Cancer Center, and Seahorse Biosciences Academic Core Facility, Medical University of South Carolina. We thank Dr. Allen J. Ebens at Genentech Inc. and Novartis Oncology for providing the Pim kinase inhibitors used in this study.
This work was supported by the NIH P30-CA138313, DOD W81XWH-08-PCRP-IDA, R01 CA1732000, and American Cancer Society Institutional Research Grant awarded to the Hollings Cancer Center, Medical University of South Carolina.
Footnotes
The authors disclose no potential conflicts of interest.
References
- 1.Verbeek S, van Lohuizen M, van der Valk M, Domen J, Kraal G, Berns A. Mice bearing the E mu-myc and E mu-pim-1 transgenes develop pre-B-cell leukemia prenatally. Mol Cell Biol. 1991;11:1176–1179. doi: 10.1128/mcb.11.2.1176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.van Lohuizen M, Verbeek S, Krimpenfort P, Domen J, Saris C, Radaszkiewicz T, et al. Predisposition to lymphomagenesis in pim-1 transgenic mice: cooperation with c-myc and N-myc in murine leukemia virus-induced tumors. Cell. 1989;56:673–682. doi: 10.1016/0092-8674(89)90589-8. [DOI] [PubMed] [Google Scholar]
- 3.Adam M, Pogacic V, Bendit M, Chappuis R, Nawijn MC, Duyster J, et al. Targeting PIM kinases impairs survival of hematopoietic cells transformed by kinase inhibitor-sensitive and kinase inhibitor-resistant forms of Fms-like tyrosine kinase 3 and BCR/ABL. Cancer Res. 2006;66:3828–3835. doi: 10.1158/0008-5472.CAN-05-2309. [DOI] [PubMed] [Google Scholar]
- 4.Agrawal S, Koschmieder S, Baumer N, Reddy NG, Berdel WE, Muller-Tidow C, et al. Pim2 complements Flt3 wild-type receptor in hematopoietic progenitor cell transformation. Leukemia. 2008;22:78–86. doi: 10.1038/sj.leu.2404988. [DOI] [PubMed] [Google Scholar]
- 5.Kim KT, Baird K, Ahn JY, Meltzer P, Lilly M, Levis M, et al. Pim-1 is up-regulated by constitutively activated FLT3 and plays a role in FLT3-mediated cell survival. Blood. 2005;105:1759–1767. doi: 10.1182/blood-2004-05-2006. [DOI] [PubMed] [Google Scholar]
- 6.Wang J, Anderson PD, Luo W, Gius D, Roh M, Abdulkadir SA. Pim1 kinase is required to maintain tumorigenicity in MYC-expressing prostate cancer cells. Oncogene. 2012;31:1794–1803. doi: 10.1038/onc.2011.371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wang JB, Erickson JW, Fuji R, Ramachandran S, Gao P, Dinavahi R, et al. Targeting mitochondrial glutaminase activity inhibits oncogenic transformation. Cancer Cell. 2010;18(3):207–19. doi: 10.1016/j.ccr.2010.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Beharry Z, Mahajan S, Zemskova M, Lin YW, Tholanikunnel BG, Xia Z, et al. The Pim protein kinases regulate energy metabolism and cell growth. Proc Natl Acad Sci USA. 2011;108:528–533. doi: 10.1073/pnas.1013214108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Soucek L, Whitfield JR, Sodir NM, Masso-Valles D, Serrano E, Karnezis AN, et al. Inhibition of Myc family proteins eradicates KRas-driven lung cancer in mice. Genes Dev. 2013;27:504–513. doi: 10.1101/gad.205542.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tran PT, Fan AC, Bendapudi PK, Koh S, Komatsubara K, Chen J, et al. Combined Inactivation of MYC and K-Ras oncogenes reverses tumorigenesis in lung adenocarcinomas and lymphomas. PLoS One. 2008;3:e2125. doi: 10.1371/journal.pone.0002125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Compere SJ, Baldacci P, Sharpe AH, Thompson T, Land H, Jaenisch R. The ras and myc oncogenes cooperate in tumor induction in many tissues when introduced into midgestation mouse embryos by retroviral vectors. Proc Natl Acad Sci USA. 1989;86:2224–2228. doi: 10.1073/pnas.86.7.2224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ying H, Kimmelman AC, Lyssiotis CA, Hua S, Chu GC, Fletcher-Sananikone E, et al. Oncogenic Kras maintains pancreatic tumors through regulation of anabolic glucose metabolism. Cell. 2012;149:656–670. doi: 10.1016/j.cell.2012.01.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gaglio D, Metallo CM, Gameiro PA, Hiller K, Danna LS, Balestrieri C, et al. Oncogenic K-Ras decouples glucose and glutamine metabolism to support cancer cell growth. Mol Syst Biol. 2011;7:523. doi: 10.1038/msb.2011.56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Son J, Lyssiotis CA, Ying H, Wang X, Hua S, Ligorio M, et al. Glutamine supports pancreatic cancer growth through a KRAS-regulated metabolic pathway. Nature. 2013;496:101–105. doi: 10.1038/nature12040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Weinberg F, Hamanaka R, Wheaton WW, Weinberg S, Joseph J, Lopez M, et al. Mitochondrial metabolism and ROS generation are essential for Kras-mediated tumorigenicity. Proc Natl Acad Sci USA. 2010;107:8788–8793. doi: 10.1073/pnas.1003428107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mitsushita J, Lambeth JD, Kamata T. The superoxide-generating oxidase Nox1 is functionally required for Ras oncogene transformation. Cancer Res. 2004;64:3580–3585. doi: 10.1158/0008-5472.CAN-03-3909. [DOI] [PubMed] [Google Scholar]
- 17.Osthus RC, Shim H, Kim S, Li Q, Reddy R, Mukherjee M, et al. Deregulation of glucose transporter 1 and glycolytic gene expression by c-Myc. J Biol Chem. 2000;275:21797–21800. doi: 10.1074/jbc.C000023200. [DOI] [PubMed] [Google Scholar]
- 18.Gao P, Tchernyshyov I, Chang TC, Lee YS, Kita K, Ochi T, et al. c-Myc suppression of miR-23a/b enhances mitochondrial glutaminase expression and glutamine metabolism. Nature. 2009;458:762–765. doi: 10.1038/nature07823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Liu W, Le A, Hancock C, Lane AN, Dang CV, Fan TW, et al. Reprogramming of proline and glutamine metabolism contributes to the proliferative and metabolic responses regulated by oncogenic transcription factor c-MYC. Proc Natl Acad Sci USA. 2012;109:8983–8988. doi: 10.1073/pnas.1203244109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Menssen A, Hydbring P, Kapelle K, Vervoorts J, Diebold J, Luscher B, et al. The c-MYC oncoprotein, the NAMPT enzyme, the SIRT1-inhibitor DBC1, and the SIRT1 deacetylase form a positive feedback loop. Proc Natl Acad Sci USA. 2012;109:E187–E196. doi: 10.1073/pnas.1105304109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.DeNicola GM, Karreth FA, Humpton TJ, Gopinathan A, Wei C, Frese K, et al. Oncogene-induced Nrf2 transcription promotes ROS detoxification and tumorigenesis. Nature. 2011;475:106–109. doi: 10.1038/nature10189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wang X, Magnuson S, Pastor R, Fan E, Hu H, Tsui V, et al. Discovery of novel pyrazolo[1,5-a]pyrimidines as potent pan-Pim inhibitors by structure- and property-based drug design. Bioorg Med Chem Lett. 2013;23:3149–3153. doi: 10.1016/j.bmcl.2013.04.020. [DOI] [PubMed] [Google Scholar]
- 23.Lu J, Zavorotinskaya T, Dai Y, Niu XH, Castillo J, Sim J, et al. Pim2 is required for maintaining multiple myeloma cell growth through modulating TSC2 phosphorylation. Blood. 2013;122:1610–1620. doi: 10.1182/blood-2013-01-481457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Garcia PD, Langowski JL, Wang Y, Chen M, Castillo J, Fanton C, et al. Pan-PIM kinase inhibition provides a novel therapy for treating hematologic cancers. Clin Cancer Res. 2014;20:1834–1845. doi: 10.1158/1078-0432.CCR-13-2062. [DOI] [PubMed] [Google Scholar]
- 25.Gomez-Duran A, Pacheu-Grau D, Martinez-Romero I, Lopez-Gallardo E, Lopez-Perez MJ, Montoya J, et al. Oxidative phosphorylation differences between mitochondrial DNA haplogroups modify the risk of Leber's hereditary optic neuropathy. Biochim Biophys Acta. 2012;1822:1216–1222. doi: 10.1016/j.bbadis.2012.04.014. [DOI] [PubMed] [Google Scholar]
- 26.Bhalla K, Hwang BJ, Dewi RE, Ou L, Twaddel W, Fang HB, et al. PGC1alpha promotes tumor growth by inducing gene expression programs supporting lipogenesis. Cancer Res. 2011;71:6888–6898. doi: 10.1158/0008-5472.CAN-11-1011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ziegler M. New functions of a long-known molecule Emerging roles of NAD in cellular signaling. Eur J Biochem. 2000;267:1550–1564. doi: 10.1046/j.1432-1327.2000.01187.x. [DOI] [PubMed] [Google Scholar]
- 28.Santidrian AF, Matsuno-Yagi A, Ritland M, Seo BB, LeBoeuf SE, Gay LJ, et al. Mitochondrial complex I activity and NAD+/NADH balance regulate breast cancer progression. J Clin Invest. 2013;123:1068–1081. doi: 10.1172/JCI64264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gorrini C, Harris IS, Mak TW. Modulation of oxidative stress as an anticancer strategy. Nat Rev Drug Discov. 2013;12:931–947. doi: 10.1038/nrd4002. [DOI] [PubMed] [Google Scholar]
- 30.Gaetani GF, Galiano S, Canepa L, Ferraris AM, Kirkman HN. Catalase and glutathione peroxidase are equally active in detoxification of hydrogen peroxide in human erythrocytes. Blood. 1989;73:334–339. [PubMed] [Google Scholar]
- 31.Yang WS, SriRamaratnam R, Welsch ME, Shimada K, Skouta R, Viswanathan VS, et al. Regulation of ferroptotic cancer cell death by GPX4. Cell. 2014;156:317–331. doi: 10.1016/j.cell.2013.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Podsypanina K, Politi K, Beverly LJ, Varmus HE. Oncogene cooperation in tumor maintenance and tumor recurrence in mouse mammary tumors induced by Myc and mutant Kras. Proc Natl Acad Sci USA. 2008;105:5242–5247. doi: 10.1073/pnas.0801197105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sinn E, Muller W, Pattengale P, Tepler I, Wallace R, Leder P. Coexpression of MMTV/v-Ha-ras and MMTV/c-myc genes in transgenic mice: synergistic action of oncogenes in vivo. Cell. 1987;49:465–475. doi: 10.1016/0092-8674(87)90449-1. [DOI] [PubMed] [Google Scholar]
- 34.Li F, Wang Y, Zeller KI, Potter JJ, Wonsey DR, O'Donnell KA, et al. Myc stimulates nuclearly encoded mitochondrial genes and mitochondrial biogenesis. Mol Cell Biol. 2005;25:6225–6234. doi: 10.1128/MCB.25.14.6225-6234.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.de Groof AJ, te Lindert MM, van Dommelen MM, Wu M, Willemse M, Smift AL, et al. Increased OXPHOS activity precedes rise in glycolytic rate in H-RasV12/E1A transformed fibroblasts that develop a Warburg phenotype. Mol Cancer. 2009;8:54. doi: 10.1186/1476-4598-8-54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Telang S, Lane AN, Nelson KK, Arumugam S, Chesney J. The oncoprotein H-RasV12 increases mitochondrial metabolism. Mol Cancer. 2007;6:77. doi: 10.1186/1476-4598-6-77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Pitkanen S, Robinson BH. Mitochondrial complex I deficiency leads to increased production of superoxide radicals and induction of superoxide dismutase. J Clin Invest. 1996;98:345–351. doi: 10.1172/JCI118798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Diaz F, Garcia S, Padgett KR, Moraes CT. A defect in the mitochondrial complex III, but not complex IV, triggers early ROS-dependent damage in defined brain regions. Hum Mol Genet. 2012;21:5066–5077. doi: 10.1093/hmg/dds350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Peek CB, Affinati AH, Ramsey KM, Kuo HY, Yu W, Sena LA, et al. Circadian clock NAD+ cycle drives mitochondrial oxidative metabolism in mice. Science. 2013;342:1243417. doi: 10.1126/science.1243417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Mikkers H, Nawijn M, Allen J, Brouwers C, Verhoeven E, Jonkers J, et al. Mice deficient for all PIM kinases display reduced body size and impaired responses to hematopoietic growth factors. Mol Cell Biol. 2004;24:6104–6115. doi: 10.1128/MCB.24.13.6104-6115.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Cai H, Memarzadeh S, Stoyanova T, Beharry Z, Kraft AS, Witte ON. Collaboration of Kras and Androgen Receptor Signaling Stimulates EZH2 Expression and Tumor-Propagating Cells in Prostate Cancer. Cancer Res. 2012;72:4672–4681. doi: 10.1158/0008-5472.CAN-12-0228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Song JH, Kraft AS. Pim kinase inhibitors sensitize prostate cancer cells to apoptosis triggered by Bcl-2 family inhibitor ABT-737. Cancer Res. 2012;72:294–303. doi: 10.1158/0008-5472.CAN-11-3240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Song JH, Bellail A, Tse MC, Yong VW, Hao C. Human astrocytes are resistant to Fas ligand and tumor necrosis factor-related apoptosis-inducing ligand-induced apoptosis. J Neurosci. 2006;26:3299–3308. doi: 10.1523/JNEUROSCI.5572-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Langley RJ, Tsalik EL, van Velkinburgh JC, Glickman SW, Rice BJ, Wang C, et al. An integrated clinico-metabolomic model improves prediction of death in sepsis. Sci Transl Med. 2013;5:195ra95. doi: 10.1126/scitranslmed.3005893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Weiner J, 3rd, Parida SK, Maertzdorf J, Black GF, Repsilber D, Telaar A, et al. Biomarkers of inflammation, immunosuppression and stress with active disease are revealed by metabolomic profiling of tuberculosis patients. PLoS One. 2012;7:e40221. doi: 10.1371/journal.pone.0040221. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.