

Abstract
Rearrangements of the anaplastic lymphoma kinase (ALK) have been described in multiple malignancies, including non-small cell lung cancer (NSCLC). ALK fusions have gain of function properties while activating mutations in wild-type ALK can also occur within the tyrosine kinase domain. ALK rearrangements define a new molecular subtype of NSCLC that is exquisitely sensitive to ALK inhibition. Crizotinib, an orally available small molecule ATP-mimetic compound which was originally designed as a MET inhibitor, was recognized to have “off-target” anti-ALK activity and has been approved in the USA for the treatment of patients with ALK-positive NSCLC. Chromosomal rearrangements involving the ROS1 receptor tyrosine kinase have also been recently described in NSCLC, while crizotinib is currently under clinical trial in this molecular subset of NSCLC patients. The basic approaches of any computer aided drug design work in terms of structure and ligand based drug design. Details of each of these approaches should be covered with an emphasis on utilizing both in order to develop multi-targeted small-molecule kinase inhibitors. Such multi-targeted tyrosine kinase inhibitors can have antiproliferative activity against both ROS1and ALK rearranged NSCLC. Herein, we highlight the importance of targeting these proteins and the advances in optimizing more potent and selective ALK and ROS1 kinase inhibitors.
Key Words: Anaplastic lymphoma kinase (ALK), drug design, kinase inhibitors, non-small cell lung cancer, proto-oncogene tyrosine-protein kinase ROS (ROS1)
Lung cancer remains the leading cause of cancer mortality in the world with non-small cell lung cancer (NSCLC) accounting for 80% of cases (1). Conventional chemotherapeutic regimens only marginally improve the outcome of NSCLC patients at advanced stages of disease, with median survival time less than one year after diagnosis (2). Protein kinase activation by somatic mutation or chromosomal alteration is a common mechanism of tumorigenesis. The discovery of a number of these molecular alterations underlying lung cancer has led to uniquely targeted therapies with specific inhibitor drugs such as erlotinib and gefitinib for mutations in the epidermal growth factor receptor (EGFR), or crizotinib for the gene translocation resulting in the echinoderm microtubule associated protein like 4 (EML4)-anaplastic lymphoma kinase (ALK) oncogene (3-5). EML4-ALK was the first targetable fusion oncokinase to be identified in 4-6% of lung adenocarcinomas. EML4-ALK generates a transforming tyrosine kinase with as many as nine different variants identified (6,7) and represents a novel molecular target in a small subset of NSCLCs. Patients with EML4-ALK positive tumors are characteristically younger age, female, and never to light smokers (5,8,9). The fusion gene has been observed predominantly in adenocarcinomas (4-7%) (5,9). Based on data from a phase I clinical trial which showed an overall response rate of 57% and a probability of progression-free survival at 6 months of 72%, crizotinib has been approved by the U.S. Food and Drug Administration for the treatment of NSCLC patients with ALK transforming rearrangements (5).
Similarly, proto-oncogene tyrosine-protein kinase ROS (ROS1) is an orphan receptor tyrosine kinase (RTK) that forms fusions and defines another clinically actionable oncogenic driver mutation in NSCLC (10). It has been recently reported that approximately 1.4% of NSCLCs harbor ROS1 rearrangements (11,12). Of the ROS1 fusion-positive tumors, 30% are known to harbor a recurrent translocation t[5;6][q32;q22], which creates the CD74 molecule, major histocompatibility complex, class II invariant chain (CD74)-ROS fusion kinase (13). In fact, ROS1 is evolutionarily related to ALK. Patients with ROS1 rearrangements are also significantly younger, more likely to be never-smokers and are more often diagnosed with the histological subtype of adenocarcinoma with wide distribution of tumor grade (11). Although ROS1 shares only 49% amino acid sequence homology with ALK in the kinase domains, several ALK inhibitors have demonstrated in vitro inhibitory activity against ROS1 (14). Recently, a report from investigators at the Massachusetts General Hospital Cancer Center has showed that ROS1-driven tumors can be treated with crizotinib and describes the remarkable response of one patient to crizotinib treatment (11). Interestingly, in this study ROS1 rearrangements were found to be mutually exclusive to ALK rearrangements (11). Preliminary results of a phase I trial of ROS1-positive advanced-stage NSCLC patients treated with crizotinib reported a response rate of 57% and a disease control rate of 79% at 8 weeks (15).
The discovery of new selective and potent inhibitors of ALK and ROS1 kinase raises the importance of using these drugs as a new method for treatment of ALK- and ROS1-derived lung cancer. This review focuses on the importance of targeting these proteins and describes the advances in optimizing more potent and selective ALK and ROS1 kinase inhibitors that have an optimal pharmacokinetic profile and the capacity to inhibit acquired resistant mutations. We aim to stimulate interest and encourage of researchers from different disciplines to learn about new therapeutic avenues following the development of compounds targeting ALK and ROS1 kinases with the aim of increasing survival to these lethal forms of lung cancer.
Structural insights and computational simulations
Receptor Tyrosine kinases (RTK) are transmembrane glycoproteins where the domain responsible to the tyrosine kinase activity is located in the cytoplasm. Although extracellular domain shows remarkable structural differences between TK families, the intracellular region is sensibly conserved.
Although a few years ago there was no resolved three-dimensional structure of ALK, similarity between its sequence permitted to predict its folding from a known RTK structure used as a template, using homology models. Thus, the human ALK receptor was modeled from mouse c-Abl (16), activated insulin receptor tyrosine kinase (InsR) (17,18) or insulin-like growth factor-1 receptor (IGF-1R) (19).
Fortunately, recently some crystal structures of the catalytic domain of ALK have been reported in literature at different resolution levels. All of them are available in the Protein Data Bank (PDB) (20) with ID entries 3L9P, 3LCS, 3LCT (Figure 1) (21).
Figure 1.
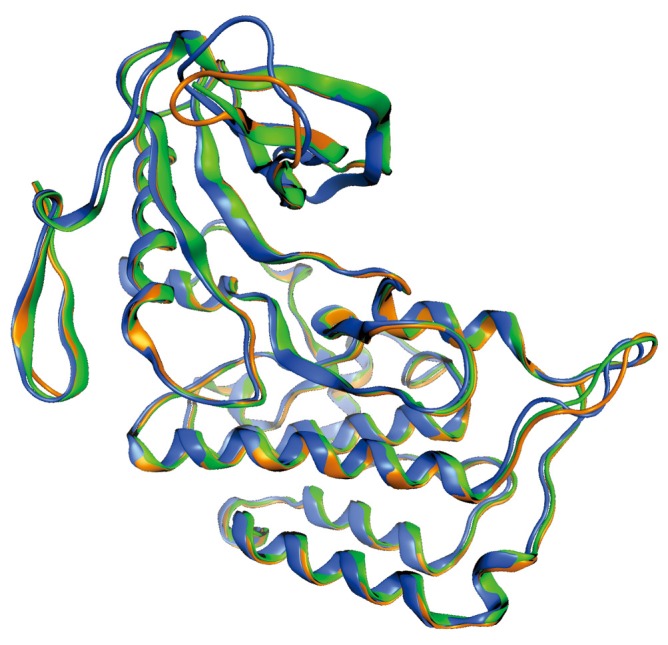
Superposition of the three structures available in the PDB of ALK’s catalytic domain. Structure has been colored according to its resolution: 3LCT (2.10Å) in orange, 3LCS (1.95Å) in blue and 3L9P (1.80Å) in green. The loop regions show the main differences between structures.
Once the three-dimensional structure of the ALK receptor is available, biological processes related to its structure can be studied virtually, as for example substrate affinity (22), receptor autoactivation or resistant mutations. It can even be used as a structural template in order to predict the structure of RTK homologues by homology modeling (23).
However, crystallographic data should be taken carefully: the combination of crystallographic and biochemical studies reveals that the active conformation of ALK protein requires the phosphorylation of specific residues (24,25) and some ALK crystal structures published to date correspond to unphosphorylated proteins (26).
Additionally, some ALK molecular structures have also been resolved including a bound ligand (Table 1). These co-crystallized structures reveal the active site of the protein against one giving compound and describe the interaction within the protein-ligand complex. This information is relevant to assist structure-aided molecular design of ALK inhibitors (i.e. Structure-Based Drug Design, SBDD), providing valuable information about how to improve ALK selectivity.
Table 1. List of the available ligands co-crystallized with ALK receptor.
| Molecular Structure | Ligand name | PDB ID (resolution) | Reference |
|---|---|---|---|
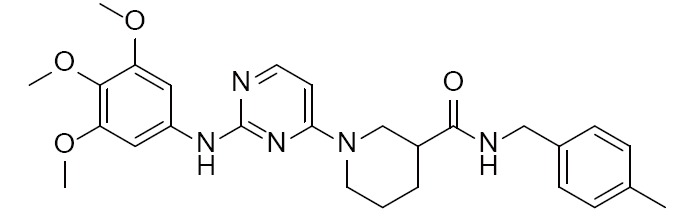 |
Piperidine-carboxamide | 4DCE (2.03Å) | (27) |
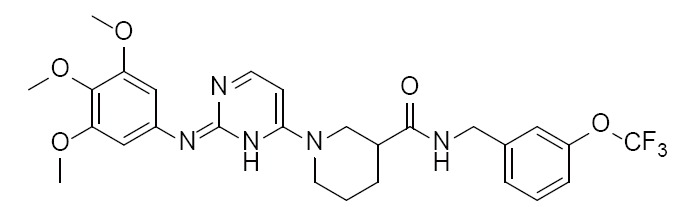 |
Piperidine-carboxamide | 4FNZ (2.60Å) | (26) |
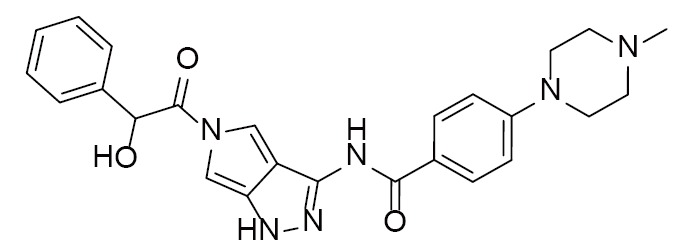 |
PHA-E429 | 2XBA (1.95Å) | (22) |
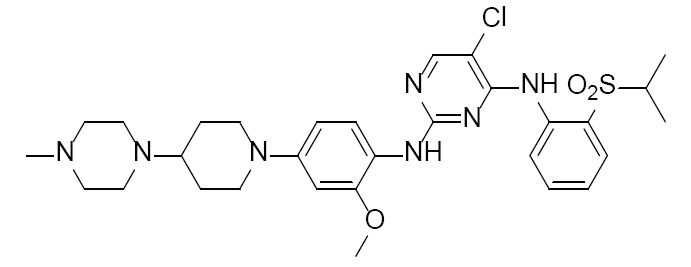 |
NVP-TAE684 | 2XB7 (2.50Å) | (22) |
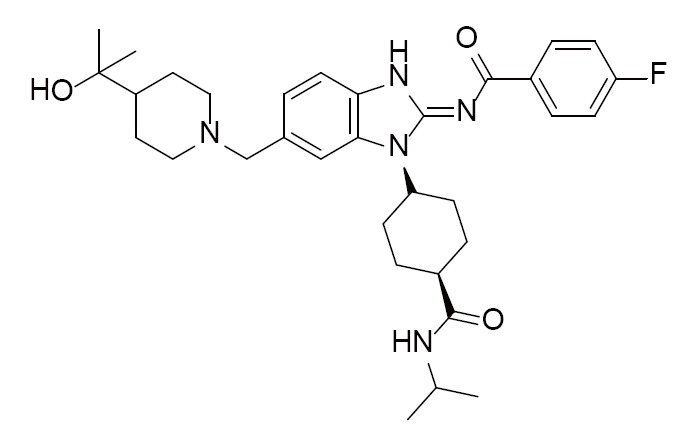 |
Acyliminobenzimidazole inhibitor 36 | 4FOD (2.00Å) | (28) |
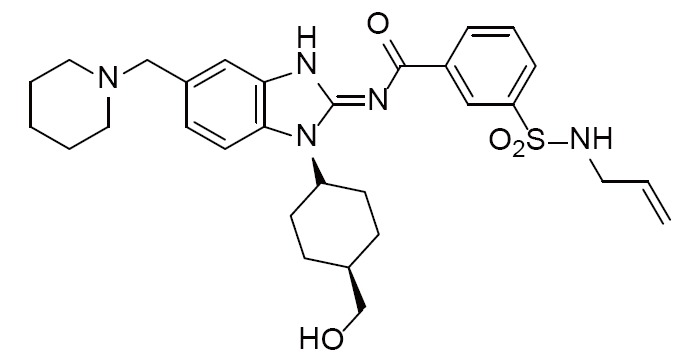 |
Acyliminobenzimidazole inhibitor 1 | 4FOB (1.90Å) | (28) |
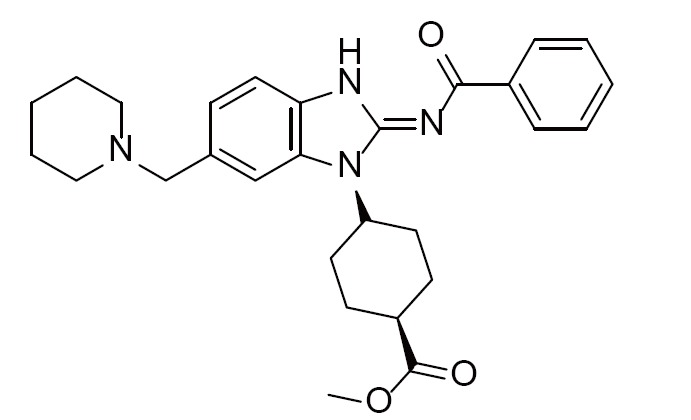 |
Acyliminobenzimidazole inhibitor 2 | 4FOC (1.70Å) | (28) |
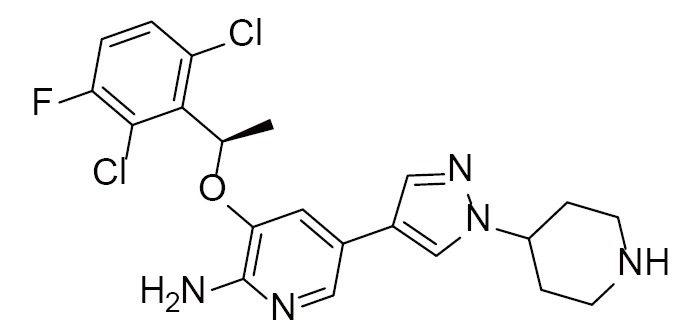 |
Crizotinib | 2XP2(1.90Å) | (29) |
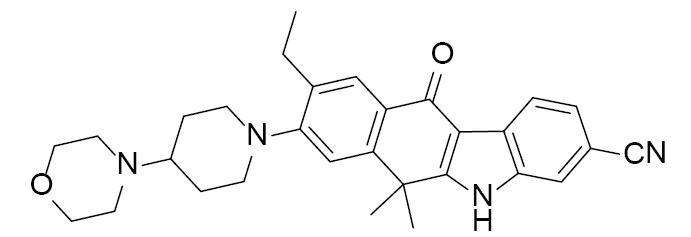 |
CH5424802 | 3AOX(1.75Å) | (30) |
From SBDD models rises the first approved drug for the treatment of ALK-positive NSCLC, crizotinib (29). By the time diaminopyrimidine (DAP) derivatives were consolidated as bioactive motives, great efforts were done in order to increase its selectivity (31), to optimize their activity (32) or to propose new scaffolds that mimic them. Evaluation of receptor-ligand interactions using docking techniques has become the most preferred SBDD method to study small-molecule ALK inhibitors, not only specific compounds as Novartis NVP-TAE684 (17) but also families including pyridine (33), pyrrolotriazine (19), 2-acyliminobenzimidazole (28), and tetrahydropyrido-[2,3-b]pyrazine (34) derivatives. Besides specific chemical families, commercial or public chemical libraries can also be screened using docking techniques in order to identify new candidates (18,33).
Most of the SBDD results agree to consider the targeting of the ATP binding site of the tyrosine kinase domain as a good approach to design RTK inhibitors (35). The analysis of docking results may help to understand the inhibition mechanism, according to the interaction between candidates and the hydrophobic region and/or the gatekeeper. This helps to identify e.g., that preferred interactions of 2-aminopyridines within the ATP binding pocket include hydrogen bonds of pyridine and amino nitrogens with residues close to the gatekeeper (i.e. Met1184 and Glu1182) (34).
Ligand-based drug design (LBDD) has also been applied in the development of new ALK inhibitors. Structure-Activity Relationship (SAR) models correlate the chemical structure of inhibitors with their biological activity. They have been applied to screen series of piperidine carboxamides (27), 2-acyliminobenzimidazoles (28), macrocycles (36), 7-amino-1,3,4,5-tetrahydrobenzo[b]azepin-2-ones (37), 2,3,4,5-tetrahydro-benzo[d]azepines (38), 2,7-disubstituted pyrrolo[2,1-f][1,2,4]triazines (39), diaminocyclohexane methanesulfonamides (40) or tetracyclic derivatives (41,42), tetrahydropyrido[2,3-b]pyrazines (34), 3,5-diamino-1,2,4-triazole ureas (43), aryloxy oxo pyrimidinones (44), identifying which substituents confer high ALK-response and proposing chemical modifications of hit compounds.
Far from what it seems, SBDD and LBDD are not exclusive. Most of published LBDD studies include modeling of candidates with the receptor using docking. LBDD and SBDD information can be combined to create pharmacophore models where chemical features (identified from LBDD methods) are used to generate structural keys that active ALK inhibitors must fulfill, taking into account receptor-ligand interactions (identified from SBDD methods) (19,45).
Since mechanisms to drug resistance have been experimentally related to mutations in the ALK amino acid sequence, many efforts have been done in order to obtain resolved crystal structures of ALK mutants (Table 2).
Table 2. List of resolved structures of mutated ALK receptor.
| Mutation | PDB ID | Reference |
|---|---|---|
| F1174L | 2YJR (1.90Å) | McTigue M, 2012, unpublished data |
| F1174L catalytic domain | 4FNW (1.75Å) | (26) |
| C1156Y | 2YJS | McTigue M, 2012, unpublished data |
| L1196M | 2YHV | McTigue M, 2012, unpublished data |
| L1196M+crizotinib | 2YFX | McTigue M, 2012, unpublished data |
| R1275Q catalytic domain | 4FNX (1.70Å) | (26) |
| R1275Q cd + benzoxazole | 4FNY (2.45Å) | (26) |
Ligand co-crystallizations are also available in mutated structures (mainly F1174L and R1275Q) (26), highlighting structural changes responsible of such behavior. Although resistance can be well correlated with structural changes in some cases (e.g., ALKR1275Q) (46), other mutations exhibit a more subtle behavior.
It is well know that ALKL1196M mutants are crizotinib resistant (47). However, it is interesting to note that the comparison between co-crystal structure of crizotinib with ALKwt (2XP2) (29) and ALKL1196M (2YFX, McTigue M, 2012, unpublished data) reveals that L1196M mutation has little effect on crizotinib’s binding mode (Figure 2).
Figure 2.
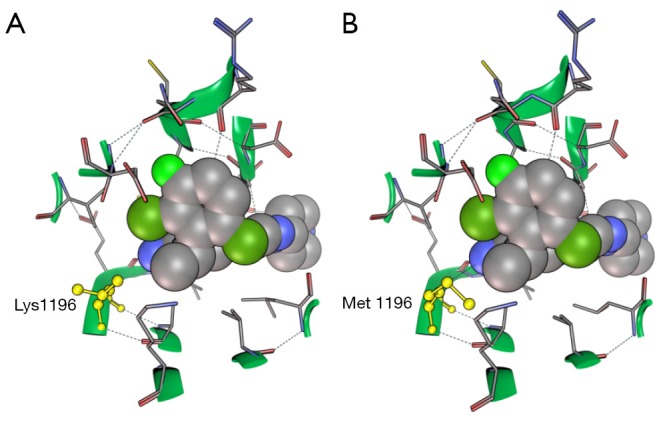
Molecular structure of crizotinib co-crystalized with (A) ALKwt receptor, PDB ID: 2XP2; and (B) ALKL1196M mutant, PDB ID: 2YFX. Although Lys-Met exchange the binding mode of crizotinib remains unchanged. Structures analysis has been performed and rendered using MOE software (48).
It has been recently demonstrated that at least four mutations could be involved in crizotinib’s resistance (Figure 3): L1196M acts as a gatekeeper and modifies crizotinib’s binding site by steric interference (49); G1202R and S1206Y are both located in the solvent-exposed region near to the binding site but G1202R blocks sterically the binding site whereas S1206Y could destabilize electrostatic interaction (50); 1151T insertion is furthest from N terminus Cα-helix but confers a high-level crizotinib resistance probably derived from the modification of ATP affinity (51). This knowledge has been used to design new compounds against gatekeeper mutants (52).
Figure 3.
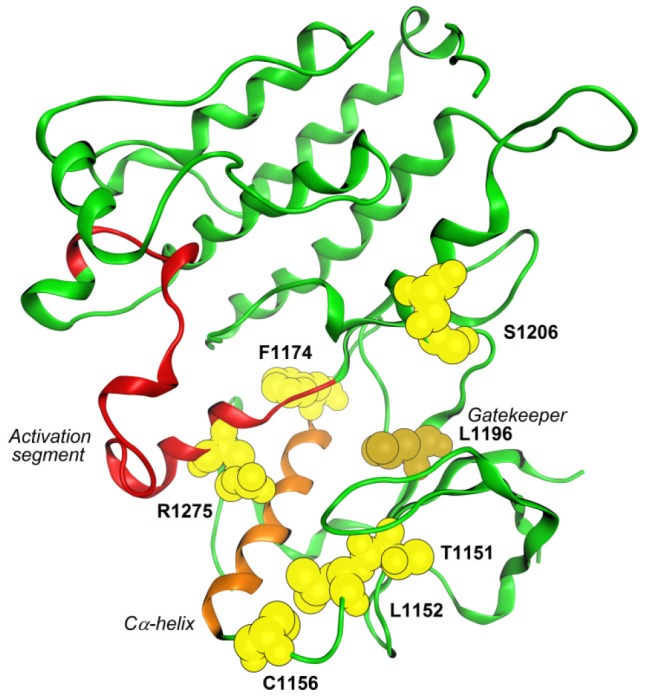
Graphical representation of key mutations related with ALK resistance
However, other mutations have also been identified. Most of them are abutting the Cα-helix and activation loop, becoming responsible for the modification of the kinase active site. On the basis of the crystal structures, these mutations can be related with formation or alteration of hydrogen bonds (e.g., L1152R and C1156Y mutations) (53) or conformational changes (e.g., F1174L induces structural changes leading to an increment of ATP affinity which requires of irreversible inhibitors to block it) (35).
Thus, ligand co-crystallizations can not only help to the understanding of the structural basis of mutations related to drug resistance, but also to identify the binding mode of the second generation ALK inhibitors (30). When no crystallographic data is available, binding mode of new inhibitors with ALK mutants are computationally modeled using docking techniques (54) or molecular dynamic simulations (55). These interaction models provide a useful tool to screen drug candidates (after their validation with experimental evidences).
However, specificity for one receptor could be undesirable in a so complex biological context. For this reason, promiscuous inhibitors (e.g., dual inhibitors) have become gradually more interesting. When a set or library of knowing active ligands for one target is available, the specificity for the second target can be predicted by screening the previous library applying either SBDD or LBDD (56). In this respect, the ALK inhibitor TAE684 has been experimentally evaluated on ROS1, given the high homology between ALK and ROS1 human receptors (57-59).
Unfortunately, tridimensional structure of ROS1 receptor is not available and studies of ligand-receptor interactions have been always performed using ROS1 homology models using IGF-1R (60) or ALK itself (14). Homology models can be later used to study molecular interactions with ligands using docking methods in order to elucidate their binding mode (60).
In contrast to homology models, other studies consider the structurally related MET receptor as a reference to find inhibitors which are able to interact with kinase domain. Given the receptor, homology docking studies on MET receptors could be therefore extended to structurally similar receptors like ROS1 (61). In fact, ALK inhibitors can also be used on ROS1 according to their similarity (62).
ALK and ROS1 small molecule inhibitors under development
In the last ten years a lot of efforts have been devoted to the development of compounds active against the ALK and ROS1 kinases. The principal reported inhibitors, the companies involved in the development, the core scaffold present in such compounds, the mutation against which the compounds are effective, their off-targets, and the clinical stage achieved are summarized in Table 3. On the other hand, the structures of the disclosed compounds are included in Figure 4.
Table 3. List of ALK inhibitors under development.
| Chemical scaffold | Therapeutics | Company | ALK activity | ALK secondary mutations | Other targets | Clinical stage |
|---|---|---|---|---|---|---|
| Aminopyridine | Crizotinib PF-02341066 Xalkori® | Pfizer | 24 nM | -- | c-MET, ROS1 | FDA approved |
| Dianilinopyridine | NVP-TAE684 | Novartis | <10 nM | L1196M | IR, IGF-1R, FLT3, TIE2, LRRK2, ROS1 | Not a clinical candidate |
| F1174L | ||||||
| Structure undisclosed | AP26113 | ARIAD | 0.62 nM | L1196M | Multiple kinases ROS1 |
Phase I/II |
| F1174C | ||||||
| I1171T | ||||||
| F1245C | ||||||
| E1210K | ||||||
| S1206R | ||||||
| G1269S | ||||||
| Structure undisclosed | X-276 | Xcovery | -- | -- | -- | preclinical |
| Aminopyridazine | X-376 | Xcovery | -- | -- | -- | preclinical |
| Structure undisclosed Aminopyridazine |
X-396 | Xcovery | <0.4 nM | L1196M | -- | Phase I |
| C1156Y | ||||||
| Tetracyclic indole | CH5424802 AF-802 |
Chugai | 1.9 nM | L1196M | GAK, LTK | Phase I/II |
| F1174L | ||||||
| R1275Q | ||||||
| C1156Y | ||||||
| Pyrrolopyrimidine | GSK1838705A | GlaxoSmithKline | 0.5 nM | -- | IR | Preclinical |
| Triazinediamine | ASP3026 | Astellas | -- | -- | ROS1 | Phase I |
| Indolocarbazole | CEP-14083 | Cephalon | 2 nM | -- | IR, VEGFR2, TIE2, DLK | Not a clinical candidate |
| Indolocarbazole | CEP-14513 | Cephalon | 4 nM | -- | IR, VEGFR2, TIE2, DLK | Not a clinical candidate |
| Tetrahydropyrazine | -- | Cephalon | 10 nM | -- | -- | preclinical |
| Diaminopyridine | CEP-28122 | Cephalon | 1.9 nM | F1174L | -- | Phase I |
| R1275Q | ||||||
| Structure undisclosed | CEP-37440 | Cephalon/Teva | -- | -- | Phase I | |
| Pyrrolotriazine | Compound 32 | Cephalon | 6 nM | -- | -- | Preclinical |
| Tetraazatetracyclo-docosanonaene | Macrocycle 2m | Cephalon | 0.5 nM | -- | -- | Preclinical |
| Pyrrolopyrazole | PHA-E429 | Nerviano Medical | 91 nM | -- | Mutiple kinases | Preclinical |
| Indazole | NMS-E628 | Nerviano Medical | 55 nM | L1196M | IGF-1R, Aurora B | Preclinical |
| C1156Y | ||||||
| Structure undisclosed pyridone | CRL151104A | Chembridge St Jude |
9.75 nM | F1174L | -- | Preclinical |
| R1275Q | ||||||
| Pyridone | Pyridone 1 | Chembridge St Jude |
380 nM | -- | -- | Preclinical |
| Structure undisclosed | WZ-5-126 | -- | 3.4 nM | -- | -- | Preclinical |
| Structure undisclosed | LDK378 | Novartis | 0.15 nM | -- | ROS1 | Phase I |
| 2,4-pyrimidinediamine | 3-39 | Novartis | -- | -- | -- | Preclinical |
| Pyridoisoquinoline | F91873 and F91874 | Institut de Recherche Pierre Fabre | -- | -- | Multiple kinases | Preclinical |
| Structure undisclosed | TSR-011 | Tesaro | -- | -- | -- | Preclinical |
c-MET, hepatocyte growth factor receptor; IR, insulin receptor; IGF-1R, insulin-like growth factor 1 receptor; FLT3, FMS-like tyrosine kinase 3; TIE2, angiopoietin-1 receptor; LRRK2, leucine-rich repeat kinase 2; GAK, cyclin G-associated kinase; LTK, leukocyte tyrosine kinase; VEGFR2, vascular endothelial growth factor receptor 2; DLK, dual leucine zipper kinase
Figure 4.

Structures of reported ALK and ROS1 inhibitors.
ALK inhibition
One of the first discovered ALK inhibitors was NVP-TAE684, a small molecule with a dianilinopyridine scaffold which targets competitively the ATP in its binding site. Firstly, it was showed that NVP-TAE684 could block growth in cell lines and in a mouse model of ALCL (17). Besides to inhibit the proliferation of neuroblastoma and NSCLC cell lines, this ALK inhibitor reduced tumors expressing variants of ALK or EML4-ALK fusion proteins, confirming the oncogenic activity of the fusion kinase and consequently the therapeutic potential of targeted inhibitors (62,63). Despite several studies have reported the effectiveness of NVP-TAE684 against tumors induced by constitutively active ALK or against cell lines with ALK translocations and point mutations, this compound is not currently in any clinical trial (64-68).
Crizotinib (PF-02341066), which was originally developed to inhibit hepatocyte growth factor receptor (c-MET) but a few time later showed inhibitory activity against ALK, is an ATP-competitive small molecule like NVP-TAE684 (69,70). Crizotinib, with an aminopyridine as a core, was described for first time in 2007 and only three years later were reported the first promising clinical trials in NSCLC patients with EML4-ALK fusion genes (5). Phase I study concluded the benefits of the treatment with crizotinib of patients with advanced EML4-ALK-positive NSCLC. Among the 119 patients enrolled, after the treatment with crizotinib, 2 displayed a total recovery, 69 had a partial response and 31 were considered to have stable disease (71). The result of this inhibitory activity and the response of patients to the treatment with crizotinib, motivated its FDA approval under the trade name of Xalkori® (Pfizer). A Subsequent study in phase III trials reported crizotinib resistance in EML4-ALK positive NSCLC patients with some ALK mutations, especially secondary mutations, indicating a 64% overall survival in response to crizotinib treatment of EML4-ALK positive NSCLC patients after two years (49,53,72-74).
After the clinical trials of crizotinib, ALK was established as a drug target in cases of NSCLC, but the discovery of resistance related to mutations created the need to develop a second-generation of ALK inhibitors with the capability to overcome mutation-mediated drug resistance. Some pharmaceutical companies and research groups have reported different new promising candidate drugs to inhibit mutated ALK.
One of these second-generation ALK inhibitors is AP26113, whose chemical structure has not been disclosed but it is believed to be a member of a family compounds described in a patent of ARIAD based on a diaminopyrimidine structure bearing pendant phosphinoyl groups (75). This product is a multikinase inhibitor which shows more selectivity and inhibitory potency against ALK (IC50 =0.62 nM) than crizotinib (IC50 =3.6 nM). Apart from showing growth inhibition against EML4-ALK and nucleophosmin-ALK fusion gene (NPM-ALK) positive cells, the most interesting feature of AP26113 is that could inhibit some EML4-ALK mutated forms, therefore it could be a useful alternative in cases of crizotinib resistance. Currently, AP26113 is in phase I/II clinical trials (67,76-78).
In order to inhibit ALK mutations, another family of compounds was developed by Xcovery including X-276, X-376 and X-396 (79,80). There is not so much information in the literature about X-276 but it is considered a more selective and potent ALK inhibitor than crizotinib (81). X-376 and X-396 are members of the same family based on aminopyridazine scaffold, despite X-396 showed better results than X-376. X-396 was approximately 10-fold more potent than crizotinib in front of H3122 (EML4-ALK positive E13; A20 NSCLC), H2228 (EML4-ALK positive E6a/b; A20 NSCLC), SU-DHL-1 (NPM-ALK positive), SY5Y (ALKF1174L) cell lines. As an example, in H3122 cell line, X-396 showed an IC50 value of 15 nM H3122 (EML4-ALK positive NSCLC) cell lines against 180 nM of crizotinib. Furthermore X-396 displayed good inhibition in crizotinib resistant cell lines (Ba/F3-EML4-ALKL1196M, IC50 =106 nM; Ba/F3-EML4-ALKC1156Y, IC50 =48 nM). Thus, X-396 initiated phase I clinical trials in June 2012 (82).
CH5424802, also known as AF-802, is a tetracyclic indole which has a high ALKwt inhibitory activity in in vitro assays (IC50 =1.9 nM) as well in front of mutated ALKs, such as ALKL1196M (Ki =1.56 nM), ALKF1174L (IC50 =1.0 nM) and ALKR1275Q (IC50 =3.5 nM). These results were also reproduced in vivo with the treatment in different cell lines: H2228 (EML4-ALK positive E6a/b;A20; IC50 =53 nM), KARPAS-299 (NPM-ALK positive ALCL; IC50 =3.0 nM), SR-786 (IC50=6.9 nM), NB-1 (ALK amp, IC50 =4.5 nM); KELLY (IC50 =62 nM) and Ba/F3 (EML4-ALKL1196M) that allowed to show that CH5424802 is a potent inhibitor for a therapy with capacity to overcome the acquired resistance to crizotinib (30,41,83). Due to these promising results, CH5424802 is in phase I/II of clinical trials.
GSK1838705A, developed by GlaxoSmithKline and currently in preclinical phase, contains a pyrrolopyrimidine scaffold and has showed to be selective IGF-1R, insulin receptor (IR) and ALK inhibitor (IC50 =1.2, 2 and 0.5 nM respectively). Furthermore, the inhibition of the proliferation of different ALCL cell lines, such as L-82 (IC50 =24 nM), SUP-M2 (IC50 =28 nM), SU-DHL-1 (IC50 =31 nM), Karpas-299 (IC50 =52 nM) and SR-786 (IC50 =88 nM), has also been described. Besides the potent inhibition of ALK by GSK1838705A, such compound also inhibit cell lines harboring ALK fusion genes in different ALCL cell lines expressing NPM-ALK (EC50 =24-88 nM) and in H2228 NSCLC cells expressing EML4-ALK (IC50 =191 nM). In addition, it was proved that GSK1838705A inhibits the EML4-ALK phosphorylation (84,85).
There are not so many details about ASP3026, a triazinediamine developed by Astellas which is in Phase I in clinical trials. This compound showed potent and selective activity against EML4-ALK driven tumors with gatekeeper mutation, therefore it is able to overcome crizotinib resistance (86).
Based on the structure of two natural products, staurosporine and 7-hydroxystaurosporine which are able to inhibit ALK (IC50 =150 nM and 5 µM respectively in the presence of 30 µM ATP in an ELISA-based ALK assay) (87), Cephalon developed some compounds targeted to inhibit ALK. CEP-14083 and CEP-14513 have showed ATP-competitive activity in ALK, displaying IC50 values in enzymatic assays of 2 and 4 nM, respectively, and 10-30 nM in cellular assays (88). The capability of CEP-14083 to inhibit cell lines and animal models harboring ALK alterations was also described (89). Furthermore, CEP-14083 is able to inhibit other kinases, such as IR, vascular endothelial growth factor receptor 2 (VEGFR2), angiopoietin-1 receptor (TIE2), and dual leucine zipper kinase (DLK) but, due to unfavorable physicochemical properties, CEP-14083 and CEP-14513 were discarded for in vivo studies (88). Then, Cephalon developed a second generation of ALK inhibitors with different scaffolds, some of them being currently in preclinical studies. Thus, they reported tetrahydropyrazines with IC50 around 10 nM and 150 nM in enzyme and in Karpas-299 cell line, respectively (34). They also described 2,4-diaminopyrimidines, the most representative compound is CEP-28122, showing a high selectivity (600-fold with respect to IR, a closely related kinase family member) and a high potency (IC50 =1.9 and 20 nM in enzyme and in Karpas-299 cell line, respectively). CEP-28122 also induced growth inhibition in NPM-ALK positive ALCL and EML4-ALK positive NSCLC tumor xenografts in mice. In addition, inhibited growth of neuroblastoma cell lines harboring ALK activating mutants, such as F1174L in NB-1643 cells and R1275Q in SH-SY5Y cells, but not in cell lines which expresses ALKwt (SKNAS cells) (32,37,90). Cephalon also described pyrrolotriazines among which compound 32 displayed an IC50 =6 nM in an enzymatic assay and 100 nM against NPM-ALK cells, showing high selectivity in a test against 256 kinases (39). Finally, they reported tetraazatetracyclo docosanonaene macrocycles, Macrocycle 2m showing a high inhibitory potency in vitro (IC50 =0.5 nM) as well as in a cell-based assay (IC50 =10 nM) and high selectivity (173-fold with respect to IR) (36).
PHA-E429 is a compound developed by Nerviano with a pyrrolopyrazole scaffold which is considered as a multikinase inhibitor that also is able to inhibit ALK (IC50 =91 nM) (51,91). NMS-E628, an indazole compound, developed by the same company, inhibits ALK (IC50 =55 nM against NMP-ALK cells) but also IGF-1R and Aurora B (92). NMS-E628 has showed a high efficiency inhibiting the growth of ALCL cells and NSCLC cells bearing EML4-ALK (H2228) rearrangement and in addition was able to overcome the L1196M and C1156Y mediated TKI resistance (93,94).
CRL151104A, developed by ChemBridge Research Laboratories and St Jude Children’s Research Hospital, whose structure is not available, is a third-generation ATP competitor pyridine compound with capacity to block the cellular phosphorylation. This compound showed a high activity against ALK in vitro (IC50 =9.75 nM) and also in an in vivo assay (IC50 ≤100 nM and 2.5 µM against NPM-ALK positive and NPM-ALK negative cells, respectively). CRL151104A has also demonstrated the capability to overcome F1174L and R1275Q ALK mutations in neuroblastoma cell lines (95). There is not so much information about a pyridone compound developed by ChemBridge Research Laboratories and St Jude Children’s Research Hospital which is able to inhibit ALK in an enzymatic assay (IC50 =380 nM) and in cell-based assays (IC50 =18.0 µM and 750 nM against BaF3/NPM-ALK and Karpas-299/NPM-ALK ALCL cell lines, respectively) (33).
WZ-5-126 is a potent ALK small molecule inhibitor with an IC50 of 3.4 nM in vitro, capable of inhibiting the growth of two ALK positive NSCLC cell lines (62).
Novartis has also been involved in the research of ALK inhibitors such as LDK-378, a potent and selective candidate (IC50 =0.15 nM). This compound, in Phase I trials nowadays, is able to inhibit the growth of genetically abnormal ALK-driven NSCLC tumors (96). Furthermore, compound 3-39 described in a Novartis patent, including a 2,4-pyrimidinediamine scaffold, showed brilliants results against EML4-ALK transgenic mouse models (63,97).
On the other hand, F91873 and F91874 are pyridoisoquinolines with multikinase inhibitory activity described by Institut de Recherche Pierre Fabre which are able to inhibit ALK and probably behave as non-ATP competitors (98).
Another ALK inhibitor in preclinical development is TSR-011, developed by Tesaro, but unfortunately the information about it is very scarce (99).
Finally, some other compounds have been reported that are not able to directly inhibit ALK but are able to inhibit the tumor growth in ALK-driven NSCLC models, such as EML4-ALKL1196M, therefore being capable to overcome crizotinib resistance. Two examples of such compounds are the Hsp90 inhibitors retaspimycin (IPI-504) and ganetespib (STA-9090) (67,100-102).
ROS1 inhibitors
Treatments targeting EGFR, in cases of NSCLC patients in which mutant ROS1 kinase is expressed, might be partially or totally ineffective. The lack of knowledge about ROS1 renders important to discover selective compounds in order to confirm theoretical speculations (103).
Some kinase inhibitors were assayed against ROS1 and one of them, staurosporine, showed high inhibitory activity (IC50 =0.9 nM) (61). On the other hand, several heterocyclic compounds, such as AST-487, PP 2, AG 1487, PDGFR I-III and D-64406, showed moderate to low inhibitory activities (IC50 =1,700, 5,200, 13,600, 48,000 and 365,000 nM respectively) (103-105).
Due to the high homology between the kinase domains of ROS1 and ALK, some ALK inhibitors were assayed against ROS1-driven cells and tumors. NVP-TAE684 showed in vitro activity against HCC78 cell lines expressing ROS1 and inhibition of signaling downstream of ROS1 inducing apoptosis in BaF3/FIG-ROS positive cells (IC50 =10 nM) (57,79). Furthermore, a computational study of NVP-TAE684 showed its higher affinity for the ROS1 kinase domain with respect to the ALK kinase domain (14).
Apart from being the first ALK inhibitor approved by FDA, crizotinib is also able to inhibit ROS1 with an IC50 value of 1.7 nM in an in vitro assay. This high inhibition in an enzymatic-based assay was not confirmed in a cell-based assay (IC50 =1.4 µM against the HCC78 ROS1-rearranged NSCLC cell line) (79). The inhibition of the ROS1 phosphorylation by crizotinib in the HCC78 cell line in a moderate manner was also described (106). Although only 2% of NSCLC cases harbor ROS1 rearrangements, it is described the case of a patient having a ROS1 positive tumor (without EGFR mutations nor ALK rearrangements) who did not responded to erlotinib treatment (an EGFR inhibitor) but was totally recovered in 12 weeks after crizotinib treatment. This case is the proof that ROS1 is a prime target for the development of new inhibitors for the treatment of NSCLC (11).
Some other ALK inhibitors were assayed against ROS1 with promising results. Thus AP26113 (IC50 =1.9 nM) (67) and WZ-5-126 (IC50 =8.2 nM) (51), together with crizotinib and ASP3026, have entered clinical trials (59).
Finally, there are only two pyrazole ROS1 selective compounds (KIST301072 and KIST301080), which showed good ROS1 inhibitory activity (IC50 =199 and 209 nM, respectively) when tested against 45 kinases (60,106,107).
Conclusions
Traditionally, depending on the type of tumor, therapeutic approaches include different combinations of surgery, radiation therapy, and chemotherapy. However, alternative therapies using receptor tyrosine kinase (RTK) as targets started to be introduced in the beginning of this century. In the case of lung cancer, such approach has led to discovery of a number of these targeted therapies with specific inhibitor drugs such as erlotinib and gefitinib for mutations in the epidermal growth factor receptor (EGFR). However, in a 4-7% of the of lung adenocarcinomas these treatments are ineffective due to the presence of ALK and/or ROS1 rearrangements. This problem has been partially overcome due to the development of crizotinib (Xalkori®, Pfizer) but the discovery of some crizotinib resistant ALK mutations has forced the research of new inhibitors. Furthermore, the implication of ROS1 protooncogene in this kind of tumors has rendered the situation even more complicated. The present review has tried to show the importance of ALK and ROS1 as combined targets for the development of new treatments for non-small cell lung cancer (NSCLC) when the most common approaches fail and the efforts carried out in this field to date.
Acknowledgements
Disclosure: The authors declare no conflict of interest.
References
- 1.Siegel R, Naishadham D, Jemal A.Cancer statistics, 2012. CA Cancer J Clin 2012;62:10-29. [DOI] [PubMed] [Google Scholar]
- 2.Schiller JH, Harrington D, Belani CP, et al. Comparison of four chemotherapy regimens for advanced non-small-cell lung cancer. N Engl J Med 2002;346:92-8. [DOI] [PubMed] [Google Scholar]
- 3.Mok TS, Wu YL, Thongprasert S, et al. Gefitinib or carboplatin-paclitaxel in pulmonary adenocarcinoma. N Engl J Med 2009;361:947-57. [DOI] [PubMed] [Google Scholar]
- 4.Rosell R, Carcereny E, Gervais R, et al. Erlotinib versus standard chemotherapy as first-line treatment for European patients with advanced EGFR mutation-positive non-small-cell lung cancer (EURTAC): a multicentre, open-label, randomised phase 3 trial. Lancet Oncol 2012;13:239-46. [DOI] [PubMed] [Google Scholar]
- 5.Kwak EL, Bang YJ, Camidge DR, et al. Anaplastic lymphoma kinase inhibition in non-small-cell lung cancer. N Engl J Med 2010;363:1693-703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Soda M, Choi YL, Enomoto M, et al. Identification of the transforming EML4-ALK fusion gene in non-small-cell lung cancer. Nature 2007;448:561-6. [DOI] [PubMed] [Google Scholar]
- 7.Zhang X, Zhang S, Yang X, et al. Fusion of EML4 and ALK is associated with development of lung adenocarcinomas lacking EGFR and KRAS mutations and is correlated with ALK expression. Mol Cancer 2010;9:188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wong DW, Leung EL, So KK, et al. The EML4-ALK fusion gene is involved in various histologic types of lung cancers from nonsmokers with wild-type EGFR and KRAS. Cancer 2009;115:1723-33. [DOI] [PubMed] [Google Scholar]
- 9.Takahashi T, Sonobe M, Kobayashi M, et al. Clinicopathologic features of non-small-cell lung cancer with EML4-ALK fusion gene. Ann Surg Oncol 2010;17:889-97. [DOI] [PubMed] [Google Scholar]
- 10.Rikova K, Guo A, Zeng Q, et al. Global survey of phosphotyrosine signaling identifies oncogenic kinases in lung cancer. Cell 2007;131:1190-203. [DOI] [PubMed] [Google Scholar]
- 11.Bergethon K, Shaw AT, Ou SH, et al. ROS1 rearrangements define a unique molecular class of lung cancers. J Clin Oncol 2012;30:863-70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Takeuchi K, Soda M, Togashi Y, et al. RET, ROS1 and ALK fusions in lung cancer. Nat Med 2012;18:378-81. [DOI] [PubMed] [Google Scholar]
- 13.Jun HJ, Johnson H, Bronson RT, et al. The oncogenic lung cancer fusion kinase CD74-ROS activates a novel invasiveness pathway through E-Syt1 phosphorylation. Cancer Res 2012;72:3764-74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ou SH, Tan J, Yen Y, et al. ROS1 as a ‘druggable’ receptor tyrosine kinase:lessons learned from inhibiting the ALK pathway. Expert Rev Anticancer Ther 2012;12:447-56. [DOI] [PubMed] [Google Scholar]
- 15.Shaw AT, Camidge DR, Engelman JA, et al. Clinical activity of crizotinib in advanced non-small cell lung cancer (NSCLC) harboring ROS1 gene rearrangement. J Clin Oncol 2012;30:abstr 7508.
- 16.Gunby RH, Ahmed S, Sottocornola R, et al. Structural Insights into the ATP Binding Pocket of the Anaplastic Lymphoma Kinase by Site-Directed Mutagenesis, Inhibitor Binding Analysis, and Homology Modeling. J Med Chem 2006;49:5759-68. [DOI] [PubMed] [Google Scholar]
- 17.Galkin AV, Melnick JS, Kim S, et al. Identification of NVP-TAE684, a potent, selective, and efficacious inhibitor of NPM-ALK. Proc Natl Acad Sci U S A 2007;104:270-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Okamoto M, Kojima H, Saito N, et al. Virtual screening and further development of novel ALK inhibitors. Bioorg Med Chem 2011;19:3086-95. [DOI] [PubMed] [Google Scholar]
- 19.Ott GR, Wells GJ, Thieu TV, et al. 2,7-disubstituted-pyrrolo[2,1-f][1,2,4]triazines:new variant of an old template and application to the discovery of anaplastic lymphoma kinase (ALK) inhibitors with in vivo antitumor activity. J Med Chem 2011;54:6328-41. [DOI] [PubMed] [Google Scholar]
- 20.Berman HM, Westbrook J, Feng Z, et al. The Protein Data Bank. Nucleic Acids Res 2000;28:235-42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lee CC, Jia Y, Li N, et al. Crystal structure of the ALK (anaplastic lymphoma kinase) catalytic domain. Biochem J 2010;430:425-37. [DOI] [PubMed] [Google Scholar]
- 22.Bossi RT, Saccardo MB, Ardini E, et al. Crystal Structures of Anaplastic Lymphoma Kinase in Complex with ATP Competitive Inhibitors. Biochemistry 2010;49:6813-25. [DOI] [PubMed] [Google Scholar]
- 23.Park H, Chi O, Kim J, et al. Identification of novel inhibitors of tropomyosin-related kinase A through the structure-based virtual screening with homology-modeled protein structure. J Chem Inf Model 2011;51:2986-93. [DOI] [PubMed] [Google Scholar]
- 24.Donella-Deana A, Marin O, Cesaro L, et al. Unique substrate specificity of anaplastic lymphoma kinase (ALK):development of phosphoacceptor peptides for the assay of ALK activity. Biochemistry 2005;44:8533-42. [DOI] [PubMed] [Google Scholar]
- 25.Tartari CJ, Gunby RH, Coluccia AM, et al. Characterization of some molecular mechanisms governing autoactivation of the catalytic domain of the anaplastic lymphoma kinase. J Biol Chem 2008;283:3743-50. [DOI] [PubMed] [Google Scholar]
- 26.Epstein LF, Chen H, Emkey R, et al. The R1275Q neuroblastoma mutant and certain ATP-competitive inhibitors stabilize alternative activation loop conformations of anaplastic lymphoma kinase. J Biol Chem 2012;287:37447-57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bryan MC, Whittington DA, Doherty EM, et al. Rapid development of piperidine carboxamides as potent and selective anaplastic lymphoma kinase inhibitors. J Med Chem 2012;55:1698-705. [DOI] [PubMed] [Google Scholar]
- 28.Lewis RT, Bode CM, Choquette DM, et al. The discovery and optimization of a novel class of potent, selective, and orally bioavailable anaplastic lymphoma kinase (ALK) inhibitors with potential utility for the treatment of cancer. J Med Chem 2012;55:6523-40. [DOI] [PubMed] [Google Scholar]
- 29.Cui JJ, Tran-Dubé M, Shen H, et al. Structure Based Drug Design of Crizotinib (PF-02341066), a Potent and Selective Dual Inhibitor of MesenchymalEpithelial Transition Factor (c-MET) Kinase and Anaplastic Lymphoma Kinase (ALK). J Med Chem 2011;54:6342-63. [DOI] [PubMed] [Google Scholar]
- 30.Sakamoto H, Tsukaguchi T, Hiroshima S, et al. CH5424802, a selective ALK inhibitor capable of blocking the resistant gatekeeper mutant. Cancer Cell 2011;19:679-90. [DOI] [PubMed] [Google Scholar]
- 31.Baskaran C, Ramachandran M.Computational molecular docking studies on anticancer drugs. Asian Pac Trop Dis 2012;2:S734-S738. [Google Scholar]
- 32.Gingrich DE, Lisko JG, Curry MA, et al. Discovery of an orally efficacious inhibitor of anaplastic lymphoma kinase. J Med Chem 2012;55:4580-93. [DOI] [PubMed] [Google Scholar]
- 33.Li R, Xue L, Zhu T, et al. Design and synthesis of 5-aryl-pyridone-carboxamides as inhibitors of anaplastic lymphoma kinase. J Med Chem 2006;49:1006-15. [DOI] [PubMed] [Google Scholar]
- 34.Milkiewicz KL, Weinberg LR, Albom MS, et al. Synthesis and structure-activity relationships of 1,2,3,4-tetrahydropyrido[2,3-b]pyrazines as potent and selective inhibitors of the anaplastic lymphoma kinase. Bioorg Med Chem 2010;18:4351-62. [DOI] [PubMed] [Google Scholar]
- 35.Ardini E, Galvani A.ALK Inhibitors, a Pharmaceutical Perspective. Front Oncol 2012;2:17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Breslin HJ, Lane BM, Ott GR, et al. Design, synthesis, and anaplastic lymphoma kinase (ALK) inhibitory activity for a novel series of 2,4,8,22-tetraazatetracyclo[14.3.1.13,7.19,13]docosa-1(20),3(22),4,6,9(21),10,12,16,18-nonaene macrocycles. J Med Chem 2012;55:449-64. [DOI] [PubMed] [Google Scholar]
- 37.Ott GR, Tripathy R, Cheng M, et al. Discovery of a Potent Inhibitor of Anaplastic Lymphoma Kinase with in Vivo Antitumor Activity. ACS Med Chem Lett 2010;1:493-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Mesaros EF, Burke JP, Parrish JD, et al. Novel 2,3,4,5-tetrahydro-benzo[d]azepine derivatives of 2,4-diaminopyrimidine, selective and orally bioavailable ALK inhibitors with antitumor efficacy in ALCL mouse models. Bioorg Med Chem Lett 2011;21:463-6. [DOI] [PubMed] [Google Scholar]
- 39.Mesaros EF, Thieu TV, Wells GJ, et al. Strategies to mitigate the bioactivation of 2-anilino-7-aryl-pyrrolo[2,1-f][1,2,4]triazines:identification of orally bioavailable, efficacious ALK inhibitors. J Med Chem 2012;55:115-25. [DOI] [PubMed] [Google Scholar]
- 40.Zificsak CA, Theroff JP, Aimone LD, et al. Methanesulfonamido-cyclohexylamine derivatives of 2,4-diaminopyrimidine as potent ALK inhibitors. Bioorg Med Chem Lett 2011;21:3877-80. [DOI] [PubMed] [Google Scholar]
- 41.inoshita K, Ono Y, Emura T, et al. Discovery of novel tetracyclic compounds as anaplastic lymphoma kinase inhibitors. Bioorg Med Chem Lett 2011;21:3788-93. [DOI] [PubMed]
- 42.Kinoshita K, Asoh K, Furuichi N, et al. Design and synthesis of a highly selective, orally active and potent anaplastic lymphoma kinase inhibitor (CH5424802). Bioorg Med Chem 2012;20:1271-80. [DOI] [PubMed] [Google Scholar]
- 43.Deng X, Wang J, Zhang J, et al. Discovery of 3,5-Diamino-1,2,4-triazole Ureas as Potent Anaplastic Lymphoma Kinase Inhibitors. ACS Med Chem Lett 2011;2:379-84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Slavish PJ, Price JE, Jiang Q, et al. Synthesis of an aryloxy oxo pyrimidinone library that displays ALK-selective inhibition. Bioorg Med Chem Lett 2011;21:4592-6. [DOI] [PubMed] [Google Scholar]
- 45.Xie HZ, Lan H, Pan YL, et al. Identification of novel anaplastic lymphoma kinase (ALK) inhibitors using a common feature pharmacophore model derived from known ligands crystallized with ALK. Chem Biol Drug Des 2013;81:175-84. [DOI] [PubMed] [Google Scholar]
- 46.Bresler SC, Wood AC, Haglund EA, et al. Differential inhibitor sensitivity of anaplastic lymphoma kinase variants found in neuroblastoma. Sci Transl Med 2011;3:108ra114. [DOI] [PMC free article] [PubMed]
- 47.Shiao H, Chiang NJ, Hsieh HP. Anaplastic Lymphoma Kinase (ALK) Inhibitors: New Cancer Breakthroughs for Lung Cancer. J Cancer Res Pract 2011;27:143-56. [Google Scholar]
- 48.Chemical Computing Group Inc.; Molecular Operating Environment (MOE), 2012.10;1010 Sherbooke St. West, Suite #910, Montreal, QC, Canada, H3A 2R7, 2012.
- 49.Choi YL, Soda M, Yamashita Y, et al. EML4-ALK mutations in lung cancer that confer resistance to ALK inhibitors. N Engl J Med 2010;363:1734-9. [DOI] [PubMed] [Google Scholar]
- 50.Katayama R, Shaw AT, Khan TM, et al. Mechanisms of acquired crizotinib resistance in ALK-rearranged lung Cancers. Sci Transl Med 2012;4:120ra17. [DOI] [PMC free article] [PubMed]
- 51.Bossi RT, Saccardo MB, Ardini E, et al. Crystal structures of anaplastic lymphoma kinase in complex with ATP competitive inhibitors. Biochemistry 2010;49:6813-25. [DOI] [PubMed] [Google Scholar]
- 52.Katayama R, Khan TM, Benes C, et al. Therapeutic strategies to overcome crizotinib resistance in non-small cell lung cancers harboring the fusion oncogene EML4-ALK. Proc Natl Acad Sci U S A 2011;108:7535-40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Sasaki T, Koivunen J, Ogino A, et al. A novel ALK secondary mutation and EGFR signaling cause resistance to ALK kinase inhibitors. Cancer Res 2011;71:6051-60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Lu L, Ghose AK, Quail MR, et al. ALK mutants in the kinase domain exhibit altered kinase activity and differential sensitivity to small molecule ALK inhibitors. Biochemistry 2009;48:3600-9. [DOI] [PubMed] [Google Scholar]
- 55.Sun HY, Ji FQ. A molecular dynamics investigation on the crizotinib resistance mechanism of C1156Y mutation in ALK. Biochem Biophys Res Commun 2012;423:319-24. [DOI] [PubMed] [Google Scholar]
- 56.Tripathy R, McHugh RJ, Ghose AK, et al. Pyrazolone-based anaplastic lymphoma kinase (ALK) inhibitors:control of selectivity by a benzyloxy group. Bioorg Med Chem Lett 2011;21:7261-4. [DOI] [PubMed] [Google Scholar]
- 57.Gu TL, Deng X, Huang F, et al. Survey of tyrosine kinase signaling reveals ROS kinase fusions in human cholangiocarcinoma. PLoS One 2011;6:e15640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Doebele RC, Camidge DR. Targeting ALK, ROS1, and BRAF kinases. J Thorac Oncol 2012;7:S375-S376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Chin LP, Soo RA, Soong R, et al. Targeting ROS1 with anaplastic lymphoma kinase inhibitors:a promising therapeutic strategy for a newly defined molecular subset of non-small-cell lung cancer. J Thorac Oncol 2012;7:1625-30. [DOI] [PubMed] [Google Scholar]
- 60.El-Deeb IM, Park BS, Jung SJ, et al. Design, synthesis, screening, and molecular modeling study of a new series of ROS1 receptor tyrosine kinase inhibitors. Bioorg Med Chem Lett 2009;19:5622-6. [DOI] [PubMed] [Google Scholar]
- 61.Peach ML, Tan N, Choyke SJ, et al. Directed discovery of agents targeting the Met tyrosine kinase domain by virtual screening. J Med Chem 2009;52:943-51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.McDermott U, Iafrate AJ, Gray NS, et al. Genomic Alterations of Anaplastic Lymphoma Kinase May Sensitize Tumors to Anaplastic Lymphoma Kinase Inhibitors. Cancer Res 2008;68:3389-95. [DOI] [PubMed] [Google Scholar]
- 63.Soda M, Takada S, Takeuchi K, et al. A mouse model for EML4-ALK-positive lung cancer. Proc Natl Acad Sci U S A 2008;105:19893-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Wang YW, Tu P, Lin K, et al. Identification of oncogenic point mutations and hyperphosphorylation of anaplastic lymphoma kinase in lung cancer. Neoplasia 2011;13:704-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.George RE, Sanda T, Hanna M, et al. Activating mutations in ALK provide a therapeutic target in neuroblastoma. Nature 2008;455:975-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Cerchietti L, Damm-Welk C, Vater I, et al. Inhibition of anaplastic lymphoma kinase (ALK) activity provides a therapeutic approach for CLTC-ALK-positive human diffuse large B cell lymphomas. PLoS One 2011;6:e18436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Katayama R, Khan TM, Benes C, et al. Therapeutic strategies to overcome crizotinib resistance in non-small cell lung cancers harboring the fusion oncogene EML4-ALK. Proc Natl Acad Sci U S A 2011;108:7535-40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Duijkers FA, Gaal J, Meijerink JP, et al. Anaplastic lymphoma kinase (ALK) inhibitor response in neuroblastoma is highly correlated with ALK mutation status, ALK mRNA and protein levels. Cell Oncol (Dordr) 2011;34:409-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Christensen JG, Zou HY, Arango ME, et al. Cytoreductive antitumor activity of PF-2341066, a novel inhibitor of anaplastic lymphoma kinase and c-Met, in experimental models of anaplastic large-cell lymphoma. Mol Cancer Ther 2007;6:3314-22. [DOI] [PubMed] [Google Scholar]
- 70.Zou HY, Li Q, Lee JH, et al. An Orally Available Small-Molecule Inhibitor of c-Met, PF-2341066, Exhibits Cytoreductive Antitumor Efficacy through Antiproliferative and Antiangiogenic Mechanisms. Cancer Res 2007;67:4408-17. [DOI] [PubMed] [Google Scholar]
- 71.Camidge DR, Bang Y, Kwak EL, et al. Progression-free survival (PFS) from a phase I study of crizotinib (PF-02341066) in patients with ALK-positive non-small cell lung cancer (NSCLC). J Clin Oncol 2011;29:abstr 2501.
- 72.Sasaki T, Okuda K, Zheng W, et al. The neuroblastoma-associated F1174L ALK mutation causes resistance to an ALK kinase inhibitor in ALK-translocated cancers. Cancer Res 2010;70:10038-43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Sasaki T, Rodig SJ, Chirieac LR, et al. The biology and treatment of EML4-ALK non-small cell lung cancer. Eur J Cancer 2010;46:1773-80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Shaw AT, Yeap BY, Solomon BJ, et al. Effect of crizotinib on overall survival in patients with advanced non-small-cell lung cancer harbouring ALK gene rearrangement:a retrospective analysis. Lancet Oncol 2011;12:1004-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Wang Y, Huang W, Liu S, et al. inventors. Ariad Pharmaceuticals I, USA, assignee. Preparation of phosphorus derivatives as kinase inhibitors. WO2009143389. Patent Application Date: May 21, 2009.
- 76.Shakespeare WC, Fantin VR, Wang F, et al. Discovery of potent and selective orally active inhibitors of anaplastic lymphoma kinase (ALK). American Association for cancer research annual meeting (AACR) 2009;abstr 3738. [Google Scholar]
- 77.Rivera VM, Anjum R, Wang F, et al. Efficacy and pharmacodynamic analysis of AP26113, a potent and selective orally active inhibitor of anaplastic lymphoma kinase (ALK). Cancer Res 2010;70:abstr 3623.
- 78.Zhang S, Wang F, Keats J, et al. AP26113, a potent ALK inhibitor, overcomes mutations in EML4-ALK that confer resistance to PF-02341066 (PF1066). Cancer Res 2010;70:abstr LB-298.
- 79.Lovly CM, Heuckmann JM, de Stanchina E, et al. Insights into ALK-Driven Cancers Revealed through Development of Novel ALK Tyrosine Kinase Inhibitors. Cancer Res 2011;71:4920-31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Liang C, Li Z. inventors. Xcovery I, USA, assignee. Substituted pyridazine carboxamides as kinase inhibitors. WO2009154769. Patent Application Date: June 18, 2009.
- 81.Lovly CM, de Stanchina E, Liang C, et al. Preclinical development of a selective, potent small molecule ALK inhibitor. Cancer Res 2010;70:abstr 1788.
- 82.Xcovery. Targeted therapeutics. Available online: http://www.xcovery.com/alk (accessed Mar 13, 2013).
- 83.Kinoshita K, Kobayashi T, Asoh K, et al. 9-Substituted 6,6-Dimethyl-11-oxo-6,11-dihydro-5H-benzo[b]carbazoles as Highly Selective and Potent Anaplastic Lymphoma Kinase Inhibitors. J Med Chem 2011;54:6286-94. [DOI] [PubMed] [Google Scholar]
- 84.Sabbatini P, Korenchuk S, Rowand JL, et al. GSK1838705A inhibits the insulin-like growth factor-1 receptor and anaplastic lymphoma kinase and shows antitumor activity in experimental models of human cancers. Mol Cancer Ther 2009;8:2811-20. [DOI] [PubMed] [Google Scholar]
- 85.Chamberlain SD, Redman AM, Wilson JW, et al. Optimization of 4,6-bis-anilino-1H-pyrrolo[2,3-d]pyrimidine IGF-1R tyrosine kinase inhibitors towards JNK selectivity. Bioorg Med Chem Lett 2009;19:360-4. [DOI] [PubMed] [Google Scholar]
- 86.Kuromitsu S, Mori M, Shimada I, et al. Antitumor activities of ASP3026 against EML4-ALK-dependent tumor models. Mol Cancer Ther 2011;10:abstr A227.
- 87.Gunby RH, Tartari CJ, Porchia F, et al. An enzyme-linked immunosorbent assay to screen for inhibitors of the oncogenic anaplastic lymphoma kinase. Haematologica 2005;90:988-90. [PubMed] [Google Scholar]
- 88.Wan W, Albom MS, Lu L, et al. Anaplastic lymphoma kinase activity is essential for the proliferation and survival of anaplastic large-cell lymphoma cells. Blood 2006;107:1617-23. [DOI] [PubMed] [Google Scholar]
- 89.Ambrogio C, Martinengo C, Voena C, et al. NPM-ALK oncogenic tyrosine kinase controls T-Cell identity by transcriptional regulation and epigenetic silencing in lymphoma cells. Cancer Res 2009;69:8611-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Allwein SP, Roemmele RC, Haley JJ, et al. Development and Scale-Up of an Optimized Route to the ALK Inhibitor CEP-28122. Org Process Res Dev 2012;16:148-55. [Google Scholar]
- 91.Fancelli D, Moll J, Varasi M, et al. 1,4,5,6-Tetrahydropyrrolo[3,4-c]pyrazoles:Identification of a Potent Aurora Kinase Inhibitor with a Favorable Antitumor Kinase Inhibition Profile. J Med Chem 2006;49:7247-51. [DOI] [PubMed] [Google Scholar]
- 92.Pulford K, Morris SW, Turturro F. Anaplastic lymphoma kinase proteins in growth control and cancer. J Cell Physiol 2004;199:330-58. [DOI] [PubMed] [Google Scholar]
- 93.Ardini E, Menichincheri M, Banfi P, et al. In vitro and in vivo activity of NMS-E628 against ALK mutations resistant to Xalkori. Mol Cancer Ther 2011;10:abstr A232.
- 94.Ardini E, Menichincheri M, De Ponti C, et al. Characterization of NMS-E628, a small molecule inhibitor of anaplastic lymphoma kinase with antitumor efficacy in ALK-dependent lymphoma and non-small cell lung cancer models. Mol Cancer Ther 2009;8:abstr A243.
- 95.Webb TR, Slavish J, George RE, et al. Anaplastic lymphoma kinase:role in cancer pathogenesis and small-molecule inhibitor development for therapy. Expert Rev Anticancer Ther 2009;9:331-56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Mehra R, Camidge DR, Sharma S, et al. First-in-human phase I study of the ALK inhibitor LDK378 in advanced solid tumors. J Clin Oncol 2012;30:abstr 3007.
- 97.Garcia-echeverria C, Kanazawa T, Kawahara E, et al. Inventors. Novartis A.-G. S, Novartis Pharma G.m.b.H. and IRM LLC, assignees. Preparation of 2,4-pyrimidinediamines useful in the treatment of neoplastic diseases, inflammatory and immune system disorders. WO2005016894. Patent Application Date: August 13, 2004.
- 98.Kruczynski A, Mayer P, Marchand A, et al. Antitumor activity of pyridoisoquinoline derivatives F91873 and F91874, novel multikinase inhibitors with activity against the anaplastic lymphoma kinase. Anti-Cancer Drugs 2009;20:364-72. [DOI] [PubMed] [Google Scholar]
- 99.Tesaro. Pipeline. Available online: http://tesarobio.com/pipeline/ (accessed Mar 13, 2013).
- 100.Sequist LV, Gettinger S, Senzer NN, et al. Activity of IPI-504, a novel heat-shock protein 90 inhibitor, in patients with molecularly defined non-small-cell lung cancer. J Clin Oncol 2010;28:4953-60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Chen Z, Sasaki T, Tan X, et al. Inhibition of ALK, PI3K/MEK, and HSP90 in Murine Lung Adenocarcinoma Induced by EML4-ALK Fusion Oncogene. Cancer Res 2010;70:9827-36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.El-Hariry I, Proia D, Vukovic V. inventors. Synta Pharmaceuticals Corp. U, assignee. Treating cancer with heat-shock protein-90 (HSP90) inhibitory compounds such as ganetespib. WO2013006864. Patent Application Date:July 9, 2012.
- 103.El-Deeb IM, Yoo KH, Lee SH. ROS receptor tyrosine kinase:a new potential target for anticancer drugs. Med Res Rev 2010;31:794-818. [DOI] [PubMed] [Google Scholar]
- 104.araman MW, Herrgard S, Treiber DK, et al. A quantitative analysis of kinase inhibitor selectivity. Nat Biotechnol 2008;26:127-32. [DOI] [PubMed]
- 105.Banerjee P.Recognition of c-ROS oncogene inhibition with a FRET-based receptor construct. MURJ 2004;10:52-7. [Google Scholar]
- 106.Park BS, El-Deeb IM, Yoo KH, et al. Design, synthesis and biological evaluation of new potent and highly selective ROS1-tyrosine kinase inhibitor. Bioorg Med Chem Lett 2009;19:4720-3. [DOI] [PubMed] [Google Scholar]
- 107.Park BS, El-deeb IM, Yoo KH, et al. Synthesis and biological activity of new 4-(pyridin-4-yl)-(3-methoxy-5-methylphenyl)-1H-pyrazoles derivatives as ROS receptor tyrosine kinase inhibitors. Bull Korean Chem Soc 2012;33:3629-34. [Google Scholar]