

Abstract
Medulloblastoma (MB) is a highly malignant brain tumor that occurs primarily in children. Although surgery, radiation and high-dose chemotherapy have led to increased survival, many MB patients still die from their disease, and patients who survive suffer severe long-term side effects as a consequence of treatment. Thus, more effective and less toxic therapies for MB are critically important. Development of such therapies depends in part on identification of genes that are necessary for growth and survival of tumor cells. Survivin is an inhibitor of apoptosis protein (IAP) that regulates cell cycle progression and resistance to apoptosis, is frequently expressed in human MB, and when expressed at high levels predicts poor clinical outcome. Therefore, we hypothesized that Survivin may play a critical role in growth and survival of MB cells and that targeting it may enhance MB therapy. Here we show that Survivin is overexpressed in tumors from patched (Ptch) mutant mice, a model of Sonic hedgehog (SHH)-driven MB. Genetic deletion of survivin in Ptch mutant tumor cells significantly inhibits proliferation and causes cell cycle arrest. Treatment with small molecule antagonists of Survivin impairs proliferation and survival of both murine and human MB cells. Finally, Survivin antagonists impede growth of MB cells in vivo. These studies highlight the importance of Survivin in SHH-driven MB, and suggest that it may represent a novel therapeutic target in patients with this disease.
Keywords: medulloblastoma, survivin, sonic hedgehog, targeted therapy, animal model, brain tumor
Introduction
Medulloblastoma (MB) is the most common malignant brain tumor in children(1). Intensive therapy – including surgery, cranio-spinal radiation and high dose chemotherapy – has improved 5-year survival rates(2), but almost a third of MB patients still die from their disease, and survivors suffer severe long term side effects that affect their quality of life(3). Thus, safer and more effective therapies are needed for this disease.
In recent years, targeted therapies have begun to be evaluated in patients with MB. For example, with the recognition that a subset of MBs results from mutations in the Sonic Hedgehog (SHH) pathway(4), antagonists of Smoothened (SMO), an activator of the pathway, have advanced into clinical trials for the disease. Although initial reports suggested that these agents can inhibit tumor growth(5), as with many targeted therapies, resistance develops quickly(6). Moreover, patients who have SHH pathway mutations downstream of SMO do not respond to these agents at all(7). Thus, even for SHH-driven MBs, additional approaches are necessary.
Finding novel therapeutic targets for MB depends on identification of genes that are critical for tumor growth and survival. SURVIVIN (also known as baculoviral inhibitor of apoptosis repeat-containing 5, or BIRC5), is a member of the IAP family that regulates both cell cycle progression and cell survival(8, 9). SURVIVIN is expressed in many human cancers, and its expression is correlated with poor clinical outcome in non-small cell lung cancer (NSCLC)(10), breast cancer(11), leukemia(12, 13), neuroblastoma(14) and glioma(15). Because of its high expression in tumors and minimal expression in most adult tissues(16), a number of approaches have been developed to inhibit its expression and function. These include small molecule antagonists, antisense oligonucleotides, and immunotherapy(17-21). Some of these approaches are in clinical trials for numerous cancers, including prostate cancer(22), melanoma(23, 24), breast cancer(25), and NSCLC(25, 26).
SURVIVIN has not been studied extensively in the context of MB. Although some studies have suggested that elevated expression is linked to poor prognosis(27-29), little is known about its role in MB growth and survival. Using an animal model of SHH-driven MB, we now show that Survivin is highly expressed in tumors and absent from normal adult cerebellum. Moreover, through both genetic deletion and pharmacological inhibition we demonstrate that Survivin is critical for proliferation and survival of mouse and human SHH driven MB cells. Finally, Survivin antagonists impair growth of MB in vivo, highlighting the potential of Survivin as a therapeutic target in patients with MB.
Results
Survivin is highly expressed in medulloblastomas from Ptch mutant mice
To determine whether Survivin could represent a target in SHH driven MB, we isolated RNA from Ptch mutant tumors and examined survivin expression using real time PCR. High levels of survivin were detected in all tumors and in granule neuron precursors (GNPs), the progenitors from which these tumors are thought to arise(30) (Figure 1A). Importantly, expression could not be detected in normal adult cerebellum. Similar results were seen when Survivin protein was examined by immunoblotting (Figure 1B). Staining of tissue sections revealed Survivin expression in the nuclei of tumor cells (abrogated by blocking peptide (Figure 1D)), and minimal staining in normal adult cerebellum (Figure 1C-F). These data indicate that Survivin is highly expressed in Ptch mutant tumors, raising the possibility that it might play an important role in tumor growth or maintenance.
Figure 1. Survivin is expressed in Ptch mutant tumors.
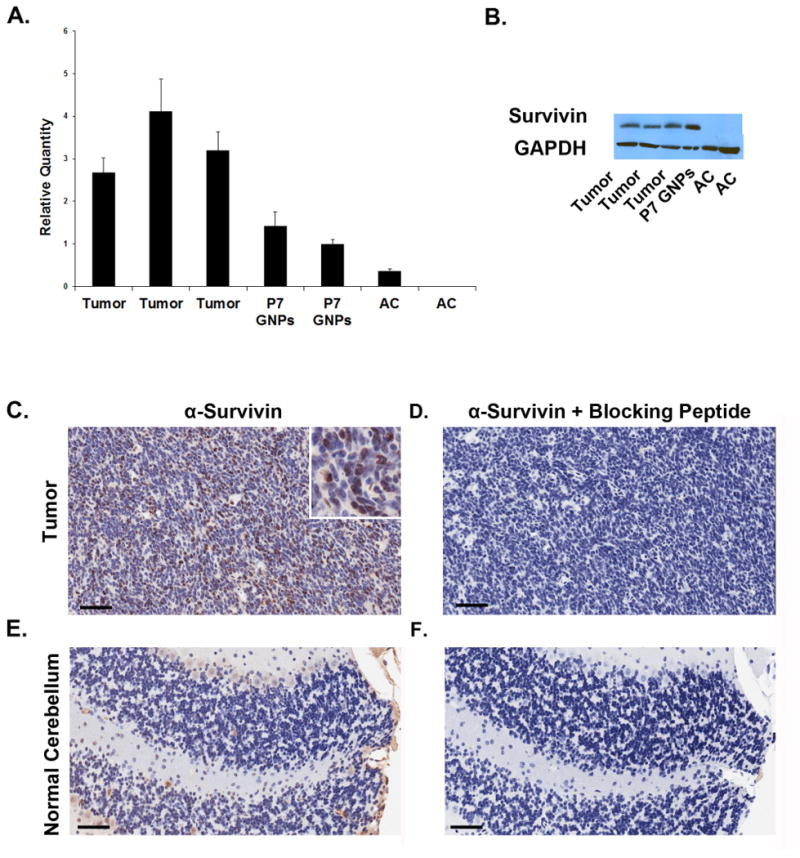
RNA and protein from Ptch mutant MB cells, P7 GNPs and adult cerebellum were analyzed for Survivin expression using real time PCR (A) and by western blotting (B). Survivin is highly expressed in tumors and GNPs, but not in adult cerebellum. Error bars in (A) represent 95% confidence interval calculated using sum of the squares method (p<0.02 by ANOVA and post hoc student's t-test). (C-F) Tissue sections from Ptch mutant tumor and normal adult cerebellum were stained with anti-Survivin antibodies alone (C,E) or with anti-Survivin antibodies that were pre-incubated for 30 min with Survivin blocking peptide (D,F). Survivin is highly expressed in tumor cells with minimal expression in adult cerebellum. Inset in (C) is 4x magnification of positive staining. Scale bars represent 50 μM. Data are representative of 3 experiments.
Survivin is critical for MB cell proliferation and cell cycle progression
To investigate the importance of Survivin for growth of MB cells, we first utilized a genetic approach. Survivinfl/fl mice(31), in which the survivin gene is flanked by loxP sites, were crossed with Ptch+/- mice, to generate tumors in which survivin can be deleted by Cre recombinase. We confirmed efficient deletion of survivin by isolating tumor cells from Survivinfl/fl;Ptch+/- (SP) mice and infecting them with Cre retroviruses. After 48hrs, survivin expression was significantly reduced (by 82%) in Cre-infected cells compared to control (GFP-infected) cells (Figure 2A). We then looked at the effect of survivin loss on proliferation. After Cre-mediated deletion of survivin from SP tumor cells, thymidine incorporation was decreased by almost 90% (Figure 2B). Importantly, when tumor cells from SurvivinWT mice were infected with Cre viruses, there was no appreciable difference in proliferation compared to control cells (Figure 2C), indicating that the decreased thymidine incorporation observed in SP tumor cells was not due to non-specific toxicity of the Cre virus. To address whether loss of survivin affects cell cycle progression, we isolated cells from SP tumors, infected them with Cre or GFP viruses, and performed cell cycle analysis (Figure 2D,E). survivin deletion led to a marked accumulation of cells in the G2/M phases of the cell cycle (39% of Cre-infected cells vs. 9.5% of control cells in G2/M). Together, these data demonstrate that Survivin is necessary for proliferation and cell cycle progression of MB cells.
Figure 2. Loss of Survivin causes decreased proliferation and cell cycle arrest.
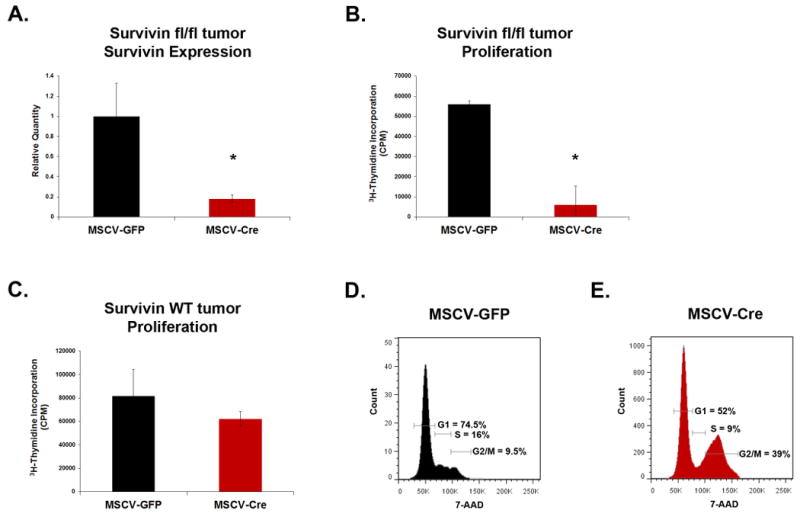
(A-B) Cells were isolated from Survivinfl/fl;Ptch+/- tumors and infected with Cre- or GFP retroviruses for 48 hr. (A) GFP+ cells were isolated by flow cytometry and survivin mRNA expression analyzed by RT-qPCR (n=2). Cre causes loss of survivin expression (p<0.02). (B) Cells were pulsed with 3H-thymidine for 12 hr, harvested, and analyzed for incorporation. Loss of survivin leads to decreased tumor cell proliferation (p<0.001). Data are representative of 5 experiments. (C) Cells were isolated from Ptch+/- tumor (wild type for survivin), infected with Cre- or GFP viruses for 48 hr, and collected after 12 hr pulse with 3H-thymidine to measure incorporation. Infection with Cre virus alone does not significantly impair proliferation (p>0.1). Data in (A-C) represent mean +/- standard deviation (SD) and are representative of 4 experiments. (D, E) Cells from Survivinfl/fl;Ptch+/- tumors were infected with virus as described above (D. GFP, E. Cre virus) and stained with 7-AAD for cell cycle analysis by flow cytometry. survivin deletion causes accumulation of cells in G2/M. Data are representative of 4 experiments and cell cycle percentages based on live cell gates (excluded subG1). p values calculated using student's t-test.
Survivin antagonists inhibit MB cell proliferation and promote apoptosis
Given the importance of Survivin for MB proliferation, we hypothesized that pharmacological agents that inhibit Survivin expression or function might interfere with tumor growth. To test this, we obtained several small molecule Survivin antagonists: YM155 is an inhibitor of survivin transcription(32), whereas S12 and LLP3 bind directly to Survivin protein and interfere with its function(33, 34). To test the ability of YM155 to inhibit survivin expression in Ptch mutant MB cells, we treated cells with the drug for 48hrs, isolated RNA and performed qRT-PCR for survivin. YM155 markedly decreased survivin expression even at a concentration of 10 nM (Supp. Figure 1A). Similarly, loss of Survivin was detected at the protein level using western blotting (Supp Figure 1B). These data suggest that YM155 effectively inhibits survivin expression in Ptch mutant MB cells. To test the effects of Survivin antagonists on MB growth, we treated tumor cells with these agents and analyzed the percentage of cells expressing the proliferation marker Ki67. Consistent with our genetic results, inhibition of Survivin using either YM155 or S12 caused a significant decrease in the number of Ki67+ cells compared to treatment with vehicle (DMSO) (Figure 3A-D). Additionally, we saw a dose dependent decrease in thymidine incorporation after treatment with YM155, S12, or LLP3 (Figure 3E-F and Supp. Figure 2). These data suggest that Survivin antagonists effectively inhibit MB growth in vitro.
Figure 3. Survivin antagonists inhibit proliferation.
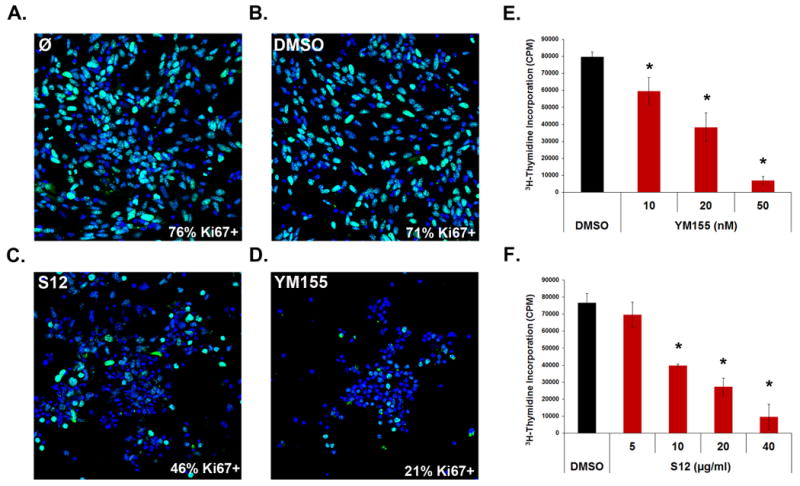
(A-D) Tumor cells were plated on chamber slides and treated for 24 hr with control media (A), 0.1% DMSO (B), 10 μg/ml S12 (C), or 50 nM YM155 (D). Cells were stained with anti-Ki67 antibodies (green) to mark proliferating cells and DAPI (blue) to label cell nuclei. Very few cells were Ki67+ after treatment with Survivin antagonists compared to controls (p<0.0001 for YM155 and S12). Data are representative of 3 experiments. (E-F) Ptch mutant tumor cells were treated with multiple doses of YM155 (E) or S12 (F) for 48 hr and pulsed with 3H-thymidine for 12 hr to measure proliferation. Treatment with either antagonist decreased proliferation in a dose-dependent manner (p< 0.02 for all YM155 and S12 doses except 5μg/ml S12, which was not significant (NS)). Ki67+ percentages in (A) were averaged from 6 images for each treatment. Data in (E-F) represent mean +/- SD and are representative of 6 experiments. Stats were calculated by ANOVA and post hoc student's t-tests.
To determine whether Survivin antagonists also cause cell cycle arrest in MB cells, we performed cell cycle analysis. After 24hrs, cells treated with S12 showed a significant accumulation in G2/M (56%) compared to cells treated with vehicle (12%) (Figure 4A,C). G2/M accumulation was also seen at 36h (52%) (Figure 4B,D). In contrast to S12, YM155 decreased the percentage of cells in G2/M (7%), with a concomitant increase in S phase (from 15 to 20% at 24hr) (Figure 4E-H). Notwithstanding these differences, our data demonstrate that both Survivin antagonists alter normal cell cycle progression of MB cells.
Figure 4. Survivin antagonists alter cell cycle progression and promote apoptosis.
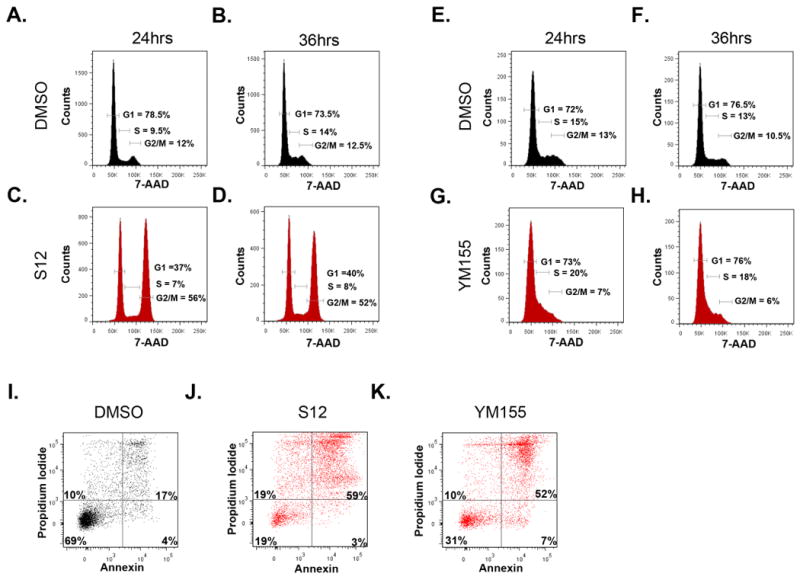
(A-H) Ptch mutant tumor cells were treated with either DMSO (A,B,E,F), 20 μg/ml S12 (C,D), or 100 nM YM155 (G,H) and stained with 7-AAD for cell cycle analysis after 24 hr (A,C,E,G) or 36 hr (B,D,F,H). YM155 decreased the percentage of cells in G2/M, while S12 treatment caused an accumulation of cells in G2/M. Data represent 4 (A-D) and 6 (E-H) experiments and percentages based on live cell gates (excluded subG1). (I-K) Tumor cells were treated with DMSO (I), 20 μg/ml (J), or 100nM YM155 (K) for 36 hr, then collected and stained with Propidium Iodide (PI) and Annexin-V for FACS analysis. The percentage of apoptotic cells was significantly higher after antagonist treatment compared to control. Data represent 6 independent experiments.
In addition to its role in regulating cell cycle progression, Survivin has been suggested to function as an inhibitor of apoptosis. To determine if Survivin inhibition promotes MB cell apoptosis, tumor cells were treated with Survivin antagonists and then stained with Annexin-V and propidium iodide (PI). As shown in Figure 4E-G, antagonists increased the percentage of apoptotic (AnnexinV+) tumor cells from 21% (after DMSO treatment) to 62% (S12) or 59% (YM155). These data show that Survivin antagonists are not merely cytostatic, but can promote apoptosis of tumor cells as well.
To verify that Survivin antagonists were not inducing non-specific toxicity, we tested their effects on GNPs, which express survivin (see Figure 1), and post-mitotic neurons, which do not. Treatment of GNPs with YM155 or S12 caused a dose dependent increase in the percentage of dead cells (as measured by EthD1 staining). In contrast, survival of post-mitotic neurons was not affected by treatment with Survivin antagonists (Supp. Fig 3A,B). These data suggest that Survivin antagonists induce death of Survivin-expressing cells but are not toxic to normal cerebellar neurons.
Survivin antagonists cooperate with radiation and SHH antagonists
Among the major drawbacks of current MB therapy are the devastating side effects of radiation(35, 36). Since recent studies have suggested that YM155 can enhance the efficacy of radiotherapy against NSCLC(37), we hypothesized that combining radiation and Survivin antagonists might be effective for MB as well. To test this, we performed thymidine incorporation assays on tumor cells treated with Survivin antagonists for 24hr followed by exposure to varying doses of radiation (Figure 5A). Treatment with sub-optimal doses of YM155 and S12 alone resulted in a small decrease in proliferation. The combination of Survivin antagonists and 0.25 grey (Gy) radiation markedly decreased tumor cell proliferation compared to radiation alone. The level of inhibition achieved with the combination treatment was equivalent to that achieved by doubling the radiation dose. These data suggest that Survivin antagonists can enhance the effects of radiation on MB cells, and raise the possibility that these agents might allow reduction in the doses of radiation used for therapy.
Figure 5. Survivin antagonists cooperate with radiation and LDE225 SHH antagonists.
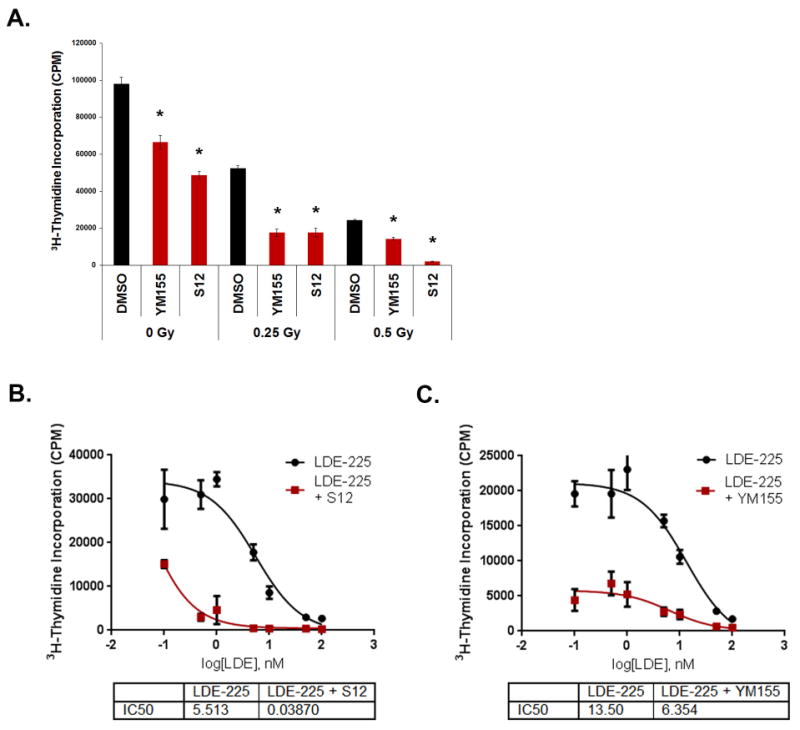
A) Tumor cells were treated with either 0.1% DMSO, 50 nM YM155, or 10 μg/ml S12 for 24 hr, and then irradiated with 0, 0.25, or 0.5 gray (Gy). After 24 hr, cells were pulsed with 3H-thymidine and assayed for incorporation. Treatment with antagonists enhanced sensitivity of tumor cells to radiation (p<0.02 for all treatments, calculated by 2 way ANOVA to identify radiation dose by treatment interaction, split by radiation dose, with post hoc student's t-tests) Data are representative of 5 experiments. (B,C) Tumor cells were plated in 96 well plates and treated with LDE225 alone (at the indicated concentrations) or in combination with 20 nM YM155 (B) or 10 μg/ml S12 (C). Cells were pulsed with 3H-thymidine after 48 hr and harvested to assay levels of incorporation. Combination treatment of LDE with either S12 or YM155 significantly lowered the IC50 compared to LDE alone (p< 0.01 by student's t-test). Data are representative of 4 (B) and 3 (C) experiments. All data represent mean +/- SD.
In addition to conventional therapy, targeted therapies have begun to be evaluated for treatment MB. For SHH-associated tumors, NVP-LDE225 a small molecule antagonist of SMO, has shown some efficacy in animal models as well as in patients(38, 39). To test whether Survivin antagonists increase the efficacy of SHH antagonists, we treated Ptch mutant tumor cells with various concentrations of LDE225 alone or in combination with 10μg/ml S12 (Figure 5B). LDE225 alone had an IC50 of 5.5 nM (range: 3-8.5 nM) while the combination of LDE225 and S12 markedly decreased the IC50 to 0.04 nM (range: 0.04-2 nM). Similarly, exposure to 20 nM YM155 decreased the IC50 of LDE225 (from 13.5nM to 6.4nM). These data suggest that Survivin antagonists significantly enhance growth inhibition by LDE.
Survivin antagonists inhibit growth of human SHH-driven MB cells
The studies described above focused on murine SHH-associated MB. To determine whether human SHH-driven MB cells also respond to Survivin antagonists, we used patient-derived xenografts (PDXs) from SHH-driven tumors. Real time PCR analysis revealed that PDX cells express high levels of survivin (Supplementary Figure 4). Treatment of PDX cells with Survivin antagonists in vitro led to significant decreases in 3H-thymidine incorporation (Figure 6A-C). Tumor cells were also treated with the SMO antagonist LDE225. Notably, the PDX line with a mutation upstream of SMO7 (DMB-012) was responsive to LDE225 (Figure 6A, left panel and ref(40)) whereas the lines with mutations downstream of SMO(40),(41) (RCMB-018 and ICb-984MB) were resistant (Figure 6B,C). In contrast, all three lines responded robustly to YM155 and high dose S12. These data suggest that Survivin antagonists can inhibit the growth of human SHH-driven MBs and that they may be useful for treating tumors that are resistant to SMO antagonists.
Figure 6. Survivin antagonists inhibit proliferation of human SHH-driven MB cells.
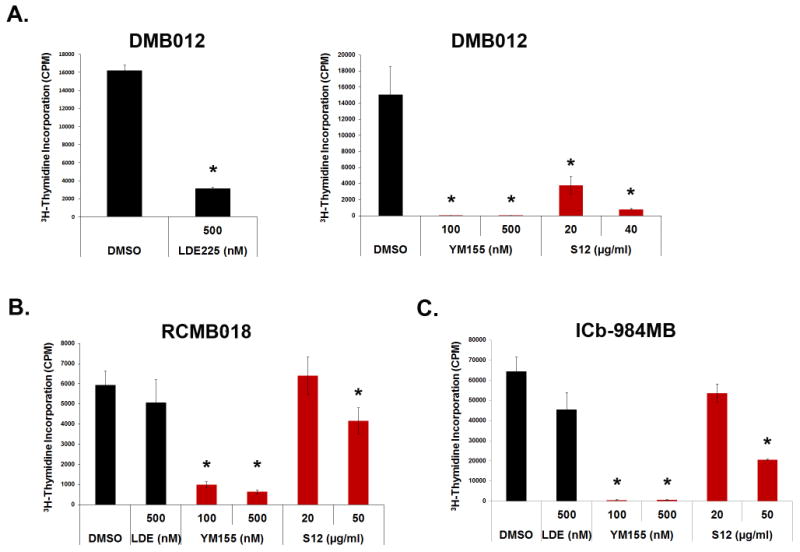
Cells isolated from LDE225-sensitive xenograft DMB012 (A) and LDE-insensitive xenografts RCMB018 (B) and ICb-984MB (C) were treated for 48 hr with DMSO, LDE225, YM155 or S12, and analyzed for thymidine incorporation following a 12-16 hr pulse. All tumor cells were sensitive to YM155 inhibition and high dose S12 treatment (DMB012 p<0.01 for all doses including LDE225, RCM018 and ICb-984MB p<0.03 for YM155 and 10μg/ml S12 while LDE and 20μg/ml S12 were not significant with p>0.08). All stats were calculated by ANOVA and post hoc student's t-test. Data represent mean +/- SD and are representative of 3 experiments.
Survivin antagonists inhibit MB growth in vivo
Given the ability of Survivin antagonists to inhibit proliferation and promote apoptosis of tumor cells in vitro, we questioned whether inhibiting Survivin could prevent tumor growth in vivo. Unfortunately, published reports (60) as well as our own preliminary studies suggested that these antagonists do not accumulate in the brain or intracranial tumors. Therefore, Ptch mutant tumors were implanted into the flanks of Nu/Nu mice and treated with YM155 or vehicle. Intratumoral injections of YM155 significantly decreased tumor growth compared to treatment with vehicle (Fig7A). Tumors harvested after 6 weeks of treatment were much smaller than those in the vehicle treated mice (Fig7B, C). We also tested whether systemic treatment with YM155 could inhibit tumor growth in vivo; since YM155 has a short half-life, we used osmotic pumps for delivery(20, 32, 42). Nu/Nu mice bearing Ptch mutant flank tumors were implanted with micro-osmotic pumps to continuously infuse YM155 or vehicle (saline) for 3 weeks. Tumors in mice with YM155-containing pumps grew significantly less than those in mice with vehicle pumps (Fig 7D-F). These data suggest that Survivin antagonists can potently inhibit MB growth in vivo.
Figure 7. YM155 inhibits growth of Ptch mutant tumor cells in vivo.
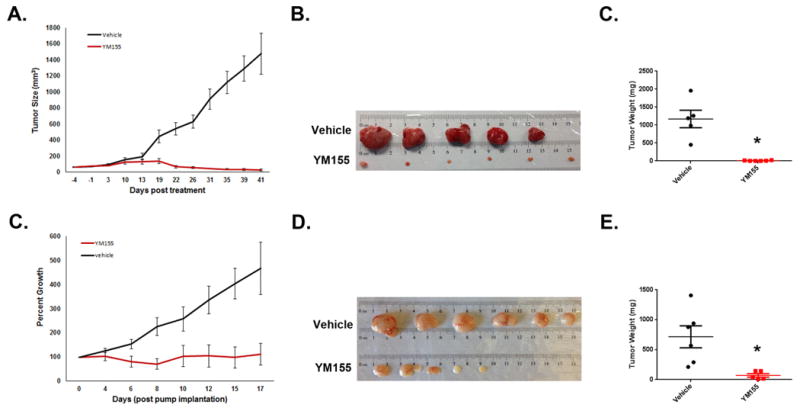
(A-F) Tumor cells were suspended in GFR matrigel (1:1 with media) and implanted in the flanks of Nude mice. When tumors reached ∼100mm3, mice were split into two cohorts and treatment was started. For (A-C), tumors were treated with vehicle (20% DMSO in saline) or YM155 (20 μM) via intratumoral injection twice a week (vehicle n=5, YM155 n=6). For (D-F), mice were treated with vehicle (saline) or YM155 (10 mg/kg/day) via micro-osmotic pump (vehicle n=6, YM155 n=5). Caliper measurements were made twice a week to monitor tumor growth (A,D) and resulting tumors were collected, photographed (B,E), and weighed (C,F). Experiments were repeated 2 (A-C) and 3 times (D-F) respectively. Both intra-tumoral and systemic treatment with YM155 decreased tumor size over time compared to vehicle control. (p<0.02 for IT and pump YM155 tumor weight by ANOVA with post hoc student's t-test). Error bars represent SEM.
Discussion
Previous studies have shown that Survivin is expressed in human MB, and that its expression is correlated with poor outcome. To study the role of Survivin in MB growth and survival, we used a mouse model of MB driven by activation of the SHH pathway. Our studies demonstrate that Survivin is highly expressed in SHH-driven MB and that genetic and pharmacologic inhibition of Survivin impedes growth of MB cells in vitro and in vivo.
Our initial studies demonstrated that Survivin is expressed at high levels in Ptch mutant MB cells and not in normal adult cerebellum, suggesting that it might represent a cancer-selective target. To test the functional importance of Survivin in MB cells, we used Cre viruses to delete Survivin from Survivinf/flx;Ptch+/- tumor cells. Loss of Survivin resulted in markedly decreased proliferation and arrest in the G2/M phases of the cell cycle. These results are consistent with the function of Survivin as a member of the chromosomal passenger complex, which is critical for alignment of chromosomes during metaphase and for successful cell cleavage(43, 44). In addition, previous studies have shown that loss of Survivin leads to aberrant mitosis, centrosome amplification and failed cytokinesis in a number of cancers, including glioma and cervical cancer(45, 46). Thus, in MB as in other tumors, Survivin seems to be critical for normal proliferation and cell cycle progression.
To evaluate Survivin's utility as a therapeutic target, we used small molecule antagonists. Consistent with our genetic studies, Survivin antagonists significantly decreased proliferation and altered cell cycle progression. Interestingly, while S12 treatment caused accumulation of cells in G2/M phase (similar to survivin deletion), YM155 caused cells to accumulate in S-phase. This observation is consistent with previous studies that demonstrated YM155 treatment can cause a loss of new DNA synthesis and concomitant stall in S phase(47). This discrepancy between S12 and YM155 could be due to differences in the kinetics or degree of Survivin inhibition induced by these drugs. Alternatively, it could result from differences in the mechanisms by which the drugs act: whereas S12 binds directly to Survivin protein(33), YM155 inhibits survivin expression by disrupting ILF3/NF110 complexes(48, 49) or Sp1 binding at the survivin promoter(50). Thus, in addition to potently decreasing survivin expression (see Ref #32 and sup fig 1), YM155 may also alter expression of other genes that regulate cell cycle progression (e.g. cyclin D1, p27) (51, 52), and thereby cause arrest at earlier stages of the cycle. Nonetheless, the fact that genetic deletion and several small molecule inhibitors all interfere with cell cycle progression in Ptch mutant tumor cells strongly supports the notion that Survivin is required for this process in SHH-driven MB.
In addition to their effects on the cell cycle, Survivin antagonists also increased the percentage of MB cells undergoing apoptosis. It remains unclear whether loss of Survivin function causes apoptosis directly or as a consequence of cell cycle arrest. Analysis of Annexin+ cells after treatment with Survivin antagonists showed that apoptosis is not detectable until 36hr, whereas cell cycle inhibition is observed by 12-24hr. These results are consistent with the notion that apoptosis occurs secondary to cell cycle arrest. The fact that Survivin antagonists not only inhibit proliferation but also cause death of tumor cells may make them potent therapeutic agents for MB.
Previous studies of glioma and other cancers have suggested that targeting Survivin can enhance the effects of radiation and chemotherapy(20, 37, 53-58). In agreement with these data, we found that Survivin antagonists significantly enhanced the sensitivity of MB cells to radiation. Combining low dose radiation (0.25 grey) with S12 or YM155 was as effective at inhibiting tumor growth as doubling the dose of radiation. These data suggest that addition of Survivin antagonists may allow children to be treated with lower doses of radiation without decreasing therapeutic benefits. The approach could markedly reduce treatment-related side effects and improve the long term quality of life of MB patients.
A major advance in treatment of SHH driven MB has been the development of SMO antagonists. However, one drawback of these agents is the swift acquisition of resistance to the drug and tumor recurrence. Our studies show that combining LDE225 and Survivin antagonists significantly enhanced inhibition of proliferation compared to either drug alone. Although in these studies we focused on co-treatment, it would be interesting to look at the ability of Survivin antagonists to overcome LDE225 resistance. In this context, it is notable that in non-small cell lung cancer, downregulation of Survivin by either siRNA or treatment with YM155 reverses Erlotinib resistance(59). It is also important to note that LDE225 and other SHH antagonists work at the level of SMO, and are thus ineffective for patients with mutations downstream in the SHH pathway(7). We show that human PDX cells with such downstream mutations are still sensitive to inhibition by Survivin antagonists. In addition, our preliminary studies suggest that that YM155 can inhibit growth of non-SHH-associated MB cells (data not shown). These studies raise the possibility that Survivin may be a therapeutic target for a broad spectrum of MB patients.
Lastly, we have shown that YM155 can inhibit tumor growth in vivo, either by direct intratumoral injection or systemic administration. These data strongly suggest that targeting Survivin could be an effective approach for treating MB. Unfortunately, pharmacokinetic studies by our lab and others ((60) and data not shown) suggest that the Survivin antagonists we have tested do not show significant accumulation in the brain or in intracranial tumors. Thus, chemical modification of these agents, or alternative modes of delivery (e.g. convection-enhanced(61, 62) or intrathecal delivery(63, 64)) may be necessary to make these agents useful for treatment of MB and other brain tumors.
Materials and Methods
Mice
Survivinfl/fl (31), Ptcfl/fl (65), Math1-Cre-ER(66)' Ptch +/-(67), and Math1CreER;ptcfl/fl (30) (MERP) mice have been described previously. P4 MERP pups were gavaged with 0.8g/40μl of tamoxifen (T-5648, Sigma, St. Louis, MO) in corn oil to generate tumors. Tumors from Ptch+/- and MERP mice were used for experiments. To allow for deletion of survivin in tumor cells, Survivinfl/fl mice were crossed with Ptch+/- mice to generate the Survivin fl/fl; Ptch+/- (SP) line. CD-1 Nu/Nu mice were from Charles River Laboratories (Wilmington, MA). P7 wild type C57BL/6 pups were obtained from the SBMRI Animal Facility. All mice were maintained in the Animal Facility, and experiments were performed in accordance with national guidelines and regulations, and with the approval of the SBMRI Institutional Animal Care and Use Committee.
Cell isolation and in vitro culture
GNPs were isolated from P7 cerebellum and tumors from adult cerebellum as previously described(30, 68). Briefly, tissue was digested in a solution containing 10 U/ml papain (Worthington Biochemical Corporation, Lakewood, NJ) and 250 U/ml DNase (Sigma), and triturated to obtain a single-cell suspension. Cells were spun through a 35-65% Percoll gradient (GE Healthcare Uppsala, Sweden) to purify GNPs and tumor cells. Cells were cultured in NB/NS-21 media (Neurobasal media, 1 mM sodium pyruvate, 2 mM L-glut, penicillin/streptomycin and NS-21 supplement, plus 1% FBS (Invitrogen Grand Island, NY)) on growth factor-reduced (GFR) matrigel-coated plates (1:50 in NB/NS-21, BD Biosciences, San Diego, CA).
Real-Time PCR
For analysis of survivin expression, mRNA was isolated from cells and tissues using an RNAeasy Plus Mini kit (QIAGEN Inc, Valencia, CA). One-step qRT-PCR reactions were performed in triplicate using QuantiTech RT mix (QIAGEN) on the Bio-Rad C1000 Thermocycler and CFX96 system (Bio-Rad Laboratories, Hercules, CA). Duplicate reactions were prepared without reverse transcriptase to confirm the absence of genomic DNA contamination. Relative gene expression was calculated using the ΔΔCT method and normalized to Actin. 95% confidence intervals for each sample were calculated using the sum of the squares method. To evaluate the efficiency of survivin deletion by Cre infection, SP tumor cells were isolated, infected with Cre-IRES-GFP or GFP retroviruses for 48hrs, sorted for GFP expression, and analyzed as outlined above.
Immunohistochemistry
For staining of paraffin-embedded tissue, animals were perfused with PBS followed by 4% paraformaldehyde (PFA, Affymetrix, cat# 19943). Cerebella were removed, fixed in 4% PFA overnight and delivered to the SBMRI Histology Shared Resource for embedding, antigen retrieval, and staining. Sections were stained either with anti-Survivin antibodies (Cell Signaling Technology Cat#2808S, Danvers, MA) alone or anti-Survivin antibodies pre-incubated with Survivin blocking peptides (Cell Signaling Technology Cat #1037).
Cell Lysis and Western-blotting
To evaluate Survivin expression, tumor cells, GNPs and adult cerebellum were lysed in RIPA buffer (Millipore, Billerica, MA). Protein was quantitated using the Bio-Rad protein assay. Equal amounts of protein were separated by SDS-PAGE, blocked with 5% BSA (Sigma) in Tris-buffered saline with 0.1% Tween-20 (TBST), and stained with anti-Survivin or GAPDH antibodies overnight (1:1000, Cell Signaling Technology Cat# 2808S, 5174) followed by anti-rabbit HRP-conjugated secondary antibody (1:2000 Cell Signaling technology, Cat# 7074S). Proteins were visualized by incubating with Pierce ECL plus (Thermo Fisher Scientific, Rockford, IL). To evaluate Survivin expression after YM155 treatment, tumor cells were plated on 6-well plates (6-8M cells/well) and treated with YM155 or DMSO (Fisher Scientific Inc. San Diego, CA) at 1 μM for 24hrs and processed as described above.
Analysis of proliferation, cell cycle and apoptosis
To analyze the effects of Survivin loss, cells were isolated from SP tumors and infected with Cre-IRES-GFP or GFP retroviruses (MSCV, 1:5 in media). To assess the effects of pharmacological inhibition of Survivin, Ptch mutant tumor cells were treated with YM155 (Selleck Chemicals), S12(33), LLP3(34), or DMSO (Fisher Scientific) at the indicated concentrations.
Ki67 staining
Cells were plated on GFR matrigel-coated chamber slides at 0.2M cells/well and treated with DMSO or antagonists for 24hrs. Cells were fixed in 4% PFA, permeabilized with 0.1% Triton X-100 in PBS (Aqua Solutions Deer Park, TX) and blocked with 10% goat serum (Jackson ImmunoResearch Laboratories, Inc. West Grove, PA) before staining with anti-Ki67 (BD Biosciences Cat# 556003) and 4′,6-diamidino-2-phenylindole (DAPI, Invitrogen). Six representative images from each treatment were collected using the Zeiss LSM-700 confocal and Ki67+ percentages calculated using ImageJ software (NIH).
Thymidine incorporation
Cells were plated (2 × 105 cells/well) in GFR-matrigel coated 96 well plates and treated with either virus or antagonists for 48hrs in triplicate wells before being pulsed with methyl-[3H]thymidine (GE Healthcare, Piscataway, NJ, USA). After 12-16 hr, cells were harvested using a Mach III manual harvester 96 (Tomtec, Hamden, CT, USA), and incorporated radioactivity was quantitated using a Wallac MicroB microplate scintillation counter (Perkin Elmer, Waltham, MA, USA).
Cell cycle analysis
Cells were plated in GFR-matrigel coated 48 well plates at 0.4M cells/well, infected with virus or treated with antagonists, and collected at various time points. Cells were fixed and stained using the FITC BrdU Flow Kit (BD Biosciences) and 7-Aminoactinomycin (7-AAD) according to the manufacturer's instructions. Analysis was performed using a FACSCanto II flow cytometer (BD Biosciences) and FlowJo v.7.6.4 software (Tree Star, Inc., Ashland, OR).
Apoptosis
Tumor cells were plated on 24-well plates at 1M cells/well and treated with Survivin antagonists or infected with viruses for 36 hr. Cells were collected by incubating with papain solution and resuspended in 100 ul of Annexin-binding buffer containing 5μL of AnnexinV conjugate (Annexin-FITC or Annexin-567, both Invitrogen) and 1μl Propidium iodide (PI, 1.0mg/ml stock, Invitrogen). Cells were analyzed using a FACSCanto II and FlowJo v.7.6.4 software.
Live/Dead Assay
To address toxicity of YM155, GNPs were isolated from WT P7 pups and plated at 0.2 × 106 cells/well in 2 96 well plates coated with GFR-matrigel for each experiment. One plate was maintained in proliferation media consisting of NB/NS-21 and Sonic hedgehog (SHH)-containing supernatant (1:5 in media) and treated with either DMSO or various doses of YM155 and S12 with each condition in triplicate wells. After 48hrs, cell viability was analyzed using the LIVE/DEAD Viability/Cytotoxicity Kit (Invitrogen). Briefly, cells were stained with 4μM EthD1 for 40 min and fluorescence emission (645 nM) was measured using a TECAN infiniteM200 Microplate reader (Morrisville, NC). The second plate was maintained in differentiation media (NB/NS-21 containing 25mM glucose and 25mM potassium chloride) for 5 days to produce post-mitotic neurons. Cells were then treated with DMSO or corresponding doses of YM155 and cell viability was evaluated after 48hrs.
Radiation and LDE225 treatment
To measure effects of inhibitors in combination with radiation, tumor cells were plated in 96-well plates at a density of 0.2 × 106 cells per well and cultured in the presence of DMSO, 50 nM YM155, or 10 ug/ml S12. After 24hrs, cells were subjected to 0, 0.25, or 0.5 Gy radiation using a Gammacell 40 Exactor (Low-dose cesium 137 irradiator, Best Theratronics Ltd., Ottawa, Ontario, Canada). Cells were cultured for an additional 24 hr, and [methyl-3H]thymidine assays were performed as described above.
To measure effects of inhibitors in combination with the SHH antagonist NVP-LDE225 (Selleck Chemicals, S2151), tumor cells were plated in 96 well plates at 0.2 × 106 cells/well and cultured with increasing doses of LDE225 or a single dose of Survivin antagonist (10μg/ml S12, 20nM YM255) alone or in combination with LDE225 as indicated. Cells were cultured for 48hrs and [methyl-3H]thymidine assays were performed as described above.
Human tumor isolation, propagation, and classification
Human MB tissue for patient-derived xenografts was obtained from surgical resection of tumors at Duke University Medical Center (Durham, NC), Rady Children's Hospital (San Diego, CA) or Texas Children's Cancer Center (Houston, TX). All procedures using human tissue were approved by the Institutional Review Boards of the respective institutions. Upon retrieval, the tissue was mechanically dissociated into a single-cell suspension, then immediately injected into the cerebella of NSG mice. When mice showed signs of MB, tumors were again dissociated into single-cell suspensions and re-transplanted back into the cerebella of naïve hosts to establish a propagated line for each patient-derived xenograft. Molecular classification of human tumors was previously described(7, 40, 41)
Flank tumor implantation and in vivo antagonist treatment
Cells isolated from tumors were re-suspended 1:1 in NB/NS-21 media and GFR-matrigel. 100μl of cell suspension (6-7 × 106 cells) was injected subcutaneously into the flanks of 5-8 week old CD-1 Nu/Nu mice. Tumor growth was monitored using calipers and tumor volume calculated using the formula 0.52*length*width2. Treatment was initiated when tumors reached ∼100 mm3. For intratumoral injections, tumors were injected twice a week with YM155 (20 μM final concentration) or vehicle (20% DMSO in saline). For systemic treatments, mice were treated with 10 mg/kg/day YM155 or saline by subcutaneous micro-osmotic pump (Alzet, model D2004). Experimental treatments were continued until control tumors reached maximum size of 2000mm3, at which point tumors were collected for analysis.
Statistics
Unless otherwise indicated, statistics were calculated by ANOVA with post hoc student's t-test. p values of less than 0.05 were considered significant and marked with asterisks where appropriate.
Supplementary Material
Supplementary Figure 1. YM155 inhibits survivin expression in MB cells. Ptch mutant tumor cells were treated with DMSO (D) or YM155 (Y) for 48 hours and analyzed for expression of survivin by real time PCR (A) and by western blotting after 24 hr. (B) YM155 dose in (B) is 1μM. YM155 decreases Survivin expression at both the RNA and protein level. Data are representative of 3 experiments.
Supplementary Figure 2. LLP3 inhibits proliferation of Ptch mutant tumor cells. (A) Ptch mutant tumor cells were treated with DMSO or multiple doses of LLP3 for 48 hr and pulsed with 3H-thymidine for 12 hr to measure proliferation. Treatment decreased proliferation in a dose-dependent manner (p<0.05 by ANOVA with post hoc student's ttest). Data represent mean +/- SD and are representative of 3 experiments.
Supplementary Figure 3. Survivin antagonists kill GNPs but not do not affect survival of post-mitotic neurons. Granule Neuron Precursors (GNPs) were isolated from P7 wild type cerebella, split into two treatment groups, and plated in either SHH-containing media (A) or differentiation media (B). Cells in SHH-containing media were treated immediately with DMSO, YM155, or S12 for 48 hr and incubated with ethidium homodimer1 (EthD1) to mark dead/dying cells (A). Cells in differentiation media were cultured for 5 days, followed by treatment with DMSO, YM155, or S12 for 48 hr and incubation with EthD1 to mark dead/dying cells (B). Both S12 and YM155 caused GNP cell death at high doses (p<0.01 for high doses. NS for 50nM YM155 and 10μg/ml S12), but did not kill PMNs (P>0.2, NS for all doses). Data represent mean +/- SD and are representative of 4 independent experiments.
Supplementary Figure 4. Survivin is highly expressed in patient derived xenografts of MB. RNA was isolated from human fetal cerebellum (hFC), adult cerebellum (hAC), and PDX tumors DMB012, DMB018, and ICb-984MB and analyzed for Survivin expression using real time PCR. Survivin is highly expressed in PDX tumors. Expression was not detected in hAC above background (RQ < 0.05). Data are plotted relative to hFC and error bars represent 95% confidence interval calculated using sum of the squares method.
Acknowledgments
The authors appreciate the support of the Sanford-Burnham Shared Resources, including Amy Cortez, Jonna Hurtado, and Yoav Altman (Flow Cytometry); Robbin Newlin (Histopathology); Danielle McAnally, Michael Vicchiarelli and Arianna Mangravita-Novo (Chemical Genomics); Lili Lacarra, Adriana Charbono and Kenny Venegas (Animal facility); and Anthony Pinkerton and members of the Medicinal Chemistry Resource for generation of S12 and LLP3. We are also grateful to Ana Miletic-Sedy and Robert Rickert for sharing Survivinfl/fl mice; and to Robert Margolis and Shirley Markant for helpful discussions. Thank you to the Flanders Institute for Biotechnology (VIB), Belgium, for allowing use of the survivin mice. EMC is supported by grants from the Canadian Institutes for Health Research (CIHR), the Natural Sciences and Engineering Research Council of Canada (NSERC). He holds a Tier 1 Canada Research Chair (CRC) in Endothelial Cell Biology. MIG is supported by grant 5R01CA08948. This work was supported by NCI award number 5P30CA030199 and R01 CA122759 and by the Cedars-Sinai/Sanford-Burnham Grant Program for Cancer Research. RWR is the recipient of a Research Leadership Award from the California Institute for Regenerative Medicine (CIRM LA1-01747.
Footnotes
Conflict of Interest: The authors declare no conflict of interest.
References
- 1.Packer RJ, Cogen P, Vezina G, Rorke LB. Medulloblastoma: clinical and biologic aspects. Neuro-oncology. 1999;1(3):232–50. doi: 10.1215/15228517-1-3-232. Epub 2001/09/12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Packer RJ, Vezina G. Management of and prognosis with medulloblastoma: therapy at a crossroads. Archives of neurology. 2008;65(11):1419–24. doi: 10.1001/archneur.65.11.1419. Epub 2008/11/13. [DOI] [PubMed] [Google Scholar]
- 3.Fossati P, Ricardi U, Orecchia R. Pediatric medulloblastoma: toxicity of current treatment and potential role of protontherapy. Cancer treatment reviews. 2009;35(1):79–96. doi: 10.1016/j.ctrv.2008.09.002. Epub 2008/11/04. [DOI] [PubMed] [Google Scholar]
- 4.Northcott PA, Dubuc AM, Pfister S, Taylor MD. Molecular subgroups of medulloblastoma. Expert review of neurotherapeutics. 2012;12(7):871–84. doi: 10.1586/ern.12.66. Epub 2012/08/03. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rudin CM, Hann CL, Laterra J, Yauch RL, Callahan CA, Fu L, et al. Treatment of medulloblastoma with hedgehog pathway inhibitor GDC-0449. The New England journal of medicine. 2009;361(12):1173–8. doi: 10.1056/NEJMoa0902903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Amakye D, Jagani Z, Dorsch M. Unraveling the therapeutic potential of the Hedgehog pathway in cancer. Nature medicine. 2013;19(11):1410–22. doi: 10.1038/nm.3389. Epub 2013/11/10. [DOI] [PubMed] [Google Scholar]
- 7.Kool M, Jones DT, Jager N, Northcott PA, Pugh TJ, Hovestadt V, et al. Genome Sequencing of SHH Medulloblastoma Predicts Genotype-Related Response to Smoothened Inhibition. Cancer cell. 2014;25(3):393–405. doi: 10.1016/j.ccr.2014.02.004. Epub 2014/03/22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Altieri DC. Survivin, cancer networks and pathway-directed drug discovery. Nature reviews Cancer. 2008;8(1):61–70. doi: 10.1038/nrc2293. Epub 2007/12/14. [DOI] [PubMed] [Google Scholar]
- 9.Wheatley SP, McNeish IA. Survivin: a protein with dual roles in mitosis and apoptosis. Int Rev Cytol. 2005;247:35–88. doi: 10.1016/S0074-7696(05)47002-3. Epub 2005/12/14. [DOI] [PubMed] [Google Scholar]
- 10.Huang LN, Wang DS, Chen YQ, Zhao CL, Gong BL, Jiang AB, et al. Expression of survivin and patients survival in non-small cell lung cancer: a meta-analysis of the published studies. Molecular biology reports. 2013;40(2):917–24. doi: 10.1007/s11033-012-2132-8. Epub 2012/10/16. [DOI] [PubMed] [Google Scholar]
- 11.Yamashita S, Masuda Y, Kurizaki T, Haga Y, Murayama T, Ikei S, et al. Survivin expression predicts early recurrence in early-stage breast cancer. Anticancer Res. 2007;27(4C):2803–8. Epub 2007/08/19. [PubMed] [Google Scholar]
- 12.Park E, Gang EJ, Hsieh YT, Schaefer P, Chae S, Klemm L, et al. Targeting survivin overcomes drug resistance in acute lymphoblastic leukemia. Blood. 2011;118(8):2191–9. doi: 10.1182/blood-2011-04-351239. Epub 2011/07/01. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Adida C, Recher C, Raffoux E, Daniel MT, Taksin AL, Rousselot P, et al. Expression and prognostic significance of survivin in de novo acute myeloid leukaemia. British journal of haematology. 2000;111(1):196–203. doi: 10.1046/j.1365-2141.2000.02328.x. Epub 2000/11/25. [DOI] [PubMed] [Google Scholar]
- 14.Fangusaro JR, Caldas H, Jiang Y, Altura RA. Survivin: an inhibitor of apoptosis in pediatric cancer. Pediatric blood & cancer. 2006;47(1):4–13. doi: 10.1002/pbc.20805. Epub 2006/03/15. [DOI] [PubMed] [Google Scholar]
- 15.Shirai K, Suzuki Y, Oka K, Noda SE, Katoh H, Itoh J, et al. Nuclear survivin expression predicts poorer prognosis in glioblastoma. Journal of neuro-oncology. 2009;91(3):353–8. doi: 10.1007/s11060-008-9720-4. Epub 2008/10/28. [DOI] [PubMed] [Google Scholar]
- 16.Adida C, Crotty PL, McGrath J, Berrebi D, Diebold J, Altieri DC. Developmentally regulated expression of the novel cancer anti-apoptosis gene survivin in human and mouse differentiation. The American journal of pathology. 1998;152(1):43–9. Epub 1998/01/09. [PMC free article] [PubMed] [Google Scholar]
- 17.Carrasco RA, Stamm NB, Marcusson E, Sandusky G, Iversen P, Patel BK. Antisense inhibition of survivin expression as a cancer therapeutic. Molecular cancer therapeutics. 2011;10(2):221–32. doi: 10.1158/1535-7163.MCT-10-0756. Epub 2011/01/11. [DOI] [PubMed] [Google Scholar]
- 18.Ling X, Cao S, Cheng Q, Keefe JT, Rustum YM, Li F. A novel small molecule FL118 that selectively inhibits survivin, Mcl-1, XIAP and cIAP2 in a p53-independent manner, shows superior antitumor activity. PloS one. 2012;7(9):e45571. doi: 10.1371/journal.pone.0045571. Epub 2012/10/03. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Nagaraj S, Pisarev V, Kinarsky L, Sherman S, Muro-Cacho C, Altieri DC, et al. Dendritic cell-based full-length survivin vaccine in treatment of experimental tumors. J Immunother. 2007;30(2):169–79. doi: 10.1097/01.cji.0000211329.83890.ba. Epub 2007/05/02. [DOI] [PubMed] [Google Scholar]
- 20.Nakahara T, Yamanaka K, Hatakeyama S, Kita A, Takeuchi M, Kinoyama I, et al. YM155, a novel survivin suppressant, enhances taxane-induced apoptosis and tumor regression in a human Calu 6 lung cancer xenograft model. Anti-cancer drugs. 2011;22(5):454–62. doi: 10.1097/CAD.0b013e328344ac68. Epub 2011/03/11. [DOI] [PubMed] [Google Scholar]
- 21.Zhu K, Qin H, Cha SC, Neelapu SS, Overwijk W, Lizee GA, et al. Survivin DNA vaccine generated specific antitumor effects in pancreatic carcinoma and lymphoma mouse models. Vaccine. 2007;25(46):7955–61. doi: 10.1016/j.vaccine.2007.08.050. Epub 2007/10/16. [DOI] [PubMed] [Google Scholar]
- 22.Tolcher AW, Quinn DI, Ferrari A, Ahmann F, Giaccone G, Drake T, et al. A phase II study of YM155, a novel small-molecule suppressor of survivin, in castration-resistant taxane-pretreated prostate cancer. Annals of oncology : official journal of the European Society for Medical Oncology / ESMO. 2012;23(4):968–73. doi: 10.1093/annonc/mdr353. Epub 2011/08/24. [DOI] [PubMed] [Google Scholar]
- 23.Becker JC, Andersen MH, Hofmeister-Muller V, Wobser M, Frey L, Sandig C, et al. Survivin-specific T-cell reactivity correlates with tumor response and patient survival: a phase-II peptide vaccination trial in metastatic melanoma. Cancer immunology, immunotherapy : CII. 2012;61(11):2091–103. doi: 10.1007/s00262-012-1266-9. Epub 2012/05/09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lewis KD, Samlowski W, Ward J, Catlett J, Cranmer L, Kirkwood J, et al. A multi-center phase II evaluation of the small molecule survivin suppressor YM155 in patients with unresectable stage III or IV melanoma. Investigational new drugs. 2011;29(1):161–6. doi: 10.1007/s10637-009-9333-6. Epub 2009/10/16. [DOI] [PubMed] [Google Scholar]
- 25.Tanioka M, Nokihara H, Yamamoto N, Yamada Y, Yamada K, Goto Y, et al. Phase I study of LY2181308, an antisense oligonucleotide against survivin, in patients with advanced solid tumors. Cancer chemotherapy and pharmacology. 2011;68(2):505–11. doi: 10.1007/s00280-010-1506-7. Epub 2010/11/17. [DOI] [PubMed] [Google Scholar]
- 26.Talbot DC, Ranson M, Davies J, Lahn M, Callies S, Andre V, et al. Tumor survivin is downregulated by the antisense oligonucleotide LY2181308: a proof-of-concept, first-in-human dose study. Clin Cancer Res. 2010;16(24):6150–8. doi: 10.1158/1078-0432.CCR-10-1932. Epub 2010/11/03. [DOI] [PubMed] [Google Scholar]
- 27.Haberler C, Slavc I, Czech T, Gelpi E, Heinzl H, Budka H, et al. Histopathological prognostic factors in medulloblastoma: high expression of survivin is related to unfavourable outcome. Eur J Cancer. 2006;42(17):2996–3003. doi: 10.1016/j.ejca.2006.05.038. Epub 2006/09/26. [DOI] [PubMed] [Google Scholar]
- 28.Fangusaro JR, Jiang Y, Holloway MP, Caldas H, Singh V, Boue DR, et al. Survivin, Survivin-2B, and Survivin-deItaEx3 expression in medulloblastoma: biologic markers of tumour morphology and clinical outcome. British journal of cancer. 2005;92(2):359–65. doi: 10.1038/sj.bjc.6602317. Epub 2005/01/19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Li XN, Shu Q, Su JM, Adesina AM, Wong KK, Perlaky L, et al. Differential expression of survivin splice isoforms in medulloblastomas. Neuropathology and applied neurobiology. 2007;33(1):67–76. doi: 10.1111/j.1365-2990.2006.00782.x. Epub 2007/01/24. [DOI] [PubMed] [Google Scholar]
- 30.Yang ZJ, Ellis T, Markant SL, Read TA, Kessler JD, Bourboulas M, et al. Medulloblastoma can be initiated by deletion of Patched in lineage-restricted progenitors or stem cells. Cancer cell. 2008;14(2):135–45. doi: 10.1016/j.ccr.2008.07.003. Epub 2008/08/12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Xing Z, Conway EM, Kang C, Winoto A. Essential role of survivin, an inhibitor of apoptosis protein, in T cell development, maturation, and homeostasis. J Exp Med. 2004;199(1):69–80. doi: 10.1084/jem.20031588. Epub 2003/12/31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Nakahara T, Takeuchi M, Kinoyama I, Minematsu T, Shirasuna K, Matsuhisa A, et al. YM155, a novel small-molecule survivin suppressant, induces regression of established human hormone-refractory prostate tumor xenografts. Cancer Res. 2007;67(17):8014–21. doi: 10.1158/0008-5472.CAN-07-1343. Epub 2007/09/07. [DOI] [PubMed] [Google Scholar]
- 33.Berezov A, Cai Z, Freudenberg JA, Zhang H, Cheng X, Thompson T, et al. Disabling the mitotic spindle and tumor growth by targeting a cavity-induced allosteric site of survivin. Oncogene. 2012;31(15):1938–48. doi: 10.1038/onc.2011.377. Epub 2011/09/06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Guvenc H, Pavlyukov MS, Joshi K, Kurt H, Banasavadi-Siddegowda YK, Mao P, et al. Impairment of glioma stem cell survival and growth by a novel inhibitor for Survivin-Ran protein complex. Clin Cancer Res. 2013;19(3):631–42. doi: 10.1158/1078-0432.CCR-12-0647. Epub 2012/12/20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Blomstrand M, Brodin NP, Munck Af Rosenschold P, Vogelius IR, Sanchez Merino G, Kiil-Berthlesen A, et al. Estimated clinical benefit of protecting neurogenesis in the developing brain during radiation therapy for pediatric medulloblastoma. Neuro-oncology. 2012;14(7):882–9. doi: 10.1093/neuonc/nos120. Epub 2012/05/23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mulhern RK, Palmer SL, Merchant TE, Wallace D, Kocak M, Brouwers P, et al. Neurocognitive consequences of risk-adapted therapy for childhood medulloblastoma. J Clin Oncol. 2005;23(24):5511–9. doi: 10.1200/JCO.2005.00.703. Epub 2005/08/20. [DOI] [PubMed] [Google Scholar]
- 37.Iwasa T, Okamoto I, Suzuki M, Nakahara T, Yamanaka K, Hatashita E, et al. Radiosensitizing effect of YM155, a novel small-molecule survivin suppressant, in non-small cell lung cancer cell lines. Clin Cancer Res. 2008;14(20):6496–504. doi: 10.1158/1078-0432.CCR-08-0468. Epub 2008/10/18. [DOI] [PubMed] [Google Scholar]
- 38.Pan S, Wu X, Jiang J, Gao W, Wan Y, CHeng D, Han D, Lie J, Englund NP, Wang Y, Peukert S, Miller-Moslin K, Yuan J, Guo R, Matsumoto M, Vattay A, Jiang Y, Tsao J, Sun F, Pferdekamper AC, Dodd S, Tuntland T, Maniara W, Kelleher JF, Y-m Y, Warmuth M, Williams J, Dortsch M. Discovery of NVP-LDE225, a potent and selective smoothened antagonist. ACS Med Chem Lett. 2010;1:130–4. doi: 10.1021/ml1000307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Rodon J, Tawbi HA, Thomas AL, Stoller RG, Turtschi CP, Baselga J, et al. A Phase I, Multicenter, Open-Label, First-in-Human, Dose-Escalation Study of the Oral Smoothened Inhibitor Sonidegib (LDE225) in Patients with Advanced Solid Tumors. Clin Cancer Res. 2014;20(7):1900–9. doi: 10.1158/1078-0432.CCR-13-1710. Epub 2014/02/14. [DOI] [PubMed] [Google Scholar]
- 40.Markant SL, Esparza LA, Sun J, Barton KL, McCoig LM, Grant GA, et al. Targeting sonic hedgehog-associated medulloblastoma through inhibition of Aurora and Polo-like kinases. Cancer Res. 2013;73(20):6310–22. doi: 10.1158/0008-5472.CAN-12-4258. Epub 2013/09/27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zhao X, Liu Z, Yu L, Zhang Y, Baxter P, Voicu H, et al. Global gene expression profiling confirms the molecular fidelity of primary tumor-based orthotopic xenograft mouse models of medulloblastoma. Neuro-oncology. 2012;14(5):574–83. doi: 10.1093/neuonc/nos061. Epub 2012/03/31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Nakahara T, Kita A, Yamanaka K, Mori M, Amino N, Takeuchi M, et al. Broad spectrum and potent antitumor activities of YM155, a novel small-molecule survivin suppressant, in a wide variety of human cancer cell lines and xenograft models. Cancer science. 2011;102(3):614–21. doi: 10.1111/j.1349-7006.2010.01834.x. Epub 2011/01/06. [DOI] [PubMed] [Google Scholar]
- 43.Ruchaud S, Carmena M, Earnshaw WC. Chromosomal passengers: conducting cell division. Nat Rev Mol Cell Biol. 2007;8(10):798–812. doi: 10.1038/nrm2257. Epub 2007/09/13. [DOI] [PubMed] [Google Scholar]
- 44.Yue Z, Carvalho A, Xu Z, Yuan X, Cardinale S, Ribeiro S, et al. Deconstructing Survivin: comprehensive genetic analysis of Survivin function by conditional knockout in a vertebrate cell line. J Cell Biol. 2008;183(2):279–96. doi: 10.1083/jcb.200806118. Epub 2008/10/22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Saito T, Hama S, Izumi H, Yamasaki F, Kajiwara Y, Matsuura S, et al. Centrosome amplification induced by survivin suppression enhances both chromosome instability and radiosensitivity in glioma cells. British journal of cancer. 2008;98(2):345–55. doi: 10.1038/sj.bjc.6604160. Epub 2008/01/16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Rosa J, Canovas P, Islam A, Altieri DC, Doxsey SJ. Survivin modulates microtubule dynamics and nucleation throughout the cell cycle. Mol Biol Cell. 2006;17(3):1483–93. doi: 10.1091/mbc.E05-08-0723. Epub 2006/01/13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Arora R, Shuda M, Guastafierro A, Feng H, Toptan T, Tolstov Y, et al. Survivin is a therapeutic target in Merkel cell carcinoma. Sci Transl Med. 2012;4(133):133ra56. doi: 10.1126/scitranslmed.3003713. Epub 2012/05/11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Yamauchi T, Nakamura N, Hiramoto M, Yuri M, Yokota H, Naitou M, et al. Sepantronium bromide (YM155) induces disruption of the ILF3/p54(nrb) complex, which is required for survivin expression. Biochem Biophys Res Commun. 2012;425(4):711–6. doi: 10.1016/j.bbrc.2012.07.103. Epub 2012/07/31. [DOI] [PubMed] [Google Scholar]
- 49.Nakamura N, Yamauchi T, Hiramoto M, Yuri M, Naito M, Takeuchi M, et al. Interleukin enhancer-binding factor 3/NF110 is a target of YM155, a suppressant of survivin. Molecular & cellular proteomics : MCP. 2012;11(7):M111 013243. doi: 10.1074/mcp.M111.013243. Epub 2012/03/24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Cheng Q, Ling X, Haller A, Nakahara T, Yamanaka K, Kita A, et al. Suppression of survivin promoter activity by YM155 involves disruption of Sp1-DNA interaction in the survivin core promoter. International journal of biochemistry and molecular biology. 2012;3(2):179–97. Epub 2012/07/10. [PMC free article] [PubMed] [Google Scholar]
- 51.Tao YF, Lu J, Du XJ, Sun LC, Zhao X, Peng L, et al. Survivin selective inhibitor YM155 induce apoptosis in SK-NEP-1 Wilms tumor cells. BMC cancer. 2012;12:619. doi: 10.1186/1471-2407-12-619. Epub 2012/12/27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Grinstein E, Jundt F, Weinert I, Wernet P, Royer HD. Sp1 as G1 cell cycle phase specific transcription factor in epithelial cells. Oncogene. 2002;21(10):1485–92. doi: 10.1038/sj.onc.1205211. Epub 2002/03/16. [DOI] [PubMed] [Google Scholar]
- 53.Chakravarti A, Zhai GG, Zhang M, Malhotra R, Latham DE, Delaney MA, et al. Survivin enhances radiation resistance in primary human glioblastoma cells via caspase-independent mechanisms. Oncogene. 2004;23(45):7494–506. doi: 10.1038/sj.onc.1208049. Epub 2004/08/25. [DOI] [PubMed] [Google Scholar]
- 54.Morrison DJ, Hogan LE, Condos G, Bhatla T, Germino N, Moskowitz NP, et al. Endogenous knockdown of survivin improves chemotherapeutic response in ALL models. Leukemia : official journal of the Leukemia Society of America, Leukemia Research Fund, UK. 2012;26(2):271–9. doi: 10.1038/leu.2011.199. Epub 2011/08/17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Yamanaka K, Nakahara T, Yamauchi T, Kita A, Takeuchi M, Kiyonaga F, et al. Antitumor activity of YM155, a selective small-molecule survivin suppressant, alone and in combination with docetaxel in human malignant melanoma models. Clin Cancer Res. 2011;17(16):5423–31. doi: 10.1158/1078-0432.CCR-10-3410. Epub 2011/07/09. [DOI] [PubMed] [Google Scholar]
- 56.Olie RA, Simoes-Wust AP, Baumann B, Leech SH, Fabbro D, Stahel RA, et al. A novel antisense oligonucleotide targeting survivin expression induces apoptosis and sensitizes lung cancer cells to chemotherapy. Cancer Res. 2000;60(11):2805–9. Epub 2000/06/13. [PubMed] [Google Scholar]
- 57.Grossman D, Altieri DC. Drug resistance in melanoma: mechanisms, apoptosis, and new potential therapeutic targets. Cancer metastasis reviews. 2001;20(1-2):3–11. doi: 10.1023/a:1013123532723. Epub 2002/02/08. [DOI] [PubMed] [Google Scholar]
- 58.Rodel F, Frey B, Leitmann W, Capalbo G, Weiss C, Rodel C. Survivin antisense oligonucleotides effectively radiosensitize colorectal cancer cells in both tissue culture and murine xenograft models. International journal of radiation oncology, biology, physics. 2008;71(1):247–55. doi: 10.1016/j.ijrobp.2008.02.011. Epub 2008/04/15. [DOI] [PubMed] [Google Scholar]
- 59.Okamoto K, Okamoto I, Hatashita E, Kuwata K, Yamaguchi H, Kita A, et al. Overcoming erlotinib resistance in EGFR mutation-positive non-small cell lung cancer cells by targeting survivin. Molecular cancer therapeutics. 2012;11(1):204–13. doi: 10.1158/1535-7163.MCT-11-0638. Epub 2011/11/15. [DOI] [PubMed] [Google Scholar]
- 60.Minematsu T, Sonoda T, Hashimoto T, Iwai M, Oppeneer T, Felder L, et al. Pharmacokinetics, distribution and excretion of YM155 monobromide, a novel small-molecule survivin suppressant, in male and pregnant or lactating female rats. Biopharmaceutics & drug disposition. 2012;33(3):160–9. doi: 10.1002/bdd.1781. Epub 2012/03/01. [DOI] [PubMed] [Google Scholar]
- 61.Hall WA, Sherr GT. Convection-enhanced delivery of targeted toxins for malignant glioma. Expert opinion on drug delivery. 2006;3(3):371–7. doi: 10.1517/17425247.3.3.371. Epub 2006/04/28. [DOI] [PubMed] [Google Scholar]
- 62.Kunwar S, Prados MD, Chang SM, Berger MS, Lang FF, Piepmeier JM, et al. Direct intracerebral delivery of cintredekin besudotox (IL13-PE38QQR) in recurrent malignant glioma: a report by the Cintredekin Besudotox Intraparenchymal Study Group. J Clin Oncol. 2007;25(7):837–44. doi: 10.1200/JCO.2006.08.1117. Epub 2007/03/01. [DOI] [PubMed] [Google Scholar]
- 63.Brown MT, Coleman RE, Friedman AH, Friedman HS, McLendon RE, Reiman R, et al. Intrathecal 131I-labeled antitenascin monoclonal antibody 81C6 treatment of patients with leptomeningeal neoplasms or primary brain tumor resection cavities with subarachnoid communication: phase I trial results. Clin Cancer Res. 1996;2(6):963–72. Epub 1996/06/01. [PubMed] [Google Scholar]
- 64.Serwer LP, James CD. Challenges in drug delivery to tumors of the central nervous system: an overview of pharmacological and surgical considerations. Advanced drug delivery reviews. 2012;64(7):590–7. doi: 10.1016/j.addr.2012.01.004. Epub 2012/02/07. [DOI] [PubMed] [Google Scholar]
- 65.Ellis T, Smyth I, Riley E, Graham S, Elliot K, Narang M, et al. Patched 1 conditional null allele in mice. Genesis. 2003;36(3):158–61. doi: 10.1002/gene.10208. Epub 2003/07/23. [DOI] [PubMed] [Google Scholar]
- 66.Machold R, Fishell G. Math1 is expressed in temporally discrete pools of cerebellar rhombic-lip neural progenitors. Neuron. 2005;48(1):17–24. doi: 10.1016/j.neuron.2005.08.028. Epub 2005/10/06. [DOI] [PubMed] [Google Scholar]
- 67.Goodrich LV, Milenkovic L, Higgins KM, Scott MP. Altered neural cell fates and medulloblastoma in mouse patched mutants. Science. 1997;277(5329):1109–13. doi: 10.1126/science.277.5329.1109. Epub 1997/08/22. [DOI] [PubMed] [Google Scholar]
- 68.Oliver TG, Read TA, Kessler JD, Mehmeti A, Wells JF, Huynh TT, et al. Loss of patched and disruption of granule cell development in a pre-neoplastic stage of medulloblastoma. Development. 2005;132(10):2425–39. doi: 10.1242/dev.01793. Epub 2005/04/22. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure 1. YM155 inhibits survivin expression in MB cells. Ptch mutant tumor cells were treated with DMSO (D) or YM155 (Y) for 48 hours and analyzed for expression of survivin by real time PCR (A) and by western blotting after 24 hr. (B) YM155 dose in (B) is 1μM. YM155 decreases Survivin expression at both the RNA and protein level. Data are representative of 3 experiments.
Supplementary Figure 2. LLP3 inhibits proliferation of Ptch mutant tumor cells. (A) Ptch mutant tumor cells were treated with DMSO or multiple doses of LLP3 for 48 hr and pulsed with 3H-thymidine for 12 hr to measure proliferation. Treatment decreased proliferation in a dose-dependent manner (p<0.05 by ANOVA with post hoc student's ttest). Data represent mean +/- SD and are representative of 3 experiments.
Supplementary Figure 3. Survivin antagonists kill GNPs but not do not affect survival of post-mitotic neurons. Granule Neuron Precursors (GNPs) were isolated from P7 wild type cerebella, split into two treatment groups, and plated in either SHH-containing media (A) or differentiation media (B). Cells in SHH-containing media were treated immediately with DMSO, YM155, or S12 for 48 hr and incubated with ethidium homodimer1 (EthD1) to mark dead/dying cells (A). Cells in differentiation media were cultured for 5 days, followed by treatment with DMSO, YM155, or S12 for 48 hr and incubation with EthD1 to mark dead/dying cells (B). Both S12 and YM155 caused GNP cell death at high doses (p<0.01 for high doses. NS for 50nM YM155 and 10μg/ml S12), but did not kill PMNs (P>0.2, NS for all doses). Data represent mean +/- SD and are representative of 4 independent experiments.
Supplementary Figure 4. Survivin is highly expressed in patient derived xenografts of MB. RNA was isolated from human fetal cerebellum (hFC), adult cerebellum (hAC), and PDX tumors DMB012, DMB018, and ICb-984MB and analyzed for Survivin expression using real time PCR. Survivin is highly expressed in PDX tumors. Expression was not detected in hAC above background (RQ < 0.05). Data are plotted relative to hFC and error bars represent 95% confidence interval calculated using sum of the squares method.