

Abstract
CYP11A1 hydroxylates the side chain of vitamin D3 (D3) in a sequential fashion [D3→20S(OH)D3→20,23(OH)2D3→ 17,20,23(OH)3D3], in an alternative to the classical pathway of activation [D3→25(OH)D3→1,25(OH)2D3]. The products/intermediates of the pathway can be further modified by the action of CYP27B1. The CYP11A1-derived products are biologically active with functions determined by the lineage of the target cells. This pathway can operate in epidermal keratinocytes. To further define the role of these novel secosteroids we tested them for protective effects against UVB-induced damage in human epidermal keratinocytes, melanocytes and HaCaT keratinocytes, cultured in vitro. The secosteroids attenuated ROS, H2O2 and NO production by UVB-irradiated keratinocytes and melanocytes, with an efficacy similar to 1,25(OH)2D3, while 25(OH)D3 had lower efficacy. These attenuations were also seen to some extent for the 20(OH)D3 precursor, 20S-hydroxy-7-dehydrocholesterol. These effects were accompanied by upregulation of genes encoding enzymes responsible for defence against oxidative stress. Using immunofluorescent staining we observed that the secosteroids reduced the generation cyclobutane pyrimidine dimers in response to UVB and enhanced expression of p53 phosphorylated at Ser-15, but not at Ser-46. Additional evidence for protection against DNA damage in cells exposed to UVB and treated with secosteroids was provided by the Comet assay where DNA fragmentation was markedly reduced by 20(OH)D3 and 20,23(OH)2D3. In conclusion, novel secosteroids that can be produced by the action of CYP11A1 in epidermal keratinocytes have protective effects against UVB radiation.
Keywords: hydroxyvitamin D, epidermal keratinocytes, epidermal melanocytes, UVB, DNA damage, oxidative stress
1. Introduction
The ultraviolet radiation (UVR) spectrum of solar light induces significant damage to the epidermis, which is tightly connected to the production of reactive oxygen species (ROS). These ROS, which include hydrogen peroxide (H2O2) and nitric oxide (NO), can cause oxidative damage and reduction of the important antioxidant, glutathione (GSH), as well as inhibition of DNA repair and genotoxic and mutagenic effects [1]. The mutagenic and genotoxic effects of UVB are related to its absorption of wave-lengths in the range of 280–320 nm, particularly by the pyrimidine bases [2], resulting in the formation of cyclobutane pyrimidine dimers (CPD) [3]. CPDs are DNA lesions produced in human skin mainly from exposure to UVB [4]. Tumor suppressor protein p53 mediates the cellular response to stress by activating cell-cycle arrest or apoptosis [5]. Thus, gamma and UV irradiation, hypoxia and some chemicals induce DNA damage and the activation of p53 [6]. The extent of DNA damage and the elevation in p53 levels are proportional [6]. Thus, p53 expression increases with UVB exposure and it initiates growth arrest and repair of damaged DNA [5, 7]. Phosphorylation plays an important role in determining the activity of p53 including its ability to bind to DNA [8, 9]. Phosphorylation of p53 at Ser-15 and Ser-20 promotes accumulation and activation of p53 and DNA repair, while p53 phosphorylation at Ser-46 closely regulates apoptosis following DNA damage [10]. Some endogenous regulators such as melatonin promote phosphorylation of p53 at Ser-15, thus activating p53 and consecutively inhibiting cell growth, preventing accumulation of damaged DNA and promoting antitumor activity [11, 12].
The human epidermis is the main site of photo-induced formation of D3 from 7-dehydrocholesterol (7DHC), representing the most fundamental reaction in photobiology [13–15]. D3 is hydroxylated in positions C25 and C1 at both systemic (liver and kidney) and local (epidermis) levels to produce 1,25(OH)2D3 [14, 16, 17]. 1,25(OH)2D3, in addition to regulating calcium metabolism, has important pleiotropic effects that include stimulation of differentiation and inhibition of proliferation of cells of different lineage, anti-cancer properties, stimulation of innate immunity, and inhibition of adaptive immunity and inflammation [14, 16–21]. In the skin it plays a significant role in the formation of the epidermal barrier and the functioning of the adnexal structures including hair follicles, and has a wide variety of ameliorating effects on skin cancer, and proliferative and inflammatory cutaneous diseases [14, 16, 18, 20, 22–24]. Most recently, it was reported that active forms of D3 prevent, attenuate, or even reverse UVB-induced cell death and DNA damage in skin cells [25–31]. Unfortunately, due to its calcemic (toxic) effect, chronic therapeutic use of D3 at pharmacological doses is severely limited, which forms a significant barrier to the use of classical forms of D3 including 1,25(OH)2D3. However, the discovery of an alternative pathway of vitamin D activation initiated by CYP11A1 which produces novel secosteroids which are biologically active but non-calcemic [32], offers promise for therapeutic applications.
The novel pathways of vitamin D metabolism initiated by the action of CYP11A1 are: D3→ 20S(OH)D3 → 20,23(OH)2D3 → 17,20,23(OH)3D3 and D2 → 20S(OH)D2 → 17,20(OH)2D2 →17,20,24(OH)3D2 [32–40]. Our studies indicate that these pathways operate in vivo with CYP 27B1 being capable of hydroxylating some of the products in the 1α-position [41, 42]. In tissues with high expression of CYP11A1, 20S(OH)D3 is the main metabolite and is more abundant than 25(OH)D3 [41]. Similarly, tissues expressing low levels of CYP11A1 such as skin [43, 44] can also produce 20S(OH)D and its hydroxyderivatives [41, 42]. The initial metabolite, 20S(OH)D3, is the major product of the pathway, and recently we detected it in human serum based on its mass spectrum and identical HPLC retention time to authentic standard [41]. 20,23(OH)2D3 is the second major metabolite of the pathway [41]. Both secosteroids demonstrate biological potency, equal or higher than that of 1,25(OH)2D3, with anti-proliferative, pro-differentiation and anti-inflammatory activities on epidermal keratinocytes, melanocytes, melanoma cells and dermal fibroblasts [32, 45–49]. Importantly, 20S(OH)D3 is noncalcemic and non-toxic at the highest pharmacological doses tested in rats (3 μg/kg), and in mice (30–60 μg/kg)[50, 51], which are up to 100-fold higher than doses of 1,25(OH)2D3 or its precursor, 25(OH)D3, that are toxic.
In this presentation we provide evidence that novel secosteroidal compounds produced by CYP11A1 can act as protective agents against UVB-induced damage.
2. Materials and Methods
2.1. Source of steroids and secosteroids
7-Dehydrocholesterol, vitamin D3, 25(OH)D3, and 1,25(OH)2D3 were purchased from Sigma-Aldrich (St. Louis, MO). 20(OH)D3 was produced either by chemical synthesis of 20-hydroxy-7-dehydrocholesterol (20(OH)7DHC) with subsequent photolytic transformation to secosteroidal structures [52], or biochemically from vitamin D3 using bovine CYP11A1 [33, 34]. 20,23(OH)2D3 was also produced from vitamin D3 using bovine CYP11A1 [34]. These compounds were purified by TLC and reverse-phase HPLC. Their structures have previously been determined by NMR [33, 34, 52]. Ethanol was used as a dilutent and a vehicle, and stock solutions of secosteroids were prepared as described previously [49, 53].
2.2. Human skin and human primary skin cells
The use of human tissue and cells was approved by the UTHSC Institutional Review Boards as an exempt protocol #4. Human skin samples were obtained from the Regional One Health Center (foreskin) and the Methodist University Hospital (adult skin), Memphis, TN. Human epidermal neonatal keratinocytes or melanocytes were isolated from foreskin of African-American donors as previously described [38, 48, 49].
2.3. Cell culture
HaCaT keratinocytes were grown in Dulbecco’s minimal essential media (DMEM) supplemented with 5% charcoal-treated fetal bovine serum (FBS) (Atlanta Biologicals, Lawrenceville, GA, USA) [47, 54]. Epidermal keratinocytes were grown in keratinocyte growth media (KGM) supplemented with keratinocyte growth factors (KGF) (Lonza Walkersville Inc, Walkersville, MD), while epidermal melanocytes were grown in MBM-4 with MGM-4 medium containing 0.5% charcoal-treated FBS [38, 55]. Keratinocytes and melanocytes in their third passages were plated either on 96-well plates (ROS assays) or 24-well plates (other experiments, unless indicated below) to reach confluence or semiconfluence, respectively [56]. Cultures were treated with secosteroids or other listed compounds for 24 h before UVB exposure and for various times after UVB exposure. For UVB exposures the media were replaced with phosphate buffered saline (PBS) [38].
2.4. Exposure of cells to UVB
The UVB irradiation was performed with a Biorad UV transluminator 2000 (Bio-RAD Laboratories, Hercules, CA, USA) as previously described [57, 58]. Cultures were irradiated (from the bottom of the plates) with doses of 0, 25, 50, 75, or 200 mJ/cm2, respectively. The irradiance spectrum of the UV incidence upon the cells is presented in Figure 1.
Figure 1.

Spectral irradiance of the Biorad Model 2000 ultraviolet transilluminator. (A), irradiance operating in the normal (—) and preparative (·····) modes. (B), the spectrum of UV passed through the plastic culture petri dish. The UV irradiance spectra were measured using an Optronic spectroradiometer, model 754.
2.5. Determination of Cell viability assay
HaCaT keratinocytes were plated in 96-well plates and after reaching confluence they were exposed to different doses of UVB. Following this, graded concentrations of the steroids and secosteroids being tested were added from ethanol stocks with the corresponding ethanol concentrations serving as controls. After 44 h of incubation with the compounds, 20 μl of MTS/PMS solution (Promega, Madison, WI) were added to the cells. Four hours later, absorbance was recorded at 490 nm using an ELISA plate reader. The number of viable cells was measured in six replicates
2.6. Measurement of Reactive oxygen species (ROS)
The assays were performed as described previously [56]. Briefly, cells were plated onto 96-well plates and treated with secosteroids, or ethanol vehicle (dilution ≤1:1,000) for 1 or 24 h. After preincubation with CM-H2DCFDA dye, cells were irradiated with UVB as above. After 3 or 4 h of incubation cells were washed with PBS and the generation of ROS was determined by measuring the fluorescence with excitation at 480 nm and emission at 528 nm, using a SpectraMax M2e (Molecular Devices, Sunnyvale, CA).
2.7. Measurement of nitric oxide (NO−) production
Nitrite (NO−) levels were measured using Griess reagent (1% sulfanilamide-0.1% N-1-naphthyl-ethylenediamine dihydrochloride in 2.5% phosphoric acid) (Sigma, St. Louis, MO) as previously described [56, 59]. Confluent (keratinocytes) or semiconfluent (melanocytes) cultures were treated with secosteroids for 24 h then irradiated (see above). After post-irradiation incubation for 5 or 30 min cells were harvested by scraping and further centrifuged at 3000 g for 5 min as described [56]. Supernatants were collected and mixed with equal amounts of Griess reagent. The generation of NO was determined by measuring the absorbance at 540 nm of purple azo compound formed from the reaction between nitrates formed in samples and Griess reagent, with calculations performed as described previously [56, 59].
2.8. Measurement of H2O2 concentrations
Hydrogen peroxide (H2O2) levels were measured using a luminescence method [56, 60]. Cells were treated as described above for 24 h, media were changed to PBS and cultures were irradiated with UVB of different intensities (see above). The generation of H2O2 was determined by measuring the luminescence of luminol (Sigma, St. Louis, MO) by H2O2 that is released by cells from 5 min until 1 h after UV exposure, as described before, using a Turner Luminometer (TD20/20) (Promega, Madison, WI). The specificity of the reaction was determined by treating separate UV-irradiated cells with 300 units/mL of catalase (Sigma, St. Louis, MO), which degraded H2O2 to H2O and O2. Data are presented as concentration of H2O2 (pmol) per 0.1 million cells and further as a percentile of control (ethanol treated cells) [56].
2.9. Measurement of Reduced glutathione (GSH) concentrations
Reduced glutathione (GSH) levels were measured using a fluorometric method [56, 59]. Briefly, cells were treated with secosteroids for 24 h, irradiated with the UVB of different intensities (see above), further incubated for 1 h in culture media then harvested following trypsinization [56]. Cells were washed once with 1x PBS-EDTA, pH 8.0, centrifuged and cell pellets were resuspended in ice cold 5% meta-phosphoric acid (MPA) (Sigma, St. Louis, MO), sonicated, and centrifuged again at 12,000 rpm. After collection of the supernatant, aliquots were taken for protein determination. The remaining supernatant was mixed with 1x PBS-EDTA buffer and OPAME (o-phthaldialdehyde (Sigma, St. Louis, MO) in methanol (Fisher, Pittsburgh, PA) and borate buffer (potassium-tetraborate, Sigma, St Lois, MO). These samples were aliquoted into 96-well plate, incubated for 15 min at RT and reduced GSH levels were determined by measuring the fluorescence with excitation at 350 nm and emission at 420 nm, using a SpectraMax M2e (Molecular Devices, Sunnyvale, CA). Data are presented as a percentile of control (EtOH- treated cells).
2.10. Measurement of CPD dimers levels
Cyclobutane pyrimidine dimers (CPDs) were measured using immunofluorescence as described previously [56]. Briefly, keratinocytes or melanocytes were plated onto chamber slides, treated with secosteroids or ethanol vehicle (dilution 1:1,000) for 24 h prior UVB exposure, then a further 3 h after the exposure. Following the manufacturer’s protocol, cells were stained with anti-CPD antibody (clone TDM2) (Cosmo Bio Co Ltd., Tokyo, Japan) (dil 1:100) and subsequently processed as described previously. Images of the immunofluorescence were recorded and analysed using ImageJ software (NIH free download).
2.11. Immunofluorescent measurement of p53
Keratinocytes or melanocytes were plated onto chamber slides in duplicates. Cells were treated with secosteroids at a range of concentrations (see figure legends) for 24 h prior UVB exposure (see above) then for 3 h after exposure for detection of p53 or phospho-p53 Ser-46, or 12 h after exposure for detection of phosphor-p53 Ser-15 [11, 56]. Immunofluorescence analysis was performed as described previously [26, 56]. Briefly, cells were fixed in 4% paraformaldehyde (PFA) for 10 min at room temperature and washed three times with 0.1% Triton X-100 (BioRad, Hercules, CA) in PBS to permebealize membranes. Blocking was performed in 10% FBS in PBS for 1 h at RT after quenching of endogenous peroxidase with 1% H2O2 (BioRad, Hercules, CA) in PBS. Cells were incubated with primary antibodies diluted in blocking buffer (1:100), overnight at 4°C. Primary antibodies were either anti-p53 (ab4060, Cell Signaling, Danvers, MA), anti-phospho p53 Ser-15 (9284 Cell Signaling, Danvers, MA) or anti-p53 (phosphor S-46) (ab131348, Abcam, Cambridge, MA). The following day cells were washed and incubated with secondary antibody, Alexa-Fluor 488 goat anti-rabbit IgG (Invitrogen Molecular Probes, Eugene, Oregon, USA) diluted in blocking buffer (1:100), for 1 h at RT. After washing with PBS, nuclei were stained red with Vectashield mounting media with propidium iodide (Vector Laboratories, Burlingame, CA). Images of stained cells were recorded with a fluorescence microscope and analysed using ImageJ software (NIH free download).
2.12. Comet assays
Cells were plated in 12-well plates and cultured until confluent. Then the medium was replaced with PBS and cells were exposed to UVB at 200 mJ/cm2. PBS was removed and replaced with fresh medium containing 100 nM secosteroids and cells further incubated for 3 h at 37°C. After detaching, cells were counted and used for the comet assay. The comet assay was performed following the manufacturer’s protocol (Trevigen, Gaithersburg, MD) as previously described [56, 61]. Cell suspensions at a density of 1x105/ml were combined with molten 1.2% low-melting-point agarose, diluted 1:10, placed onto two frosted slides precoated with 0.6% normal agarose, and incubated at 4°C for 30 min. Cells were further digested in lysis solution at 4°C overnight. DNA fragments resulting from strand breaks were separated by electrophoresis using alkaline electrophoreses solution (200 mM NaOH, 1 mM EDTA, pH>13) in a horizontal gel electrophoresis slide tray (Comet-10, Thistle Scientific, UK). DNA fragments were exposed to alkaline unwinding for 20 min at room temperature after which electrophoresis was performed at 25 V for 35 min. Following neutralization with 0.4 M Tris, pH7, comets were visualized with propidium iodine (Sigma-Aldrich, St. Louis, MO). The slides were examined and images were captured using a Nikon fluorescent microscope and the Leica digital DM 4000b fluorescent microscope equipped with image analysis software. At least 60 comet images were taken for each condition. DNA damage was measured by the tail length [62] using TriTrek Comet Score version 1.5.
2.13. Reverse transcription (RT) and quantitative polymerase chain reaction (qPCR)
Human keratinocytes were isolated from neonatal foreskins, from donors of Afican-American descent, as described previously [48, 49]. Cells were cultured in KBM media supplemented with KGF (Lonza). Cells in their 3rd passage were treated with 0.1 μM 1,25(OH)2D3, 20S(OH)D3, 25(OH)D3, 20,23(OH)2D3 or 20S(OH)7DHC, or ethanol vehicle, for 24 h. Cells were harvested and RNA isolated using the Absolutely RNA Miniprep kit (Stratagen) as previously described [63].
Reverse transcription was performed using the Transcriptor First Strand cDNA Synthesis kit (Roche) using 1 μg of RNA per reaction. Primers were designed using the Universal Probe Library software (UPL Roche, Germany or as previously described [64, 65] and are shown in Table 1. Real-time PCR (qPCR) data were generated from input cDNA using a Cyber Green Mix (n = 3) that was amplified using standard settings on a Roche LC480 LightCycler. Gene expression was normalized using β-actin by the DDCp method. Changes in gene expression are presented as a fold change ±SD, as indicated.
Table 1.
Sequences of the primers used for qPCR
| Genes | Description | Primers |
|---|---|---|
| β-actin | β-actin | L 5′-CCAACCGCGAGAAGATGA-3′ R 5′-CCAGAGGCGTACAGGGATAG-3′ |
| CAT | catalase | L 5′-CGTGCTGAATGAGGAACAGA-3′ R 5′-AGTCAGGGTGGACCTCAGTG-3′ |
| Cu/Zn-SOD | superoxide dismutase | L 5′-GGCAAAGGTGGAAATGAAGA-3′ R 5′-GGGCCTCAGACTACATCCAA-3′ |
| Mn-SOD | superoxide dismutase | L 5′-GCTCATGCTTGAGACCCAAT-3′ R 5′-CACCCGATCTCGACTGATTT-3′ |
| NRF2 | the transcription factor NF-E2-related factor 2 | L 5′-TTCTGTTGCTCAGGTAGCCCC -3′ R 5′-TCAGTTTGGCTTCTGGACTTGG -3′ |
2.14. Statistical analysis
Data are presented as means ± SD and were analysed using the Student’s t-test or ANOVA with appropriate post hoc test (for more than two groups), Dunnett’s test using Microsoft Excel and Prism 4.00 (GraphPad Software, San Diego, CA). Statistically significant differences are denoted in the figures and corresponding figure legends. Data are presented as a percentile of the control ethanol-treated cells.
3. Results and Discussion
3.1. Overview on synthesis of 20S(OH)D3 and 20,23(OH)2D3 in epidermal keratinocytes
We have provided ex-vivo evidence that tissues expressing CYP11A1, including epidermal keratinocytes, are able to metabolize vitamin D3 producing 20S(OH)D3 as the major metabolite and 20,23(OH)2D3 the second major metabolite of the pathway [41]. Smaller amounts of other mon-, di- and tri-hydroxy-derivatives of D3 are also produced [41]. In the present follow-up study we have confirmed the ability of HaCaT keratinocytes to produce 20S(OH)D3 from D3 precursor (Fig. 2A and B). Using LC-MS/MS in the ESI mode we also detected the monohydroxy D3 transition for m/z 401[M+H]+→383[M+H-H2O]+ at a retention time corresponding to 20(OH)D3, in samples of extracted human epidermis (Fig. 2C). While the first result is confirmatory, the latter indicates that 20S(OH)D3 is present in the human epidermis in vivo.
Figure 2.
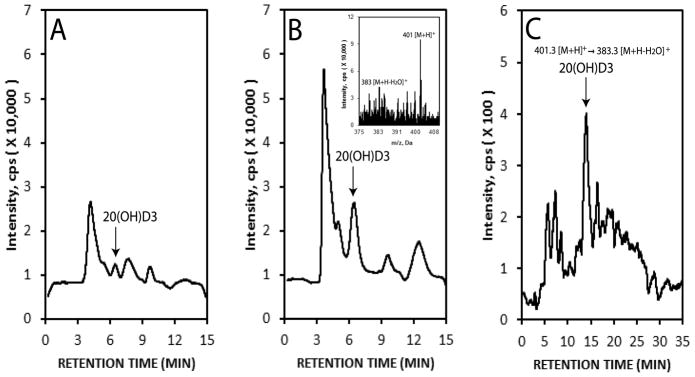
Detection of 20S(OH)D3 in human keratinocytes. HaCaT epidermal were incubated with 50 μM vitamin D for 0 (A) or 18 h (B) and extracted for LC/MS analysis. Untreated human epidermis was similarly extracted and analysed (C). The production of 20S(OH)D3 in cultured HaCaT keratinocytes was determined by LC/MS-SIM with m/z = 383 [M+H-H2O]+ (insert in B). The epidermal extract was analysed by LC-MS/MS by ESI with MRM for the transition m/z 401[M+H]+→383[M+H-H2O]+ (C). The arrow shows the retention time corresponding to 20S(OH)D3 standard. The extraction procedures and conditions for LC/MS have been described previously [41].
We have previously established a chemical route for the synthesis of 20S(OH)D3 with 20S(OH)7DHC serving as an intermediary product [52], and synthesized the C-20 epimer with the R configuration (20R(OH)D3) [66]. Armed with these synthetic analogs and with the information listed above, plus the known ability of classical vitamin D3 metabolites to protect epidermal keratinocytes against UVB induced damage (reviewed in [25, 28]), we tested the protective effects of non-calcemic 20S(OH)D3 and 20,23(OH)2D3 against UVB-induced damage in comparison to 1,25(OH)2D3 and related sterols, in human epidermal keratinocytes and melanocytes.
3.2. Anti-oxidative responses of cells to novel vitamin D analogs
In a preliminary experiment, we used the MTS test on HaCaT keratinocytes to compare the protective effects of 20S(OH)D3, its enantiomer 20R(OH)D3, and precursors 20S(OH)7DHC and D3 against UVB-induced cell death. Only 20S(OH)D3 attenuated cell death induced by UVB (Fig. 3). The lack of a detectable effect of 20R(OH)D3 at identical concentrations to 20S(OH)D3 is consistent with the reported lower potency of the R versus S enantiomer in previous biological testing [66]. Therefore, the naturally occurring 20S(OH)D3 enantiomer was selected for subsequent experiments.
Figure 3.
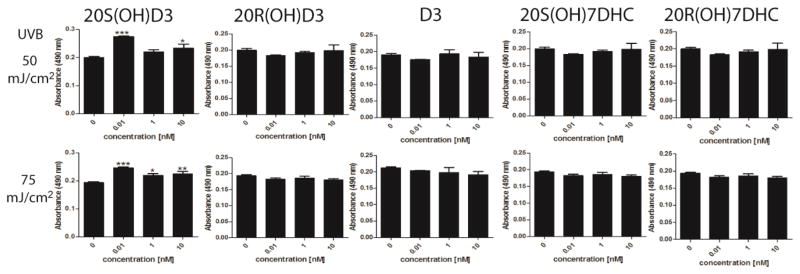
The effects of 20S(OH)D3, 20R(OH)D3, D3, 20S(OH)7DHC, and 20R(OH)7DHC on attenuation of HaCaT cell death induced by UVB. Confluent cultures of HaCaT keratinocytes were irradiated with 0, 25, 50, or 75 mJ/cm2 doses of UVB and then treated with graded concentrations of the steroids and secosteroids. After 48 h, cell viability was measured with the MTS test. Data were analyzed using ANOVA, * p<0.05, ** p<0.01, and *** p<0.001.
Using HaCaT keratinocytes we tested the protective effects of steroids and secosteroids against UVB-induced production of ROS (Fig. 4). Initially we tested whether 20S(OH)D3 and 20,23(OH)2D3, in comparison to 1,25(OH)2D3, 20S(OH)7DHC, 7-hydroxycholestrol (7(OH)C) and cholesterol sulphate, could attenuate ROS production induced by 200 mJ/cm2 of UVB (Fig. 4A and B). The last two cholesterol derivatives were included because of the recent detection of retinoic acid orphan receptors (ROR)α and γ in human keratinocytes [67], for which these compounds serve as ligands. With the exception of 7(OH)C and cholesterol sulphate, all the hydroxy-derivatives of vitamin D3 tested, as well as 20S(OH)7DHC, attenuated UVB-induced ROS production with significant protection being observed at concentrations of both 100 nM and 1 nM (Fig. 4A and B). The protective effects of 100 nM 20S(OH)D3, 20,23(OH)2D3 and 20S(OH)7DHC were also examined in comparison to 1,25(OH)2D3, 25(OH)D3 and D3 using lower UVB doses of 25 and 50 mJ/cm2 (Fig. 4B and C). The vitamin D hydroxy-derivatives, but not D3 itself or 20S(OH)7DHC, attenuated ROS production with 25(OH)D3 having significant action only at 50 mJ//cm2. The use of vitamin hydroxy-derivatives and their precursors at a concentration of 100 nM in this and following experiments was rationalized by our earlier observations that this concentration was most optimal for measuring their phenotypic effects in keratinocytes and melanocytes [38, 45, 47–49].
Figure 4.

The effects of 1,25(OH)2D3, 25(OH)D3, 20S(OH)D3, 20,23(OH)2D3, 20S(OH)7DHC, D3, cholesterol sulphate, or 7(OH)cholesterol on the attenuation of UVB-induced ROS generation. HaCaT keratinocytes were given dosed of UVB of 200 (A and B), 25 (C) or 50 (D) mJ/cm2. Cells were treated with above compounds or the ethanol vehicle for 1 h (C, D) or 24 h (A, B) prior to the UV irradiation and for a further 4 h (A, B) or 3 h (C, D) post-UVB exposure, at the concentration 1 nM (A) or 100 nM (B,C,D). Data are presented as % of control [mean ±SD (n=6)] and analysed using the student t-test, * p<0.05, ** p<0.01, *** p<0.001.
To test the ability of vitamin D hydroxy-derivatives and 20S(OH)7DHC to scavenge H2O2 produced in response to UVR, HaCaT keratinocytes were incubated with these compounds for 24 h before UVB exposure (Fig. 5). Cells were irradiated with UVB doses of 25 or 50 mJ/cm2. Supernatants were collected after 30 min of irradiation to measure the concentration of H2O2 released from the cells. While there was no H2O2 released by non-irradiated cells, keratinocytes produced H2O2 in response to UVB irradiation as reported before [1], and this production was UVB dose- and time- dependent with a peak after 30 min as described previously [56]. Treatment of cells with 20S(OH)D3, 20,23(OH)2D3, 20S(OH)7DHC, 1,25(OH)2D3 or 25(OH)D3 significantly reduced H2O2 concentrations in cell supernatants following UVB exposure, although the effect of 25(OH)D3 was not significant at the higher dose (50 mJ/cm2) (Fig. 5). At 25 mJ/cm2, 20S(OH)D3, 20,23(OH)2D3 and 1,25(OH)2D3 exhibited a significantly stronger inhibition of H2O2 release than 25(OH)D3 (Fig. 5A), and at 50 mJ/cm2 dose 20S(OH)D3 showed stronger inhibition than 1,25(OH)2D3 (Fig. 5B).
Figure 5.

1,25(OH)2D3, 25(OH)D3, 20S(OH)D3, 20,23(OH)2D3 and 20S(OH)7DHC reduce levels of H2O2 produced by UVB-irradiated HaCaT keratinocytes. Keratinocytes were treated with the compounds above (100 nM) or ethanol vehicle for 24 h and then irradiated with 25 (A) or 50 (B) mJ/cm2 UVB. H2O2 produced by keratinocytes was determined 30 min after UVB irradiation. The dose dependent stimulation of H2O2 production by UVB is shown as an insert. Data are presented as % of control [mean ±SD (n=3)] and were analysed using the t-test, * p<0.05, ** p<0.01, *** p<0.001.
The effect of UVB on H2O2 release by human melanocytes 5 and 30 min after UVB exposure is shown in Fig. 6A and B. There was a statistically significant inhibition of H2O2 release by 100 nM 20S(OH)D3 at 25 and 50 mJ/cm2 5 min after exposure (Fig. 6A), and at 50 and 75 mJ/cm2 30 min after exposure (Fig. 6B). Using the same passages of human melanocytes we also tested the protective effect of 20S(OH)D3 on UVB-induced production of NO (Fig. 6C and D). Cells were incubated with 100 nM 20S(OH)D3 for 24 h before replacing media with PBS and irradiating with UVB doses of 25, 50, or 75 mJ/cm2. Treatment of cells with 20S(OH)D3 before UVB irradiation significantly reduced NO production at 25 and 50 mJ/cm2 both after both 5 and 30 min of exposure (Fig. 6C and D).
Figure 6.
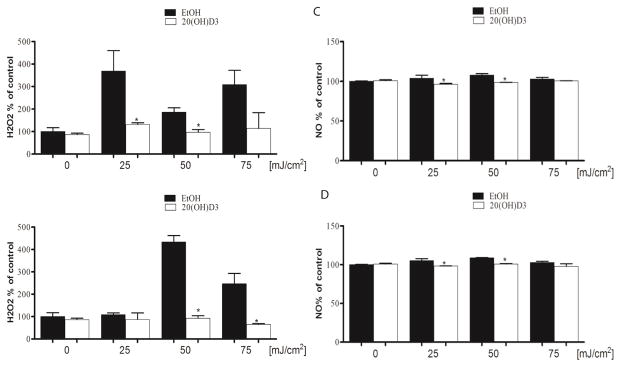
20S(OH)D3 reduces H2O2 and NO production induced by UVB irradiation of melanocytes. Melanocytes were treated with 100 nM 20S(OH)D3 or ethanol vehicle for 24 h and subsequently irradiated with 0, 25 50, or 75 mJ/cm2 of UVB. H2O2 (A, B) or NO (C, D) production by cells was determined 5 min (A, C)) and 30 min (B, D) after UVB irradiation.. Data are presented as % of control [mean ±SD (n=3] and analysed using the t- test, * p<0.05.
Using the same experimental design we also tested the protective effects of other novel secosteroids against UVB-induced NO release. It is known that keratinocytes produce NO in response to UVB irradiation [1] in a UVB-dose dependent manner (Fig 7, insert). The production of NO occurs shortly after irradiation, reaching a peak at 30 min and returns to low levels after 1 h [56]. Treatment of cells with 20S(OH)D3, 20,23(OH)2D3, 20S(OH)7DHC, 1,25(OH)2D3 or 25(OH)D3 before UVB irradiation reduced NO production. Of note, the inhibition of NO release by 20(OH)D3, 20,23(OH)2D3, 1,25(OH)2D3 and 20(OH)7DHC was stronger in comparison to 25(OH)D3 at the higher dose of UVB, e.g., 50 mJ/cm2 (p<0.05)(Fig. 7B).
Figure 7.

1,25(OH)2D3, 25(OH)D3, 20S(OH)D3, 20,23(OH)2D3, and 20S(OH)7DHC reduce levels of NO produced by UVB-irradiation of HaCaT keratinocytes. Keratinocytes were treated with the above compounds or ethanol vehicle for 24 h then irradiated with 25 (A) or 50 (B) mJ/cm2 UVB. NO produced by keratinocytes was determined 30 min after UVB irradiation. The dose dependent stimulation of NO production by UVB is shown as an insert. Data are presented as % of control [mean ±SD (n=3)] and were analysed using the t-test, * p<0.05, ** p<0.01, *** p<0.001. The inhibitory effect of 20,23(OH)2D3 on NO production at 50 mJ/cm2 was stronger than that of 1,25(OH)2D3 (p<0.05).
Exposure to UVB causes an immediate reduction of intracellular GSH [68]. Previously, we documented that melatonin and its metabolites can attenuate UVB-induced reduction of GSH in human epidermal keratinocytes [56]. Using a similar experimental design, we tested the effects of 20S(OH)D3, 20,23(OH)2D3, 20S(OH)7DHC, 1,25(OH)2D3 and 25(OH)D3 on the relative GSH content after treatment of keratinocytes for 24 h prior to UVB exposure (Fig. 8). The level of reduced GSH decreased with the exposure to UVB in a dose-dependent manner (Fig 8A, insert). At 25 and 50 mJ/cm2, 20S(OH)D3, 20,23(OH)2D3, 1,25(OH)2D3 and 25(OH)D3, and 20S(OH)7DHC only at 25 mJ/cm2, significantly attenuated the reduction of reduced GSH after 1 h of UVB exposure (Fig. 8).
Figure 8.

1,25(OH)2D3, 25(OH)D3, 20S(OH)D3, 20,23(OH)2D3, and 20S(OH)7DHC attenuate inhibition of GSH levels induced by UVB in epidermal keratinocytes. HaCaT keratinocytes were treated with the above compounds (100 nM) or ethanol vehicle for 24 h and then irradiated with 25 (A) or 50 mJ/cm2 UVB (B). GSH produced in keratinocytes was determined 1 h after UVB irradiation. The differences in GSH production under different UVB intensities are shown as an insert. Data are presented as % of control [mean ±SD (n=3)] and analysed using the t- test, * p<0.05, ** p<0.01, *** p<0.001.
Since the above data clearly show that the secosteroids tested have a protective effect against UVB-induced oxidative damage, we tested whether they could stimulate the expression of genes encoding enzymes involved in protection against oxidative stress. Increases in the expression of genes for catalase and both forms of SOD were observed following the incubation of human epidermal keratinocytes for 24 h with 100 nM 20S(OH)D3, 20,23(OH)2D3, 20S(OH)7DHC or 25(OH)D3, but not with 1,25(OH)2D3, (Fig. 9A-C). In addition, the expression of NRF2, a master regulator of oxidative responses [69] and a protector of genomic stability [70], was also upregulated by 20S(OH)7DHC, 20S(OH)D3 and 1,25(OH)2D3, but not by 25(OH)D3 or 20,23(OH)2D3 (Fig. 9C)
Figure 9.

The effects of 1,25(OH)2D3, 25(OH)D3, 20S(OH)D3, 20,23(OH)2D3 and 20S(OH)7DHC on the expression of genes mediating the anti-oxidative stress response in human epidermal (HEKn) keratinocytes. Cells were treated with 100 nM of the above compounds for 24 h before RNA isolation. A: catalase (CAT), B: Cu/Zn superoxide dismutase (Cu/Zn-SOD), C: Mn superoxide dismutase (Mn-SOD), D: the transcription factor NF-E2-related factor 2 (NRF2). Gene expression was normalized using B-actin by the DDCp method. Changes in gene expression are presented as a fold change (mean ±SD, n=3) and analysed using the t- test, * p<0.05, ** p<0.01, *** p<0.001; **** p<0.0001.
UVB-induced pathology in the skin includes oxidative DNA damage and the production of reactive oxygen species (ROS), including hydrogen peroxide and nitric oxide, which change intracellular levels of glutathione [1, 2, 25]. UVB also induces transformation of 7DHC to vitamin D3 [13, 14], which in the epidermis is further hydroxylated to 1,25(OH)2D3 [14, 19]. Experimental evidence is accumulating that 1,25(OH)2D3 can protect epidermal keratinocytes against oxidative damage induced by UVB [25–28], as well as against epidermal skin cancers that are induced by UVB [18, 29–31]. The present data show that that CYP11A1-derived noncalcemic vitamin D hydroxy-derivatives, 20S(OH)D3 and 20,23(OH)2D3, also inhibit UVB-induced production of ROS, H2O2, NO and attenuate the reduction of intracellular levels of GSH, being at least as effective as 25(OH)D3 and 1,25(OH)2D3. These results imply that the novel secosteroids should play a role in protection against UVB induced skin cancer, that can be similar to the role proposed for 1,25(OH)2D3 [25, 27, 29, 31]. The pharmacological advantage of novel CYP11A1-derived hydroxyderivatives of vitamin D over classical 1,25(OH)2D3 is that they are non-calcemic and non-toxic at supra-physiological doses [50, 51, 53], while having inhibitory activity on melanomas [38, 45, 63].
An interesting finding of this study is the ability of 20S(OH)7DHC to inhibit H2O2 and NO production induced by UVB as well as cause significant stimulation of the expression of genes encoding CAT, both types of SOD and NRF2, which was unexpected. While the majority of protective effects against oxidative stress and attendant skin cancerogenesis by 1,25(OH)2D3 [25, 29] and novel 20S(OH)D3 and 20,20(OH)2D3 [38, 71] are expected to be mediated by the VDR, a target receptor for 20S(OH)7DHC has not been identified. Good candidates are ROR or LXR receptors; however, this hypothesis remains to be tested experimentally using transgenic mice. Also, non-receptor mediated induction of anti-oxidative actions should be considered, since our previous studies showed that 7DHC significantly attenuated oxidative protein modification more effectively than cholesterol, vitamin D3 and D2, and surprisingly 7DHC was more effective than GSH or melatonin [72]. The latter is recognised as a potent anti-oxidant also operating in the skin [73, 74]. Non-genomic receptors have been proposed for the UVB-protective effects of 1,25(OH)2D3 [27]. For the CYP11A1 generated secosteroids, our molecular modelling predicts poor binding to the non-genomic (alternative) pocket of the VDR [71]. However, RORα and γ could represent alternative receptors to mediate some of these effects, since 20S(OH)D3 and 20,23(OH)2D3 can also act through these receptors [67].
3.3. Attenuation of DNA damage induced by UVB
DNA serves as a chromophore for UVB [2] which causes DNA damage primarily by the formation of CPD [3]. P53 plays an important role in the repair of UV-induced DNA damage [5, 75]. After exposure to UVB or oxidative stress, phosphorylated p53 accumulates in the nucleus. The most important phosphorylation sites on p53 are Ser-15 which promotes accumulation and activation of p53 and DNA repair, and Ser-46 which regulates apoptosis following DNA damage [10]. Previously it was shown that hormonal factors such as melatonin can induce the p53-dependent DNA damage response through stimulation of phosphorylation at Ser-15 but not at Ser-46 [56, 76, 77].
We therefore tested whether post-treatment of normal epidermal keratinocytes and melanocytes with classical 1,25(OH)2D3 and novel 20S(OH)D3 could increase phosphorylation of p53 at Ser-15 or Ser-46, and reduce CPD levels following UVB exposure (Fig. 10). Previously we have shown that phosphorylation of Ser-46 in keratinocytes occurs earlier after UVB exposure (3 h), while phosphorylation of Ser-15 occurs later (12 h after UVB exposure) [56]. Therefore, these times after exposure to UVB were evaluated in the current experiments. The vitamin D analogues tested did not significantly change the expression of p53 or p53 phosphorylated at Ser-46 (data not shown), which is similar to the lack of effect of melatonin on these markers after exposure to UVB [56]. However, 1,25(OH)2D3 and 20S(OH)D3 stimulated p53 phosphorylation at Ser-15 in keratinocytes (Fig. 10A) and melanocytes (Fig. 10C), with significant effects seen at UVB doses of 25 and 50 mJ/cm2 for keratinocytes (Fig. 10A) and 25 mJ/cm2 for melanocytes (Fig. 10C).
Figure 10.

Treatment of keratinocytes or melanocytes with 1,25(OH)2D3 or 20S(OH)D3 decreases CPD levels and increase p53 phosphorylation at Ser-15 following UVB exposure. Keratinocytes (A,B) and melanocytes (C,D) were treated with 100 nM 1,25(OH)2D3 or 20S(OH)D3 for 24 h prior to UVB exposure. Cells were exposed to UVB intensities of 25, 50, or 75 mJ/cm2 and immediately treated again with 100 nM 1,25(OH)2D3, 20(OH)D3, or vehicle control for 3 h for detection of CPDs (B,D) or for 12 h for detection of phosphorylated p53 at Ser-15 (S15) (A,C). Cells were fixed and stained with anti-CPD antibody (green) (B, D inserts) or with anti-phosphorylated p53S15 antibody (A, C inserts). Stained cells were imaged with a fluorescence microscope and fluorescence intensity was analysed using ImageJ software, and data are analysed using Graph Pad. Data are presented as % of control [mean ±SD (n=6)] and analysed using t- test, * p<0.05, ** p<0.01, *** p<0.001, **** p<0.0001.
In complementary experiments, UVB-irradiated keratinocytes (Fig. 10B) or melanocytes (Fig. 10D) were immediately treated after UV exposure with 1,25(OH)2D3 or 20S(OH)D3 for 3 h, and CPD-positive cells were evaluated with CPD-specific antibody. CPD-positive cells were not detected in untreated cells (Fig. 10B and D). CPD-staining intensity of UVB-irradiated cells was weaker in cells treated with 1,25(OH)2D3 or 20S(OH)D3 (Fig. 10B, D), as compared to untreated cells. These effects were again dependent on the UVB dose with significant inhibitory effects of 1,25(OH)2D3 or 20S(OH)D3 in keratinocytes at doses of 25 and 50 mJ/cm2 (Fig. 10B), and in melanocytes at all does tested (25, 50 and 75 mJ/cm2) (Fig. 9D).
To further analyse the extent of the DNA damage, we employed the Comet assay which allows the DNA of damaged cells to migrate from intact DNA in agarose under an electric field, and uses fluorescent microscopy for visualisation of the DNA migration. We observed extensive DNA damage when melanocytes were exposed to UVB (Fig. 11, insert). There was a significant difference (p<0.0001) between UVB-induced DNA damage in control cells (non-treated) versus cells treated with 1,25(OH)2D3, 20S(OH)D3 or 20,23(OH)2D3, as measured by the mean comet tail moment (Fig. 11). Also, 20,23(OH)2D3 and 20S(OH)D3 exhibited a statistically stronger inhibitory effect on DNA damage that 1,25(OH)2D3, with 20S(OH)D3 showing the greatest efficacy.
Figure 11.

1,25(OH)2D3, 20S(OH)D3, and 20,23(OH)2D3 inhibit DNA damage induced by UVB as shown by the Comet assay. Epidermal melanocytes were treated with the above secosteroids for 3 h following UV-irradiation (200 mJ/cm2). Photographs (upper panel) show Comets for treated and non-treated cells. UVB caused strand breaks in DNA which migrated in the electrophoretic field to form comet tails. Tail moments wert used to measure DNA damage. Data were analysed using the student t-test and labelled as **** p<0.0001.
In summary, our studies using the immunofluorescent staining technique indicate that 20S(OH)D3, 20,23(OH)2D3 and 1,25(OH)2D3 (the latter serving as a positive control because of previous demonstrations of its protection against DNA damage in keratinocytes [25, 28]) can protect keratinocytes and melanocytes against UVB-induced damage as illustrated by CPD generation and by results of the Comet assay. The enhancement of the expression of p53 phosphorylated at Ser-15, but not at Ser-46 and down-regulation of CPD formation suggest that the vitamin D analogs may enhance the DNA repair capacity of melanocytes and keratinocytes. Thus, this study on the protective effects of secosteroids against UVB-induced DNA damage not only complements the excellent studies by Mason and coworkers [25–28], but also identifies novel non-calcemic CYP11A1-derived vitamin D derivatives, and the 20(OH)D3 precursor 20S(OH)7DHC, as protectors against UV induced DNA damage and oxidative stress, not only in human keratinocytes but also in melanocytes. The highest efficacy is seen for CYP11A1-derived 20S(OH)D3 and 20,23(OH)2D3, and classical 1,25(OH)2D3, with both precursors 25(OH)D3 and 20S(OH)7DHC having less efficacy or showing differential effects.
In conclusion, novel CYP11A1-derived and classical secosteroids which are endogenously produced in keratinocytes, protect both epidermal keratinocytes and melanocytes against UVB-induced oxidative stress and DNA damage.
4. Concluding remarks
Skin, being exposed to environmental stressors, has developed sophisticated systems to deal with noxious factors [78, 79]. The UVB spectrum of solar radiation represents a major cutaneous stressor, with the skin pigmentary system [80, 81] protecting against its deleterious effects. Most recently, it was proposed that the local melatoninergic anti-oxidative system (MAS) [82] and the cutaneous serotoninergic/melatoninergic system [74] protect the skin against UVB induced damage. The data coming from the laboratories of Dr Mason and Dr Bikle indicate that vitamin D3, generated through the action of UVB and hydroxylated to its active form, 1,25(OH)2D3 [25–28], acts via the VDR [19, 29, 30] to protect the skin against the damage induced by UVB. We have discovered novel secosteroidogenic pathway(s) operating in vivo [41, 42, 83, 84], for which the major products also protect keratinocytes and melanocytes against UVB-induced oxidative stress and DNA damage, as shown in this study. Of note is the expression of the enzyme that initiates and regulates these novel secosteroidogenic pathways, CYP11A1, which is also be upregulated by UVB [85, 86]. Therefore, we propose that the activities of the classical (producing 1,25(OH)2D3) and novel (producing 20(OH)D3 and 20,23(OH)2D3) secosteroidogenic enzyme systems regulate the protection of the human epidermis against UVB exposure by attenuating DNA- and metabolic-damage via several mechanisms. These include the reduction in H2O2 and NO levels, elevation of GSH levels, enhancement of DNA repair, and promotion of keratinocytes and melanocytes survival in a context dependent fashion. Thus the secosteroidal molecules of which production in the skin is induced by solar radiation, are activated to secure skin homeostasis under the sun.
Highlights.
We detected 20-hydroxyvitamin D3 in the human epidermis
20-Hydroxy- and 20,23-dihydroxyvitamin D3 attenuate oxidative stress in keratinocytes and melanocytes
20-Hydroxy-7-dehydrocholesterol attenuates oxidative stress in keratinocytes and melanocytes
20-Hydroxyvitamin D3 attenuates UVB induced DNA damage to a similar degree as 1,25-dihydroxyvitamin D3
Acknowledgments
The work was supported by NIH grants 2R01AR052190-A6, 1RO1ES024132-01A1, R21 AR066505-01A1 and 1R01AR056666-01A2 to AS, 1R21AR063242-01A1, 1S10OD010678-01, and 1S10RR026377-01 to WL and the University of Western Australia (RCT). PW and SF were supported by the International Federation of Medical Student’s Association (IFMSA) sponsored Research Exchange at UTHSC.
Footnotes
Conflict of Interest:
The authors declare no conflict of interest
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Heck DE, Vetrano AM, Mariano TM, Laskin JD. UVB light stimulates production of reactive oxygen species: unexpected role for catalase. J Biol Chem. 2003;278:22432–22436. doi: 10.1074/jbc.C300048200. [DOI] [PubMed] [Google Scholar]
- 2.Wondrak GT, Roberts MJ, Cervantes-Laurean D, Jacobson MK, Jacobson EL. Proteins of the extracellular matrix are sensitizers of photo-oxidative stress in human skin cells. J Invest Dermatol. 2003;121:578–586. doi: 10.1046/j.1523-1747.2003.12414.x. [DOI] [PubMed] [Google Scholar]
- 3.Pfeifer GP, Besaratinia A. UV wavelength-dependent DNA damage and human non-melanoma and melanoma skin cancer. Photochem Photobiol Sci. 2012;11:90–97. doi: 10.1039/c1pp05144j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mouret S, Baudouin C, Charveron M, Favier A, Cadet J, Douki T. Cyclobutane pyrimidine dimers are predominant DNA lesions in whole human skin exposed to UVA radiation. Proc Natl Acad Sci U S A. 2006;103:13765–13770. doi: 10.1073/pnas.0604213103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chen X, Chen J, Gan S, Guan H, Zhou Y, Ouyang Q, Shi J. DNA damage strength modulates a bimodal switch of p53 dynamics for cell-fate control. BMC Biol. 2013;11:73. doi: 10.1186/1741-7007-11-73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Levine AJ. p53, the cellular gatekeeper for growth and division. Cell. 1997;88:323–331. doi: 10.1016/s0092-8674(00)81871-1. [DOI] [PubMed] [Google Scholar]
- 7.Campbell C, Quinn AG, Angus B, Farr PM, Rees JL. Wavelength specific patterns of p53 induction in human skin following exposure to UV radiation. Cancer Res. 1993;53:2697–2699. [PubMed] [Google Scholar]
- 8.Milczarek GJ, Martinez J, Bowden GT. p53 Phosphorylation: biochemical and functional consequences. Life Sci. 1997;60:1–11. doi: 10.1016/s0024-3205(96)00479-1. [DOI] [PubMed] [Google Scholar]
- 9.Steegenga WT, van der Eb AJ, Jochemsen AG. How phosphorylation regulates the activity of p53. J Mol Biol. 1996;263:103–113. doi: 10.1006/jmbi.1996.0560. [DOI] [PubMed] [Google Scholar]
- 10.Oda K, Arakawa H, Tanaka T, Matsuda K, Tanikawa C, Mori T, Nishimori H, Tamai K, Tokino T, Nakamura Y, Taya Y. p53AIP1, a potential mediator of p53-dependent apoptosis, and its regulation by Ser-46-phosphorylated p53. Cell. 2000;102:849–862. doi: 10.1016/s0092-8674(00)00073-8. [DOI] [PubMed] [Google Scholar]
- 11.Santoro R, Marani M, Blandino G, Muti P, Strano S. Melatonin triggers p53Ser phosphorylation and prevents DNA damage accumulation. Oncogene. 2012;31:2931–2942. doi: 10.1038/onc.2011.469. [DOI] [PubMed] [Google Scholar]
- 12.Slominski RM, Reiter RJ, Schlabritz-Loutsevitch N, Ostrom RS, Slominski AT. Melatonin membrane receptors in peripheral tissues: distribution and functions. Mol Cell Endocrinol. 2012;351:152–166. doi: 10.1016/j.mce.2012.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Holick MF, Clark MB. The photobiogenesis and metabolism of vitamin D. Fed Proc. 1978;37:2567–2574. [PubMed] [Google Scholar]
- 14.Holick MF. Vitamin D: A millenium perspective. J Cell Biochem. 2003;88:296–307. doi: 10.1002/jcb.10338. [DOI] [PubMed] [Google Scholar]
- 15.Holick MF, Tian XQ, Allen M. Evolutionary importance for the membrane enhancement of the production of vitamin D3 in the skin of poikilothermic animals. Proc Natl Acad Sci U S A. 1995;92:3124–3126. doi: 10.1073/pnas.92.8.3124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bikle DD. Vitamin D: newly discovered actions require reconsideration of physiologic requirements. Trends Endocrinol Metab. 2010;21:375–384. doi: 10.1016/j.tem.2010.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bikle DD. Vitamin D and the skin. J Bone Miner Metab. 2010;28:117–130. doi: 10.1007/s00774-009-0153-8. [DOI] [PubMed] [Google Scholar]
- 18.Bikle DD. Vitamin D receptor, UVR, and skin cancer: a potential protective mechanism. J Invest Dermatol. 2008;128:2357–2361. doi: 10.1038/jid.2008.249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bikle DD. Vitamin D metabolism and function in the skin. Mol Cell Endocrinol. 2011;347:80–89. doi: 10.1016/j.mce.2011.05.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Holick MF. Vitamin D deficiency. N Engl J Med. 2007;357:266–281. doi: 10.1056/NEJMra070553. [DOI] [PubMed] [Google Scholar]
- 21.Plum LA, DeLuca HF. Vitamin D, disease and therapeutic opportunities. Nat Rev Drug Discov. 2010;9:941–955. doi: 10.1038/nrd3318. [DOI] [PubMed] [Google Scholar]
- 22.Elias PM. Structure and function of the stratum corneum extracellular matrix. J Invest Dermatol. 2012;132:2131–2133. doi: 10.1038/jid.2012.246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Indra AK, Castaneda E, Antal MC, Jiang M, Messaddeq N, Meng X, Loehr CV, Gariglio P, Kato S, Wahli W, Desvergne B, Metzger D, Chambon P. Malignant transformation of DMBA/TPA-induced papillomas and nevi in the skin of mice selectively lacking retinoid-X-receptor alpha in epidermal keratinocytes. J Invest Dermatol. 2007;127:1250–1260. doi: 10.1038/sj.jid.5700672. [DOI] [PubMed] [Google Scholar]
- 24.Trojanowska M. Cellular and molecular aspects of vascular dysfunction in systemic sclerosis. Nat Rev Rheumatol. 2010;6:453–460. doi: 10.1038/nrrheum.2010.102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Dixon KM, Tongkao-On W, Sequeira VB, Carter SE, Song EJ, Rybchyn MS, Gordon-Thomson C, Mason RS. Vitamin d and death by sunshine. Int J Mol Sci. 2013;14:1964–1977. doi: 10.3390/ijms14011964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gupta R, Dixon KM, Deo SS, Holliday CJ, Slater M, Halliday GM, Reeve VE, Mason RS. Photoprotection by 1,25 dihydroxyvitamin D3 is associated with an increase in p53 and a decrease in nitric oxide products. J Invest Dermatol. 2007;127:707–715. doi: 10.1038/sj.jid.5700597. [DOI] [PubMed] [Google Scholar]
- 27.Dixon KM, Norman AW, Sequeira VB, Mohan R, Rybchyn MS, Reeve VE, Halliday GM, Mason RS. 1alpha,25(OH)(2)-vitamin D and a nongenomic vitamin D analogue inhibit ultraviolet radiation-induced skin carcinogenesis. Cancer Prev Res (Phila) 2011;4:1485–1494. doi: 10.1158/1940-6207.CAPR-11-0165. [DOI] [PubMed] [Google Scholar]
- 28.Song EJ, Gordon-Thomson C, Cole L, Stern H, Halliday GM, Damian DL, Reeve VE, Mason RS. 1alpha,25-Dihydroxyvitamin D3 reduces several types of UV-induced DNA damage and contributes to photoprotection. J Steroid Biochem Mol Biol. 2013;136:131–138. doi: 10.1016/j.jsbmb.2012.11.003. [DOI] [PubMed] [Google Scholar]
- 29.Bikle DD, Elalieh H, Welsh J, Oh D, Cleaver J, Teichert A. Protective role of vitamin D signaling in skin cancer formation. J Steroid Biochem Mol Biol. 2013;136:271–279. doi: 10.1016/j.jsbmb.2012.09.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Demetriou SK, Ona-Vu K, Teichert AE, Cleaver JE, Bikle DD, Oh DH. Vitamin D receptor mediates DNA repair and is UV inducible in intact epidermis but not in cultured keratinocytes. J Invest Dermatol. 2012;132:2097–2100. doi: 10.1038/jid.2012.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Jiang YJ, Teichert AE, Fong F, Oda Y, Bikle DD. 1alpha,25(OH)2-dihydroxyvitamin D3/VDR protects the skin from UVB-induced tumor formation by interacting with the beta-catenin pathway. J Steroid Biochem Mol Biol. 2013;136:229–232. doi: 10.1016/j.jsbmb.2012.09.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Slominski AT, Kim TK, Li W, Yi AK, Postlethwaite A, Tuckey RC. The role of CYP11A1 in the production of vitamin D metabolites and their role in the regulation of epidermal functions. J Steroid Biochem Mol Biol. 2013 doi: 10.1016/j.jsbmb.2013.10.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Slominski A, Semak I, Zjawiony J, Wortsman J, Li W, Szczesniewski A, Tuckey RC. The cytochrome P450scc system opens an alternate pathway of vitamin D3 metabolism. FEBS J. 2005;272:4080–4090. doi: 10.1111/j.1742-4658.2005.04819.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tuckey RC, Li W, Zjawiony JK, Zmijewski MA, Nguyen MN, Sweatman T, Miller D, Slominski A. Pathways and products for the metabolism of vitamin D3 by cytochrome P450scc. FEBS J. 2008;275:2585–2596. doi: 10.1111/j.1742-4658.2008.06406.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Slominski A, Semak I, Wortsman J, Zjawiony J, Li W, Zbytek B, Tuckey RC. An alternative pathway of vitamin D metabolism. Cytochrome P450scc (CYP11A1)-mediated conversion to 20-hydroxyvitamin D2 and 17,20-dihydroxyvitamin D2. FEBS J. 2006;273:2891–2901. doi: 10.1111/j.1742-4658.2006.05302.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Nguyen MN, Slominski A, Li W, Ng YR, Tuckey RC. Metabolism of vitamin D2 to 17,20,24-trihydroxyvitamin D2 by cytochrome p450scc (CYP11A1) Drug Metab Dispos. 2009;37:761–767. doi: 10.1124/dmd.108.025619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tuckey RC, Li W, Shehabi HZ, Janjetovic Z, Nguyen MN, Kim TK, Chen J, Howell DE, Benson HA, Sweatman T, Baldisseri DM, Slominski A. Production of 22-hydroxy metabolites of vitamin D3 by cytochrome p450scc (CYP11A1) and analysis of their biological activities on skin cells. Drug Metab Dispos. 2011;39:1577–1588. doi: 10.1124/dmd.111.040071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Slominski AT, Kim TK, Janjetovic Z, Tuckey RC, Bieniek R, Yue J, Li W, Chen J, Nguyen MN, Tang EK, Miller D, Chen TC, Holick M. 20-Hydroxyvitamin D2 is a noncalcemic analog of vitamin D with potent antiproliferative and prodifferentiation activities in normal and malignant cells. Am J Physiol Cell Physiol. 2011;300:C526–541. doi: 10.1152/ajpcell.00203.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Tang EK, Chen J, Janjetovic Z, Tieu EW, Slominski AT, Li W, Tuckey RC. Hydroxylation of CYP11A1-derived products of vitamin D3 metabolism by human and mouse CYP27B1. Drug Metab Dispos. 2013;41:1112–1124. doi: 10.1124/dmd.113.050955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Guryev O, Carvalho RA, Usanov S, Gilep A, Estabrook RW. A pathway for the metabolism of vitamin D3: unique hydroxylated metabolites formed during catalysis with cytochrome P450scc (CYP11A1) Proc Natl Acad Sci U S A. 2003;100:14754–14759. doi: 10.1073/pnas.2336107100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Slominski AT, Kim TK, Shehabi HZ, Semak I, Tang EK, Nguyen MN, Benson HA, Korik E, Janjetovic Z, Chen J, Yates CR, Postlethwaite A, Li W, Tuckey RC. In vivo evidence for a novel pathway of vitamin D(3) metabolism initiated by P450scc and modified by CYP27B1. FASEB J. 2012;26:3901–3915. doi: 10.1096/fj.12-208975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Slominski AT, Kim TK, Shehabi HZ, Tang EK, Benson HA, Semak I, Lin Z, Yates CR, Wang J, Li W, Tuckey RC. In vivo production of novel vitamin D2 hydroxy-derivatives by human placentas, epidermal keratinocytes, Caco-2 colon cells and the adrenal gland. Mol Cell Endocrinol. 2014;383:181–192. doi: 10.1016/j.mce.2013.12.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Slominski A, Ermak G, Mihm M. ACTH receptor, CYP11A1, CYP17 and CYP21A2 genes are expressed in skin. J Clin Endocrinol Metab. 1996;81:2746–2749. doi: 10.1210/jcem.81.7.8675607. [DOI] [PubMed] [Google Scholar]
- 44.Slominski A, Zjawiony J, Wortsman J, Semak I, Stewart J, Pisarchik A, Sweatman T, Marcos J, Dunbar CC, Tuckey RC. A novel pathway for sequential transformation of 7-dehydrocholesterol and expression of the P450scc system in mammalian skin. Eur J Biochem. 2004;271:4178–4188. doi: 10.1111/j.1432-1033.2004.04356.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Slominski A, Janjetovic Z, Kim T-K, Wright AC, Greese LN, Riney SJ, Nguyen MN, Tuckey RC. Novel vitamin D hydroxyderivatives inhibit melanoma growth and show differential effects on normal melanocytes. Anticancer Res. 2012;32 [PMC free article] [PubMed] [Google Scholar]
- 46.Slominski A, Janjetovic Z, Tuckey RC, Nguyen MN, Bhattacharya KG, Wang J, Li W, Jiao Y, Gu W, Brown M, Postlethwaite AE. 20S-Hydroxyvitamin D3, noncalcemic product of CYP11A1 action on vitamin D3, exhibits potent antifibrogenic activity in vivo. J Clin Endocrinol Metab. 2013;98:E298–303. doi: 10.1210/jc.2012-3074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zbytek B, Janjetovic Z, Tuckey RC, Zmijewski MA, Sweatman TW, Jones E, Nguyen MN, Slominski AT. 20-Hydroxyvitamin D3, a product of vitamin D3 hydroxylation by cytochrome P450scc, stimulates keratinocyte differentiation. J Invest Dermatol. 2008;128:2271–2280. doi: 10.1038/jid.2008.62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Janjetovic Z, Zmijewski MA, Tuckey RC, DeLeon DA, Nguyen MN, Pfeffer LM, Slominski AT. 20-Hydroxycholecalciferol, product of vitamin D3 hydroxylation by P450scc, decreases NF-kappaB activity by increasing IkappaB alpha levels in human keratinocytes. PLoS One. 2009;4:e5988. doi: 10.1371/journal.pone.0005988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Janjetovic Z, Tuckey RC, Nguyen MN, Thorpe EM, Jr, Slominski AT. 20,23-dihydroxyvitamin D3, novel P450scc product, stimulates differentiation and inhibits proliferation and NF-kappaB activity in human keratinocytes. J Cell Physiol. 2010;223:36–48. doi: 10.1002/jcp.21992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wang J, Slominski A, Tuckey RC, Janjetovic Z, Kulkarni A, Chen J, Postlethwaite AE, Miller D, Li W. 20-hydroxyvitamin D inhibits proliferation of cancer cells with high efficacy while being non-toxic. Anticancer Res. 2012;32:739–746. [PMC free article] [PubMed] [Google Scholar]
- 51.Chen J, Wang J, Kim TK, Tieu EW, Tang EK, Lin Z, Kovacic D, Miller DD, Postlethwaite A, Tuckey RC, Slominski AT, Li W. Novel vitamin D analogs as potential therapeutics: metabolism, toxicity profiling, and antiproliferative activity. Anticancer Res. 2014;34:2153–2163. [PMC free article] [PubMed] [Google Scholar]
- 52.Li W, Chen J, Janjetovic Z, Kim TK, Sweatman T, Lu Y, Zjawiony J, Tuckey RC, Miller D, Slominski A. Chemical synthesis of 20S-hydroxyvitamin D3, which shows antiproliferative activity. Steroids. 2010;75:926–935. doi: 10.1016/j.steroids.2010.05.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Slominski AT, Janjetovic Z, Fuller BE, Zmijewski MA, Tuckey RC, Nguyen MN, Sweatman T, Li W, Zjawiony J, Miller D, Chen TC, Lozanski G, Holick MF. Products of vitamin D3 or 7-dehydrocholesterol metabolism by cytochrome P450scc show anti-leukemia effects, having low or absent calcemic activity. PLoS One. 2010;5:e9907. doi: 10.1371/journal.pone.0009907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Slominski A, Pisarchik A, Zbytek B, Tobin DJ, Kauser S, Wortsman J. Functional activity of serotoninergic and melatoninergic systems expressed in the skin. J Cell Physiol. 2003;196:144–153. doi: 10.1002/jcp.10287. [DOI] [PubMed] [Google Scholar]
- 55.Kim TK, Kleszczynski K, Janjetovic Z, Sweatman T, Lin Z, Li W, Reiter RJ, Fischer TW, Slominski AT. Metabolism of melatonin and biological activity of intermediates of melatoninergic pathway in human skin cells. FASEB J. 2013;27:2742–2755. doi: 10.1096/fj.12-224691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Janjetovic Z, Nahmias ZP, Hanna S, Jarrett SG, Kim TK, Reiter RJ, Slominski AT. Melatonin and its metabolites ameliorate ultraviolet B-induced damage in human epidermal keratinocytes. J Pineal Res. 2014;57:90–102. doi: 10.1111/jpi.12146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Fischer TW, Zbytek B, Sayre RM, Apostolov EO, Basnakian AG, Sweatman TW, Wortsman J, Elsner P, Slominski A. Melatonin increases survival of HaCaT keratinocytes by suppressing UV-induced apoptosis. J Pineal Res. 2006;40:18–26. doi: 10.1111/j.1600-079X.2005.00273.x. [DOI] [PubMed] [Google Scholar]
- 58.Pisarchik A, Slominski AT. Alternative splicing of CRH-R1 receptors in human and mouse skin: identification of new variants and their differential expression. FASEB J. 2001;15:2754–2756. doi: 10.1096/fj.01-0487fje. [DOI] [PubMed] [Google Scholar]
- 59.Tobon-Velasco JC, Vazquez-Victorio G, Macias-Silva M, Cuevas E, Ali SF, Maldonado PD, Gonzalez-Trujano ME, Cuadrado A, Pedraza-Chaverri J, Santamaria A. S-allyl cysteine protects against 6-hydroxydopamine-induced neurotoxicity in the rat striatum: involvement of Nrf2 transcription factor activation and modulation of signaling kinase cascades. Free Radic Biol Med. 2012;53:1024–1040. doi: 10.1016/j.freeradbiomed.2012.06.040. [DOI] [PubMed] [Google Scholar]
- 60.Kadekaro AL, Kavanagh R, Kanto H, Terzieva S, Hauser J, Kobayashi N, Schwemberger S, Cornelius J, Babcock G, Shertzer HG, Scott G, Abdel-Malek ZA. alpha-Melanocortin and endothelin-1 activate antiapoptotic pathways and reduce DNA damage in human melanocytes. Cancer Res. 2005;65:4292–4299. doi: 10.1158/0008-5472.CAN-04-4535. [DOI] [PubMed] [Google Scholar]
- 61.Hosni-Ahmed A, Barnes JD, Wan J, Jones TS. Thiopurine methyltransferase predicts the extent of cytotoxicty and DNA damage in astroglial cells after thioguanine exposure. PLoS One. 2011;6:e29163. doi: 10.1371/journal.pone.0029163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Duan J, Duan J, Zhang Z, Tong T. Irreversible cellular senescence induced by prolonged exposure to H2O2 involves DNA-damage-and-repair genes and telomere shortening. Int J Biochem Cell Biol. 2005;37:1407–1420. doi: 10.1016/j.biocel.2005.01.010. [DOI] [PubMed] [Google Scholar]
- 63.Janjetovic Z, Brozyna AA, Tuckey RC, Kim TK, Nguyen MN, Jozwicki W, Pfeffer SR, Pfeffer LM, Slominski AT. High basal NF-kappaB activity in nonpigmented melanoma cells is associated with an enhanced sensitivity to vitamin D3 derivatives. Br J Cancer. 2011;105:1874–1884. doi: 10.1038/bjc.2011.458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Fischer TW, Kleszczynski K, Hardkop LH, Kruse N, Zillikens D. Melatonin enhances antioxidative enzyme gene expression (CAT, GPx, SOD), prevents their UVR-induced depletion, and protects against the formation of DNA damage (8-hydroxy-2′-deoxyguanosine) in ex vivo human skin. J Pineal Res. 2013;54:303–312. doi: 10.1111/jpi.12018. [DOI] [PubMed] [Google Scholar]
- 65.Zhang T, Kimura Y, Jiang S, Harada K, Yamashita Y, Ashida H. Luteolin modulates expression of drug-metabolizing enzymes through the AhR and Nrf2 pathways in hepatic cells. Arch Biochem Biophys. 2014;557:36–46. doi: 10.1016/j.abb.2014.05.023. [DOI] [PubMed] [Google Scholar]
- 66.Lu Y, Chen J, Janjetovic Z, Michaels P, Tang EK, Wang J, Tuckey RC, Slominski AT, Li W, Miller DD. Design, synthesis, and biological action of 20R-hydroxyvitamin D3. J Med Chem. 2012;55:3573–3577. doi: 10.1021/jm201478e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Slominski AT, Kim TK, Takeda Y, Janjetovic Z, Brozyna AA, Skobowiat C, Wang J, Postlethwaite A, Li W, Tuckey RC, Jetten AM. RORalpha and ROR gamma are expressed in human skin and serve as receptors for endogenously produced noncalcemic 20-hydroxy- and 20,23-dihydroxyvitamin D. FASEB J. 2014;28:2775–2789. doi: 10.1096/fj.13-242040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Larsson P, Andersson E, Johansson U, Ollinger K, Rosdahl I. Ultraviolet A and B affect human melanocytes and keratinocytes differently. A study of oxidative alterations and apoptosis. Exp Dermatol. 2005;14:117–123. doi: 10.1111/j.0906-6705.2005.00238.x. [DOI] [PubMed] [Google Scholar]
- 69.Nguyen T, Nioi P, Pickett CB. The Nrf2-antioxidant response element signaling pathway and its activation by oxidative stress. J Biol Chem. 2009;284:13291–13295. doi: 10.1074/jbc.R900010200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Oh DH, Rigas D, Cho A, Chan JY. Deficiency in the nuclear-related factor erythroid 2 transcription factor (Nrf1) leads to genetic instability. FEBS J. 2012;279:4121–4130. doi: 10.1111/febs.12005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Kim TK, Wang J, Janjetovic Z, Chen J, Tuckey RC, Nguyen MN, Tang EK, Miller D, Li W, Slominski AT. Correlation between secosteroid-induced vitamin D receptor activity in melanoma cells and computer-modeled receptor binding strength. Mol Cell Endocrinol. 2012;361:143–152. doi: 10.1016/j.mce.2012.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Slominski A, Kim TK, Zmijewski MA, Janjetovic Z, Li W, Chen J, Kusniatsova EI, Semak I, Postlethwaite A, Miller D, Zjawiony J, Tuckey RC. Novel vitamin D photoproducts and their precursors in the skin. Dermato-Endocrinology. 2013;5:1–13. doi: 10.4161/derm.23938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Fischer TW, Sweatman TW, Semak I, Sayre RM, Wortsman J, Slominski A. Constitutive and UV-induced metabolism of melatonin in keratinocytes and cell-free systems. FASEB J. 2006;20:1564–1566. doi: 10.1096/fj.05-5227fje. [DOI] [PubMed] [Google Scholar]
- 74.Slominski A, Wortsman J, Tobin DJ. The cutaneous serotoninergic/melatoninergic system: securing a place under the sun. FASEB J. 2005;19:176–194. doi: 10.1096/fj.04-2079rev. [DOI] [PubMed] [Google Scholar]
- 75.Hall PA, McKee PH, Menage HD, Dover R, Lane DP. High levels of p53 protein in UV-irradiated normal human skin. Oncogene. 1993;8:203–207. [PubMed] [Google Scholar]
- 76.Santoro R, Mori F, Marani M, Grasso G, Cambria MA, Blandino G, Muti P, Strano S. Blockage of melatonin receptors impairs p53-mediated prevention of DNA damage accumulation. Carcinogenesis. 2013;34:1051–1061. doi: 10.1093/carcin/bgt025. [DOI] [PubMed] [Google Scholar]
- 77.Mediavilla MD, Cos S, Sanchez-Barcelo EJ. Melatonin increases p53 and p21WAF1 expression in MCF-7 human breast cancer cells in vitro. Life Sci. 1999;65:415–420. doi: 10.1016/s0024-3205(99)00262-3. [DOI] [PubMed] [Google Scholar]
- 78.Slominski AT, Zmijewski MA, Skobowiat C, Zbytek B, Slominski RM, Steketee JD. Sensing the environment: regulation of local and global homeostasis by the skin’s neuroendocrine system. Adv Anat Embryol Cell Biol. 2012;212:v, vii, 1–115. doi: 10.1007/978-3-642-19683-6_1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Slominski AT, Zmijewski MA, Zbytek B, Tobin DJ, Theoharides TC, Rivier J. Key Role of CRF in the Skin Stress Response System. Endocr Rev. 2013;34:827–884. doi: 10.1210/er.2012-1092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Slominski A, Zmijewski MA, Pawelek J. L-tyrosine and L-dihydroxyphenylalanine as hormone-like regulators of melanocyte functions. Pigment Cell Melanoma Res. 2012;25:14–27. doi: 10.1111/j.1755-148X.2011.00898.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Slominski A, Tobin DJ, Shibahara S, Wortsman J. Melanin pigmentation in mammalian skin and its hormonal regulation. Physiol Rev. 2004;84:1155–1228. doi: 10.1152/physrev.00044.2003. [DOI] [PubMed] [Google Scholar]
- 82.Fischer TW, Slominski A, Zmijewski MA, Reiter RJ, Paus R. Melatonin as a major skin protectant: from free radical scavenging to DNA damage repair. Exp Dermatol. 2008;17:713–730. doi: 10.1111/j.1600-0625.2008.00767.x. [DOI] [PubMed] [Google Scholar]
- 83.Slominski AT, Zmijewski MA, Semak I, Sweatman T, Janjetovic Z, Li W, Zjawiony JK, Tuckey RC. Sequential metabolism of 7-dehydrocholesterol to steroidal 5,7-dienes in adrenal glands and its biological implication in the skin. PLoS One. 2009;4:e4309. doi: 10.1371/journal.pone.0004309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Slominski AT, Kim TK, Chen J, Nguyen MN, Li W, Yates CR, Sweatman T, Janjetovic Z, Tuckey RC. Cytochrome P450scc-dependent metabolism of 7-dehydrocholesterol in placenta and epidermal keratinocytes. Int J Biochem Cell Biol. 2012;44:2003–2018. doi: 10.1016/j.biocel.2012.07.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Skobowiat C, Dowdy JC, Sayre RM, Tuckey RC, Slominski A. Cutaneous hypothalamic-pituitary-adrenal axis homolog: regulation by ultraviolet radiation. Am J Physiol Endocrinol Metab. 2011;301:E484–493. doi: 10.1152/ajpendo.00217.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Skobowiat C, Nejati R, Lu L, Williams RW, Slominski AT. Genetic variation of the cutaneous HPA axis: an analysis of UVB-induced differential responses. Gene. 2013;530:1–7. doi: 10.1016/j.gene.2013.08.035. [DOI] [PMC free article] [PubMed] [Google Scholar]