

Abstract
Angiogenesis is the formation of new blood vessels from preexisting vascular network that plays an important role in the tumor growth, invasion and metastasis. Anti-angiogenesis targeting tyrosine kinases such as vascular endothelial growth factor receptor 2 (VEGFR2) and platelet derived growth factor receptor β (PDGFRβ) constitutes a successful target for the treatment of cancer. In this work, molecular docking studies of three bioflavanoid such as indigocarpan, mucronulatol, indigocarpan diacetate and two diterpenes namely erythroxydiol X and Y derived from Indigofera aspalathoides as PDGFRβ and VEGFR2 inhibitors were performed using computational tools. The crystal structures of two target proteins were retrieved from PDB website. Among the five compounds investigated, indigocarpan exhibited potent binding energy ΔG = -7.04 kcal/mol with VEGFR2 and ΔG = -4.82 with PDGFRβ compared to commercially available anti-angiogenic drug sorafenib (positive control). Our results strongly suggested that indigocarpan is a potent angiogenesis inhibitor as ascertained by its potential interaction with VEGFR2 and PDGFRβ. This hypothesis provides a better insight to control metastasis by blocking angiogenesis.
Keywords: VEGFR2, PDGFRβ, Angiogenesis, Lipinski rules, Autodock, Indigocarpan
Background
Receptor tyrosine kinases (RTKs) constitutes a large family of growth factors all of which contain an integral protein tyrosine kinase (PTK). Growths factors like PDGF, VEGF, EGF, FGF and IGF-1 belong to RTKs are involved in regulating cellular growth and development [1]. Angiogenesis is the sprouting of new blood vessels from existing vasculature, is a crucial process in tumor growth and metastasis. This process is critical in normal physiological development but excessive angiogenesis is a common cause in a wide range of pathologies, most notably cancer [2]. During tumor growth, transformed cells secrete a group of pro-angiogenic proteins such as vascular endothelial growth factors (VEGFs), fibroblast growth factors (FGFs) and platelet derived growth factors (PDGFs) [3]. Among the many proangiogenic factors, VEGF and PDGF have been identified as the most important regulator of tumor angiogenesis [4]. These proteins stimulate endothelial cell proliferation, migration and vascular remodeling, which contribute to tumor neovascularization. An enriched blood supply provides the tumor with nutrients for further growth and facilitates invasion and metastasis [5].
Retardation of angiogenesis process by interfering these signaling cascades have been emerged as one of the most potential new approaches for the treatment of cancers [6]. Out of the three VEGF receptors, vascular endothelial growth factor receptor 2 (VEGFR2) is reported to be the most important regulator of cancer angiogenesis, mediating the majority of angiogenic effects of VEGF-A [7]. VEFG receptor activation depends on PDGF upon activation of PDGF receptor, VEGF-A binding to VEGFR2 promotes receptor dimerization, tyrosine kinase activation and trans-autophosphorylation of specific tyrosine residues within the cytoplasmic domain [8]. Plateletderived growth factors (PDGFs) represent a group of growth factors consists of five different disulphide-linked dimers, PDGF-AA, -BB,-AB, -CC and -DD that act through the two receptors PDGFRα and PDGFRβ [9]. After receptor activation, several intracellular pathways are stimulated; leading to cell proliferation and several other crucial processes that play a significant role in angiogenesis [10]. Blocking of PDGF receptor beta and VEGFR2 retards angiogenesis, vascular maturation and cell proliferation leading to tumor regression. Bevacizumab (Avastin), Sunitinib malate and Sorafenib, are inhibitors of VEGFR2 and PDGFRβ approved by FDA [11].
The medicinal plant Indigofera aspalathoides (Family- Fabaceace) found in South India and Srilanka and is traditionally used for treating various skin disorders and tumors. Phytochemical and Pharmacological studies have been investigated much due to its anti-cancerous and antioxidant property [12]. The major bioactive compounds are indigocarpan, indigocarpan diacetate, mucronulatol, erythroxydiol X and erythroxydiol Y [13]. Thus, plant derived natural bioactive compounds can be a better way to find a new potential anti-PDGF and VEGF agents with less side effects to control metastasis by angiogenesis through interfering tyrosine kinases. Many reports are available on phytochemistry, and pharmacological action of I. aspalathoides, the main bioactive compounds and their mechanism of action to control the angiogenesis not been reported. In this perspective, in the present work I. aspalathoides׳s key metabolites indigocarpan, indigocarpan diacetate, mucronulatol, erythroxydiol X and erythroxydiol Y were studied for their inhibitory activity on PDGF and VEGF receptor tyrosine phosphorylation. Different cheminformatics approaches like target identification, active site prediction, drug likeliness properties, ADMET properties, drug metabolism, biological activity, molecular docking of selected phytoligands with VEGFR2 and PDGFRβ were studied.
Methodology
Hardware and Software used:
All the computational studies were executed by the PC windows 7 ultimate with Intel Core i5 microprocessor, 4 GB memory and 32 Bit operating system. We used biological databases such as PubChem, PDB (Protein Data Bank) and PharmMapper server. Online tools such as Molinspiration, Osiris property explorer, admetSAR, Online pass server and Meta print 2D and software׳s like Autodock 4.2, Chemdraw Ultra 6.0 were used.
Potential therapeutic target identification:
The precise identification of drug target was performed by PharmMapper server (http://59.78.96.61/pharmmapper). It is a freely accessed web server designed to identify potential target candidates for the given plant derived small molecules by means of reverse pharmacophore mapping approach. This model is supported by a large repertoire of pharmacophore database composed of more than 7000 receptor based pharmacophore models that are extracted from Target Bank, BindingDB, Drug Bank and Potential Drug Target Database. A strategy algorithm of sequential combination of triangle hashing and genetic algorithm was designed to solve the molecule pharmacophore best fitting task. The possibility of potential interaction between compounds derived from I. aspalathoides and protein tyrosine kinase receptors (PDGFRβ & VEGFR2) were studied using this server. The SDF format was submitted to the pharmMapper server to find out fit score. The target set was limited to human targets, and all parameters were kept as default [14].
Active Site prediction:
The active sites of selected target proteins were identified by using CASTp server (Computed Atlas of Surface Topography of proteins) (http://cast.engr.uic.edu). It detects all the feasible pockets in the protein structure. It measures analytically the area and volume of each pocket and cavity, both in solvent accessible surface (SA, Richards׳ surface) and molecular surface (MS, Connolly׳s surface) [15]. The first pocket was chosen as the biologically most favorable active site for docking studies.
Preparation of phyto-ligand:
The 3D structure of the ligand molecule Mucronulatol was retrieved from pubchem ( http: // pubchem.ncbi.nlm.nih.gov/pccompound) in SDF format, and converted to PDB format using Accryl Discovery studio visualizer 3.5 software. Other ligands were drawn by using Chemdraw ultra 6.0 and are shown in (Figure 1).
Figure 1.
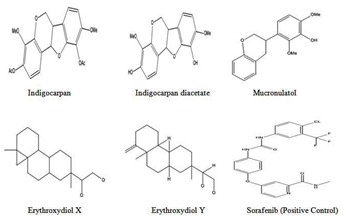
Structure of various phytoligands reported in I.aspalathoides
Preparation of target protein:
Two targets VEGFR2 (PDB ID: 3VHE) and PDGFRβ (PDB ID: 3MJG) retrieved from PDB website were viewed by discovery studio visualizer 3.5 (Figure 2) and used for docking simulation. The crystal structures of target proteins were retrieved from RCSB protein data bank ( http://www.rcsb.org/pdb/home/home.do). The VEGFR2 protein was resolved by X-ray diffraction method with a resolution factor of 1.55 Å, R value 0.186 and other target protein PDGFRβ was resolved by X-Ray diffraction method with a resolution factor 2.30 Å, R value 0.237. After obtaining the pdb format of proteins, they were processed by removing native ligand and crystalline water from the structure and subjected for docking studies.
Figure 2.
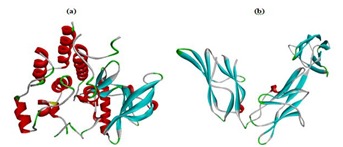
The three dimensional structure of selected target proteins a) VEGFR2 – 3VHE; b) PDGFRβ (Only X Chain – 3MJG)
In silico pharmacokinetics analysis:
a) Molinspiration:
Molecular descriptors and drug likeliness properties of compounds were analyzed using the tool Molinspiration server (http://www.molinspiration.com), based on Lipinski Rules of five [16]. The rule states that most “druglike“ molecules must have log P≤ 5, molecular weight ≤ 500, number of hydrogen bond acceptors ≤ 10, and number of hydrogen bond donors ≤ 5. Molecules violating more than one of these rules may have problems with oral bioavailability. Molinspiration supports for calculation of important molecular properties such as (LogP, polar surface area, number of hydrogen bond donors and acceptors), as well as prediction of bioactivity score for the most important drug targets (GPCR ligands, kinase inhibitors, ion channel modulators, enzymes and nuclear receptors [17]. TPSA was used to calculate the percentage of absorption (%ABS) using the equation reported by [18]. Percentage of absorbance = 109 − 0.345 × TPSA.
b) admetSAR Predictions:
The pharmacokinetic properties such as Absorption, Distribution, Metabolism, Excretion and the Toxicity of the compounds can be predicted using admetSAR (http://www.admetexp.org) database. In admetSAR, web based query tools incorporating a molecular build-in interface enable the database to be queried by SMILES and structural similarity search. It provides the latest and most comprehensive manually curated data for diverse chemicals associated with known ADMET profiles (admetSAR@LMMD) [19].
c) Toxicity risk assessment:
To identify the any undesirable toxic properties of our compounds, Osiris Property Explorer ( http://www.organicchemistry.org/prog/peo/) was used. The prediction was based on the functional group similarity for the query molecules with the in vitro and in vivo validated compounds present in this database. The toxic properties such as mutagenic, tumorogenic, irritant, reproductive effects, drug- relevant properties [c Log P, Log S (Solubility)], molecular weight, and overall drug-score were calculated. The results were visualized using different color codes. Green color shows less toxic, orange color shows the mid and red color shows high tendency of toxicity.
Metabolic site prediction by MetaPrint2D:
MetaPrint2D is a tool for predicting the sites of a molecule that are most likely to undergo Phase I metabolism, based on their similarity to known and unknown sites of metabolism to be metabolized [20]. It builds based on a database of atom environments found in molecules known to undergo metabolic transformation, such as the data found in the Symyx(R) (previously MDL), Metabolite database http://www. symyx. com, which contains over 80,000 metabolic transformations of xenobiotics, curated from reports in scientific literature. This software was used on the web platform (http: //wwwmetaprint2d. ch.cam.ac.uk / metaprint2d/), by uploading the SMILES string of compounds.
Molecular docking Simulation:
To validate drug- target association, the molecular docking stimulation was performed on active compounds with PDGFRβ & VEGFR2 by Autodock software (version 4.2) by employing Lamarckian genetic algorithm [21]. The receptor was kept rigid, while ligands were set flexible to rotate and explore most probable binding poses. All the torsional bonds of ligands were set free by Ligand module in AutoDock Tool (ADT). The Grid was set at the centre of active site pocket, which covers all the residues present inside the active site pocket with 60×60×60 points in x, y, z direction and 14.631, -9.472, -24.167 grid centre for VEGFR2. Similarly another active site pocket with 65×65×65 points in x, y, z direction and 1.583, -2.444, -5.305 grid centre was set for PDGFRβ. The parameters were saved as grid parameter file (.gpf) and followed by Autogrid run. In third step of docking, parameter files (.dpf) were prepared in which genetic algorithm was selected, the value of which remains as default. These values determine the optimal run parameter which depends upon the nature of ligand molecules and proteins (receptor). Fifty generations were set for each GA run and each run with a population size of 150. A maximum mutation rate is 0.02 and crossing over rates of 0.08 were used to generate docking trails for subsequent generation. This was followed by saving the parameters as docking parameter file (.dpf) and finally subjected to autodock run. The results generated were visualized in Discovery studio visualizer 3.5. The interactions were studied in terms of minimum binding energy (Kcal/mol), Ki (Inhibition constant) value (µM), and number of hydrogen bonds and stacking interaction formed between the active site residues of macromolecule and ligand.
Biological activity spectrum (BAS):
BAS of a compound represents the complex of pharmacological effects, physiological and biochemical mechanisms of action, specific toxicity (mutagenicity, carcinogenicity, teratogenicity, and embryotoxicity) which can be revealed in compound׳s interaction with biological system. BAS describes the intrinsic properties of the compound depended on its structural particularities [22]. The set of pharmacological effects, mechanisms of action, and specific toxicities that might be exhibited by a particular compound in its interaction with biological entities are predicted by PASS and it is termed as “biological activity spectrum” of the compound [23]. PASS uses Sdffile (.sdf) or MOLfile (.mol) formats as an external source of structure and activity data (http://www.mdli.com). Their values vary from 0.000 to 1.000. Only those activity types for which Pa > Pi were considered possible.
Discussion
Possible drug target prediction:
In the present study, the bioactive compounds from I. aspalathoides were used to find out the possibility of selected putative angiogenic targets based on the high fit score using PharmMapper Server. The results were shown in Table 1 (see supplementary material). Annotations of these putative targets were carried out to derive their association to the proposed anticancer mechanisms. Further exploratory studies on the binding postures of bioactive principles of I. aspalathoides with its therapeutic targets were carried out to validate the outcomes of the docking simulation. The pharmMapper results revealed that the selected phytoligands have significant interaction with VEGFR2 protein, while none of the compounds interact with PDGFRβ protein. However, VEGFR2 activation depends on PDGFRβ stimulation by growth factor PDGF-BB and it was supported our docking. PDGFRβ closely associated with VEGFR2 protein in their signaling pathway [24]. Mucronulatol had highest fit score value 3.196 followed by indigocarpan 3.113. Lowest fit score value 2.866 was noticed for Erythroxydiol X. This result suggests that mucronulatol and indigocarpan can be considered as a better insight to tyrosine kinase inhibitor. Further exploratory studies on the binding postures of bioactive principles of I. aspalathoides with its therapeutic targets were carried out to validate the outcomes of the docking simulation.
Active site identification:
The prominent binding site of proteins VEGFR2 and PDGFRβ was evaluated through CASTp server with ideal parameters (Figure 3). CASTp calculation showed the surface accessible pockets as well as interior inaccessible cavities of VEGFR2 and PDGFRβ. In VEGFR2 protein, all 38 binding pockets were characterized to obtain the residues around probe radius 1.4Å. The green color represents amino acid residues involved in configuration of binding pockets which is ranging from ASP814-PHE1047. Similarly all 33 binding pockets of PDGFRβ protein was characterized to obtain residues around the probe radius 1.4Å. The green color represents amino acid residues involved in configuration of binding pockets which is ranging from GLU63-ASN298.
Figure 3.

Binding pocket identification by CASTp server. (a,c) Shows the binding sites of PDGFRβ and VEGFR2 protein respectively, and (b,d) Green color boxes highlights the amino acid residues present in the binding site.
Molinspiration Calculation:
The CLogP (octanol / water partition co efficient) was calculated by the methodology developed by Molinspiration as a sum of fragment based contributions and correlation factors. The molecular descriptors of five compounds given in Table 2 (see supplementary material) were tested to Lipinski׳s rule of five, interestingly all the ligands which we selected have molecular weight in the range of 292 − 400 (< 500). Low molecular weight drug molecules (<500) are easily transported, diffuse and absorbed as compared to heavy molecules. Molecular weight is an important aspect in therapeutic drug action; If it increases beyond certain limit, the bulkiness of the compounds also increases correspondingly, which in turn affects the drug action [25]. Number of hydrogen bond acceptors (O and N atoms) and number of hydrogen bond donors (NH and OH) in the tested compounds were found to be within Lipinski׳s limit range from 7-2 & 3-2 i.e. less than 10 and 5 respectively.
Lipophilicity (log P value) and TPSA values are two important properties for the prediction of per oral bioavailability of drug molecules [26]. Permeability property of compounds were analyzed, the calculated log P value of five compounds was ranging from 3.381 to 4.759 (<5), which is the acceptable limit for the drugs to be able to penetrate through biomembranes. Topological Polar Surface Area (TPSA) was calculated as described by [17]. O- and N- centered polar fragments were considered. TPSA has shown to be a very good descriptor characterizing drug absorption, including intestinal absorption, bioavailability, Caco-2 permeability and BBB penetration.
The highest degree of lipophilicity was found with all the compounds which are an indication for good lipid solubility that will help the drug to interact with the membranes. TPSA was calculated from the surface areas that are occupied by oxygen and nitrogen atoms and by hydrogen atoms attached to them. Thus, the TPSA is closely related to the hydrogen bonding potential of a compound [27]. In our study, all ligands exhibited 77% to 95% absorption, indicates good bioavailability by oral route. Good bioavailability is more likely for compounds with ≤10 rotatable bonds and TPSA of ≤ 140 Å [28]. As the number of rotatable bonds increases, the molecule becomes more flexible and more adaptable for efficient interaction with a particular binding pocket. Interestingly, all the five compounds have 3-6 rotatable bonds and flexible.
Drug likeliness property of five compounds against GPCR ligand, ion channel modulator, kinase inhibitor, nuclear receptor ligand, protease inhibitor and enzyme inhibitory activity were studied and summarized in Table 3 (see supplementary material). The molecule having bioactivity score more than 0.00 is likely to possess considerable biological activities, values -0.50 to 0.00 are expected to be moderately active and if score is less than -0.50, it is presumed to be inactive [29]. The results of the present study demonstrated that the investigated compounds were biologically active and produced the physiological actions by interacting with GPCR ligands, nuclear receptor ligands, inhibit protease and other enzymes. GPCR ligand-based signaling cascade was used for the development of a new functional drug with increased binding selectivity profile and less undesirable effects. Though bioactivity score for GPCR ligand was found to be >0.00 for all tested compounds, but the highest score 0.24 was observed for indigocarpan closely followed by the compound Erythroxydiol X (0.22). Ion channel modulators allowed the movement of charged particles across cell membranes and are important therapeutic targets which are modulated by a range of therapeutic drugs. Bioactivity score for ion channel modulator activity was in between 0.00 and -0.50. Similar results were obtained for all five compounds showed score value of >-0.50. Kinase inhibitors for development of selective inhibitors that can block or modulate diseased signaling pathways are considered a promising approach for drug development [30]. Bioactivity scores for nuclear receptor ligand, protease inhibitor and enzyme inhibition was found to be in the range of 0.38 - 0.68, 0.02 - 0.35 and 0.51- 0.51 respectively.
admetSAR prediction:
The ADMET (Absorption, Distribution, Metabolism, Excretion and Toxicity) properties of the target compounds were calculated using admetSAR as described by [31]. Blood-Brain Barrier (BBB) penetration, HIA (Human Intestinal Absorption), Caco-2 cell permeability and AMES test were calculated. The predicted ADMET data were summarized in Table 4 (see supplementary material). The cytochrome P450 super family plays an important role in drug metabolism and clearance in the liver, and the most important isoforms are CYP1A2, CYP2A6, CYP2C9, CYP2C19, CYP2D6, CYP2E1, and CYP3A4 [32]. Thus, inhibition of cytochrome P450 isoforms might cause drug-drug interactions in which co-administered drugs fail to metabolized and accumulate to toxic levels [33]. The analysis showed ROS to be a substrate for P-glycoprotein, which effluxes drugs and various compounds to undergo further metabolism and clearance. If P-glycoprotein is induced, drugs in the medication would be transported out of the cells at a greater rate and could lead to therapeutic failure because the drug concentration would be lower than expected [34]. Therefore, dosage control and knowledge of co-administered drugs might be considered to reduce therapeutic failure. Based on the predicted values of admetSAR, all the selected phytoligands are able to penetrate to BBB, Caco-2 and absorbed by human intestines. Furthermore, all the compounds did not show any acute toxicity and mutagenic effect with respect to the AMES test data.
Biological activity predictions:
In order to find out the possible biological activity of selected bioactive constituents were obtained by using PASS online server. The set of pharmacological effects, mechanisms of action, and specific toxicities, that might be exhibited by a particular compound in its interaction with biological entities, and which is predicted by PASS, is termed the “BAS” of this compound [24]. The Pa and Pi values vary from 0 to 1, and Pa + Pi < 1, since these probabilities are calculated independently. Pa and Pi can be considered to be measures of the compound under study belonging to the classes of active and inactive compounds respectively.
The PASS prediction results denoted that the highest Pa value than Pi value occurred for antineoplastic and thereby it obviously showed the anticancerous potential of selected compounds evaluated in Table 5 (see supplementary material). All the five compounds showed anti-neoplastic property, and the values ranges from 0.342-0.67. However, among the five mucronulatol showed a strong Pa value while compared to erythroxydiol X &Y. These compounds may block metastasis by blocking angiogenesis by interfering VEGFR2 and PDGFRβ as evidenced by docking studies.
Toxicity prediction by Osiris:
Structure based design is now fairly routine but many potential drugs fail to reach the clinic because of ADME/Tox liabilities. One important class of enzymes, responsible for many ADMET problems, is the cytochromes P450. Inhibition of these or production of unwanted metabolites can result in many adverse drug reactions. Toxicity risks (mutagenicity, tumorogenicity, irritation, reproduction) and physico-chemical properties (cLogP, solubility, drug-likeness and drug-score) of selected compounds were calculated by Osiris and their results were shown in Table 6 (see supplementary material). In the present study, drug-likeliness property and toxicity were studied using Osiris tools and indicated no Toxicity risks (mutagenicity, tumorogenicity, irritation, reproduction) and revealed a good score as compared to positive control Sorafenib.
Drug solubility is an important factor that affects the movement of a drug from the site of administration into the blood. It is known that insufficient solubility of drug can leads to poor absorption [35]. All the compounds shown good soluble while compared to sorafenib. The Drug-Score values were in the range of 0.08 to 0.85. The drug likeliness is another important parameter in drug development. Because drug like molecules exhibit favorable absorption, distribution, metabolism, excretion, toxicological (ADMET) parameters. Currently, there are many approaches to assess a compound drug-likeness based on topological descriptors, fingerprints of molecular drug-likeness structure keys, clogP and molecular weight [36]. Toxicity risk includes mutagenic, tumoregenic, irritant, reproductive effective parameters and green color which represents the drug conforming property. Interestingly, all our compounds appeared as green color, which clearly indicates no toxicity (Figure 4).
Figure 4.
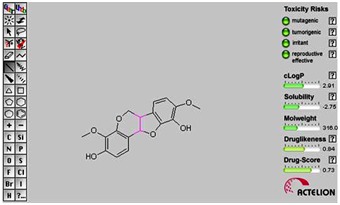
Osiris property prediction of lead compound (Indigocarpan). It indicates that there are no toxicity risks (mutagenicity, tumorogenicity, irritating effect, reproductive effect).
Metaprint2D:
MetaPrint2D is a fast, efficient and accurate predictor of both the sites and products of metabolism in small molecule drugs using circular fingerprints and substrate/product ratios [37]. The atoms indicated in red color would be metabolized high followed by medium orange color, low green color, and very low is not colored. Our MetaPrint2D predicted that the various oxygen and methoxy group were most likely to be metabolized (colored in red) in the flavonoid and diterbenes, followed by the group colored in orange and then by the groups marked in green. Although all the compounds have metabolic site, indigocarpan have more metabolic site than other compounds (Figure 5). In the case of lead compound indigocarpan, the carbon atom no 4 and 9th position showed good metabolic site and hydroxyl group showed moderate metabolic site. Molecular docking studies can be particularly useful for gaining selectivity and steric information about potential compounds, which can be used to predict their sites of metabolism and possible toxic metabolites [38].
Figure 5.

Plot of Metaprint 2D predictions. Site of metabolism; the atoms in red color that most will be metabolized are colored according to the likelihood of a metabolic site; High: red, Medium: orange, Low: green, very low is not colored, and No data: grey. NOR indicates the Normalized Occurrence Ratio; a high NOR indicates a more frequently reported site of metabolism in the metabolite database.
Molecular Docking Simulations:
The molecular interaction leading the ligand from the surface of the protein to the active site reveals the cytotoxicity of the flavonoids which have strong affinities towards the target proteins that were examined in the docking analysis. Among the five selected bioactive compounds listed in Table 7 & Table 8 (see supplementary material), indigocarpan, an isoflavonoid compound were found to consistently have lower binding energies and showed higher interaction with proteins VEGFR2 and PDGFRβ. Docking results showed that the bioactive compound indigocarpan has the lowest binding energy of -7.04 Kcal/mol, lowest ligand efficiency value of -0.31 Kcal/mol, lowest inhibitory constant of 6.88µM with VEGFR2 protein. It showed lowest binding energy of -4.82 Kcal/mol, lowest ligand efficiency value of -0.21 Kcal/mol and lowest inhibitory constant of 293.55 µM were observed for PDGFRβ protein. This suggests that therapies that target a wide range of RTKs will provide a more effective and long term anti-angiogenic effects than those that target a limited numbers of growth factor receptors [39].
a) Binding affinity of phytoligands with receptor tyrosine kinases:
The activation of VEGFR2 kinase is known to be an ATPconsuming process. The ATP-binding site of VEGFR2 is located between the N-terminal lobe and C-terminal lobe within the catalytic domain. Many kinase inhibitors act as ATP minetics and compete with the cellular ATP for the binding with the ATP binding site and subsequently suppressing the receptor autophosphorylation [40]. It has been previously reported that the ATP binding site of VEGFR2 is mainly constituted with residues such as LEU 868, GLU 883, LYS 885, GLU 915, PHE 916, CYS 917, LYS 918, PHE 919, GLY 920, ASN 921, LEU 926, ARG 927, SER 1035, ASP 1044 and LYS 1053 [41]. In our present study, as shown in (Figure 6c) it has been viewed that indigocarpan stably locate at the ATP binding pocket near the hinge region. Hydrogen bonding interaction makes the lead compound Indigocarpan (C-3 position of hydroxyl group in ring A) interacts with the (Sulfur and basic amino acid group of the residues) main chain of Cys 919 and Lys920 at a distance of 1.85Å & 2.47Å and it has Vander Waals interaction with residues such as Phe 918, Gly 841, Asn 923, Val 848, Phe 1047, Lys 868, Asp 1046, Cys 1045, Val 899, Val 916, Leu 1035, Leu 840, and Ala 866. These interactions mediate the binding of indigocarpan to the ATP binding site of VEGFR2 and hence inhibit the function of VEGFR2.
Figure 6.
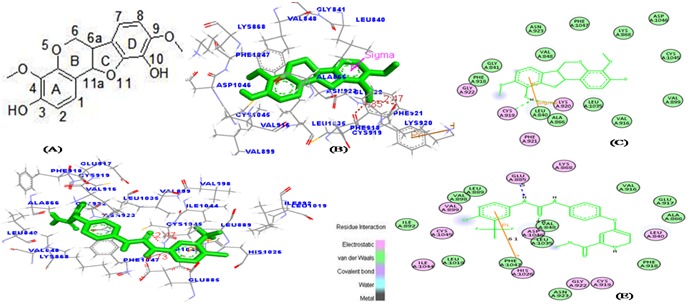
The favorable binding portion of indigocarpan with lowest binding free energy in the ATP-binding site of VEGFR2 (PDB ID: 3VHE) as analyzed by molecular docking study. (a) 2D structure of Indigocarpan, (b) The three dimensional diagram displays the interaction of indigocarpan (the green stick) to the ATP binding site of VEGFR2 with the labeled amino acid residues CYS 919 and LYS 920 which significantly contributed to the binding. (c) The two dimensional diagram shows the interactions of indigocarpan to the amino acid residues. (d, e) denotes the binding mode of Sorafenib with VEGFR2. Similarly, colors of the residues indicate the forms of interactions as follows: van der Waals forces, green; polarity, magenta. Green arrow represents Hbonding with the amino acid main chain.
Similarly, PDGFRβ as shown in (Figure 7b) has hydrogen bonding interaction with indigocarpan (C-9th position of hydroxyl group and the corresponding phenyl ring D) could interact with the (oxygen atom) main chain of Thr88 (1.89Å), THR86 (2.00Å). THR86 and THR88 are found to be the major active residues for the inhibition of PDGFRβ. Vander Waals interaction occurs with residues Met 65, Pro 69 and Leu 90. The presence of a methoxy group at C3 & C9 position seemed to be an important structural requirement for the cytotoxic activity of the indigocarpan compound compared with other compounds. Specifically, the presence of methoxy group in the benzofuran ring which enables the ligand to fit within the ATP binding residues of PDGFRβ to inhibit its activity. From this it is evident that Indigocarpan may act as potent tyrosine kinase inhibitor than Sorafenib (positive control) in respect of its binding affinity and inhibitory activity against receptor tyrosine kinases.
Figure 7.
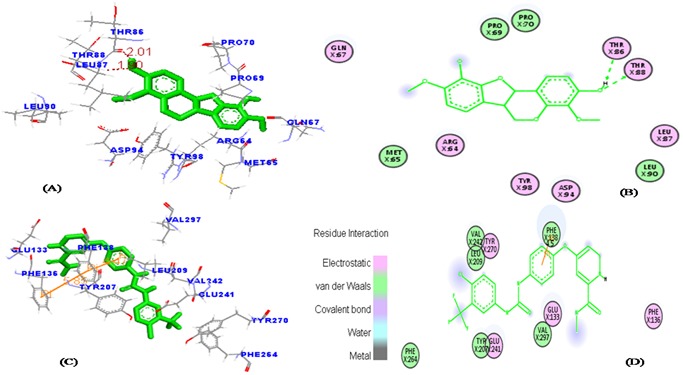
The favorable binding position of indigocarpan with lowest binding free energy of PDGFRβ (PDB ID: 3MJG) as analyzed by molecular docking study. (a) The three-dimensional diagram displays the interaction of indigocarpan (green stick) with PDGFRβ labeled amino acid residues of THR86 and THR88 which significantly contributed to the binding. (b) The two dimensional diagram shows the interactions of indigocarpan to the amino acid residues in the ATP- binding site. (c & d) denotes the binding mode of Sorafenib with PDGFRβ. Similarly, colors of the residues indicate the forms of interactions as follows: van der Waals forces, green; polarity, magenta; Green arrow represents H-bonding with the amino acid main chain.
Conclusion
Among the five compounds, indigocarpan has potent inhibitory activity against angiogenic targets namely PDGFRβ and VEGFR2. Targeting VEGFR2 and PDGFRβ, which simultaneously targets endothelial cells and pericytes, acts as a potent anti-vascular strategy including endothelial cell apoptosis and tumor blood vessel regression. Future studies will have to address the stability of protein-ligand complex by molecular dynamic simulation. However, experimental studies will also have to address the interaction of indigocarpan with targets.
Supplementary material
Acknowledgments
Dr. Kannan thanks Science and Engineering Research Board (SERB) for financial assistance (No. SB/YS/LS-148/2013), New Delhi, Government of India.
Footnotes
Citation:Paramashivam et al, Bioinformation 11(2): 073-084 (2015)
References
- 1.Panday A, et al. J Med Chem. 2002;45:3772. doi: 10.1021/jm020143r. [DOI] [PubMed] [Google Scholar]
- 2.Hanahan D, Weinberg RA. Cell. 2011;144:646. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 3.Carmeliet P, Jain RK. Nature. 2011;473:298. doi: 10.1038/nature10144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ferrara N, et al. Biochem Biophys Res Commun. 2005;333:328. doi: 10.1016/j.bbrc.2005.05.132. [DOI] [PubMed] [Google Scholar]
- 5.Carmeliet P, et al. Oncology. 2005;69:4. doi: 10.1159/000088478. [DOI] [PubMed] [Google Scholar]
- 6.Kim JH, et al. Mol vis. 2009;15:1868. [PMC free article] [PubMed] [Google Scholar]
- 7.Neufeld G, et al. FASEB J. 1999;13:9. [PubMed] [Google Scholar]
- 8.Koch S, et al. Biochem J. 2011;437:169. doi: 10.1042/BJ20110301. [DOI] [PubMed] [Google Scholar]
- 9.Heldin CH, et al. Arch Biochem Biophys. 2002;398:284. doi: 10.1006/abbi.2001.2707. [DOI] [PubMed] [Google Scholar]
- 10.Tallquist M, Kazlauskas A. Cytokine Growth Factor Rev. 2004;15:205. doi: 10.1016/j.cytogfr.2004.03.003. [DOI] [PubMed] [Google Scholar]
- 11.Murukesh N, et al. Br J Cancer. 2010;102:8. doi: 10.1038/sj.bjc.6605483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Boopala bhagavan N, et al. J Pharm Res. 2013;6:350. [Google Scholar]
- 13.Selvam C, et al. Tetrahedron Lett. 2004;45:4311. [Google Scholar]
- 14.Liu X, et al. Nucleic Acids Res. 2010;38:W609. doi: 10.1093/nar/gkq300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dundas J, et al. Nucleic Acids Res. 2006;34:W116. doi: 10.1093/nar/gkl282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lipinski CA, et al. Adv Drug Deliv Rev. 2001;46:3. doi: 10.1016/s0169-409x(00)00129-0. [DOI] [PubMed] [Google Scholar]
- 17.Ertl P, et al. J Med Chem. 2000;43:3714. doi: 10.1021/jm000942e. [DOI] [PubMed] [Google Scholar]
- 18.Zhao YH, et al. Pharm Res. 2002;19:1446. doi: 10.1023/a:1020444330011. [DOI] [PubMed] [Google Scholar]
- 19.Cheng F, et al. J Chem Inf Model. 2012;52:3099. doi: 10.1021/ci300367a. [DOI] [PubMed] [Google Scholar]
- 20.Carlsson L, et al. BMC Bioinformatics. 2010;11:362. doi: 10.1186/1471-2105-11-362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Morris GM, et al. J Comput Chem. 1998;19:1639. [Google Scholar]
- 22.Lagunin A, et al. Bioinformation. 2000;16:747. [Google Scholar]
- 23.Poroikov VV, et al. J Chem Inf Comput Sci. 2003;43:228. doi: 10.1021/ci020048r. [DOI] [PubMed] [Google Scholar]
- 24.Liao X, et al. Plos one. 2013;8:e82294. doi: 10.1371/journal.pone.0082294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Srimai V, et al. Med Chem Res. 2013;22:5314. [Google Scholar]
- 26.Chang LC, et al. J Med Chem. 2004;47:6529. doi: 10.1021/jm049448r. [DOI] [PubMed] [Google Scholar]
- 27.Clark DE. J Pharm Sci. 1999;88:807. doi: 10.1021/js9804011. [DOI] [PubMed] [Google Scholar]
- 28.Veber DF, et al. J Med Chem. 2002;45:2615. doi: 10.1021/jm020017n. [DOI] [PubMed] [Google Scholar]
- 29.Verma A. Asian Pac J Trop Biomed. 2012;2:S1735. doi: 10.1016/S2221-1691(12)60212-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chatterjee S, et al. Bioinformation. 2012;8:1195. doi: 10.6026/97320630081195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Cheng F, et al. J Chem Inf Model. 2012;52:3099. doi: 10.1021/ci300367a. [DOI] [PubMed] [Google Scholar]
- 32.Vasanthanathan P, et al. Drug Metab Dispos. 2009;37:658. doi: 10.1124/dmd.108.023507. [DOI] [PubMed] [Google Scholar]
- 33.Lynch T, Price A. Am Fam Physician. 2007;76:391. [PubMed] [Google Scholar]
- 34.Levin GM. Curr Psychiatr. 2012;11:38. [Google Scholar]
- 35.Muegge I. Med Res Rev. 2003;23:302. doi: 10.1002/med.10041. [DOI] [PubMed] [Google Scholar]
- 36.Tetko IV. Drug Discov Today. 2006;11:700. doi: 10.1016/j.drudis.2006.06.013. [DOI] [PubMed] [Google Scholar]
- 37.Boyer S, et al. J Chem Inf Model. 2007;47:583. doi: 10.1021/ci600376q. [DOI] [PubMed] [Google Scholar]
- 38.Fraga AGM, et al. 2007 In: 30th Reunião Anual da Sociedade Brasileira de Química, Águas de Lindóia - SP, Abstracts of Papers. Sociedade Brasileira de Química, [Google Scholar]
- 39.Erber R, et al. FASEB J. 2004;18:338. doi: 10.1096/fj.03-0271fje. [DOI] [PubMed] [Google Scholar]
- 40.Noble ME, et al. Science. 2004;303:1800. doi: 10.1126/science.1095920. [DOI] [PubMed] [Google Scholar]
- 41.Yang Y, et al. Bioorg Med Chem Lett. 2010;20:6653. doi: 10.1016/j.bmcl.2010.09.014. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.