

Glycogen storage disease type III (GSD III) is an autosomal recessive metabolic disorder that is caused by the deficiency of the glycogen-debranching enzyme. Presenting problems often include hepatomegaly, elevated transaminases, hyperlipidemia, mild hypoglycemia, and failure to thrive. We describe a patient who developed marked elevation in serum transaminases (alanine aminotransferase [ALT] > 2000 IU/L), without evidence of significant hepatocellular necrosis or liver dysfunction.
A 17-month-old girl presented to our emergency department with acute vomiting and diarrhea. Physical examination revealed hepatosplenomegaly (liver and spleen 6 and 2 cm below the costal margins, respectively). Her weight and height were at the 75th percentile. Laboratory tests showed elevated transaminases: ALT 408 IU/L (reference range <40 IU/L), aspartate aminotransferase (AST) 328 IU/L (reference range <40 IU/L), gamma-glutamyl transpeptidase (gGTP) 53 IU/L (reference range 8–78 IU/L), and total bilirubin 0.3 mg/dL (reference range <1.0 mg/dL). Initial workup revealed unremarkable acetaminophen level, creatine phosphokinase (CPK), coagulation studies, hepatitis virus panel, thyroid function tests, ceruloplasmin, α-1 antitrypsin phenotype, auto-immune studies, and liver ultrasound.
Although the symptoms of acute gastroenteritis improved, her transaminases remained elevated. Additional history revealed that the patient had episodes of shakiness, crankiness, and vomiting in the morning before eating breakfast. Fasting laboratory studies revealed hypoglycemia (glucose 55 mg/dL [reference range 70–110 mg/dL] and hyperlipidemia (triglyceride 316 mg/dL [reference range 28–153 mg/dL] and cholesterol 188 mg/dL [reference range 75–200]). Given the clinical picture, a suspicion for GSD III arose, so genetic studies of peripheral blood DNA were undertaken.
Although genetic studies were pending, the patient experienced a sudden spike in transaminases on routine testing (ALT 2104 IU/L, AST 1441 IU/L) without any signs of illness or other laboratory abnormalities (total bilirubin 0.5 mg/dL, direct bilirubin 0.2 mg/dL (0.1–0.4), gGTP 142 IU/L, prothrombin time 12.6 seconds (11.5–16.1 seconds), albumin 4.2 g/dL (3.8–5.4 g/dL), and CPK 71 IU/L (29–168 IU/L). A liver biopsy was performed to exclude other etiologies aside from GSD III for this elevation of transaminases. The biopsy revealed enlarged hepatocytes with abundant glycogen-rich material (Fig. 1), and enzymatic analysis of liver tissue revealed absent debranching enzyme activity (0 μmol/min/g tissue [control 0.31 ± 0.1 μmol/min/g tissue]), supporting the diagnosis of GSD III. Of note, there was minimal evidence of hepatocellular injury or inflammation. Subsequently, genetic studies revealed heterozygous mutations in the amylo-1,6-glucosidase (AGL) gene, including a nonsense mutation, c.2590C>T (R864X), previously reported to be causative for GSD type III (1–3), and a potentially pathological missense mutation in a highly conserved tyrosine residue, c.1484A>G (Y495C), not previously associated with GSD type III. During the ensuing 2 years, her serum transaminase levels remained elevated between 300 and 500 IU/L (Fig. 2). Her other biochemical markers of liver disease have remained unremarkable, and she has not developed any clinical stigmata of advanced liver disease.
FIGURE 1.
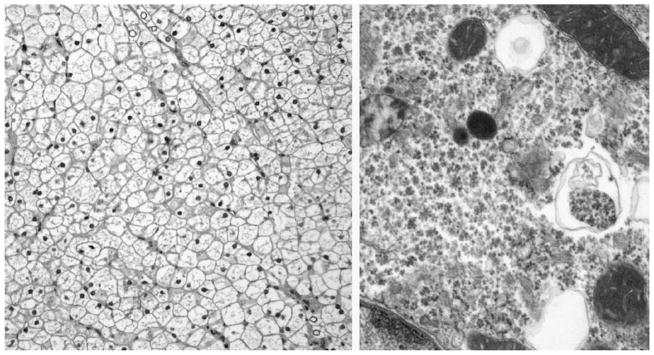
Microscopic appearance of GSD III. Left, The hepatocytes are plant-like with clear cytoplasm and prominent cell membranes. The nuclei are small and uniform. Scattered glycogenated nuclei are seen. No cholestasis or significant inflammation is present (hematoxylin and eosin stain, original magnification ×20). Right, Hepatocyte with vast pools of particulate cytoplasmic glycogen. Rare lysosomes filled with glycogen are also seen (original magnification ×58,000).
FIGURE 2.

Clinical course of GSD III. Serum transaminase levels over time are shown.
DISCUSSION
The glycogen storage diseases, or glycogenoses, encompass several inherited diseases that result in abnormalities in the various enzymes that are responsible for the synthesis or degradation of glycogen. Like the majority of the glycogenoses, GSD III is an autosomal recessive metabolic disorder. It is caused by the deficiency of the glycogen-debranching enzyme with 2 different catalytic enzyme functions. This leads to an interruption in glycogenolysis, resulting in an accumulation of the partially degraded glycogen, phosphorylase limit dextrin, in affected tissues.
GSD III can be separated into 4 types depending on which tissues and enzyme functions are affected. GSD types IIIa and IIIb are the most common and lack both enzyme functions. Type IIIa may affect liver and muscle tissue, including cardiac and skeletal muscle. Type IIIb exclusively involves the liver, and muscle strength is preserved. Types IIIc and IIId are rare, and display a deficient activity of only 1 of 2 functions of the debranching enzyme (4). The estimated incidence is 1 case per 100,000 live births in North America; almost 80% of the GSD III cases in North America are type IIIa (5).
Patients affected by either subtype present with symptoms along a spectrum and can present from infancy to adulthood. More severe cases are usually seen in infancy with hepatomegaly, hypoglycemia, and failure to thrive. Children with GSD III typically grow slowly and may have delayed onset of puberty (4). Although hypotonia and myopathy are typically not seen until after adolescence, recent case reports have described children as young as 17 months old with difficulty walking as one of the presenting complaints (5,6). If cardiomyopathy is present secondary to limit dextrin deposits, then it has an appearance similar to that of idiopathic hypertrophic cardiomyopathy on echocardiography (4). Some mild cases may go unrecognized until adulthood when they manifest as asymptomatic hepatomegaly, occult liver disease, or severe myopathy.
The diagnosis of GSD III should begin with clinical suspicion in a child with hepatomegaly, mild-to-moderate transaminase elevation, hyperlipidemia, and intolerance to fasting. Serum creatine kinase or aldolase may be used as markers of muscle involvement, but normal values do not rule out myopathy. Bernier et al (7) found that hypertriglyceridemia and hypercholesterolemia were more common in children younger than 3 years and that hypertriglyceridemia correlated negatively with age. A more definitive diagnosis of GSD III necessitates assay of AGL activity in liver tissue. Alternatively, sequence analysis of genomic DNA often reveals mutations predictive of a disease state (6). There have been more than 60 mutations reported in AGL, with mutations varying among ethnic groups, particularly when accounting for frequency of consanguinity (8). In this era of genetic studies, the role of liver biopsy seems less essential, although mutations of unclear significance may require confirmation by tissue-derived enzymology.
Elevations in serum transaminases are commonly reported in GSD III, although there are limited descriptions of the degree of elevation and the natural history of the laboratory abnormalities. In the present case, the degree of elevation seemed atypical, raising the issue of a secondary cause for the liver disease that necessitated a liver biopsy; that biopsy yielded biochemical data supporting the diagnosis. One of the best descriptions of longitudinal analysis of serum enzyme activities is from Coleman et al (9), who studied 13 patients with GSD III, some from infancy to 15 years of age. In that report it was found that serum transaminases spiked (ALT levels <1000 IU/L) at about 2 years old in 3 of the patients, a pattern similar to that observed in our patient. Subsequently, levels declined somewhat in childhood, with a more impressive decline occurring in puberty. The continued elevation of enzyme activities during childhood correlated inconsistently with diet, childhood growth rate, or hypoglycemic episodes. It is notable that our patient’s other laboratory results were normal, including bilirubin, albumin, gGTP, coagulation panel, and CPK, and they remained normal during the ensuing 2 years. This argues in favor of isolated hepatocellular-based release of transaminases.
Additional factors can contribute to the serum transaminase elevation in GSD III. Kimura et al (10) reported a case of an 18-month-old girl with GSD whose transaminases were moderately elevated at diagnosis age 10 months. With concurrent mononucleosis, her ALT was 1122 IU/L and AST was 2778 IU/L. This case demonstrates the increase that can be seen in a patient with GSD III with concurrent infection. Bhuiyan et al described the laboratory findings of 26 confirmed patients with GSD III in Saudi Arabia (11). They found that ALT ranged from normal (<35) to 2250 IU/L and AST ranged from normal (<45) to 3255 IU/L. The authors, however, do not comment at what point in the disease course these transaminases were measured, but do suggest that the lower values reflect the beneficial effect of a high L-alanine or high-protein diet; this may indicate that some of the higher values could have occurred at diagnosis. In addition, all of the patients had elevated CPK levels, which suggests that muscle injury accounts for some of the transaminase elevation (11,12). Zimakas and Rodd (5) described a 17-month-old Inuit with an AST at presentation of 4210 and an ALT of 2655 IU/L. This patient had cardiomyopathy despite an initially normal CPK and had marked elevation in serum alkaline phosphatase and γ-glutamyl transpeptidase (4210 and 886 IU/L, respectively), the latter suggesting an additional liver-based disease process.
The reason for the impressive elevation in transaminases in patients with GSD III is unclear. It is necessary to first rule out muscle involvement and/or concurrent illness as mentioned. In our case, the pathology was rather benign, with no features of significant inflammation, necrosis, or progressive disease. Focal bridging fibrosis was a suggestion, however, of ongoing low-grade hepatocellular insult. Another hypothesis for the elevation could be related to the presence of glycogen in the hepatocytes. In glycogenoses, the hepatocyte appears swollen because of glycogen deposition, which displaces organelles—perhaps subcellular nonlytic injury leads to leakage or altered processing of transaminases. It is intriguing that in this case, despite during 2 years of persistent elevation of transaminases >300 IU/L, there is no biochemical or clinical evidence of progressive liver disease. This finding is consistent with the fact that GSD III rarely leads to end-stage liver disease during childhood and is an uncommon indication for liver transplantation (13).
In conclusion, transaminase elevation in GSD III can include rather marked and isolated increases especially in the first few years of life. The exact pathogenesis of the elevation is unclear but is not typically associated with progressive liver disease as seen in other circumstances. In the current era of relatively rapid and lower-cost genetic testing, the role of liver biopsy in the diagnosis of GSD III may be limited.
References
- 1.Shen J, Bao Y, Liu HM, et al. Mutations in exon 3 of the glycogen debranching enzyme gene are associated with glycogen storage disease type III that is differentially expressed in liver and muscle. J Clin Invest. 1996;98:352–7. doi: 10.1172/JCI118799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lucchiari S, Donati MA, Melis D, et al. Mutational analysis of the AGL gene: five novel mutations in GSD III patients. Hum Mutat. 2003;22:337. doi: 10.1002/humu.9177. [DOI] [PubMed] [Google Scholar]
- 3.Endo Y, Horinishi A, Vorgerd M, et al. Molecular analysis of the AGL gene: heterogeneity of mutations in patients with glycogen storage disease type III from Germany, Canada, Afghanistan, Iran, and Turkey. J Hum Genet. 2006;51:958–63. doi: 10.1007/s10038-006-0045-x. [DOI] [PubMed] [Google Scholar]
- 4.Wolfsdorf JI, Weinstein DA. Glycogen storage diseases. Rev Endocr Metab Disord. 2003;4:95–102. doi: 10.1023/a:1021831621210. [DOI] [PubMed] [Google Scholar]
- 5.Zimakas PJ, Rodd CJ. Glycogen storage disease type III in Inuit children. CMAJ. 2005;172:355–8. doi: 10.1503/cmaj.1031589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Oh SH, Park HD, Ki CS, et al. Biochemical and molecular invesitgation of two Korean patients with glycogen storgae disease type III. Clin Chem Lab Med. 2008;46:1245–9. doi: 10.1515/CCLM.2008.252. [DOI] [PubMed] [Google Scholar]
- 7.Bernier AV, Sentner CP, Correia CE, et al. Hyperlipidemia in glycogen storage disease type III: effect of age and metabolic control. J Inherit Metab Dis. 2008;31:729–32. doi: 10.1007/s10545-008-0919-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Endo Y, Fateen E, El Shabrawy M, et al. Egyptian glycogen storage disease type III—identification of six novel AGL mutations, including a large 1.5 kb deletion and a missense mutation p. L620P with subtype IIId. Clin Chem Lab Med. 2009;47:1233–8. doi: 10.1515/CCLM.2009.281. [DOI] [PubMed] [Google Scholar]
- 9.Coleman RA, Winter HS, Wolf B, et al. Glycogen debranching enzyme deficiency: long-term study of serum enzyme activities and clinical features. J Inherit Metab Dis. 1992;15:869–81. doi: 10.1007/BF01800225. [DOI] [PubMed] [Google Scholar]
- 10.Kimura T, Ikeda H, Kato M, et al. Severe hypoglycemia in a patient with glycogen storage disease type III induced by infectious mononucleosis. J Inherit Metab Dis. 2001;24:873–4. doi: 10.1023/a:1013900526628. [DOI] [PubMed] [Google Scholar]
- 11.Bhuiyan J, Al Odaib AN, Ozand PT. A simple, rapid test for the differential diagnosis of glycogen storage disease type 3. Clin Chem Acta. 2003;335:21–6. doi: 10.1016/s0009-8981(03)00234-1. [DOI] [PubMed] [Google Scholar]
- 12.Kohli R, Harris DC, Whitington PF. Relative elevations of serum alanine and aspartate aminotransferase in muscular dystrophy. J Pediatr Gastroenterol Nutr. 2005;41:121–4. doi: 10.1097/01.wno.0000161657.98895.97. [DOI] [PubMed] [Google Scholar]
- 13.Kishnani PS, Austin SL, Arn P, et al. Glycogen storage disease type III diagnosis and management guidelines. Genet Med. 2010;12:446–63. doi: 10.1097/GIM.0b013e3181e655b6. [DOI] [PubMed] [Google Scholar]