

Abstract
Background. Oxidative stress plays a pivotal role in the lung injuries of critical ill patients. This study investigates the protection conferred by α 2 adrenoceptor agonist dexmedetomidine (Dex) from lung alveolar epithelial cell injury induced by hydrogen peroxide (H2O2) and the underlying mechanisms. Methods. The lung alveolar epithelial cell line, A549, was cultured and then treated with 500 μM H2O2 with or without Dex (1 nM) or Dex in combination with atipamezole (10 nM), an antagonist of α 2 receptors. Their effect on mitochondrial membrane potential (Δψ m), reactive oxygen species (ROS), and the cell cycle was assessed by flow cytometry. Cleaved-caspases 3 and 9, BAX, Bcl-2, phospho-mTOR (p-mTOR), ERK1/2, and E-cadherin expression were also determined with immunocytochemistry. Results. Upregulation of cleaved-caspases 3 and 9 and BAX and downregulation of Bcl-2, p-mTOR, and E-cadherin were found following H2O2 treatment, and all of these were reversed by Dex. Dex also prevented the ROS generation, cytochrome C release, and cell cycle arrest induced by H2O2. The effects of Dex were partially reversed by atipamezole. Conclusion. Our study demonstrated that Dex protected lung alveolar epithelial cells from apoptotic injury, cell cycle arrest, and loss of cell adhesion induced by H2O2 through enhancing the cell survival and proliferation.
1. Introduction
Oxidative stress plays a pivotal role in acute lung injury (ALI) and acute respiratory distress syndrome (ARDS), which is common in critically ill patients undergoing mechanical ventilation [1, 2]. The source of oxidative stress for lung cells may be intrinsic or extrinsic. Oxidants generated intrinsically are from the mitochondria of injured alveolar epithelial cells. Extrinsic oxidants may arise from air inhalation or generated from phagocytic cells [3, 4]. Phagocytic cells can generate free radicals which act on the cell surfaces to form nicotinamide adenine dinucleotide phosphate (NADP+), an oxidant, and this process often occurs in vascular endothelial cells and lung alveolar epithelial cells [5].
The degradation of the oxidizing products depends on the antioxidant enzymes including superoxide dismutase (SOD), catalase, glutathione peroxidase, and glutathione [6]. SOD catalyses the dismutation of the superoxide anion to hydrogen peroxide (H2O2). Catalase converts H2O2 to H2O in the presence of glutathione, which is then converted to its oxidized form, glutathione disulfide (GSSG). The presence of these antioxidant enzymes is important in maintaining a redox balance in our body and so that one can respond to oxidizing conditions which may threaten the structural and functional integrity of the vital organs including the lungs [7]. In a variety of situations such as severe liver disease, cachexia, and alimentary deficiency, the antioxidant enzymes are insufficient to clear the oxidation products [8]. Lung alveolar epithelial cells, especially type II alveolar cells, are particularly sensitive to oxidative stress [9]. Epithelial cell apoptosis is followed by remodeling processes, which consist of epithelial and fibroblast activation, cytokine production, activation of the coagulation pathway, neoangiogenesis, reepithelialization, and fibrosis [10]; therefore, lung injury is often present at the end stage of liver failure [11].
Recent studies have demonstrated that dexmedetomidine (Dex), a potent α 2 adrenergic agonist with sedative, analgesic, sympatholytic, and hemodynamic effects, can reduce systemic inflammation and improve diaphragmatic function and gas exchange in perioperative patients [12, 13]. We hypothesized that protective signaling pathways in lung cells might be activated by Dex to prevent cellular apoptosis when subjected to oxidative insult. In this study, we aim to investigate whether Dex protects the lung alveolar epithelial cells against H2O2-induced oxidative stress.
2. Material and Methods
2.1. Cell Culture and Drug Treatments
The A549 human alveolar epithelial cells (European Cell Culture Collection, UK) were cultured in RPMI 1640 (Abcam, Cambridge, UK) medium supplemented with 10% fetal bovine serum (FBS, Abcam) at 37°C in 5% CO2. Cells were challenged with 500 μM H2O2 (Sigma-Aldrich, Dorset, UK) in fresh RPMI 1640 medium without FBS for 24 hours in the control group, while, in the treatment groups, the cells were pretreated with 1 nM Dex (Orion Pharm Ltd., Newbury, Berkshire, UK) for 15 minutes, in the presence or absence of 10 nM atipamezole (Sigma Aldrich, Cambridge, UK), a synthetic α 2-adrenergic antagonist and then exposed to 500 μM H2O2 for additional 24 hours.
2.2. Immunocytochemistry
The naive or treated A549 cells were blocked with donkey serum and then incubated with the following primary antibodies: rabbit anti-human BAX, Bcl-2, cleaved-caspase 9, caspase 3, p-mTOR, ERK1/2, and E-cadherin (1 : 250, Abcam) overnight followed by fluorochrome conjugated donkey anti-rabbit secondary antibody for 1 h. The nuclei were counterstained with 4′,6-diamidino-2-phenylindole (DAPI, Invitrogen, Warrington, UK) and examined with an Olympus BX40 microphotography system (Rochester NY, USA). The mean intensity of fluorescence obtained from ten samples was analyzed with Image J software (NIH, US National Institutes of Health, Bethesda, MD).
2.3. Determination of Δψ m
The cells were labeled with 5,5′,6,6′-tetrachloro-1,1achlortetrathylbenzimidazolyl-carbocyanine iodide (JC-1) dye (e-bioscience, Ireland, UK) and assessed by flow cytometry and microscope, respectively [14]. For FACS detection, cells were detached using 0.25% trypsin and then transferred to 5 mL polystyrene tubes. After washing with FACS buffer once, the cells were incubated with 0.2 μM JC-1 in FACS for 30 minutes at 37°C and protected from light. The cells were analyzed by flow cytometry after being washed with warm FACS buffer twice. Fluorescence was measured in the FL1 (FITC) and FL2 (PE) channels, gating only living cells. The mean intensity of red fluorescence (PE) and green fluorescence (FITC) was analyzed using FlowJo 7.6.1 software (TreeStar, San Carlos, CA) and then the ratio of red/green fluorescence intensity was analyzed. A decrease in the red/green fluorescence ratio is indicative of mitochondrial depolarization. To further investigate the effect of Dex and H2O2 on the changes of cell Δψ m, cells stained with JC-1 dye were also visualized by microscope. In brief, cells were incubated with 10 μM JC-1 dye in FACS buffer for 30 minutes at 37°C after being washed with warm 0.1 M PBS once. Then, the nuclei were stained using DAPI. The fluorescence staining was examined under rhodamine (red), fluorescein (green), and cyan (blue) spectral filters with the Olympus BX40 microphotography system.
2.4. Determination of ROS Production by Flow Cytometry
ROS production was determined by 2′-7′-dichlorodihydrofluorescein diacetate (DCF) staining and the intensity of fluorescence was assessed by flow cytometry [15]. A549 cells were harvested by trypsinization and washed with FACS buffer. The cells were incubated in 2 μM DCF diluted in FACS buffer for 30 minutes at 37°C. The fluorescence was assessed by flow cytometry (FACS Calibur; Becton Dickinson, Sunnyvale, CA) and analyzed with FlowJo 7.6.1 software. Each assay included at least 10,000 gated events.
2.5. Cell Cycle Analysis by Flow Cytometry
The cell cycle was analyzed by flow cytometry as described previously [16]. The cells were fixed with 70% ethanol at 4°C overnight. After centrifugation at 2500 rpm for 10 minutes and resuspension in 500 μL PBS, 10 μL of 500 ng/L RNase and 10 μL of 40 μg/L PI were added to the cell suspension and then incubated for 10 minutes at room temperature. The fluorescence of PI uptake in cells was detected with flow cytometry and analyzed with FlowJo 7.6.1 software. Each assay included at least 10,000 gated events.
All of the above experiments have been repeated more than 8 times independently.
3. Statistical Analysis
All numerical data is presented as mean ± SD. Comparison between the treatment groups was analyzed by one-way ANOVA of variance, followed by Tukey's test (GraphPad prism 5, San Diego, CA, USA). A P value of 0.05 was considered as statistically significant.
4. Results
4.1. Effect of Dex on the Expression of Cleaved-Caspases 3 and 9, BAX, and Bcl-2 following H2O2 Challenge
Expression of apoptotic proteins cleaved-caspase 3, caspase 9, and the proapoptotic protein BAX were upregulated in the A549 cells exposed to H2O2. The expression of antiapoptotic protein Bcl-2 in A549 cells was downregulated when detected with immunocytochemistry following the challenge of 500 μM H2O2 for 24 hours, and all these effects were partially reversed by the pretreatment with 1 nM Dex. Atipamezole at a dose of 10 nM only partially reversed the effects of Dex on the expression of BAX and Bcl-2 and had no effects on that of cleaved-caspases 3 and 9 (Figure 1).
Figure 1.

Effect of Dex on the expression of cleaved-caspases 3, 9, BAX, and Bcl-2 in A549 cells. A549 cells were treated with 500 μM H2O2 for 24 hours. Cohort cultures received 1 nM Dex together with or without 10 nM atipamezole for 15 minutes and then treated with H2O2 for additional 24 hours. (a) Expression of cleaved-caspase 9 (green); (b) fluorescence intensity of cleaved-caspase 9; (c) expression of cleaved-caspase 3 (green); (d) fluorescence intensity of cleaved-caspase 3; (e) expression of BAX in A549 cells assessed by immunofluorescence staining (green); (f) mean fluorescence intensity of BAX; (g) expression of Bcl-2 in A549 cells assessed by immunofluorescent staining (green); (h) mean fluorescence intensity of Bcl-2. Scale bar = 50 μm. * P < 0.05; ** P < 0.01; *** P < 0.001; n = 10. NC: naïve control; Dex: dexmedetomidine; Ati: atipamezole; DH: A549 cells treated with H2O2 followed by Dex; ADH: A549 cells treated with H2O2 following by Dex and Ati.
4.2. Effect of Dex on the Δψ m Collapse and ROS Generation and Cytochrome C Release Induced by H2O2
Compared with that of naïve control cells, the Δψ m of mitochondria in H2O2 treated A549 cells decreased significantly, and this effect was also abolished by Dex when detected by flow cytometry and microscopy. There was no significant difference in Δψ m between Dex and Dex combined with atipamezole pretreated cells (Figure 2). The percentage of cells with high ROS production increased significantly after being challenged with 500 μM H2O2 for 24 hours when detected with flow cytometry. This was reversed by pretreatment with 1 nM Dex (percentage of DCF positive cells decreased from 43.08 ± 3.15% in H2O2 treated cells to 30.28 ± 3.49% in H2O2 combined Dex treated cells, P < 0.001). Atipamezole attenuated the effect of Dex on the prevention of ROS generation induced by H2O2 (Figures 3(a) and 3(b)). H2O2-induced an increase in the release of cytochrome C from the mitochondria in A549 cells, but there was no significant difference between H2O2 treated cells and H2O2 combined Dex treated cells (51.11 ± 6.89 in H2O2 treated cells versus 53.21 ± 6.04 in H2O2 combined Dex treated cells, P < 0.05). Pretreatment with atipamezole enhanced the release of cytochrome C compared to H2O2 and Dex combined H2O2 treated cells (Figures 3(c) and 3(d)).
Figure 2.

Effects of Dex on H2O2-induced Δψ m collapse in A549 cells. (a) Δψ m was measured by flow cytometry under FL1 (FITC) and FL2 (PE) channel after being stained with JC-1. The cells in gating areas with low ratio of red/green fluorescence intensity indicated cells with low Δψ m; (b) ratio of fluorescence intensity of JC-1 (red/green), assessed by flow cytometry. * P < 0.05, ** P < 0.01; n = 8; (c) A549 cells were staining with JC-1 and nuclei were counterstained with DAPI. The cells were examined with microscope under rhodamine (red), fluorescein (green), and cyan (blue) spectral filters. The figures are merged from the three colors. Mitochondrial depolarization was indicated by a decreased red fluorescence and increased green fluorescence. Scale bar = 50 μm. PE: phycoerythrin; FITC: fluorescein isothiocyanate.
Figure 3.

Dex reversed the ROS generation and cytochrome C release in A549 cells induced by H2O2. (a) ROS production detected by flow cytometry after DCF staining. The increase of the FITC fluorescence intensity indicated the increase in percentage of DCF positive cells; (b) percentage of the DCF positive cells, n = 8; (c) Overexpression of cytochrome C in A549 cells was not reversed by Dex and was enhanced by atipamezole, Dex treated together; (d) mean fluorescence intensity of cytochrome C. Scale bar = 50 μm. *** P < 0.001; n = 10. DCF: 2′-7′-dichlorodihydrofluorescein diacetate.
4.3. Effect of Dex on the Activation of mTOR/ERK1/2 Pathway and Cell Cycle Arrest Induced by H2O2 in A549
The activation of mTOR and ERK1/2 mediated cell proliferation/survival pathway was investigated by immunostaining to explore the effect of Dex on H2O2-induced cell proliferation and survival. Expression of p-mTOR (activated mTOR) was reduced and there was no change in ERK1/2 levels after the H2O2 challenge. Pretreatment with Dex enhanced the expression of p-mTOR and ERK1/2 compared to that in H2O2 challenged cells, which was partially reversed by atipamezole (fluorescence intensity of p-mTOR: 52.60 ± 4.84 in the DH group compared to 11.70 ± 2.11 in H2O2 group and 27.41 ± 8.09 in the ADH group, P < 0.001; ERK1/2: 29.80 ± 4.69 in the DH group compared to 15.00 ± 3.74 in H2O2 group and 22.83 ± 4.73 in the ADH group, P < 0.01) (Figures 4(a) and 4(b)).
Figure 4.

Dex prevented the downregulation of p-mTOR, ERK1/2, and cell cycle arrest induced by H2O2, which was reversed by atipamezole. (a) Expression of p-mTOR in A549 cells assessed by immunofluorescent staining (green); (b) mean fluorescence intensity of p-mTOR; (c) expression of ERK1/2 in A549 cells assessed by immunofluorescent staining (green); (d) mean fluorescence intensity of ERK1/2. Scale bar = 50 μm; n = 10; (e) cell cycle analyzed by flow cytometry. Dex prevented the G0/G1 arrest of A549 cells induced by H2O2 and attenuated by atipamezole; (f) scatter plots of mean percentage of G0/G1 cells; n = 8. ** P < 0.01, *** P < 0.001.
Flow cytometry was used to investigate the effect of Dex on cell cycle progression following H2O2 insult. Compared with naïve control cells, a greater percentage of cells were arrested at G0/G1 following the H2O2 challenge (4.43 ± 2.08% in naïve control cells versus 43.40 ± 8.99% in H2O2 treated cells, P < 0.001). Dex attenuated the cell cycle arrest at G0/G1 phase induced by H2O2 and this effect was partially abolished by 10 nM atipamezole (22.60 ± 6.62% in DH group versus 39.83 ± 10.11% in ADH group, P < 0.001) (Figures 4(e) and 4(f)).
4.4. Effect of Dex on the Expression of E-Cadherin in A549 Cells after H2O2 Insult
Expression of E-cadherin, which maintains cell adhesion and forms cell-cell junctions to bind cells together, was found to be significantly reduced following the H2O2 challenge; this was partially reversed by pretreatment with Dex (22.27 ± 4.24 in DH group compared to 17.12 ± 1.52 in H2O2 group, P < 0.01). Atipamezole reversed the inhibition of Dex on the downregulation of E-cadherin induced by H2O2 (15.77 ± 1.25 in ADH versus 22.27 ± 4.24 in the DH group, P < 0.001) (Figure 5).
Figure 5.
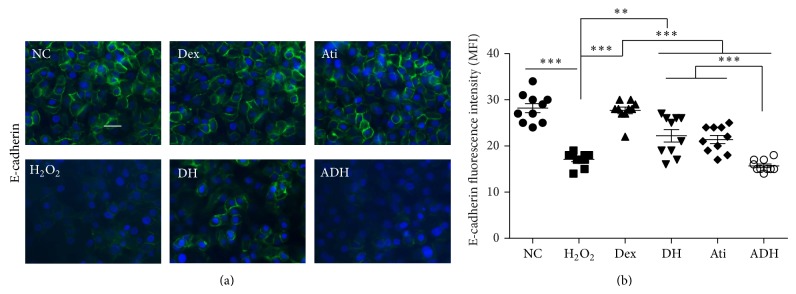
Effect of Dex on the expression of E-cadherin in A549 cells following the challenge of H2O2. (a) Expression of E-cadherin in A549 cells assessed by immunofluorescent staining (green); (b) fluorescence intensity of E-cadherin. Scale bar = 50 μm. * P < 0.01, ** P < 0.01, and *** P < 0.001. n = 10.
5. Discussion
The present study, for the first time, explores the effect of Dex, a potent α 2 adrenergic agonist, on H2O2-induced lung alveolar epithelial cell injury. Our results demonstrated that (1) cell injury and cell cycle arrest of A549 cells was found after the 24-hour H2O2 challenge; (2) Dex attenuated the cell injury and enhanced cell survival following the H2O2 insult; (3) the protective effect of Dex on H2O2-induced cell injury was partially dependent on α 2 adrenoceptor activation (Figure 6).
Figure 6.

Possible molecular mechanism of Dex-mediated protection against oxidative stress in A549 cell death. H2O2-induced A549 cell death, cell cycle arrest, and loss of cellular adhesion, which was attenuated by Dex via inhibiting the ROS production. ↓ enhanced; ⊥ attenuated. Cas 3: caspase 3; Cas 9: caspase 9.
One important aspect of ALI is the oxidative stress to the lungs mediated by ROS [4]. Biologically significant ROS include superoxide anion radical (O2 −), H2O2, hydroxyl radical (OH−), and hypohalous acids such as hypochlorous acid (HOCI) [17]. In this study, we used H2O2 to challenge the lung alveolar epithelial A549 cells and explored the relationship between oxidative stress and epithelial cell injury. Following the challenge of H2O2 on A549 cells for 24 hours, proapoptotic proteins such as cleaved-caspases 3 and 9 and BAX were upregulated and antiapoptotic protein Bcl-2 was downregulated; all of these results indicated that 500 μM H2O2 was sufficient to activate the cell intrinsic apoptotic pathway (Figure 1). Upstream of the caspase mediated apoptotic pathway and mitochondrial membrane potential (Δψ m) decreased and ROS generation accompanied by cytochrome C release increased following H2O2 challenge (Figures 2 and 3). Mitochondrial dysfunction and toxic substance accumulation in the cytoplasm activated caspase mediated programmed cell death [18]. The inhibitory effects of Dex on the upregulation of apoptotic proteins, downregulation of antiapoptotic proteins, and reduction of Δψ m identified its protective effects on oxidative stress induced cell death which at least in part is due to the activation of the α 2 adrenoceptor (Figure 2).
Cell cycle arrest in alveolar epithelial cells induced by H2O2 indicated that cell proliferation and further repair of the damaged epithelium were inhibited [19]. The mTOR/ERK1/2 proliferation/survival pathway is activated, which was enhanced by pretreatment with Dex in A549 cells following H2O2 challenge. This indicated that Dex may exert its potent effect on promoting the repair of the injured epithelium following oxidative stress (Figure 4). Cell surface adhesion molecule E-cadherin is linked internally with cytoskeletal components and provides linkage between the cell membranes and thus is essential for cell-to-cell attachment [20]. Dex enhanced the expression of E-cadherin during H2O2 challenge, indicating that Dex also protects the alveolar epithelial cells from damage at the cell junctions; this may be beneficial in maintaining the integrity of epithelial barrier [21] (Figure 5).
Dex is a potent α 2 adrenergic agonist and it also binds to imidazoline receptors recognizing the imidazoline or oxazoline structure of α 2 agents [22]. α 2 receptors have also been classified into three subtypes (A, B, and C) according to radioligand binding studies and their pharmacokinetic profiles [23]. The molecular mechanism of α 2 receptors is still not clear and it may be due to the activation of inhibitory G proteins, membrane-bound ion channels, and the nitric oxide-cGMP pathway [22]. In this study, we used a potent nonspecific antagonist of α 2 receptors, atipamezole, to block the activation of α 2 receptors, but it did not completely reverse the inhibitory effect of Dex on H2O2-induced A549 cell apoptosis and cell cycle arrest. These results indicated that imidazoline receptors may be involved in antiapoptotic effect of Dex, but the dose response relationship remains unknown. More studies are needed to determine whether atipamezole binds to imidazoline receptors.
We have clearly demonstrated the effects of Dex on oxidative stress induced lung cell injury and death in our study. However, there are some limitations which warrant further study: (1) using specific inhibitors to inhibit the signaling pathways could better explore the role of such pathways in the cell's fate; (2) other antioxidants could be used to compare its effects with those elicited by Dex; (3) Dex-mediated protective effect via its antioxidative effect is likely one of the mechanisms responsible for oxidative stress induced cell apoptosis. Further studies are required to characterize the mechanism, including its effect on antioxidant enzymes (SOD, GPX, and CAT). This protective effect must be validated in a large animal model and clinical trials.
Dex is a sedative used by intensive care units and anaesthetists to reduce the morbidity of patients under mechanical ventilation [24]. Some clinical trials have that indicated Dex is beneficial for patients due to the reduction in mechanical ventilation time [25–27]. Although not clearly understood, it is possible that less time weaning off the ventilator is associated with a reduced level of lung injuries [28]. However, caution must be taken. (1) This is an in vitro study, and further proper in vivo study is required. (2) The injurious insult is a “single” one rather than multiple challenges. Nevertheless, our data indicated that Dex represents a promising anaesthetic/sedative choice in protecting patients from ALI under oxidative stress insult, though this warrants further study.
In summary, this study demonstrated that Dex attenuated the H2O2-induced lung alveolar epithelial cell injury in vitro. Although further studies particularly in vivo studies followed by clinical trials are needed to further validate the protective effects of Dex on lung injury, its inhibitory effect on cell apoptosis and promotion of cell survival represent a promising anaesthetic/sedative choice in treating the patients with lung injury.
Acknowledgments
This work was supported by a grant from the Natural Science Foundation of Chongqing, China (no. cstc2013jcyjA1150). Jian Cui was supported by a scholarship of Chinese Society of Anesthesiology, Beijing, China.
Conflict of Interests
The authors declare that there is no conflict of interests regarding the publication of this paper.
Authors' Contribution
Jian Cui, Hailin Zhao, Chunyan Wang, and James J. Sun conducted the experiments and data analysis; Kaizhi Lu and Daqing Ma designed the experiment. All authors contributed to the preparation of the paper.
References
- 1.Ronchi C. F., Ferreira A. L. A., Campos F. J., et al. Interactive effects of mechanical ventilation, inhaled nitric oxide and oxidative stress in acute lung injury. Respiratory Physiology and Neurobiology. 2014;190(1):118–123. doi: 10.1016/j.resp.2013.10.008. [DOI] [PubMed] [Google Scholar]
- 2.Hammerschmidt S., Sandvoß T., Gessner C., Schauer J., Wirtz H. High in comparison with low tidal volume ventilation aggravates oxidative stress-induced lung injury. Biochimica et Biophysica Acta. 2003;1637(1):75–82. doi: 10.1016/s0925-4439(02)00216-8. [DOI] [PubMed] [Google Scholar]
- 3.Ciencewicki J., Trivedi S., Kleeberger S. R. Oxidants and the pathogenesis of lung diseases. The Journal of Allergy and Clinical Immunology. 2008;122(3):456–468. doi: 10.1016/j.jaci.2008.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ward P. A. Oxidative stress: acute and progressive lung injury. Annals of the New York Academy of Sciences. 2010;1203:53–59. doi: 10.1111/j.1749-6632.2010.05552.x. [DOI] [PubMed] [Google Scholar]
- 5.Chow C.-W., Abreu M. T. H., Suzuki T., Downey G. P. Oxidative stress and acute lung injury. The American Journal of Respiratory Cell and Molecular Biology. 2003;29(4):427–431. doi: 10.1165/rcmb.f278. [DOI] [PubMed] [Google Scholar]
- 6.Andrades M., Ritter C., de Oliveira M. R., Streck E. L., Fonseca Moreira J. C., Dal-Pizzol F. Antioxidant treatment reverses organ failure in rat model of sepsis: role of antioxidant enzymes imbalance, neutrophil infiltration, and oxidative stress. The Journal of Surgical Research. 2011;167(2):e307–e313. doi: 10.1016/j.jss.2009.08.005. [DOI] [PubMed] [Google Scholar]
- 7.Hatwalne M. S. Free radical scavengers in anaesthesiology and critical care. Indian Journal of Anaesthesia. 2012;56(3):227–233. doi: 10.4103/0019-5049.98760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bosoi C. R., Rose C. F. Oxidative stress: a systemic factor implicated in the pathogenesis of hepatic encephalopathy. Metabolic Brain Disease. 2013;28(2):175–178. doi: 10.1007/s11011-012-9351-5. [DOI] [PubMed] [Google Scholar]
- 9.Farcal L. R., Uboldi C., Mehn D., et al. Mechanisms of toxicity induced by SiO2 nanoparticles of in vitro human alveolar barrier: Effects on cytokine production, oxidative stress induction, surfactant proteins A mRNA expression and nanoparticles uptake. Nanotoxicology. 2013;7(6):1095–1110. doi: 10.3109/17435390.2012.710658. [DOI] [PubMed] [Google Scholar]
- 10.Kuwano K. Epithelial cell apoptosis and lung remodeling. Cellular & Molecular Immunology. 2007;4(6):419–429. [PubMed] [Google Scholar]
- 11.Assimakopoulos S. F., Gogos C., Labropoulou-Karatza C. Could antioxidants be the ‘magic pill’ for cirrhosis-related complications? A pathophysiological appraisal. Medical Hypotheses. 2011;77(3):419–423. doi: 10.1016/j.mehy.2011.05.034. [DOI] [PubMed] [Google Scholar]
- 12.Xia R., Yin H., Xia Z. Y., Mao Q. J., Chen G. D., Xu W. Effect of intravenous infusion of dexmedetomidine combined with inhalation of isoflurane on arterial oxygenation and intrapulmonary shunt during single-lung ventilation. Cell Biochemistry and Biophysics. 2013;67(3):1547–1550. doi: 10.1007/s12013-013-9659-8. [DOI] [PubMed] [Google Scholar]
- 13.Xiang H., Hu B., Li Z., Li J. Dexmedetomidine controls systemic cytokine levels through the cholinergic anti-inflammatory pathway. Inflammation. 2014;37(5):1763–1770. doi: 10.1007/s10753-014-9906-1. [DOI] [PubMed] [Google Scholar]
- 14.Cui J., Ou S., He W.-J., Du L., Zhao Y.-D., Ruan H.-Z. Prevention of extracellular ADP-induced ATP accumulation of the cultured rat spinal astrocytes via P2Y(1)-mediated inhibition of AMPK. Neuroscience Letters. 2011;503(3):244–249. doi: 10.1016/j.neulet.2011.08.045. [DOI] [PubMed] [Google Scholar]
- 15.Eruslanov E., Kusmartsev S. Identification of ROS using oxidized DCFDA and flow-cytometry. Methods in Molecular Biology. 2010;594:57–72. doi: 10.1007/978-1-60761-411-1_4. [DOI] [PubMed] [Google Scholar]
- 16.Yoshizawa-Sugata N., Masai H. Cell cycle synchronization and flow cytometry analysis of mammalian cells. Methods in Molecular Biology. 2014;1170:279–293. doi: 10.1007/978-1-4939-0888-2_13. [DOI] [PubMed] [Google Scholar]
- 17.Niki E. Do antioxidants impair signaling by reactive oxygen species and lipid oxidation products? FEBS Letters. 2012;586(21):3767–3770. doi: 10.1016/j.febslet.2012.09.025. [DOI] [PubMed] [Google Scholar]
- 18.Xiong S., Mu T., Wang G., Jiang X. Mitochondria-mediated apoptosis in mammals. Protein & Cell. 2014;5(10):737–749. doi: 10.1007/s13238-014-0089-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.O'Reilly M. A. DNA damage and cell cycle checkpoints in hyperoxic lung injury: braking to facilitate repair. American Journal of Physiology: Lung Cellular and Molecular Physiology. 2001;281(2):L291–L305. doi: 10.1152/ajplung.2001.281.2.L291. [DOI] [PubMed] [Google Scholar]
- 20.van Roy F., Berx G. The cell-cell adhesion molecule E-cadherin. Cellular and Molecular Life Sciences. 2008;65(23):3756–3788. doi: 10.1007/s00018-008-8281-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tunggal J. A., Helfrich I., Schmitz A., et al. E-cadherin is essential for in vivo epidermal barrier function by regulating tight junctions. The EMBO Journal. 2005;24(6):1146–1156. doi: 10.1038/sj.emboj.7600605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Khan Z. P., Ferguson C. N., Jones R. M. Alpha-2 and imidazoline receptor agonists. Their pharmacology and therapeutic role. Anaesthesia. 1999;54(2):146–165. doi: 10.1046/j.1365-2044.1999.00659.x. [DOI] [PubMed] [Google Scholar]
- 23.Daunt D. A., Hurt C., Hein L., Kallio J., Feng F., Kobilka B. K. Subtype-specific intracellular trafficking of α2-adrenergic receptors. Molecular Pharmacology. 1997;51(5):711–720. doi: 10.1124/mol.51.5.711. [DOI] [PubMed] [Google Scholar]
- 24.Wujtewicz M., Maciejewski D., Misiołek H., et al. Use of dexmedetomidine in the adult intensive care unit. Anaesthesiology Intensive Therapy. 2013;45(4):235–240. doi: 10.5603/AIT.2013.0045. [DOI] [PubMed] [Google Scholar]
- 25.Wanat M., Fitousis K., Boston F., Masud F. Comparison of dexmedetomidine versus propofol for sedation in mechanically ventilated patients after cardiovascular surgery. Methodist DeBakey Cardiovascular Journal. 2014;10(2):111–117. doi: 10.14797/mdcj-10-2-111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Keating G. M., Hoy S. M., Lyseng-Williamson K. A. Dexmedetomidine: a guide to its use for sedation in the US. Clinical Drug Investigation. 2012;32(8):561–567. doi: 10.2165/11209820. [DOI] [PubMed] [Google Scholar]
- 27.O’Mara K., Gal P., Wimmer J., et al. Dexmedetomidine versus standard therapy with fentanyl for sedation in mechanically ventilated premature neonates. The Journal of Pediatric Pharmacology and Therapeutics. 2012;17:252–262. doi: 10.5863/1551-6776-17.3.252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Reardon D. P., Anger K. E., Adams C. D., Szumita P. M. Role of dexmedetomidine in adults in the intensive care unit: an update. American Journal of Health-System Pharmacy. 2013;70(9):767–777. doi: 10.2146/ajhp120211. [DOI] [PubMed] [Google Scholar]