

Abstract
Neural stem cells (NSCs) and imprinted genes play an important role in brain development. On historical grounds, these two determinants have been largely studied independently of each other. Recent evidence suggests, however, that NSCs can reset select genomic imprints to prevent precocious depletion of the stem cell reservoir. Moreover, imprinted genes like the transcriptional regulator Zac1 can fine tune neuronal vs astroglial differentiation of NSCs. Zac1 binds in a sequence-specific manner to pro-neuronal and imprinted genes to confer transcriptional regulation and furthermore coregulates members of the p53-family in NSCs. At the genome scale, Zac1 is a central hub of an imprinted gene network comprising genes with an important role for NSC quiescence, proliferation and differentiation. Overall, transcriptional, epigenomic, and genomic mechanisms seem to coordinate the functional relationships of NSCs and imprinted genes from development to maturation, and possibly aging.
Keywords: Zac1, Cell fate decisions, Neural stem cells, Genomic imprinting, Igf2-H19, Dlk1, p57Kip2, Necdin, Differentiation, Imprinted gene networks
Core tip: Both neural stem cells (NSCs) and imprinted genes participate in the same developmental processes. Here, we will explore the possibility that these two processes actually interact with each other. We will exemplarily consider the role of single imprinted genes in NSC biology based on their functional relationship to the imprinted gene Zac1, which is itself at the focus of this review due to its role in directing neuronal vs astroglial differentiation of NSCs and as a central hub of an imprinted gene network comprising genes important to NSC biology.
INTRODUCTION
Neural stem cells (NSC), also known as neural precursor cells (NPC), are the common source of all neuronal and glial cells, including astrocytes and oligodendrocytes, in the developing and adult CNS. They arise from the neuroepithelial layers which line the spinal canal and forebrain ventricles at early embryonic stages and reside in circumscribed regions in the postnatal brain to produce in a spatio-temporal controlled manner a variety of cell-types. Following a series of symmetric proliferative divisions, NSCs progress to asymmetric neurogenic divisions. Hereby, the parent cell maintains the progenitor state while the daughter cell migrates to its final destination, exits from the cell cycle, differentiates and participates in the formation of complex neural networks[1]. Although the last decade has witnessed major progress on the pathways and genes coordinating NSC behavior, a potential role of imprinted genes has been largely ignored. In this review, we will consider current evidences for the general impact of imprinted genes in NSC cell fate decisions and differentiation with a particular focus on their relationship with the imprinted gene Zac1, as well as the role of Zac1 itself, which encodes a versatile transcriptional regulator.
GENOMIC IMPRINTING
Although equivalent complements of paternally and maternally expressed autosomes are transmitted from parent to offspring, select autosomal regions can be lastingly silenced as a result of their parental origin. This process takes place in the respective parental germ cells and is known as genomic imprinting. Germline-derived imprints are preserved during fertilization and somatic development with few remarkable exceptions (see below). Some 120 genes with a verified imprinting status have been identified in mouse, which are largely conserved in human and correspond to less than 1% of the genome[2]. A considerably larger number of genes with a strong bias in allele-specific expression has been recently detected in mice brains although an authentic imprinting status has still to be proven[3,4]. In this respect we note that the reported number of genes showing allele-specific expression differences in mice actually matches general estimates on tissue-specific and allele-specific differences in human gene expression (approximately 10%) based on common genetic variations (i.e., single nucleotide and copy number polymorphisms)[5]. This observation speaks against the hypothesis that most of these allelic differences originate from genomic imprinting.
Multiple molecular mechanisms govern imprinted gene expression, they include CpG methylation of specific DNA sequences, ncRNAs, alterations in chromatin structure, and posttranslational histone modifications (e.g., lysine acetylation, lysine and arginine methylation, serine phosphorylation, and covalent binding of the small peptide ubiquitin)[6,7]. Above all, DNA methylation is at the center of the imprinting process and is thought to catalyze the establishment and life-long maintenance of genomic imprints. Differentially methylated regions (DMR), which harbor CpG-rich regulatory sequences, play a critical role in determining parental allele-specific expression. With few exceptions (i.e., Zac1) imprinted genes cluster in huge, conserved chromosomal domains throughout the genome, and their well-balanced expression enables regular development from fetus to early postnatal life[8,9].
GENOMIC IMPRINTING AND THE BRAIN
Dysregulation of imprinted gene expression can elicit complex neurodevelopmental syndromes in humans, frequently associated with mental retardation [i.e., Angelman syndrome (OMIM 105830) and Prader-Willi syndrome (OMIM 176270)[10]]. Moreover, psychotic and autistic spectrum disorders possibly result from more subtle deregulation of imprinted genes. Indeed, mice harboring altered dosage of single or multiple imprinted genes showed various defects in higher brain functions ranging from learning[11] and memory formation[12] to social and nurturing behaviors[13-15].
A ROLE OF IMPRINTED GENES IN NSCS?
On a historical ground, both NSCs and imprinted genes have been studied in the context of brain development. While our insight into the role of NSCs in brain development has largely expanded over the last decade[1], the molecular targets and cellular pathways by which imprinted genes participate in brain development remain poorly defined. Importantly, the roles of NSCs and imprinted genes have been commonly investigated independently of each other although they apparently participate in the same developmental processes. Thus, new studies should explore possible reciprocal interactions between imprinted genes and NSCs during neurodevelopment. Here, we highlight recent evidences in the scientific literature on the critical role of imprinted genes in NSCs and major cellular processes they control.
INSULIN-LIKE GROWTH FACTOR 2
Insulin-like growth factor (Igf2) was the first mammalian gene shown to be maternally imprinted as a result of a differentially methylated imprinting control region (ICR) located nearby and upstream of H19, which is imprinted in the opposite direction (see below)[16].
The product of Igf2 is a potent growth factor promoting cell survival, proliferation, and differentiation by binding with high affinity to the insulin-like receptors Igf1r or Igf2r, but less efficiently to the insulin receptor (Insr). Hereby, Igf2r has no signaling function and is encoded by the paternally imprinted Igf2r gene. This antagonistic functional relationship together with the opposite imprinting of Igf2 and Igf2r originally stimulated the genetic conflict hypothesis for genomic imprinting[17].
During CNS development, Igf2 is for the most part synthesized by the choroid plexus, and released into the cerebrospinal fluid (CSF), which contacts the primary cilia and apical surfaces of cortical progenitors. There, CSF-borne Igf2 binding to the Igf1r stimulates neural precursors proliferation[18].
Neurogenesis is maintained at a low level in the adult brain in so-called neurogenic niches, which comprise the subventricular zone (SVZ) of the lateral ventricles and the subgranular zone (SGZ) of the dentate gyrus (DG) of the hippocampus. Adult neurogenesis resembles in many aspects embryonic neurogenesis and raises the possibility of an additional role of Igf2 in stemness maintenance in the mature brain. In support of this hypothesis, transcriptome analysis of the SGZ evidenced substantially higher expression of Igf2 in stem cells than in immature neurons[19]. Igf2 expression localized to radial-glial like NSCs (Nestin+, Sox2+, and Gfap+) and to a significant fraction of dividing cells (Ki67+). Interestingly, Igf2 enhances in vivo and in vitro the proliferation of NSCs isolated from the DG, but not from the SGZ, indicative of a site-specific effect. Finally, the secretion of Igf2 by Nestin+ progenitors in the external granule cell layer (EGL) potently stimulates neuronal cell proliferation whereas overexpression of Igf2 in granule neurons facilitates tumor formation in rodents[20].
In sum, these reports show that Igf2 enhances in a tissue- and age-dependent manner NSC proliferation and maintenance.
H19 ncRNA
The maternally imprinted H19 locus localizes in tandem with the oppositely imprinted Igf2 gene[16] and encodes high levels of a 2.5 kb long RNA polymerase II-derived transcript. This large intergenic non-coding RNA (lincRNA) is not involved in the imprinting process[21,22] but inhibits in vitro and in vivo tumor growth possibly due to its participation in and regulation of an imprinted gene network (IGN, see below).
Additionally, the first exon of H19 encodes a micro RNA-containing hairpin that serves as a template for the miRNA 675, which reduces Igf1r expression and Igf2-signalling in the placenta[23]. These self-restraining activities of the tandem Igf2-H19 locus are necessary for normal embryogenesis and protect against parthenogenetic development in mammals[24].
Erasure of imprinting at the Igf2-H19 DMR is found in primordial germ cells (PGS) and associates with overexpression of H19 RNAs at the expense of Igf2. This epigenetic switch-off is thought to safeguard PGS quiescence and prevent from teratoma formation[25].
A similar strategy seems to be used by very small embryonic-like stem cells (VSELs), a population of very rare early-development cells with broad differentiation potential[26]. VSELs can give rise to neurons, oligodendrocytes, and microglia among other cell types and possibly fulfill a role in physiological tissue rejuvenation and regeneration following cell damage.
In VSELs, the paternally silenced allele of select imprinted genes (i.e., Igf2-H19 and Rasgrf1) is reactivated by demethylation and results in biallelic expression. Conversely, select maternally expressed alleles (i.e., Peg1, Igfr2, and p57Kip2) undergo deactivation and silencing by DNA methylation. Overall, this cell type-specific resetting of a limited number of genomic imprints supports growth-inhibition, cellular quiescence, and preservation of the stem cell population. On the other hand, methylation at the Igf2-H19 DMR slowly increases with aging and has been suggested to facilitate increased insulin signaling and age-related depletion of the VSELs reservoir[27].
Taken together, integrate imprinting at the Igf2-H19 tandem locus critically controls growth and cell proliferation in the early embryo as well as in VSELs.
DELTA-LIKE HOMOLOG 1
The imprinted Dlk1-Dio3 domain harbors the delta-like homolog 1 (Dlk1) and type III iodothyronine deiodinase (Dio3) genes which are expressed from the paternal derived chromosome. Similarly to the Igf2-H19 locus, Dlk1 and the close by Gtl2 (gene trap locus 2, alias maternally expressed gene 3, Meg3) are imprinted in an opposite manner and locate 80 kb apart from each other. Three DMRs containing specific epigenetic signatures are hypermethylated on the paternal allele in somatic tissues[28]. Hereby, those DNA methylation marks which are deposited in the paternal germline are confined to the central DMR. This region contains tandem repeats and localizes in the intergenic region of the tail-to-head orientated Dlk1 and Gtl2 genes (Figure 1).
Figure 1.
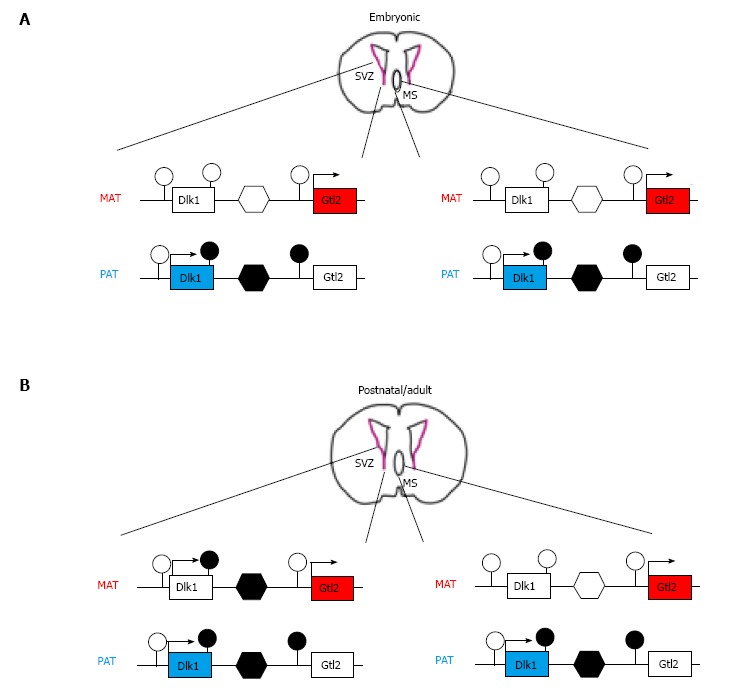
Genomic imprinting at the delta-like homolog 1 locus is reset in the subventricular zone. A: Delta-like homolog 1 (Dlk1) is monoallelically expressed from the paternal allele during development. Silencing of the maternal allele takes place in non-neuronal and various neuronal tissues such as the subventricular zone (SVZ) of the lateral ventricle (SVZ) and the medial septum (MS). At the molecular level, maternal silencing results from the absence of DNA methylation at the intergenic, germ line-controlled differentially methylated regions (DMR) (unfilled hexagon), which resides between the Dlk1 and Gtl2 genes and their associated DMRs (unfilled lollipops). Conversely, methylation at the intergenic DMR (filled hexagon), the 3’ end Dlk1 DMR and the Gtl2 DMR (filled lollipops) associates with expression from the paternal allele. The same methylation patterns are present in the SVZ (left scheme) and MS (right scheme) at embryonic ages; B: Dlk1 shows biallelic expression from postnatal day 7 onward towards adulthood in the SVZ, but not in the MS. Hereby, the maternal methylation pattern closely resembles the one on the paternal allele (i.e., methylation at the 3’end Dlk1 DMR, the intergenic DMR, but not the Gtl2 DMR). At opposite, the methylation pattern in the MS is preserved and determines monoallelic Dlk1 expression.
Dlk1 encodes a transmembrane protein and closely resembles the Notch/Delta/Serrate type family of signaling molecules. Due to minor albeit important structural differences Dlk1 is thought to compete with canonical Delta-like (DLL) ligands at the Notch receptor and to inhibit downstream signaling[29].
Dlk1 is broadly expressed in developing mouse tissues and continues to be expressed in some adult neuronal tissues (i.e., ventral striatum, septum, and ventral tegmental area) including the SVZ[29,30]. Here, Dlk1 is detected in NSCs (Nestin+, Sox+, Gfap+) and astrocytes (Sox2-, Gfap+, S100b-) localized to the germinal niche but not in differentiated parenchymal astrocytes (Gfap+, S100b+) and neuroblasts (βIII-tubulin+). NSCs mainly express the membrane-bound form of Dlk1 that is poorly active on its own but necessary for the response to secreted Dlk1 produced by nearby astrocytes.
Postnatal deletion of Dlk1 enhances NSC proliferation in the SVZ, causing the depletion of the quiescent NSC pool and reduced neurogenesis at later ages. Interestingly, this process takes place irrespectively of the parental origin of the deleted allele[30], suggesting that Dlk1 can be expressed from either parental allele. Indeed, both alleles of Dlk1 are expressed from postnatal day 7 onward in NSCs and niche astrocytes as a result of an increase in methylation at the germ line targeted ICR of the maternal allele (Figure 1). This postnatal epigenetic switch at the Dlk1 locus is confined to the neurogenic areas, whereas all other tissues continue to express Dlk1 exclusively from the paternal allele from embryogenesis to adulthood.
These findings suggest that genomic imprints at select loci (i.e., Igf2-H19 and Dlk1) are dynamically regulated in NSCs during specific developmental time windows, possibly to match the need for cell stemness vs cell differentiation.
NECDIN
Human and mouse necdin genes are maternally imprinted and localize to chromosome 15 q11.2 and a syntenic segment on chromosome 7, respectively[10], within a cluster of paternally expressed genes. Paternal deletion of this chromosomal segment in human underlies the neurodevelopmental Prader-Willi syndrome, which manifests with feeding anomalies, gross obesity, and hypogonadism[10].
Necdin (Ndn) is broadly expressed in postmitotic neurons from early embryonic to adult ages with particular high expression in the developing hypothalamus, medulla oblongata, pons, and midbrain. Knock out mice show variable neonatal lethality, reductions in oxytocin and luteinizing hormone-releasing hormone producing hypothalamic neurons, impairments in serotonergic and catecholaminergic projections, and decreased tangential migration of neocortical interneurons from the basal forebrain[31].
Ndn was originally discovered in a screen for genes induced in neurally differentiated embryonic carcinoma cells[32]. Functionally, Ndn potently inhibits proliferation in favor of differentiation by virtue of its interaction with various proteins critically involved in cell cycle progression and survival[33,34]. Similar to the retinoblastoma tumor suppressor gene, Ndn binds to the carboxyl-terminal transactivation domain of E2F1 to repress its activity and consequently cell cycle progression[33]. On the other hand, Ndn interacts with the amino-terminal transactivation domain of p53 to abolish its proapoptotic function without interfering with p53-dependent cell cycle arrest[34]. Ndn also recruits the deacetylase sirtuin 1 to promote deacetylation of p53 leading to its inactivation and protection against DNA damage induced neuronal apoptosis[35].
Furthermore, Ndn interacts and promotes the degradation of the hypoxia inducible factor-1 alpha (HIF) under normoxia[36], whereas hypoxia enhances degradation of Ndn in primary NSCs through the HIF-associated ubiquitin-proteasome system[37].
Interestingly, a growing number of reports (e.g.,[38-40]) suggest that NSC proliferation is increased under hypoxia. Accordingly, Ndn-deficient NSCs show increased proliferation and apoptosis under normoxia, but not under hypoxia, which triggers degradation of endogenous Ndn in wild-type NSCs. Moreover, Ndn null mice show higher rates of NSC proliferation and apoptosis [e.g., in the embryonic (E14.5) ganglion eminence], strengthening Ndn’s dual role in the suppression of proliferation and apoptosis[33-35].
Ndn controls additionally the proliferation of NSCs in the ventricular zone of the embryonic cortex as evidenced by an upregulation of the stem cell marker Sox2, a downregulation of the cyclin-dependent kinase inhibitor p16Ink4, and significantly increased proliferation rates in Ndn null mice[41]. Interestingly, Ndn binds in vitro and in vivo to the polycomb protein Bmi1 and counteracts Bmi1-dependent inhibition of the p16Ink4 promoter and consequently promotes NPC proliferation. Conversely, overexpression of Bmi1 prevents Ndn-mediated inhibition of E2F1-driven Cdk1 promoter activity[41].
Together, these findings suggest that Ndn controls NSC proliferation and apoptosis in an oxygen-dependent manner through interaction with various proteins driving cell proliferation (E2F1, Bmi1) and apoptosis (p53, Sirt1).
CYCLIN-DEPENDENT KINASE INHIBITOR P57KIP2
The catalytic activity of cyclin-dependent kinases (CDK) is regulated by the binding of cyclins which oscillate periodically during the cell cycle and drive the orderly progression through consecutive phases. Conversely, inhibition of these complexes by CKIs induces transient or permanent cell cycle arrest, differentiation, quiescence, senescence, or apoptosis.
The formation of early progenitors from neuroepithelial cells and the transition from proliferative symmetric to neurogenic asymmetric division is accompanied by a lengthening of the cell cycle, preferentially in G1-phase, implicating an involvement of CKIs[42].
Paternally imprinted p57Kip2 gene encodes a cyclin-dependent kinase inhibitor (CKI)[43] expressed in the VZ and SVZ, midbrain, thalamus, hypothalamus, cortical plate, septum, basal ganglia, cortex, and mantle zone of the hippocampus during development[44,45]. By means of controlling cortical progenitor cell cycle exit, p57Kip influences the migration and differentiation of neuronal precursors. Absence of p57Kip2(+/m-) during late embryogenesis and postnatal life gives rise to cortical hyperplasia. P57Kip2(+/m-) deficiency leads to an increased proliferation of radial glia cells (RGC) and intermediate precursors (IPC) and promotes re-entry into the cell cycle during corticogenesis[46]. Cell cycle analysis of RGCs and IPCs evidenced an abnormal short cell cycle length favoring precursor proliferation and aberrant migration into cortical layers.
In mice adult hippocampus, p57Kip2 deficiency causes the severe depletion of the NSC reservoir by enhancing neuronal differentiation of NSCs at early stages of life[47]. Moreover, consistent with a role of p57Kip2 in restraining NSC proliferation, neurogenic stimuli such as extensive running elicit a stronger activation of NSCs in mid-aged p57Kip2(+/m-) animals indicating that p57Kip2 might also play a critical role in the life long plasticity of brain functions[47].
In addition to their canonical role in cell cycle control, CKIs have further functions including transcriptional regulation[48]. As an example, nuclear p57Kip2 expression rises transiently during early telencephalic progenitor proliferation [embryonic day (E) 12.5] without inducing cell cycle exit. Instead, p57Kip2 interacts with the pro-neuronal basic helix-loop-helix (bHLH) factor Mash1 and blocks its transcriptional activity. As a result, p57Kip2 delays neuronal differentiation of telencephalic progenitors by antagonizing Mash1.
Taken together, p57Kip2 through inhibition of cell cycle progression and unrelated transcriptional mechanisms regulates many key processes in NSCs, including proliferation, cell cycle exit, differentiation, cell fate decisions, and stem cell quiescence in a cell type- and age-specific manner.
ZINC FINGER PROTEIN REGULATING APOPTOSIS AND CELL CYCLE ARREST
The Zac1 gene is maternally imprinted and maps on chromosome 10 in mice and chromosome 6q24 in human[49,50]. The presence of a canonical C2H2 zinc finger domain and the potent induction of apoptosis and cell-cycle arrest in transformed tumor cells inspired originally the naming “Zac1”[51].
An increased dosage of ZAC1 due to chromosomal anomalies or imprinting defects at the DMR is the most frequent genetic defect underlying transient neonatal diabetes mellitus (TNDM)[52]. This disease manifests with intrauterine growth retardation (IUGR), dehydration, hypoinsulinemia, and early-onset hyperglycemia in term, newborn infants. Transgenic mice, which harbor an extra copy of the human ZAC1 locus, display key symptoms of the human condition and show at the cellular level an impaired proliferation and maturation of β-cell progenitors[53]. In naïve pancreata, Zac1 is preferentially expressed in insulin-positive progenitor cells and increases strongly perinatally with the onset of terminal differentiation. At the same time, Zac1 confers repression following binding to specific DNA elements (see below) at the proximal promoter of the paternally imprinted Rasgrf1 gene, an important modulator of various growth factor pathways. As a result, stimulus-induced activation of mitogen-activated protein kinase and phosphoinositide 3-kinase pathways and, ultimately, insulin secretion is impaired under conditions of increased Zac1 dosage[54].
Zac1 confers transcriptional regulation either by DNA-binding[50,55-57] or as coregulator of the nuclear receptor family, in particular of those members belonging to the subgroup of steroid receptors[58], and furthermore, by coactivation of members of the p53 family[59,60]. Transcription factors typically comprise separable, modular DNA-binding and transcriptional domains; the latter confer gene activation and repression in a context-dependent manner[61]. Zac1 matches well to these criteria, whereby its diverse transcriptional functions are tightly controlled by the interaction of single zinc fingers at the level of protein-protein and protein-DNA interactions[55,62].
The N-terminal zinc finger domain, containing seven canonical C2H2 zinc fingers, is highly conserved in mouse and human Zac1 proteins and across the Zac1 gene family[63]. Mouse and human proteins diverge, however, by a central region rich in proline residues and a C-terminal cluster of glutamic acid residues both of which exist exclusively in mice. The proline-rich region together with the adjacent upstream region, termed linker region, confers potent transactivation[64]. This function is further enhanced by the simultaneous recruitment of the general coactivators p300/CBP through the C-terminus. In contrast to the separable function of multiple domains in mice, transactivation and p300 recruitment map indistinguishably to the C-terminus of human ZAC1[62].
Zac1 can bind as a monomer to GC-rich palindromic DNA-elements or as a dimer to direct and reverse repeat elements to confer transactivation. Conversely, Zac1 binding to a half-site of a repeat element causes repression[55]. Mechanistically, the zinc fingers assist in the recruitment of p300 and regulate p300’s catalytic activities in a manner dependent on the nature of the bound DNA element[62]. Hereby, single zinc fingers can participate selectively in DNA binding and/or regulation of coactivator activities.
Moreover, Zac1 can also act as a coregulator for unrelated transcription factors comprising nuclear steroid or thyroid hormone receptors or various members of the p53 family (including p53 itself, p63, and p73). All of these transcription factors bind to specific DNA elements at their target genes to critically control cell proliferation and differentiation in a cell type-specific manner[58,59]. For example, Zac1 is recruited jointly with the coactivators p300 and PCAF by the tumor suppressor p73 at the p21Cip1 promoter during early neuronal differentiation (see below). In addition to serving as a scaffold for coactivator assembly, Zac1 furthermore regulates PCAF’s catalytic functions similar to the ones of p300[65]. Overall, this close relationship between Zac1 DNA-binding and enzymatic regulation of transcription might contribute to the precise and efficient regulation of target genes.
In brief, Zac1’s transcriptional activities are coordinated by sequence-specific DNA binding or the coregulation of unrelated transcription factors.
Zac1 expression during neurodevelopment
Zac1 is highly expressed in progenitor/stem cells of neuroectodermal and mesodermal tissues during early embryogenesis[66]. Zones of active cellular proliferation, comprising the telencephalic vesicles and the infundibular recess of the third ventricle (the origin of the neurohypophysis) show robust Zac1 expression at E9.5 and E12.5. Moreover, Zac1 is also detected in mitotically active regions of the developing nervous system including the neural tube at E9.5 and the neural retina at E10.5. At later stage of neurodevelopment, high levels of Zac1 expression appeared in the mitotically active cell layers lining the VZ of the 3rd and 4th ventricle, the EGL of the cerebellum, and different neuroepithelia (e.g., infundibulum, ventral hypothalamic sulcus)[67,68].
As noted before, Zac1 expression induced apoptosis and cell cycle arrest in transformed tumor cells[50,51,56] raising the question of its function in neural progenitor cells. In this respect, the analysis of Zac1 deficient mice (Zac1+/m-) provided interesting insight into the role of Zac1 in the brain[69]. Specifically, these animals revealed a high incidence of hydrocephalus, a significant decrease in brain size, and a substantial increase in the proliferation rate of progenitor cells in the germinative zones of the dentate gyrus, the RMS of the olfactory system, and the dentate gyrus[70]. At the same time, the number of Nestin+ cells was largely unaffected, compatible with the view that Zac1 rather controls the proliferation of progenitor cells than of stem cells. Yet, a limitation to this work is the fact that Zac1 deficient mice develop IUGR, various skeletal anomalies, and postnatal lung failure pointing to additional defects at the level of single organs and whole systems that are likely to impact on brain development. As an alternative to the multilayered phenotype of Zac1 knock out mice, we conducted genome wide expression profiling (see below) in order to identify Zac1 target genes and analyzed their roles in well-defined in vitro and in vivo systems to elucidate Zac1’s function at the cellular level.
Zac1 target genes in NSCs
Pituitary adenylate cyclase activating polypeptide receptor 1: The first Zac1 target gene to be identified was the G-protein coupled receptor for the neuropeptide pituitary adenylate cyclase activating polypeptide (PACAP)[51,57,71], which controls various neuroendocrine functions in addition to its role as potent neurotrophic factor[72-74]. Mechanistically, Zac1 binds to an imperfect palindromic DNA element localized in the proximal polypeptide receptor 1 (Pac1) promoter to confer transactivation in a cell-type specific manner and to compete at the same time with the estrogen receptor dependent activation of PAC1[75].
PACAP and PAC1 are broadly expressed in the fetal brain and reduce the proliferation rate of certain neural precursor populations. At E13 in rat, PACAP can be detected hippocampus, hypothalamus, cortex, amygdala, dorsal root ganglia, and spinal cord, whereas PAC1 is expressed in the neural plate, the neuroepithelia of the mesencephalon and rhombencephalon at E9, and in the neuroepithelia of the cortex, hippocampal formation, and cerebellum at E11, and in the basal telencephalon and olfactory bulk from E13 and E16 forward, respectively[76]. PACAP induces a sharp increase in p57Kip2 expression in embryonic cortical precursors resulting in decreased CDK2 kinase activity, S-phase entry, and DNA synthesis. Furthermore, PACAP promotes the association of p57Kip2 with the kinase complex supporting its anti-mitogenic activity in neural progenitors. In accord with the specific role of p57Kip2 in cortical neurogenesis (see above), the expression levels of CDK2, cyclin E, or of the CKIs p21Cip2 and p27Kip1 remain unaffected by PACAP.
Coregulation of p53 and p73 target genes: As noted before, Zac1 can serve as a coregulator for p53 and p73 due to its scaffolding function and regulation of coactivator activities[59,65]. The p53 family consisting of p53 itself, p63, and p73 encodes transcription factors with a key role in proliferation, differentiation, apoptosis, stem cell renewal and cell fate commitment[77].
Zac1 coregulation of p53 has been originally discovered in a cervical carcinoma cell line[59] and led us to investigate its potential role in NSCs. Consistent with previous studies[78,79], we observed high levels of p53 in the undifferentiated state in ESCs which reside primarily in the cytoplasm[80] and decline upon differentiation[65]. At the same time, p53 binds to the promoter of the pluripotency genes Nanog and Oct-4[81,82], represses their transcription, and triggers the transition from self-renewal to differentiation. Although Zac1 has been reported to co-repress nuclear receptors in a cell-type specific manner, no evidence exists for a comparable role towards members of the p53 family[58,59].
In the developing brain, p53 is expressed in progenitor cells of the SVZ[83] where it induces cell-cycle arrest, DNA repair and cell death following genotoxic stress[84]. P53-deficient mice show higher cell proliferation in the SVZ and enhanced neurosphere formation in vitro[83,85] consistent with a role of p53 as negative regulator of NSC self-renewal. Moreover, p53 seems to be involved in different aspects of NSC differentiation including repression of self-renewal and promotion of gliogenesis[86,87].
Mounting experimental evidence indicates that endogenous reactive oxygen species (ROS) play a critical role in cell signaling and NSC physiology including appropriate timing of neurogenesis. In this respect, the pattern of ROS generation matches the one of p53 immunoreactivity in the developing telencephalon. Similarly to Zac1, nuclear p53 is detected in Nestin+ NSCs in E11 and E13 proliferative germinal zones, and its expression decreases towards the cortical plate[88]. Loss of p53 function causes enhanced ROS production and premature neurogenesis, which is partly reversed by reinstatement of p53 or antioxidant treatment[88].
Despite these interesting findings, a role of Zac1 in any of these p53-dependent processes is presently unknown and future studies are needed to address this topic.
P73 has been recognized for its critical role in brain development as evidenced by the highly penetrant phenotype in p73 null mice which display cortical hypoplasia, hydrocephalus, and hippocampal dysgenesis[89,90]. The hippocampal anomaly corresponds with either a complete absence or truncation of the lower blade of the neurogenic DG and an abnormal gyration of the Ammon’s horn. Isoform-specific p73 knock-out mice showed that this phenotype results in major part from the absence of the activation-proficient p73 protein (TAp73) as opposed to the activation-deficient p73 protein (ΔNp73). The latter isoform lacks Zac1 coactivation[65] and is thought to act as a potent prosurvival protein in neurons by counteracting the proapoptotic function of p53[91,92]. Therefore, it would be of interest to investigate the possibility of Zac1-dependent coregulation of TAp73 in NSC in greater depth.
Withdrawal of leukemia inhibitory factor (Lif1) potently induces neuronal differentiation of ESCs and strongly upregulates Zac1 and p73 expression concomitantly to a rapid decline in p53 mRNA and protein[65]. At the same time, p73 isoforms switch from activation-deficient DNp73, prevailing under the undifferentiated state, to activation-proficient TAp73 and caused a strong up-regulation of the p21Cip1 and p57Kip2 genes, two well-known direct p73 target genes[93]. As referred to above, DNA bound p73 recruits Zac1 to select target genes, where it serves as a scaffold for coactivator assembly and enhancement of catalytic functions. As a result, Zac1 enhances p73 transcriptional activities in a site-specific manner[65]. These findings suggest a joint role for Zac1 and p73 in inducing cell cycle exit of ESCs and in differentiation towards neural cells.
In vivo studies evidenced a reduced proliferation of neurogenic cells isolated from the E16 and E18 VZ/SVZ and a smaller size of the perinatal SVZ in p73 deficient mice when compared to controls. As a result, p73(-/-) mice suffer from a depletion of the stem cell compartment at birth pointing to a role of p73 in NSC self-renewal and maintenance[94]. This function of p73 extends also to the adult neurogenesis in the DG. At the molecular level, p73 deficiency elicits perturbations in the canonical Sox2 and Notch signaling pathways driving NSC proliferation[95]. Additionally, a reduced transcription of Hey2, a negative regulator of activator bHLH proteins, impairs the long-term maintenance of neural precursors in the absence of p73[95].
Because Zac1 is expressed in embryonic and adult neural stem cells it could potentially interact with and coregulate TAp73 functions. Presently, the actual evidence remains limited to Zac1 coactivation of TAp73 during neuronal differentiation of ECSs. However, further studies should address the role of Zac1 coregulation for NSC self-renewal, maintenance and differentiation at different developmental stages though.
Suppressor of cytokine signaling 3: Genome-wide expression profiling in a cerebellar neural stem cell line (C17.2) using a Tet-off system led to the identification of suppressor of cytokine signaling 3 (Socs3), a negative regulator of Jak/Stat3 signaling, as potential Zac1 target gene[96].
The transition from neuronal cell types to glial subtype-specific precursors is critically controlled by preset developmental programs and extracellular signals like the cytokine driven Jak/Stat3 pathway, which is largely inactive at early, neurogenic stages and takes on at later gliogenic stages, when the expression of neurogenic factors progressively declines[97].
Zac1 recognizes a cluster of GC-rich DNA elements in the proximal promoter and intronic region of the mice and human Socs3 genes to confer transcriptional activation[96].
Two radial glial-like NSC lines, derived from E15 or the adult SVZ of mice, showed a transient upregulation of Zac1 mRNA and protein upon neuronal or astroglial differentiation, whereby transactivation of Socs3 occurred solely under the latter condition indicative of a role of Socs3 as a lineage-specific target gene. Consistent with this finding, DNA methylation at the Socs3 gene decreased during astroglial differentiation but remained unaltered during neuronal differentiation. Zac1 and Socs3 are expressed simultaneously exclusively during the early stage of astroglial differentiation and associated with a strong decline in receptor activation-dependent tyrosine phosphorylation of Stat3. In agreement with these results, Zac1 and Socs3 are highly expressed in the neocortical ventricular zone at E18, which corresponds to the onset of astrogliogenesis[96].
Astroglial differentiation is triggered by various cytokines namely cardiotrophin-1, ciliary neurotrophic factor, or leukemia inhibitory factor, which activate Jak/Stat3 signaling[97-100]. Conversely, genetic deletions in this pathway (i.e., gp130, LIF and Stat3) reduce astroglial differentiation[97,101,102]. Importantly, over-expression of Socs3 inhibits Stat3 signaling and impairs astrogliogenesis[103], whereas conditional deletion of Socs3 leads to enhanced astrogliogenesis in neonatal mouse brain and primary neuroepithelial cells[100]. These findings led us to investigate whether Zac1-dependent induction of Socs3 could provide a negative feedback loop to inhibit Jak/Stat3 signaling during early astroglial differentiation of NSCs.
In accord with an inhibitory role in Jak/Stat3 signaling, Zac1 overexpression delayed astroglial differentiation, independent of a simultaneous increase in the number of cells in transition into G1 arrest[96]. Conversely, knock-down of Zac1 in NSCs facilitated astroglial differentiation and postponed cell cycle arrest. Although lengthening of the G1 phase has been suggested to initiate differentiation, this mechanism seems not to apply to Zac1’s cell cycle arrest function in NSCs and supports the self-sufficient role of Socs3 in the control of astroglial differentiation. Compatible with this view, knock-down of Socs3 in Zac1 overexpressing cells reinstated timely astroglial differentiation. Similar results were obtained in primary E18 NSCs strengthening the role of Zac1-dependent regulation of Socs3 in early astroglial differentiation of NSCs[96].
Overall, this study provides detailed insight into the molecular mechanisms by which an imprinted gene can fine tune cell-fate decisions and differentiation in NSCs and assigns to Zac1 a critical role in the prevention of precocious astroglial differentiation (Figure 2).
Figure 2.
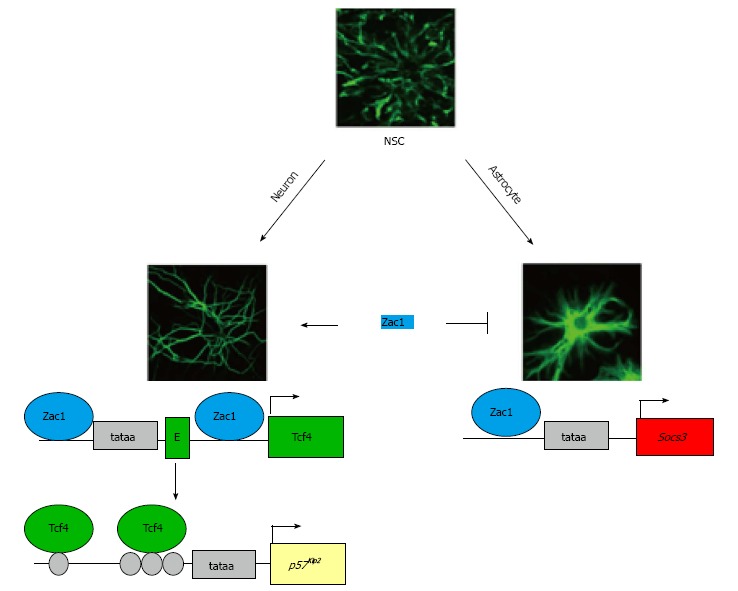
Imprinted Zac1 favors neuronal differentiation of neural stem cells. The Zac1 gene is maternally imprinted and encodes a versatile transcriptional regulator. Zac1 expression is strongly induced during neuronal and astroglial differentiation of embryonic and adult neural stem cells (NSCs). During neuronal differentiation (left scheme), Zac1 binds to GC-rich DNA binding sites at the Tcf4 promoter and first intron to confer synergistic transactivation. As a result, enhanced Tcf4 expression promotes binding to and transactivation of the cyclin kinase inhibitor p57Kip2, which induces G1 arrest. Moreover, Zac1 binds to GC-rich DNA elements at the Socs3 promoter during astroglial differentiation (right scheme). Socs3 encodes a potent inhibitor of prodifferentiative Jak/Stat3 signaling and prevents precocious astroglial differentiation. The tataa-elements are boxed in light grey and the transcriptional start sites are symbolized by arrows. The first exon in the Tcf4 locus is depicted as a green box (labeled E) and coding exons of Tcf4 and Socs3 as green and red boxes, respectively. Tcf4 binds to various E-box motifs (light grey circles) localizing to the p57Kip2 regulatory region.
Transcription factor 4: Zac1 expression robustly increased under astroglial as well as neuronal differentiation, which led us to question Zac1’s function during the latter condition[96]. The family of bHLH proteins plays a prominent role in cell fate decisions and differentiation. These proteins share the eponymic bHLH domain which is necessary for homo- or heterodimerization and for binding to a specific DNA sequence named E-box motif[104]. bHLH proteins expressed in the brain can be classified into two groups; the so-called specification factors (i.e., Math, Mash, neurogenin, and NeuroD), which are expressed in a spatiotemporally controlled manner, and their ubiquitously expressed dimerization partners, the E proteins[105,106]. Under the undifferentiated, proliferative state proneural factors are weakly expressed, prior to a rapid increase in concert with the E-proteins at the initiation of neurogenesis. E-protein family members comprise two splice variants of E2A (E12 and E47), HEB, and transcription factor 4 (Tcf4) (alias E2-2, SEF2, or ITF2), which upon heterodimerization with a specification factor, bind to the promoter of their target genes, in order to promote neurogenesis, and inhibit astrogliogenesis and NSC proliferation[107,108].
Expression profiling in a NSC line (C17.2) based on inducible Zac1 expression showed a robust upregulation of Tcf4 mRNA and protein[109]. Thereby, Zac1 bound simultaneously to the proximal promoter and first intron of Tcf4 and binding at either site was necessary to confer synergistic transactivation.
Neuronal differentiation of ESCs caused a strong Zac1 and Tcf4 upregulation and associated with Zac1 binding at the Tcf4 gene. As noted before, Zac1 upregulation following differentiation of embryonic and adult NSC lines occurred under either astroglial or neuronal differentiation, whereby induction of Socs3 was confined to the astroglial lineage. Oppositely, Zac1-dependent upregulation of Tcf4 was specific to neuronal differentiation, associated with an overall increase in active chromatin marks but no change in DNA methylation, at the Tcf4 locus[109]. Zac1-dependent Tcf4 regulation was also confined to neuronal differentiation of primary NSCs; moreover, Zac1 binding to and transactivation of the Tcf4 locus occurred exclusively during periods of neuronal differentiation in the neocortical ventricular zone[109].
Among known Tcf4 target genes, p57Kip2 is of particular interest as it is co-regulated with Zac1 in the framework of an imprinted gene network (see below), it critically controls differentiation and migration of radial glia cells, and shares with Zac1 and Tcf4 a cell cycle arrest function (see above).
Zac1 and Tcf4 are broadly expressed in neuronal progenitor cell populations during early (E11) and midneurogenesis (E15) such as in the caudal brain regions, the pallium, and the prethalamic eminence[109].
P57Kip2 expression is more localized and mapped strongly to the subpallium and peduncular hypothalamus where all three factors were detected.
Functionally, Zac1 enhances G1-cell cycle arrest in NSC lines and primary NSCs (E15) during neuronal differentiation, by inducing p57Kip2 expression through Tcf4[109].
Taken together, these results suggest a role to Zac1 in the prevention of precocious astroglial differentiation through Socs3 induction and in advancing at the same time neuronal differentiation through Tcf4 induction and Tcf4-mediated upregulation of p57Kip2 (Figure 2).
ZAC1 IMPRINTED GENE NETWORK
Recent evidence indicates that many imprinted genes might work in an integrated network of imprinted genes. In this respect, Arima et al[110] noted that Zac1 and p57Kip2 show a conspicuously similar expression pattern in mesenchymal and neuroepithelial tissues, suggesting a functional interaction between these genes. Interestingly, the Beckwith-Wiedemann syndrome (loss of p57Kip2 imprinting) and TNDM (loss of ZAC1 imprinting) represent with partly opposite phenotypes including gigantism vs IUGR or hypoglycemia vs hyperglycemia.
At the molecular level, ZAC1 binds in a methylation-sensitive manner within the promoter CpG island of LIT1 (KCNQ1OT1), which encodes a paternally expressed, anti-sense RNA thought to repress p57Kip2 in cis[16]. ZAC1 confers transactivation to LIT1 promoter constructs suggesting that ZAC1 might down-regulate p57Kip2 via LIT1 anti-sense[110]. Oppositely, we demonstrated that Zac1 upregulates p57Kip2 via Tcf4 in NSCs[109].
Some aspects of the Zac1 knock-out phenotype, such as growth retardation, perinatal death, and incomplete bone formation appear difficult to reconcile with Zac1’s antiproliferative activities[69]. A possible explanation to this puzzle is provided by a meta-analysis of microarray data, which indicates that Zac1 coordinates a network of genes that consists of a remarkable huge number of imprinted genes. These include Igf2, H19, Dlk1, Ndn, and p57Kip2, which share an important role in NSC maintenance and differentiation, among others (Grb10, Gnas, Meg3, Mest, and Sgce). Moreover, Zac1-deficient liver tissue and Zac1 overexpression experiments in neuroblastoma cells showed an opposite regulation of Igf2, H19, Dlk1, and p57Kip2. Interestingly, Zac1 bound to the downstream H19 enhancer and conferred transactivation to both the H19 and Igf2 promoters (Figure 3).
Figure 3.
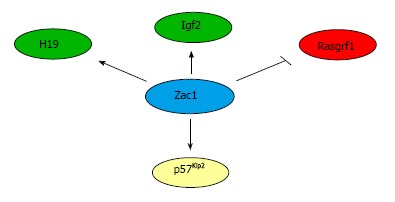
Zac1-dependent transcriptional regulation of imprinted genes. Zac1 binds to the downstream enhancer of H19 and transactivates H19 and Igf2 expression in neuronal cells. In pancreatic progenitor cell, Zac1 bind to the promoter of the paternally imprinted Rasgrf1 gene and confers repression. Following neuronal differentiation of NSCs, Zac1 induces Tcf4, which binds at the promoter of the imprinted p57Kip2 gene and enhances G1 cell cycle arrest.
More recently, Lui et al[111] showed that IGNs are possibly involved in mammalian somatic growth control. Early postnatal life is a period of fast somatic growth, which slows down with maturation and finally arrests in adulthood. A group of 11 imprinted genes involved in cell proliferation (including Zac1, Igf2, H19, Dlk1, Ndn, and p57Kip2) and part of the Zac1-IGN is expressed in multiple tissues at levels that correlate with trajectories of overall somatic growth and decrease coordinately with age. Although this study does not explicitly address the role of IGNs in NSCs, it importantly suggests that the function of IGNs is not reserved to embryonic life but potentially extends across lifespan.
Another recent study focused on gene expression profiling in murine long-term hematopoietic stem cells (LT-HSC) vs their differentiated progeny. These experiments showed that imprinted genes (including Zac1, Igf2, H19, Dlk1, Ndn, and p57Kip2) were more uniformly expressed in progenitors when compared to the differentiated counterparts[112]. Moreover, stem/progenitor cells from adult skeletal muscle and epidermis show a higher expression of these genes when compared to their differentiated derivatives.
Taken together, the Zac1-associated IGN network comprises, among others, imprinted genes that play a critical role in NSC maintenance and differentiation. Some of them, like Igf2 and H19 seem to be under the direct transcriptional control of Zac1 while others like Dlk1 might be regulated more indirectly as exemplified by Zac1-dependent upregulation of p57Kip2 via Tcf4 (Figure 3).
CONCLUSION
A wide range of genes acts in a concerted manner during brain development to regulate NSC proliferation, migration, and differentiation. Among these, imprinted genes have gained increasing apprehension in NSC biology due to their critical roles in quiescence, stemness, and cellular differentiation.
The molecular processes controlling both NSC fate and imprinted gene expression are manifold and include transcriptional regulation, epigenetic, and genomic interactions.
For instance, imprinted genes can regulate the expression or the transcriptional activities of proneural bHLH proteins in NSCs. Zac1 activates Tcf4 during neuronal differentiation of NSCs, while p57Kip2 inhibits the transcriptional activity of Mash1. Conversely, the proneuronal bHLH-protein neurogenin regulates the imprinted Dlk1-Gtl2 locus in the dorsal telencephalon[113] indicating bidirectional interactions between imprinted genes and proneuronal proteins.
Genomic imprinting defects have been originally recognized for their role in early development of the embryo and placenta[9] and more recently for postnatal life[114]. New evidences suggest an additional role of variation in genomic imprinting in the mediation of environmental exposures, which is thought to associate with less severe consequences than those resulting from loss of genomic imprinting and might represent an important component of complex traits[115]. For example, newborn of obese parents show altered DNA methylation profiles of multiple imprinted genes, which may be carried onto the next generation and confer an increased risk for metabolic diseases in adulthood[116]. Moreover, the degree of methylation of ZAC1 associates with pre- and postnatal growth in healthy infants as well as with maternal nutrition and lasts at least until the first year of life[117]. Similarly, individuals prenatally exposed to famine showed 6 decades later less DNA methylation of IGF2 compared with their unexposed, same-sex siblings[118], although variation in DNA methylation at this locus is thought to increase as a result of the aging process itself[119]. Collectively, these findings indicate the need for further studies on genetic and epigenetic variation at imprinted loci in response to environmental exposures and across lifetime.
Remarkably, in NSCs and other discrete stem cell populations, recent findings indicate that genomic imprinting can be epigenetically switched off at defined developmental time windows as shown for Igf2-H19 and Dlk1. Such temporary changes in allele-specific transcription of imprinted genes alter gene dosage in a cell-type and tissue-specific manner and are required to prevent precocious depletion of the stem cell pool. The influence of various environmental exposures on epigenetic switches in NSCs is presently unknown and might contribute to brain function and aging in individuals at risk.
Interestingly, recent literature suggests that imprinted genes do not operate in isolation, but as complex network of genes, whose expression is dynamically controlled by epigenetic mechanisms that extend from prenatal to postnatal development and possibly during aging. For instance, Zac1 is a central hub in an IGN comprising Igf2, H19, Dlk1, p57Kip2, and Ndn, which share a role in NSC maintenance and differentiation.
Though, additionally studies are necessary to explore in more detail the role of IGNs in NSCs across lifespan as well as in response to environmental exposures and to elicit their molecular basis. Collectively, a better understanding of the complex interactions governing imprinted genes expression promises new insight into the biology of NSC and associated conditions ranging from imprinting disorders to age-related diseases.
ACKNOWLEDGMENTS
We are thankful to Christoph Zimmermann for critical discussions and advice on the manuscript.
Footnotes
P- Reviewer: Frade JM, Feng Z S- Editor: Song XX L- Editor: A E- Editor: Lu YJ
Supported by Max Planck Institute of Psychiatry.
Open-Access: This article is an open-access article which was selected by an in-house editor and fully peer-reviewed by external reviewers. It is distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Peer-review started: July 25, 2014
First decision: August 28, 2014
Article in press: November 19, 2014
References
- 1.Ming GL, Song H. Adult neurogenesis in the mammalian brain: significant answers and significant questions. Neuron. 2011;70:687–702. doi: 10.1016/j.neuron.2011.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Jirtle RL. Geneimprint: Genes by species. Available from: http: //www.geneimprint.com/site/genes-by-species.
- 3.Gregg C, Zhang J, Weissbourd B, Luo S, Schroth GP, Haig D, Dulac C. High-resolution analysis of parent-of-origin allelic expression in the mouse brain. Science. 2010;329:643–648. doi: 10.1126/science.1190830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gregg C, Zhang J, Butler JE, Haig D, Dulac C. Sex-specific parent-of-origin allelic expression in the mouse brain. Science. 2010;329:682–685. doi: 10.1126/science.1190831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zhang K, Li JB, Gao Y, Egli D, Xie B, Deng J, Li Z, Lee JH, Aach J, Leproust EM, et al. Digital RNA allelotyping reveals tissue-specific and allele-specific gene expression in human. Nat Methods. 2009;6:613–618. doi: 10.1038/nmeth.1357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bhaumik SR, Smith E, Shilatifard A. Covalent modifications of histones during development and disease pathogenesis. Nat Struct Mol Biol. 2007;14:1008–1016. doi: 10.1038/nsmb1337. [DOI] [PubMed] [Google Scholar]
- 7.Kouzarides T. Chromatin modifications and their function. Cell. 2007;128:693–705. doi: 10.1016/j.cell.2007.02.005. [DOI] [PubMed] [Google Scholar]
- 8.Eggermann T, Leisten I, Binder G, Begemann M, Spengler S. Disturbed methylation at multiple imprinted loci: an increasing observation in imprinting disorders. Epigenomics. 2011;3:625–637. doi: 10.2217/epi.11.84. [DOI] [PubMed] [Google Scholar]
- 9.Constância M, Kelsey G, Reik W. Resourceful imprinting. Nature. 2004;432:53–57. doi: 10.1038/432053a. [DOI] [PubMed] [Google Scholar]
- 10.Kernohan KD, Jiang Y, Tremblay DC, Bonvissuto AC, Eubanks JH, Mann MR, Bérubé NG. ATRX partners with cohesin and MeCP2 and contributes to developmental silencing of imprinted genes in the brain. Dev Cell. 2010;18:191–202. doi: 10.1016/j.devcel.2009.12.017. [DOI] [PubMed] [Google Scholar]
- 11.Drake NM, DeVito LM, Cleland TA, Soloway PD. Imprinted Rasgrf1 expression in neonatal mice affects olfactory learning and memory. Genes Brain Behav. 2011;10:392–403. doi: 10.1111/j.1601-183X.2011.00678.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chen DY, Stern SA, Garcia-Osta A, Saunier-Rebori B, Pollonini G, Bambah-Mukku D, Blitzer RD, Alberini CM. A critical role for IGF-II in memory consolidation and enhancement. Nature. 2011;469:491–497. doi: 10.1038/nature09667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lefebvre L, Viville S, Barton SC, Ishino F, Keverne EB, Surani MA. Abnormal maternal behaviour and growth retardation associated with loss of the imprinted gene Mest. Nat Genet. 1998;20:163–169. doi: 10.1038/2464. [DOI] [PubMed] [Google Scholar]
- 14.Li L, Keverne EB, Aparicio SA, Ishino F, Barton SC, Surani MA. Regulation of maternal behavior and offspring growth by paternally expressed Peg3. Science. 1999;284:330–333. doi: 10.1126/science.284.5412.330. [DOI] [PubMed] [Google Scholar]
- 15.Champagne FA, Curley JP, Swaney WT, Hasen NS, Keverne EB. Paternal influence on female behavior: the role of Peg3 in exploration, olfaction, and neuroendocrine regulation of maternal behavior of female mice. Behav Neurosci. 2009;123:469–480. doi: 10.1037/a0015060. [DOI] [PubMed] [Google Scholar]
- 16.Edwards CA, Ferguson-Smith AC. Mechanisms regulating imprinted genes in clusters. Curr Opin Cell Biol. 2007;19:281–289. doi: 10.1016/j.ceb.2007.04.013. [DOI] [PubMed] [Google Scholar]
- 17.Moore T, Haig D. Genomic imprinting in mammalian development: a parental tug-of-war. Trends Genet. 1991;7:45–49. doi: 10.1016/0168-9525(91)90230-N. [DOI] [PubMed] [Google Scholar]
- 18.Lehtinen MK, Zappaterra MW, Chen X, Yang YJ, Hill AD, Lun M, Maynard T, Gonzalez D, Kim S, Ye P, et al. The cerebrospinal fluid provides a proliferative niche for neural progenitor cells. Neuron. 2011;69:893–905. doi: 10.1016/j.neuron.2011.01.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bracko O, Singer T, Aigner S, Knobloch M, Winner B, Ray J, Clemenson GD, Suh H, Couillard-Despres S, Aigner L, et al. Gene expression profiling of neural stem cells and their neuronal progeny reveals IGF2 as a regulator of adult hippocampal neurogenesis. J Neurosci. 2012;32:3376–3387. doi: 10.1523/JNEUROSCI.4248-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rao G, Pedone CA, Del Valle L, Reiss K, Holland EC, Fults DW. Sonic hedgehog and insulin-like growth factor signaling synergize to induce medulloblastoma formation from nestin-expressing neural progenitors in mice. Oncogene. 2004;23:6156–6162. doi: 10.1038/sj.onc.1207818. [DOI] [PubMed] [Google Scholar]
- 21.Hao Y, Crenshaw T, Moulton T, Newcomb E, Tycko B. Tumour-suppressor activity of H19 RNA. Nature. 1993;365:764–767. doi: 10.1038/365764a0. [DOI] [PubMed] [Google Scholar]
- 22.Yoshimizu T, Miroglio A, Ripoche MA, Gabory A, Vernucci M, Riccio A, Colnot S, Godard C, Terris B, Jammes H, et al. The H19 locus acts in vivo as a tumor suppressor. Proc Natl Acad Sci USA. 2008;105:12417–12422. doi: 10.1073/pnas.0801540105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Keniry A, Oxley D, Monnier P, Kyba M, Dandolo L, Smits G, Reik W. The H19 lincRNA is a developmental reservoir of miR-675 that suppresses growth and Igf1r. Nat Cell Biol. 2012;14:659–665. doi: 10.1038/ncb2521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kono T, Obata Y, Wu Q, Niwa K, Ono Y, Yamamoto Y, Park ES, Seo JS, Ogawa H. Birth of parthenogenetic mice that can develop to adulthood. Nature. 2004;428:860–864. doi: 10.1038/nature02402. [DOI] [PubMed] [Google Scholar]
- 25.Surani MA, Hayashi K, Hajkova P. Genetic and epigenetic regulators of pluripotency. Cell. 2007;128:747–762. doi: 10.1016/j.cell.2007.02.010. [DOI] [PubMed] [Google Scholar]
- 26.Ratajczak MZ, Zuba-Surma E, Wojakowski W, Suszynska M, Mierzejewska K, Liu R, Ratajczak J, Shin DM, Kucia M. Very small embryonic-like stem cells (VSELs) represent a real challenge in stem cell biology: recent pros and cons in the midst of a lively debate. Leukemia. 2014;28:473–484. doi: 10.1038/leu.2013.255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ratajczak MZ, Shin DM, Schneider G, Ratajczak J, Kucia M. Parental imprinting regulates insulin-like growth factor signaling: a Rosetta Stone for understanding the biology of pluripotent stem cells, aging and cancerogenesis. Leukemia. 2013;27:773–779. doi: 10.1038/leu.2012.322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Takada S, Paulsen M, Tevendale M, Tsai CE, Kelsey G, Cattanach BM, Ferguson-Smith AC. Epigenetic analysis of the Dlk1-Gtl2 imprinted domain on mouse chromosome 12: implications for imprinting control from comparison with Igf2-H19. Hum Mol Genet. 2002;11:77–86. doi: 10.1093/hmg/11.1.77. [DOI] [PubMed] [Google Scholar]
- 29.Falix FA, Aronson DC, Lamers WH, Gaemers IC. Possible roles of DLK1 in the Notch pathway during development and disease. Biochim Biophys Acta. 2012;1822:988–995. doi: 10.1016/j.bbadis.2012.02.003. [DOI] [PubMed] [Google Scholar]
- 30.Ferrón SR, Charalambous M, Radford E, McEwen K, Wildner H, Hind E, Morante-Redolat JM, Laborda J, Guillemot F, Bauer SR, et al. Postnatal loss of Dlk1 imprinting in stem cells and niche astrocytes regulates neurogenesis. Nature. 2011;475:381–385. doi: 10.1038/nature10229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Takumi T. The neurobiology of mouse models syntenic to human chromosome 15q. J Neurodev Disord. 2011;3:270–281. doi: 10.1007/s11689-011-9088-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Maruyama K, Usami M, Aizawa T, Yoshikawa K. A novel brain-specific mRNA encoding nuclear protein (necdin) expressed in neurally differentiated embryonal carcinoma cells. Biochem Biophys Res Commun. 1991;178:291–296. doi: 10.1016/0006-291x(91)91812-q. [DOI] [PubMed] [Google Scholar]
- 33.Taniura H, Taniguchi N, Hara M, Yoshikawa K. Necdin, a postmitotic neuron-specific growth suppressor, interacts with viral transforming proteins and cellular transcription factor E2F1. J Biol Chem. 1998;273:720–728. doi: 10.1074/jbc.273.2.720. [DOI] [PubMed] [Google Scholar]
- 34.Taniura H, Matsumoto K, Yoshikawa K. Physical and functional interactions of neuronal growth suppressor necdin with p53. J Biol Chem. 1999;274:16242–16248. doi: 10.1074/jbc.274.23.16242. [DOI] [PubMed] [Google Scholar]
- 35.Hasegawa K, Yoshikawa K. Necdin regulates p53 acetylation via Sirtuin1 to modulate DNA damage response in cortical neurons. J Neurosci. 2008;28:8772–8784. doi: 10.1523/JNEUROSCI.3052-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Moon HE, Ahn MY, Park JA, Min KJ, Kwon YW, Kim KW. Negative regulation of hypoxia inducible factor-1alpha by necdin. FEBS Lett. 2005;579:3797–3801. doi: 10.1016/j.febslet.2005.05.072. [DOI] [PubMed] [Google Scholar]
- 37.Huang Z, Fujiwara K, Minamide R, Hasegawa K, Yoshikawa K. Necdin controls proliferation and apoptosis of embryonic neural stem cells in an oxygen tension-dependent manner. J Neurosci. 2013;33:10362–10373. doi: 10.1523/JNEUROSCI.5682-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Chen HL, Pistollato F, Hoeppner DJ, Ni HT, McKay RD, Panchision DM. Oxygen tension regulates survival and fate of mouse central nervous system precursors at multiple levels. Stem Cells. 2007;25:2291–2301. doi: 10.1634/stemcells.2006-0609. [DOI] [PubMed] [Google Scholar]
- 39.Clarke L, van der Kooy D. Low oxygen enhances primitive and definitive neural stem cell colony formation by inhibiting distinct cell death pathways. Stem Cells. 2009;27:1879–1886. doi: 10.1002/stem.96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zhao T, Zhang CP, Liu ZH, Wu LY, Huang X, Wu HT, Xiong L, Wang X, Wang XM, Zhu LL, et al. Hypoxia-driven proliferation of embryonic neural stem/progenitor cells--role of hypoxia-inducible transcription factor-1alpha. FEBS J. 2008;275:1824–1834. doi: 10.1111/j.1742-4658.2008.06340.x. [DOI] [PubMed] [Google Scholar]
- 41.Minamide R, Fujiwara K, Hasegawa K, Yoshikawa K. Antagonistic interplay between necdin and Bmi1 controls proliferation of neural precursor cells in the embryonic mouse neocortex. PLoS One. 2014;9:e84460. doi: 10.1371/journal.pone.0084460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ohnuma S, Harris WA. Neurogenesis and the cell cycle. Neuron. 2003;40:199–208. doi: 10.1016/s0896-6273(03)00632-9. [DOI] [PubMed] [Google Scholar]
- 43.Zhang P, Liégeois NJ, Wong C, Finegold M, Hou H, Thompson JC, Silverman A, Harper JW, DePinho RA, Elledge SJ. Altered cell differentiation and proliferation in mice lacking p57KIP2 indicates a role in Beckwith-Wiedemann syndrome. Nature. 1997;387:151–158. doi: 10.1038/387151a0. [DOI] [PubMed] [Google Scholar]
- 44.Carey RG, Li B, DiCicco-Bloom E. Pituitary adenylate cyclase activating polypeptide anti-mitogenic signaling in cerebral cortical progenitors is regulated by p57Kip2-dependent CDK2 activity. J Neurosci. 2002;22:1583–1591. doi: 10.1523/JNEUROSCI.22-05-01583.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ye W, Mairet-Coello G, Pasoreck E, Dicicco-Bloom E. Patterns of p57Kip2 expression in embryonic rat brain suggest roles in progenitor cell cycle exit and neuronal differentiation. Dev Neurobiol. 2009;69:1–21. doi: 10.1002/dneu.20680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Mairet-Coello G, Tury A, Van Buskirk E, Robinson K, Genestine M, DiCicco-Bloom E. p57(KIP2) regulates radial glia and intermediate precursor cell cycle dynamics and lower layer neurogenesis in developing cerebral cortex. Development. 2012;139:475–487. doi: 10.1242/dev.067314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Furutachi S, Matsumoto A, Nakayama KI, Gotoh Y. p57 controls adult neural stem cell quiescence and modulates the pace of lifelong neurogenesis. EMBO J. 2013;32:970–981. doi: 10.1038/emboj.2013.50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Besson A, Dowdy SF, Roberts JM. CDK inhibitors: cell cycle regulators and beyond. Dev Cell. 2008;14:159–169. doi: 10.1016/j.devcel.2008.01.013. [DOI] [PubMed] [Google Scholar]
- 49.Piras G, El Kharroubi A, Kozlov S, Escalante-Alcalde D, Hernandez L, Copeland NG, Gilbert DJ, Jenkins NA, Stewart CL. Zac1 (Lot1), a potential tumor suppressor gene, and the gene for epsilon-sarcoglycan are maternally imprinted genes: identification by a subtractive screen of novel uniparental fibroblast lines. Mol Cell Biol. 2000;20:3308–3315. doi: 10.1128/mcb.20.9.3308-3315.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Varrault A, Ciani E, Apiou F, Bilanges B, Hoffmann A, Pantaloni C, Bockaert J, Spengler D, Journot L. hZAC encodes a zinc finger protein with antiproliferative properties and maps to a chromosomal region frequently lost in cancer. Proc Natl Acad Sci USA. 1998;95:8835–8840. doi: 10.1073/pnas.95.15.8835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Spengler D, Villalba M, Hoffmann A, Pantaloni C, Houssami S, Bockaert J, Journot L. Regulation of apoptosis and cell cycle arrest by Zac1, a novel zinc finger protein expressed in the pituitary gland and the brain. EMBO J. 1997;16:2814–2825. doi: 10.1093/emboj/16.10.2814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Aguilar-Bryan L, Bryan J. Neonatal diabetes mellitus. Endocr Rev. 2008;29:265–291. doi: 10.1210/er.2007-0029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ma D, Shield JP, Dean W, Leclerc I, Knauf C, Burcelin R Ré, Rutter GA, Kelsey G. Impaired glucose homeostasis in transgenic mice expressing the human transient neonatal diabetes mellitus locus, TNDM. J Clin Invest. 2004;114:339–348. doi: 10.1172/JCI19876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Hoffmann A, Spengler D. Transient neonatal diabetes mellitus gene Zac1 impairs insulin secretion in mice through Rasgrf1. Mol Cell Biol. 2012;32:2549–2560. doi: 10.1128/MCB.06637-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Hoffmann A, Ciani E, Boeckardt J, Holsboer F, Journot L, Spengler D. Transcriptional activities of the zinc finger protein Zac are differentially controlled by DNA binding. Mol Cell Biol. 2003;23:988–1003. doi: 10.1128/MCB.23.3.988-1003.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Bilanges B, Varrault A, Mazumdar A, Pantaloni C, Hoffmann A, Bockaert J, Spengler D, Journot L. Alternative splicing of the imprinted candidate tumor suppressor gene ZAC regulates its antiproliferative and DNA binding activities. Oncogene. 2001;20:1246–1253. doi: 10.1038/sj.onc.1204237. [DOI] [PubMed] [Google Scholar]
- 57.Ciani E, Hoffmann A, Schmidt P, Journot L, Spengler D. Induction of the PAC1-R (PACAP-type I receptor) gene by p53 and Zac. Brain Res Mol Brain Res. 1999;69:290–294. doi: 10.1016/s0169-328x(99)00116-3. [DOI] [PubMed] [Google Scholar]
- 58.Huang SM, Stallcup MR. Mouse Zac1, a transcriptional coactivator and repressor for nuclear receptors. Mol Cell Biol. 2000;20:1855–1867. doi: 10.1128/mcb.20.5.1855-1867.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Huang SM, Schönthal AH, Stallcup MR. Enhancement of p53-dependent gene activation by the transcriptional coactivator Zac1. Oncogene. 2001;20:2134–2143. doi: 10.1038/sj.onc.1204298. [DOI] [PubMed] [Google Scholar]
- 60.Rozenfeld-Granot G, Krishnamurthy J, Kannan K, Toren A, Amariglio N, Givol D, Rechavi G. A positive feedback mechanism in the transcriptional activation of Apaf-1 by p53 and the coactivator Zac-1. Oncogene. 2002;21:1469–1476. doi: 10.1038/sj.onc.1205218. [DOI] [PubMed] [Google Scholar]
- 61.Ptashne M, Gann A. Transcriptional activation by recruitment. Nature. 1997;386:569–577. doi: 10.1038/386569a0. [DOI] [PubMed] [Google Scholar]
- 62.Hoffmann A, Barz T, Spengler D. Multitasking C2H2 zinc fingers link Zac DNA binding to coordinated regulation of p300-histone acetyltransferase activity. Mol Cell Biol. 2006;26:5544–5557. doi: 10.1128/MCB.02270-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Abdollahi A. LOT1 (ZAC1/PLAGL1) and its family members: mechanisms and functions. J Cell Physiol. 2007;210:16–25. doi: 10.1002/jcp.20835. [DOI] [PubMed] [Google Scholar]
- 64.Theodoropoulou M, Stalla GK, Spengler D. ZAC1 target genes and pituitary tumorigenesis. Mol Cell Endocrinol. 2010;326:60–65. doi: 10.1016/j.mce.2010.01.033. [DOI] [PubMed] [Google Scholar]
- 65.Hoffmann A, Spengler D. A new coactivator function for Zac1’s C2H2 zinc finger DNA-binding domain in selectively controlling PCAF activity. Mol Cell Biol. 2008;28:6078–6093. doi: 10.1128/MCB.00842-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Valente T, Junyent F, Auladell C. Zac1 is expressed in progenitor/stem cells of the neuroectoderm and mesoderm during embryogenesis: differential phenotype of the Zac1-expressing cells during development. Dev Dyn. 2005;233:667–679. doi: 10.1002/dvdy.20373. [DOI] [PubMed] [Google Scholar]
- 67.Valente T, Auladell C. Expression pattern of Zac1 mouse gene, a new zinc-finger protein that regulates apoptosis and cellular cycle arrest, in both adult brain and along development. Mech Dev. 2001;108:207–211. doi: 10.1016/s0925-4773(01)00492-0. [DOI] [PubMed] [Google Scholar]
- 68.Alam S, Zinyk D, Ma L, Schuurmans C. Members of the Plag gene family are expressed in complementary and overlapping regions in the developing murine nervous system. Dev Dyn. 2005;234:772–782. doi: 10.1002/dvdy.20577. [DOI] [PubMed] [Google Scholar]
- 69.Varrault A, Gueydan C, Delalbre A, Bellmann A, Houssami S, Aknin C, Severac D, Chotard L, Kahli M, Le Digarcher A, et al. Zac1 regulates an imprinted gene network critically involved in the control of embryonic growth. Dev Cell. 2006;11:711–722. doi: 10.1016/j.devcel.2006.09.003. [DOI] [PubMed] [Google Scholar]
- 70.Valente T. Expressió de la Zac1 durant el desenvolupament de ratolí. Paper de la Zac1 en el sistema nerviós central: ISBN: 8468924865 [PhD thesis] Barcelona: Universitat de Barcelona; 2005. Available from: http://www.tdx.cat/handle/10803/840. [Google Scholar]
- 71.Hoffmann A, Ciani E, Houssami S, Brabet P, Journot L, Spengler D. Induction of type I PACAP receptor expression by the new zinc finger protein Zac1 and p53. Ann N Y Acad Sci. 1998;865:49–58. doi: 10.1111/j.1749-6632.1998.tb11162.x. [DOI] [PubMed] [Google Scholar]
- 72.Arimura A. Perspectives on pituitary adenylate cyclase activating polypeptide (PACAP) in the neuroendocrine, endocrine, and nervous systems. Jpn J Physiol. 1998;48:301–331. doi: 10.2170/jjphysiol.48.301. [DOI] [PubMed] [Google Scholar]
- 73.Li M, Arimura A. Neuropeptides of the pituitary adenylate cyclase-activating polypeptide/vasoactive intestinal polypeptide/growth hormone-releasing hormone/secretin family in testis. Endocrine. 2003;20:201–214. doi: 10.1385/ENDO:20:3:201. [DOI] [PubMed] [Google Scholar]
- 74.Mustafa T. Pituitary adenylate cyclase-activating polypeptide (PACAP): a master regulator in central and peripheral stress responses. Adv Pharmacol. 2013;68:445–457. doi: 10.1016/B978-0-12-411512-5.00021-X. [DOI] [PubMed] [Google Scholar]
- 75.Rodríguez-Henche N, Jamen F, Leroy C, Bockaert J, Brabet P. Transcription of the mouse PAC1 receptor gene: cell-specific expression and regulation by Zac1. Biochim Biophys Acta. 2002;1576:157–162. doi: 10.1016/s0167-4781(02)00303-2. [DOI] [PubMed] [Google Scholar]
- 76.Shen S, Gehlert DR, Collier DA. PACAP and PAC1 receptor in brain development and behavior. Neuropeptides. 2013;47:421–430. doi: 10.1016/j.npep.2013.10.005. [DOI] [PubMed] [Google Scholar]
- 77.Vousden KH, Lane DP. p53 in health and disease. Nat Rev Mol Cell Biol. 2007;8:275–283. doi: 10.1038/nrm2147. [DOI] [PubMed] [Google Scholar]
- 78.Sabapathy K, Klemm M, Jaenisch R, Wagner EF. Regulation of ES cell differentiation by functional and conformational modulation of p53. EMBO J. 1997;16:6217–6229. doi: 10.1093/emboj/16.20.6217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Solozobova V, Blattner C. Regulation of p53 in embryonic stem cells. Exp Cell Res. 2010;316:2434–2446. doi: 10.1016/j.yexcr.2010.06.006. [DOI] [PubMed] [Google Scholar]
- 80.Han MK, Song EK, Guo Y, Ou X, Mantel C, Broxmeyer HE. SIRT1 regulates apoptosis and Nanog expression in mouse embryonic stem cells by controlling p53 subcellular localization. Cell Stem Cell. 2008;2:241–251. doi: 10.1016/j.stem.2008.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Lin T, Chao C, Saito S, Mazur SJ, Murphy ME, Appella E, Xu Y. p53 induces differentiation of mouse embryonic stem cells by suppressing Nanog expression. Nat Cell Biol. 2005;7:165–171. doi: 10.1038/ncb1211. [DOI] [PubMed] [Google Scholar]
- 82.Qin H, Yu T, Qing T, Liu Y, Zhao Y, Cai J, Li J, Song Z, Qu X, Zhou P, et al. Regulation of apoptosis and differentiation by p53 in human embryonic stem cells. J Biol Chem. 2007;282:5842–5852. doi: 10.1074/jbc.M610464200. [DOI] [PubMed] [Google Scholar]
- 83.Meletis K, Wirta V, Hede SM, Nistér M, Lundeberg J, Frisén J. p53 suppresses the self-renewal of adult neural stem cells. Development. 2006;133:363–369. doi: 10.1242/dev.02208. [DOI] [PubMed] [Google Scholar]
- 84.Tedeschi A, Di Giovanni S. The non-apoptotic role of p53 in neuronal biology: enlightening the dark side of the moon. EMBO Rep. 2009;10:576–583. doi: 10.1038/embor.2009.89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Gil-Perotin S, Marin-Husstege M, Li J, Soriano-Navarro M, Zindy F, Roussel MF, Garcia-Verdugo JM, Casaccia-Bonnefil P. Loss of p53 induces changes in the behavior of subventricular zone cells: implication for the genesis of glial tumors. J Neurosci. 2006;26:1107–1116. doi: 10.1523/JNEUROSCI.3970-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Nagao M, Campbell K, Burns K, Kuan CY, Trumpp A, Nakafuku M. Coordinated control of self-renewal and differentiation of neural stem cells by Myc and the p19ARF-p53 pathway. J Cell Biol. 2008;183:1243–1257. doi: 10.1083/jcb.200807130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Solozobova V, Blattner C. p53 in stem cells. World J Biol Chem. 2011;2:202–214. doi: 10.4331/wjbc.v2.i9.202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Forsberg K, Wuttke A, Quadrato G, Chumakov PM, Wizenmann A, Di Giovanni S. The tumor suppressor p53 fine-tunes reactive oxygen species levels and neurogenesis via PI3 kinase signaling. J Neurosci. 2013;33:14318–14330. doi: 10.1523/JNEUROSCI.1056-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Meyer G, Cabrera Socorro A, Perez Garcia CG, Martinez Millan L, Walker N, Caput D. Developmental roles of p73 in Cajal-Retzius cells and cortical patterning. J Neurosci. 2004;24:9878–9887. doi: 10.1523/JNEUROSCI.3060-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Yang A, Walker N, Bronson R, Kaghad M, Oosterwegel M, Bonnin J, Vagner C, Bonnet H, Dikkes P, Sharpe A, et al. p73-deficient mice have neurological, pheromonal and inflammatory defects but lack spontaneous tumours. Nature. 2000;404:99–103. doi: 10.1038/35003607. [DOI] [PubMed] [Google Scholar]
- 91.Pozniak CD, Radinovic S, Yang A, McKeon F, Kaplan DR, Miller FD. An anti-apoptotic role for the p53 family member, p73, during developmental neuron death. Science. 2000;289:304–306. doi: 10.1126/science.289.5477.304. [DOI] [PubMed] [Google Scholar]
- 92.Tissir F, Ravni A, Achouri Y, Riethmacher D, Meyer G, Goffinet AM. DeltaNp73 regulates neuronal survival in vivo. Proc Natl Acad Sci USA. 2009;106:16871–16876. doi: 10.1073/pnas.0903191106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Balint E, Phillips AC, Kozlov S, Stewart CL, Vousden KH. Induction of p57(KIP2) expression by p73beta. Proc Natl Acad Sci USA. 2002;99:3529–3534. doi: 10.1073/pnas.062491899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Talos F, Abraham A, Vaseva AV, Holembowski L, Tsirka SE, Scheel A, Bode D, Dobbelstein M, Brück W, Moll UM. p73 is an essential regulator of neural stem cell maintenance in embryonal and adult CNS neurogenesis. Cell Death Differ. 2010;17:1816–1829. doi: 10.1038/cdd.2010.131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Fujitani M, Cancino GI, Dugani CB, Weaver IC, Gauthier-Fisher A, Paquin A, Mak TW, Wojtowicz MJ, Miller FD, Kaplan DR. TAp73 acts via the bHLH Hey2 to promote long-term maintenance of neural precursors. Curr Biol. 2010;20:2058–2065. doi: 10.1016/j.cub.2010.10.029. [DOI] [PubMed] [Google Scholar]
- 96.Schmidt-Edelkraut U, Hoffmann A, Daniel G, Spengler D. Zac1 regulates astroglial differentiation of neural stem cells through Socs3. Stem Cells. 2013;31:1621–1632. doi: 10.1002/stem.1405. [DOI] [PubMed] [Google Scholar]
- 97.Bonni A, Sun Y, Nadal-Vicens M, Bhatt A, Frank DA, Rozovsky I, Stahl N, Yancopoulos GD, Greenberg ME. Regulation of gliogenesis in the central nervous system by the JAK-STAT signaling pathway. Science. 1997;278:477–483. doi: 10.1126/science.278.5337.477. [DOI] [PubMed] [Google Scholar]
- 98.He F, Ge W, Martinowich K, Becker-Catania S, Coskun V, Zhu W, Wu H, Castro D, Guillemot F, Fan G, et al. A positive autoregulatory loop of Jak-STAT signaling controls the onset of astrogliogenesis. Nat Neurosci. 2005;8:616–625. doi: 10.1038/nn1440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Freeman MR. Specification and morphogenesis of astrocytes. Science. 2010;330:774–778. doi: 10.1126/science.1190928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Fukuda S, Abematsu M, Mori H, Yanagisawa M, Kagawa T, Nakashima K, Yoshimura A, Taga T. Potentiation of astrogliogenesis by STAT3-mediated activation of bone morphogenetic protein-Smad signaling in neural stem cells. Mol Cell Biol. 2007;27:4931–4937. doi: 10.1128/MCB.02435-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Bugga L, Gadient RA, Kwan K, Stewart CL, Patterson PH. Analysis of neuronal and glial phenotypes in brains of mice deficient in leukemia inhibitory factor. J Neurobiol. 1998;36:509–524. doi: 10.1002/(sici)1097-4695(19980915)36:4<509::aid-neu5>3.0.co;2-#. [DOI] [PubMed] [Google Scholar]
- 102.Nakashima K, Wiese S, Yanagisawa M, Arakawa H, Kimura N, Hisatsune T, Yoshida K, Kishimoto T, Sendtner M, Taga T. Developmental requirement of gp130 signaling in neuronal survival and astrocyte differentiation. J Neurosci. 1999;19:5429–5434. doi: 10.1523/JNEUROSCI.19-13-05429.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Cao F, Hata R, Zhu P, Ma YJ, Tanaka J, Hanakawa Y, Hashimoto K, Niinobe M, Yoshikawa K, Sakanaka M. Overexpression of SOCS3 inhibits astrogliogenesis and promotes maintenance of neural stem cells. J Neurochem. 2006;98:459–470. doi: 10.1111/j.1471-4159.2006.03890.x. [DOI] [PubMed] [Google Scholar]
- 104.Murre C, McCaw PS, Baltimore D. A new DNA binding and dimerization motif in immunoglobulin enhancer binding, daughterless, MyoD, and myc proteins. Cell. 1989;56:777–783. doi: 10.1016/0092-8674(89)90682-x. [DOI] [PubMed] [Google Scholar]
- 105.Bertrand N, Castro DS, Guillemot F. Proneural genes and the specification of neural cell types. Nat Rev Neurosci. 2002;3:517–530. doi: 10.1038/nrn874. [DOI] [PubMed] [Google Scholar]
- 106.Bhattacharya A, Baker NE. A network of broadly expressed HLH genes regulates tissue-specific cell fates. Cell. 2011;147:881–892. doi: 10.1016/j.cell.2011.08.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Ross SE, Greenberg ME, Stiles CD. Basic helix-loop-helix factors in cortical development. Neuron. 2003;39:13–25. doi: 10.1016/s0896-6273(03)00365-9. [DOI] [PubMed] [Google Scholar]
- 108.Sun Y, Nadal-Vicens M, Misono S, Lin MZ, Zubiaga A, Hua X, Fan G, Greenberg ME. Neurogenin promotes neurogenesis and inhibits glial differentiation by independent mechanisms. Cell. 2001;104:365–376. doi: 10.1016/s0092-8674(01)00224-0. [DOI] [PubMed] [Google Scholar]
- 109.Schmidt-Edelkraut U, Daniel G, Hoffmann A, Spengler D. Zac1 regulates cell cycle arrest in neuronal progenitors via Tcf4. Mol Cell Biol. 2014;34:1020–1030. doi: 10.1128/MCB.01195-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Arima T, Kamikihara T, Hayashida T, Kato K, Inoue T, Shirayoshi Y, Oshimura M, Soejima H, Mukai T, Wake N. ZAC, LIT1 (KCNQ1OT1) and p57KIP2 (CDKN1C) are in an imprinted gene network that may play a role in Beckwith-Wiedemann syndrome. Nucleic Acids Res. 2005;33:2650–2660. doi: 10.1093/nar/gki555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Lui JC, Finkielstain GP, Barnes KM, Baron J. An imprinted gene network that controls mammalian somatic growth is down-regulated during postnatal growth deceleration in multiple organs. Am J Physiol Regul Integr Comp Physiol. 2008;295:R189–R196. doi: 10.1152/ajpregu.00182.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Berg JS, Lin KK, Sonnet C, Boles NC, Weksberg DC, Nguyen H, Holt LJ, Rickwood D, Daly RJ, Goodell MA. Imprinted genes that regulate early mammalian growth are coexpressed in somatic stem cells. PLoS One. 2011;6:e26410. doi: 10.1371/journal.pone.0026410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Seibt J, Armant O, Le Digarcher A, Castro D, Ramesh V, Journot L, Guillemot F, Vanderhaeghen P, Bouschet T. Expression at the imprinted dlk1-gtl2 locus is regulated by proneural genes in the developing telencephalon. PLoS One. 2012;7:e48675. doi: 10.1371/journal.pone.0048675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Cowley M, Garfield AS, Madon-Simon M, Charalambous M, Clarkson RW, Smalley MJ, Kendrick H, Isles AR, Parry AJ, Carney S, et al. Developmental programming mediated by complementary roles of imprinted Grb10 in mother and pup. PLoS Biol. 2014;12:e1001799. doi: 10.1371/journal.pbio.1001799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Lawson HA, Cheverud JM, Wolf JB. Genomic imprinting and parent-of-origin effects on complex traits. Nat Rev Genet. 2013;14:609–617. doi: 10.1038/nrg3543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Soubry A, Murphy SK, Wang F, Huang Z, Vidal AC, Fuemmeler BF, Kurtzberg J, Murtha A, Jirtle RL, Schildkraut JM, et al. Newborns of obese parents have altered DNA methylation patterns at imprinted genes. Int J Obes (Lond) 2013:Oct 25; Epub ahead of print. doi: 10.1038/ijo.2013.193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Azzi S, Sas TC, Koudou Y, Le Bouc Y, Souberbielle JC, Dargent-Molina P, Netchine I, Charles MA. Degree of methylation of ZAC1 (PLAGL1) is associated with prenatal and post-natal growth in healthy infants of the EDEN mother child cohort. Epigenetics. 2014;9:338–345. doi: 10.4161/epi.27387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Heijmans BT, Tobi EW, Stein AD, Putter H, Blauw GJ, Susser ES, Slagboom PE, Lumey LH. Persistent epigenetic differences associated with prenatal exposure to famine in humans. Proc Natl Acad Sci USA. 2008;105:17046–17049. doi: 10.1073/pnas.0806560105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Pirazzini C, Giuliani C, Bacalini MG, Boattini A, Capri M, Fontanesi E, Marasco E, Mantovani V, Pierini M, Pini E, et al. Space/population and time/age in DNA methylation variability in humans: a study on IGF2/H19 locus in different Italian populations and in mono- and di-zygotic twins of different age. Aging (Albany NY) 2012;4:509–520. doi: 10.18632/aging.100476. [DOI] [PMC free article] [PubMed] [Google Scholar]