

Abstract
Cancers that develop after middle age usually exhibit genomic instability and multiple mutations. This is in direct contrast to pediatric tumors that usually develop as a result of specific chromosomal translocations and epigenetic aberrations. The development of genomic instability is associated with mutations that contribute to cellular immortalization and transformation. Cancer occurs when cancer-initiating cells (CICs), also called cancer stem cells, develop as a result of these mutations. In this paper, we explore how CICs develop as a result of genomic instability, including looking at which cancer suppression mechanisms are abrogated. A recent in vitro study revealed the existence of a CIC induction pathway in differentiating stem cells. Under aberrant differentiation conditions, cells become senescent and develop genomic instabilities that lead to the development of CICs. The resulting CICs contain a mutation in the alternative reading frame of CDKN2A (ARF)/p53 module, i.e., in either ARF or p53. We summarize recently established knowledge of CIC development and cellular immortality, explore the role of the ARF/p53 module in protecting cells from transformation, and describe a risk factor for genomic destabilization that increases during the process of normal cell growth and differentiation and is associated with the downregulation of histone H2AX to levels representative of growth arrest in normal cells.
Keywords: ARF/p53 module, Cancer stem cells, Cancer-initiating cells, Differentiation, Genomic instability, H2AX
Core tip: Cancer usually develops in conjunction with genomic instability and multiple genetic mutations. Only a small number of cells, called cancer-initiating cells (CICs), are the progenitors of cancerous tissue; but how genomic instability and genetic mutations prompt CICs to develop is still unclear. Recent investigations have uncovered the existence of a pathway that could be responsible. This review explores how that pathway might induce the development of CICs, the tumor suppression mechanisms that must be abrogated in order for malignancies to occur, and the role of the alternative reading frame of CDKN2A (ARF)/p53 module/p53 module in protecting normal cells from oncologic transformation.
INTRODUCTION
Somatic stem cells are responsible for the development and homeostasis of organs and other bodily tissues. Cancer-initiating cells (CICs), or cancer stem cells, are responsible for the development[1,2], metastasis, and drug resistance[3-6] of tumors. Although CIC characteristics are increasingly being well defined, how CICs develop remains unclear.
Unlike pediatric tumors, which usually develop as a result of very specific chromosomal translocations[7,8] and epigenetic aberrations[9,10], cancers that become more common around the age of 40 years exhibit extreme chromosomal instability (CIN) or microsatellite instability (MSI)[11-14]. MSI occurs in cells that do not have adequate mismatch repair systems[15-17], and CIN occurs in the presence of other types of repair deficiencies. The hereditary versions of myelodysplastic syndrome[18], breast and ovarian cancers[19-22], and skin cancers[23-25] are examples of CIN. CIN can even occur in normal senescent cells[26,27].
MSI and CIN usually do not occur together and are considered mutually exclusive. For example, 15% of colorectal cancers exhibit MSI, but most of the remainder exhibit CIN. There are also tumors that do not exhibit either MSI or CIN and result from DNA polymerase ε mutations, which interfere with proofreading functions and produce hypermutations[28,29]. Massive genomic rearrangements occur in a wide variety of cancers[30-32], and the unstable structures that result vary widely[33,34]. Since only a very small number of CICs are necessary to produce a malignancy, what causes CICs to develop in the first place is an important question[35,36].
In malignant cells, mutations in either alternative reading frame of CDKN2A (ARF) or p53 are common. The ARF/p53 module plays a major role in keeping normal cells from transforming into malignancies, so these mutations that interfere with ARF/p53 functioning show us how the barrier reactions performed by the ARF/p53 module normally work and the way genomic instability promotes the development of CICs.
EFFECTS OF GENOMIC INSTABILITY
Like cancer cells that develop because of genomic instability in vivo[37-40], cells cultured in vitro can also be transformed and/or immortalized in association with either CIN- or MSI-type genomic instabilities[26] and mutations in the ARF/p53 module[41]. Following serial proliferation, normal mouse embryonic fibroblast cells (MEFs) stop reproducing after a growth arrest command issued by the ARF/p53 module[41]. This command prevents cells from immortalizing[27,42]; but immortality can develop if the genome is destabilized[43], clearly demonstrating that genomic instability is a triggering event in cellular immortalization.
Genomic instability probably contributes to the induction of mutations in the ARF/p53 module[27,42]. In MEFs, immortalization can result if genomic instability is induced and/or mutations occur in either ARF or p53 (Figure 1), but is blocked in cells with stable genomes that are under the continuous regulation of the ARF/p53 module[27]. Healthy ARF/p53 regulation is essential for the prevention of cellular transformation and immortalization, but which functions of ARF and p53 are responsible for tumor suppression is controversial.
Figure 1.
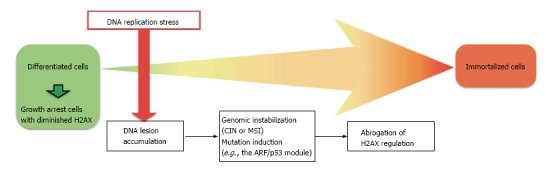
Model of cellular immortalization in association with genomic instability. As illustrated in mouse embryonic fibroblasts, immortality is induced by genomic instability and mutation of either Arf or p53. Growth arrest and the downregulation of H2AX protect mouse embryonic fibroblasts against immortalization. Because H2AX downregulation is dependent on the ARF/p53 module, cells with mutations in this module recover H2AX expression and growth activity. CIN: Chromosomal instability; MSI: Microsatellite instability.
It was generally believed that p53 suppresses tumors by inducing apoptosis and senescence[44-47], but recent studies have cast doubt on this hypothesis. For example, in multiple transgenic mouse models in which p53 cannot induce cell-cycle arrest and apoptosis after DNA damage, the number of malignancies that form as a result is not significantly higher than the rate of malignancies in normal mice[48-50]. These observations raise questions regarding the exact role of p53 in tumor suppression. One hypothesis is that p53 regulates metabolism by inhibiting glycolysis[51-53] and/or activating the mammalian target of rapamycin pathway[52,54,55].
EFFECT OF MUTATIONS IN THE ARF/P53 MODULE
Because ARF and p53 are mutated in cancer cells in a mutually exclusive manner, it is likely that the p53 functions that are essential for cancer suppression are expressed under the control of ARF[56]. Intriguingly, transgenic mice with an extra copy of Arf and p53 (super-Arf/p53 mice) exhibit both reduced rates of cancer and slower overall aging[41]. Furthermore, MEFs derived from these mice rarely immortalize spontaneously. Therefore, it is likely that the roles of ARF and p53 in cancer suppression are involved in the maintenance of homeostasis in vivo and are primarily regulated at the level of individual cells. This cellular-level protection against transformation is generally associated with growth arrest and low levels of the histone H2A variant H2AX[27,57], which is required for active cell growth[58].
The downregulation of H2AX is governed by the ARF/p53 module[27,42]. Growth-arrested cells in the liver, spleen, and other organs of healthy adult mice generally exhibit downregulated levels of H2AX, whereas immortalized/transformed cells, which have mutations in the ARF/p53 module, have normal H2AX levels and exhibit active growth[27,57].
As described above, cellular transformation is suppressed primarily by the induction of growth arrest and the downregulation of H2AX under the control of the ARF/p53 module. It is promoted by genomic instability and the mutations it produces. Unfortunately, when H2AX is downregulated, the mechanisms that repair lesions fail and this is a risk factor for the genomic instability and tumorigenesis seen in many hereditary cancers[15-25]. Despite the aforementioned protective effect of H2AX downregulation, the risk of sporadic cancer development is probably due, in large part, to a reduction in H2AX levels and the associated repair deficiencies[27]. In fact, cells without H2AX exhibit faulty homologous recombination and non-homologous end-joining during DNA repair[59,60], which results in elevated genomic instability[58].
CIC DEVELOPMENT IS DISTINCT FROM IMMORTALIZATION
Although immortalized MEFs develop in association with genomic instability and mutations in the ARF/p53 module[26,41], the resultant cells do not exhibit robust tumor-forming ability unless they are pre-transformed by oncogenes[61,62]. This observation illustrates the difference between immortalized MEFs and CICs. MEFs are differentiated cells and do not exhibit stem cell characteristics, but CICs act like stem cells in cancer-tissue development. In fact, CICs share a number of common characteristics with normal stem cells, including embryonic stem (ES) cells and induced pluripotent stem cells, but do not resemble immortalized MEFs with their high dependence on normal glycolysis[63,64], sphere-forming ability, and expressed stem cell marker genes[65-67]. Cancer tissues generally exhibit the “Warburg Effect” seen in normal stem cells that produce elevated glycolysis.
Transformation of normal stem cells into CICs after genomic destabilization
To truly understand cancer, we must discover how genomic instability promotes the development of CICs. CICs may develop via multiple pathways. A recent study using normal murine ES cells as a model showed that one of these possible pathways might be created when normal stem cells are developing under unstable genomic conditions. Under aberrant differentiation conditions, ES cells with the ability to form normal mice develop high levels of DNA lesions. These lesions induce cellular senescence and genomic instability, ultimately leading to renewed growth in cells that harbor mutations in the ARF/p53 module[68]. These cells exhibit a number of stem cell characteristics, including sphere formation and the expression of undifferentiated marker genes such as Nanog, Klf4, Oct3/4, and Sox2, even in a growth factor–poor medium containing 10% newborn bovine serum with no leukemia inhibitory factor. Furthermore, these cells possess tumor-forming ability and express c-Myc and CIC markers such as CD133, CD33, and CD34.
The fact that oncogenesis can be triggered by niche disruption supports this theory. For example, both leukemia and myelodysplasia develop after dysregulation of the stem cell niche[69], and cancers often develop from stem cells that are injected at heterotropic sites[70]. Stem cells growing in such an aberrant environment do not get what they need to maintain themselves properly and often transform into CICs[70]. Another example is embryonal carcinomas that can develop from cells transplanted from the inside of blastocysts and from primordial germ cells derailed from the migration track[71,72]. These in vivo and in vitro findings suggest that the genomic instability that leads to the development of CICs can be triggered by aberrations in the environment when stem cells are differentiating.
Because cancer development is initiated by CICs, these cells are considered promising targets for chemotherapy aimed at either killing them directly or getting them to differentiate[73-75]. However, this strategy is not always successful, partly because CICs are so adaptable and plastic[76,77]. In one study in which CICs developed from ES cells, transformation was coupled with the acquisition of plasticity. ES cells start to differentiate in response to leukemia inhibitory factor withdrawal and become senescent when differentiation conditions are aberrant; but the CICs that result re-acquire stem cell characteristics as they develop genomic instability, even under conditions in which stem cells ordinarily do not thrive and cannot be cultivated[68]. CICs that re-acquire stemness under these conditions maintain stem cell characteristics with great persistence, and it is difficult to get them to differentiate completely.
RELEVANCE OF IN VITRO MODELS TO CANCER DEVELOPMENT
Unlike in vitro malignancy transformation, in vivo cancer development has multiple steps that include abrogating organic regulation, which results in the development of benign tumors such as adenomatous polyposis coli mutations in the colon[78,79], and losing the ARF/p53 module that leads to uncontrolled growth and the development of malignancies[80,81]. Each organ in a complex animal is regulated by its own specific mechanisms that keep the organ in homeostasis, which can malfunction and allow tissues to become precancerous or malignant when the genome becomes unstable or the mutations affect the ARF/p53 module[40,56].
CONCLUSION
Genomic instability contributes to the development of CICs by directly transforming somatic stem cells, reprogramming differentiated cancer cells, and a number of other mechanisms. Several lines of evidence suggest that one of these pathways is triggered by the disruption of the niche environment (Figure 2). Maintenance of the niche environment is essential for somatic stem cell maintenance and differentiation and homeostasis (Figure 2A). The stem cell niche can be damaged by exogenous stresses that cause irregular stem cell differentiation, the accumulation of DNA damage, and the induction of senescence (Figure 2B). DNA damage often triggers genomic destabilization, which can promote the development of precancerous (Figure 2C) and cancerous (Figure 2D) lesions through the maintenance of small pockets of pluripotent stem cells that eventually become CICs.
Figure 2.
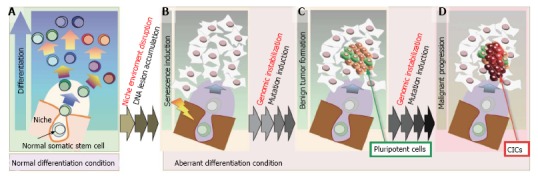
Model of cancer-initiating cell development. A: In normal tissues, stem cells are maintained in a specific niche and the niche environment is important both for stem cell maintenance and organ homeostasis; B: Niche disruption deranges the differentiation environment of stem cells, leading to the accumulation of DNA lesions and the induction of cellular senescence. Such DNA lesions trigger genomic destabilization, leading to the development of benign tumors that contain a small number of C: Pluripotent cells that eventually develop into; D: Cancer-initiating cells (CICs) that cause malignancies.
If stem cells start to differentiate under unfavorable differentiation conditions, they become senescent, but can start developing again and reacquire stem cell characteristics when conditions change. Stemness recovered under these circumstances is associated with robust plasticity and the ability of these cells to self-renew, even under conditions in which normal stem cells do not thrive. Unfortunately, such cells do not differentiate completely and remain permanently in less developed states that often lead to malignancies.
Footnotes
P- Reviewer: Freitas VM, Hasty P, Huang YH, Kiselev SL S- Editor: Song XX L- Editor: A E- Editor: Lu YJ
Supported by Funding from the National Cancer Center Research and Development Fund Grant, No. 23-C-10; Grants-in-Aid for Scientific Research MEXT KAKENHI, No. 20770136; and Grants-in-Aid for JSPS Fellows.
Open-Access: This article is an open-access article which was selected by an in-house editor and fully peer-reviewed by external reviewers. It is distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Peer-review started: July 25, 2014
First decision: August 28, 2014
Article in press: November 10, 2014
References
- 1.Perez-Losada J, Balmain A. Stem-cell hierarchy in skin cancer. Nat Rev Cancer. 2003;3:434–443. doi: 10.1038/nrc1095. [DOI] [PubMed] [Google Scholar]
- 2.Ratajczak MZ. Cancer stem cells--normal stem cells “Jedi” that went over to the “dark side”. Folia Histochem Cytobiol. 2005;43:175–181. [PubMed] [Google Scholar]
- 3.Li F, Tiede B, Massagué J, Kang Y. Beyond tumorigenesis: cancer stem cells in metastasis. Cell Res. 2007;17:3–14. doi: 10.1038/sj.cr.7310118. [DOI] [PubMed] [Google Scholar]
- 4.Lawson DA, Witte ON. Stem cells in prostate cancer initiation and progression. J Clin Invest. 2007;117:2044–2050. doi: 10.1172/JCI32810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bleau AM, Agliano A, Larzabal L, de Aberasturi AL, Calvo A. Metastatic dormancy: a complex network between cancer stem cells and their microenvironment. Histol Histopathol. 2014;29:1499–510. doi: 10.14670/HH-29.1499. [DOI] [PubMed] [Google Scholar]
- 6.Ni J, Cozzi PJ, Hao JL, Beretov J, Chang L, Duan W, Shigdar S, Delprado WJ, Graham PH, Bucci J, et al. CD44 variant 6 is associated with prostate cancer metastasis and chemo-/radioresistance. Prostate. 2014;74:602–617. doi: 10.1002/pros.22775. [DOI] [PubMed] [Google Scholar]
- 7.Carroll WL, Bhojwani D, Min DJ, Raetz E, Relling M, Davies S, Downing JR, Willman CL, Reed JC. Pediatric acute lymphoblastic leukemia. Hematology Am Soc Hematol Educ Program. 2003:102–131. doi: 10.1182/asheducation-2003.1.102. [DOI] [PubMed] [Google Scholar]
- 8.Greaves M. Childhood leukaemia. BMJ. 2002;324:283–287. doi: 10.1136/bmj.324.7332.283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Huether R, Dong L, Chen X, Wu G, Parker M, Wei L, Ma J, Edmonson MN, Hedlund EK, Rusch MC, et al. The landscape of somatic mutations in epigenetic regulators across 1,000 paediatric cancer genomes. Nat Commun. 2014;5:3630. doi: 10.1038/ncomms4630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Garcia-Manero G, Jeha S, Daniel J, Williamson J, Albitar M, Kantarjian HM, Issa JP. Aberrant DNA methylation in pediatric patients with acute lymphocytic leukemia. Cancer. 2003;97:695–702. doi: 10.1002/cncr.11090. [DOI] [PubMed] [Google Scholar]
- 11.Lengauer C, Kinzler KW, Vogelstein B. Genetic instability in colorectal cancers. Nature. 1997;386:623–627. doi: 10.1038/386623a0. [DOI] [PubMed] [Google Scholar]
- 12.Lengauer C, Kinzler KW, Vogelstein B. Genetic instabilities in human cancers. Nature. 1998;396:643–649. doi: 10.1038/25292. [DOI] [PubMed] [Google Scholar]
- 13.Shih IM, Zhou W, Goodman SN, Lengauer C, Kinzler KW, Vogelstein B. Evidence that genetic instability occurs at an early stage of colorectal tumorigenesis. Cancer Res. 2001;61:818–822. [PubMed] [Google Scholar]
- 14.Hveem TS, Merok MA, Pretorius ME, Novelli M, Bævre MS, Sjo OH, Clinch N, Liestøl K, Svindland A, Lothe RA, et al. Prognostic impact of genomic instability in colorectal cancer. Br J Cancer. 2014;110:2159–2164. doi: 10.1038/bjc.2014.133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Poulogiannis G, Frayling IM, Arends MJ. DNA mismatch repair deficiency in sporadic colorectal cancer and Lynch syndrome. Histopathology. 2010;56:167–179. doi: 10.1111/j.1365-2559.2009.03392.x. [DOI] [PubMed] [Google Scholar]
- 16.Saletti P, Edwin ID, Pack K, Cavalli F, Atkin WS. Microsatellite instability: application in hereditary non-polyposis colorectal cancer. Ann Oncol. 2001;12:151–160. doi: 10.1023/a:1008342420825. [DOI] [PubMed] [Google Scholar]
- 17.Yacoub G, Nagalla S, Aklilu M. Oncologic management of hereditary colorectal cancer. Clin Colon Rectal Surg. 2012;25:118–122. doi: 10.1055/s-0032-1313783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhou T, Hasty P, Walter CA, Bishop AJ, Scott LM, Rebel VI. Myelodysplastic syndrome: an inability to appropriately respond to damaged DNA? Exp Hematol. 2013;41:665–674. doi: 10.1016/j.exphem.2013.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Burgess M, Puhalla S. BRCA 1/2-Mutation Related and Sporadic Breast and Ovarian Cancers: More Alike than Different. Front Oncol. 2014;4:19. doi: 10.3389/fonc.2014.00019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ewald IP, Ribeiro PL, Palmero EI, Cossio SL, Giugliani R, Ashton-Prolla P. Genomic rearrangements in BRCA1 and BRCA2: A literature review. Genet Mol Biol. 2009;32:437–446. doi: 10.1590/S1415-47572009005000049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Liu Y, West SC. Distinct functions of BRCA1 and BRCA2 in double-strand break repair. Breast Cancer Res. 2002;4:9–13. doi: 10.1186/bcr417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Metcalfe KA, Finch A, Poll A, Horsman D, Kim-Sing C, Scott J, Royer R, Sun P, Narod SA. Breast cancer risks in women with a family history of breast or ovarian cancer who have tested negative for a BRCA1 or BRCA2 mutation. Br J Cancer. 2009;100:421–425. doi: 10.1038/sj.bjc.6604830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cleaver JE. Common pathways for ultraviolet skin carcinogenesis in the repair and replication defective groups of xeroderma pigmentosum. J Dermatol Sci. 2000;23:1–11. doi: 10.1016/s0923-1811(99)00088-2. [DOI] [PubMed] [Google Scholar]
- 24.Gratchev A, Strein P, Utikal J, Sergij G. Molecular genetics of Xeroderma pigmentosum variant. Exp Dermatol. 2003;12:529–536. doi: 10.1034/j.1600-0625.2003.00124.x. [DOI] [PubMed] [Google Scholar]
- 25.Friedberg EC. How nucleotide excision repair protects against cancer. Nat Rev Cancer. 2001;1:22–33. doi: 10.1038/35094000. [DOI] [PubMed] [Google Scholar]
- 26.Ichijima Y, Yoshioka K, Yoshioka Y, Shinohe K, Fujimori H, Unno J, Takagi M, Goto H, Inagaki M, Mizutani S, et al. DNA lesions induced by replication stress trigger mitotic aberration and tetraploidy development. PLoS One. 2010;5:e8821. doi: 10.1371/journal.pone.0008821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Atsumi Y, Fujimori H, Fukuda H, Inase A, Shinohe K, Yoshioka Y, Shikanai M, Ichijima Y, Unno J, Mizutani S, et al. Onset of quiescence following p53 mediated down-regulation of H2AX in normal cells. PLoS One. 2011;6:e23432. doi: 10.1371/journal.pone.0023432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yoshida R, Miyashita K, Inoue M, Shimamoto A, Yan Z, Egashira A, Oki E, Kakeji Y, Oda S, Maehara Y. Concurrent genetic alterations in DNA polymerase proofreading and mismatch repair in human colorectal cancer. Eur J Hum Genet. 2011;19:320–325. doi: 10.1038/ejhg.2010.216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.The Cancer Genome Atlas Network. Comprehensive molecular characterization of human colon and rectal cancer. Nature. 2012;487:330–337. doi: 10.1038/nature11252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lawrence MS, Stojanov P, Mermel CH, Robinson JT, Garraway LA, Golub TR, Meyerson M, Gabriel SB, Lander ES, Getz G. Discovery and saturation analysis of cancer genes across 21 tumour types. Nature. 2014;505:495–501. doi: 10.1038/nature12912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Razzak M. Genetics: new molecular classification of gastric adenocarcinoma proposed by The Cancer Genome Atlas. Nat Rev Clin Oncol. 2014;11:499. doi: 10.1038/nrclinonc.2014.138. [DOI] [PubMed] [Google Scholar]
- 32.Stephens PJ, Greenman CD, Fu B, Yang F, Bignell GR, Mudie LJ, Pleasance ED, Lau KW, Beare D, Stebbings LA, et al. Massive genomic rearrangement acquired in a single catastrophic event during cancer development. Cell. 2011;144:27–40. doi: 10.1016/j.cell.2010.11.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Pleasance ED, Cheetham RK, Stephens PJ, McBride DJ, Humphray SJ, Greenman CD, Varela I, Lin ML, Ordóñez GR, Bignell GR, et al. A comprehensive catalogue of somatic mutations from a human cancer genome. Nature. 2010;463:191–196. doi: 10.1038/nature08658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Stephens PJ, McBride DJ, Lin ML, Varela I, Pleasance ED, Simpson JT, Stebbings LA, Leroy C, Edkins S, Mudie LJ, et al. Complex landscapes of somatic rearrangement in human breast cancer genomes. Nature. 2009;462:1005–1010. doi: 10.1038/nature08645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Grichnik JM. Genomic instability and tumor stem cells. J Invest Dermatol. 2006;126:1214–1216. doi: 10.1038/sj.jid.5700240. [DOI] [PubMed] [Google Scholar]
- 36.Li L, Borodyansky L, Yang Y. Genomic instability en route to and from cancer stem cells. Cell Cycle. 2009;8:1000–1002. doi: 10.4161/cc.8.7.8041. [DOI] [PubMed] [Google Scholar]
- 37.Danes BS. Increased in vitro tetraploidy: tissue specific within the heritable colorectal cancer syndromes with polyposis coli. Cancer. 1978;41:2330–2334. doi: 10.1002/1097-0142(197806)41:6<2330::aid-cncr2820410635>3.0.co;2-x. [DOI] [PubMed] [Google Scholar]
- 38.Dutrillaux B, Gerbault-Seureau M, Remvikos Y, Zafrani B, Prieur M. Breast cancer genetic evolution: I. Data from cytogenetics and DNA content. Breast Cancer Res Treat. 1991;19:245–255. doi: 10.1007/BF01961161. [DOI] [PubMed] [Google Scholar]
- 39.Heselmeyer K, Schröck E, du Manoir S, Blegen H, Shah K, Steinbeck R, Auer G, Ried T. Gain of chromosome 3q defines the transition from severe dysplasia to invasive carcinoma of the uterine cervix. Proc Natl Acad Sci USA. 1996;93:479–484. doi: 10.1073/pnas.93.1.479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Maley CC, Galipeau PC, Li X, Sanchez CA, Paulson TG, Blount PL, Reid BJ. The combination of genetic instability and clonal expansion predicts progression to esophageal adenocarcinoma. Cancer Res. 2004;64:7629–7633. doi: 10.1158/0008-5472.CAN-04-1738. [DOI] [PubMed] [Google Scholar]
- 41.Matheu A, Maraver A, Klatt P, Flores I, Garcia-Cao I, Borras C, Flores JM, Viña J, Blasco MA, Serrano M. Delayed ageing through damage protection by the Arf/p53 pathway. Nature. 2007;448:375–379. doi: 10.1038/nature05949. [DOI] [PubMed] [Google Scholar]
- 42.Osawa T, Atsumi Y, Sugihara E, Saya H, Kanno M, Tashiro F, Masutani M, Yoshioka K. Arf and p53 act as guardians of a quiescent cellular state by protecting against immortalization of cells with stable genomes. Biochem Biophys Res Commun. 2013;432:34–39. doi: 10.1016/j.bbrc.2013.01.091. [DOI] [PubMed] [Google Scholar]
- 43.Yoshioka K, Atsumi Y, Fukuda H, Masutani M, Teraoka H. The Quiescent Cellular State is Arf/p53-Dependent and Associated with H2AX Downregulation and Genome Stability. Int J Mol Sci. 2012;13:6492–6506. doi: 10.3390/ijms13056492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Rufini A, Tucci P, Celardo I, Melino G. Senescence and aging: the critical roles of p53. Oncogene. 2013;32:5129–5143. doi: 10.1038/onc.2012.640. [DOI] [PubMed] [Google Scholar]
- 45.Vigneron A, Vousden KH. p53, ROS and senescence in the control of aging. Aging (Albany NY) 2010;2:471–474. doi: 10.18632/aging.100189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zuckerman V, Wolyniec K, Sionov RV, Haupt S, Haupt Y. Tumour suppression by p53: the importance of apoptosis and cellular senescence. J Pathol. 2009;219:3–15. doi: 10.1002/path.2584. [DOI] [PubMed] [Google Scholar]
- 47.Choisy-Rossi C, Reisdorf P, Yonish-Rouach E. Mechanisms of p53-induced apoptosis: in search of genes which are regulated during p53-mediated cell death. Toxicol Lett. 1998;102-103:491–496. doi: 10.1016/s0378-4274(98)00238-0. [DOI] [PubMed] [Google Scholar]
- 48.Brady CA, Jiang D, Mello SS, Johnson TM, Jarvis LA, Kozak MM, Kenzelmann Broz D, Basak S, Park EJ, McLaughlin ME, et al. Distinct p53 transcriptional programs dictate acute DNA-damage responses and tumor suppression. Cell. 2011;145:571–583. doi: 10.1016/j.cell.2011.03.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Li T, Kon N, Jiang L, Tan M, Ludwig T, Zhao Y, Baer R, Gu W. Tumor suppression in the absence of p53-mediated cell-cycle arrest, apoptosis, and senescence. Cell. 2012;149:1269–1283. doi: 10.1016/j.cell.2012.04.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Valente LJ, Gray DH, Michalak EM, Pinon-Hofbauer J, Egle A, Scott CL, Janic A, Strasser A. p53 efficiently suppresses tumor development in the complete absence of its cell-cycle inhibitory and proapoptotic effectors p21, Puma, and Noxa. Cell Rep. 2013;3:1339–1345. doi: 10.1016/j.celrep.2013.04.012. [DOI] [PubMed] [Google Scholar]
- 51.Blagosklonny MV. Tumor suppression by p53 without apoptosis and senescence: conundrum or rapalog-like gerosuppression? Aging (Albany NY) 2012;4:450–455. doi: 10.18632/aging.100475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Feng Z, Hu W, de Stanchina E, Teresky AK, Jin S, Lowe S, Levine AJ. The regulation of AMPK beta1, TSC2, and PTEN expression by p53: stress, cell and tissue specificity, and the role of these gene products in modulating the IGF-1-AKT-mTOR pathways. Cancer Res. 2007;67:3043–3053. doi: 10.1158/0008-5472.CAN-06-4149. [DOI] [PubMed] [Google Scholar]
- 53.Liu J, Zhang C, Feng Z. Tumor suppressor p53 and its gain-of-function mutants in cancer. Acta Biochim Biophys Sin (Shanghai) 2014;46:170–179. doi: 10.1093/abbs/gmt144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Budanov AV, Karin M. p53 target genes sestrin1 and sestrin2 connect genotoxic stress and mTOR signaling. Cell. 2008;134:451–460. doi: 10.1016/j.cell.2008.06.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Zhang XD, Qin ZH, Wang J. The role of p53 in cell metabolism. Acta Pharmacol Sin. 2010;31:1208–1212. doi: 10.1038/aps.2010.151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Matheu A, Maraver A, Serrano M. The Arf/p53 pathway in cancer and aging. Cancer Res. 2008;68:6031–6034. doi: 10.1158/0008-5472.CAN-07-6851. [DOI] [PubMed] [Google Scholar]
- 57.Atsumi Y, Inase A, Osawa T, Sugihara E, Sakasai R, Fujimori H, Teraoka H, Saya H, Kanno M, Tashiro F, et al. The Arf/p53 protein module, which induces apoptosis, down-regulates histone H2AX to allow normal cells to survive in the presence of anti-cancer drugs. J Biol Chem. 2013;288:13269–13277. doi: 10.1074/jbc.M112.402560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Bonner WM, Redon CE, Dickey JS, Nakamura AJ, Sedelnikova OA, Solier S, Pommier Y. GammaH2AX and cancer. Nat Rev Cancer. 2008;8:957–967. doi: 10.1038/nrc2523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Bassing CH, Alt FW. H2AX may function as an anchor to hold broken chromosomal DNA ends in close proximity. Cell Cycle. 2004;3:149–153. doi: 10.4161/cc.3.2.689. [DOI] [PubMed] [Google Scholar]
- 60.Celeste A, Petersen S, Romanienko PJ, Fernandez-Capetillo O, Chen HT, Sedelnikova OA, Reina-San-Martin B, Coppola V, Meffre E, Difilippantonio MJ, et al. Genomic instability in mice lacking histone H2AX. Science. 2002;296:922–927. doi: 10.1126/science.1069398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Newbold RF, Overell RW. Fibroblast immortality is a prerequisite for transformation by EJ c-Ha-ras oncogene. Nature. 1983;304:648–651. doi: 10.1038/304648a0. [DOI] [PubMed] [Google Scholar]
- 62.Land H, Parada LF, Weinberg RA. Tumorigenic conversion of primary embryo fibroblasts requires at least two cooperating oncogenes. Nature. 1983;304:596–602. doi: 10.1038/304596a0. [DOI] [PubMed] [Google Scholar]
- 63.Bunting KD. ABC transporters as phenotypic markers and functional regulators of stem cells. Stem Cells. 2002;20:11–20. doi: 10.1002/stem.200011. [DOI] [PubMed] [Google Scholar]
- 64.Kondoh H, Lleonart ME, Gil J, Wang J, Degan P, Peters G, Martinez D, Carnero A, Beach D. Glycolytic enzymes can modulate cellular life span. Cancer Res. 2005;65:177–185. [PubMed] [Google Scholar]
- 65.Leahy A, Xiong JW, Kuhnert F, Stuhlmann H. Use of developmental marker genes to define temporal and spatial patterns of differentiation during embryoid body formation. J Exp Zool. 1999;284:67–81. doi: 10.1002/(sici)1097-010x(19990615)284:1<67::aid-jez10>3.0.co;2-o. [DOI] [PubMed] [Google Scholar]
- 66.Seaberg RM, van der Kooy D. Stem and progenitor cells: the premature desertion of rigorous definitions. Trends Neurosci. 2003;26:125–131. doi: 10.1016/S0166-2236(03)00031-6. [DOI] [PubMed] [Google Scholar]
- 67.Yang YM, Chang JW. Current status and issues in cancer stem cell study. Cancer Invest. 2008;26:741–755. doi: 10.1080/07357900801901856. [DOI] [PubMed] [Google Scholar]
- 68.Fujimori H, Shikanai M, Teraoka H, Masutani M, Yoshioka K. Induction of cancerous stem cells during embryonic stem cell differentiation. J Biol Chem. 2012;287:36777–36791. doi: 10.1074/jbc.M112.372557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Raaijmakers MH, Mukherjee S, Guo S, Zhang S, Kobayashi T, Schoonmaker JA, Ebert BL, Al-Shahrour F, Hasserjian RP, Scadden EO, et al. Bone progenitor dysfunction induces myelodysplasia and secondary leukaemia. Nature. 2010;464:852–857. doi: 10.1038/nature08851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Knoepfler PS. Deconstructing stem cell tumorigenicity: a roadmap to safe regenerative medicine. Stem Cells. 2009;27:1050–1056. doi: 10.1002/stem.37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Morange M. What history tells us VII. Twenty-five years ago: the production of mouse embryonic stem cells. J Biosci. 2006;31:537–541. doi: 10.1007/BF02708404. [DOI] [PubMed] [Google Scholar]
- 72.Wylie C. Germ cells. Cell. 1999;96:165–174. doi: 10.1016/s0092-8674(00)80557-7. [DOI] [PubMed] [Google Scholar]
- 73.Sell S. Stem cell origin of cancer and differentiation therapy. Crit Rev Oncol Hematol. 2004;51:1–28. doi: 10.1016/j.critrevonc.2004.04.007. [DOI] [PubMed] [Google Scholar]
- 74.Duggal R, Minev B, Geissinger U, Wang H, Chen NG, Koka PS, Szalay AA. Biotherapeutic approaches to target cancer stem cells. J Stem Cells. 2013;8:135–149. [PubMed] [Google Scholar]
- 75.Rajan P, Srinivasan R. Targeting cancer stem cells in cancer prevention and therapy. Stem Cell Rev. 2008;4:211–216. doi: 10.1007/s12015-008-9037-x. [DOI] [PubMed] [Google Scholar]
- 76.Pinto CA, Widodo E, Waltham M, Thompson EW. Breast cancer stem cells and epithelial mesenchymal plasticity - Implications for chemoresistance. Cancer Lett. 2013;341:56–62. doi: 10.1016/j.canlet.2013.06.003. [DOI] [PubMed] [Google Scholar]
- 77.Tang DG. Understanding cancer stem cell heterogeneity and plasticity. Cell Res. 2012;22:457–472. doi: 10.1038/cr.2012.13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Boman BM, Fields JZ. An APC: WNT Counter-Current-Like Mechanism Regulates Cell Division Along the Human Colonic Crypt Axis: A Mechanism That Explains How APC Mutations Induce Proliferative Abnormalities That Drive Colon Cancer Development. Front Oncol. 2013;3:244. doi: 10.3389/fonc.2013.00244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Najdi R, Holcombe RF, Waterman ML. Wnt signaling and colon carcinogenesis: beyond APC. J Carcinog. 2011;10:5. doi: 10.4103/1477-3163.78111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Farber E. The multistep nature of cancer development. Cancer Res. 1984;44:4217–4223. [PubMed] [Google Scholar]
- 81.Vogelstein B, Kinzler KW. The multistep nature of cancer. Trends Genet. 1993;9:138–141. doi: 10.1016/0168-9525(93)90209-z. [DOI] [PubMed] [Google Scholar]