

Abstract
A case of idiopathic dilated cardiomyopathy (DCM) that is likely to be associated with LMNA mutation Arg190Pro in a heterozygote is described. The features of DCM in the patient were conduction disorders, cardiac arrhythmias, progressive heart failure and minor musculoskeletal disturbances. We consider that the mutation Arg190Pro contributes to the formation of a weak nuclear lamina and diminishes muscle mechanical stability which is critical during cardiac contraction. The case report illustrates in detail the phenotypic manifestations of the novel LMNA mutation and difficulties in management related to it.
INTRODUCTION
Idiopathic dilated cardiomyopathy (DCM) caused by mutations in the lamin A/C (LMNA) gene is often associated with conduction disorders, cardiac arrhythmias and extracardiac features with or without discrete muscle disruption, including skeletal muscle weakness and contractures [1, 2].
Mutations in the LMNA gene that encodes two major intermediate filament proteins, lamin A and C isoforms, cause a range of laminopathies including DCM associated with poor prognosis and a frequent occurrence of sudden cardiac death due to conduction defects and fatal ventricular tachyarrhythmia [3].
The conduction system abnormalities caused by LMNA mutations are severe and progress with age; about a half of patients require pacemaker implantation. Implantation of cardioverter-defibrillator (ICD) is considered effective for primary prevention of ventricular arrhythmias and sudden cardiac death in LMNA mutant patients [3, 4]. Cardiologists increasingly face complicated issues regarding optimal management, especially timing for preventive ICD.
We present a case of DCM probably associated with the novel LMNA mutation to assist into the diagnosis and management of these patients.
CASE REPORT
A 24-year-old female without a history of familial DCM was admitted with a progressive heart failure (HF). Clinical signs of HF (NYHA III) developed within 12 weeks of the initial development of dyspnea on both walking and bending forward, a feeling of palpitations, weakness and unexplained syncope.
On examination, the patient was asthenic. The patient was in slow atrial fibrillation (AF) with an intermittent complete right bundle branch block, incomplete left bundle branch block (alternating left fascicular posterior and left fascicular anterior block), mean heart rate of 52 b.p.m. and blood pressure 105/65 mmHg. Echocardiographic study revealed biventricular global dysfunction and dilatation: low left ventricular ejection fraction (LVEF) of 27%, hypokinesis of all segments, mean global strain (GS) LV −8.8%, LV end-diastolic volume (EDV) 171 ml, LV end-systolic volume (ESV) 124 ml, right ventricular (RV) EDV 122 ml, RV ESV 74 ml, right ventricular ejection fraction (RVEF) 39%, mean GS RV −9.8%. Left ventricular thickness of the posterior wall was at the upper limit of normal (10 mm). There were biatrial dilatation (left atrial volume 78 ml, right atrial volume 200 ml), mitral and tricuspid regurgitation (II degree MR, III degree TR) with the estimated mean pulmonary artery pressure (PAP) 18 mmHg.
Coronary angiography revealed no major abnormalities; however, cardiopulmonary exercise testing evaluated VO2 peak 15,5 ml/kg/min (normal score range 29–40 ml/kg/min considering age and sex). Blood serum creatine kinase concentration was 279 U/l (normal values 24–190 U/l) and the brain natriuretic peptide level was 676 pg/ml (normal range 0–50 pg/ml).
Neurological examination revealed mild hypotrophy of quadriceps muscles, hypertrophy of calf muscles but without reduced limb strength. Reflexes and nerve conduction studies were normal.
The 24 h ECG Holter monitoring detected a slow AF (mean HR 45 b.p.m., max HR 72 b.p.m.), intermittent complete heart block with HR 27 b.p.m., episodes of non-sustained ventricular tachycardia (n = 42), polymorphic ventricular extrasystoles (VEs >300/h). Patient's ECG Holter showed ventricular tachyarrhythmia which is presented in Fig. 1a and b.
Figure 1:
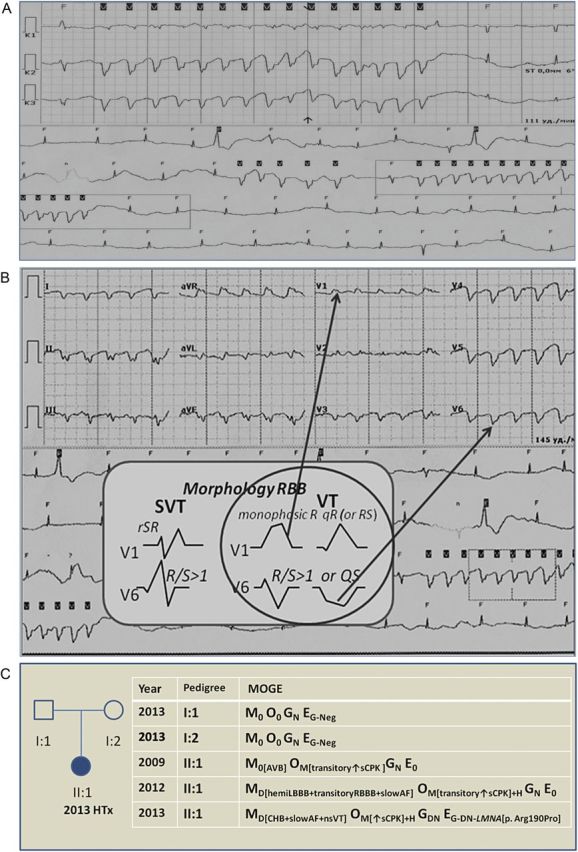
Patient's ECG HM and schematic diagram of the pedigree family. ECG HM showing slow AF, ventricular premature beats and non-sustained ventricular tachycardia (nsVT): (A) signs of monomorphic nsVT, HR 145 b.p.m.; (B) Brugada criteria show clear signs of VT in 12 lead ECG to modification EASY; (C) schematic diagram of the family illustrates evolution disease of the patient (2009–2013) and MOGE system with comprehensive summary of the clinical and genetic status of the pedigree. squares indicate male and circles female, individual affected is shown with solid symbol, unaffected with white symbols; HTx, heart transplantation; AVB, atrioventricular block; AF, atrial fibrillation; sCPK, serum creatine kinase; CHB, complete heart block.
The patient underwent implantation of a biventricular pacemaker with cardioverter-defibrillator (CRT-D) as a preventive measure prior to confirmation of the genetic lesion. After CRT-D implantation, the patient was treated with beta-blocker, ACE inhibitor, aldosterone antagonist and diuretic (daily doses: bisoprolol 3,75 mg; ramipril 5 mg; spironolactone 50 mg; torasemide 5–10 mg). After 6 months, the patient showed no evidence of cardiac remodeling (LVESV increased up to 140 ml, LVEDV up to 180 ml, LVEF decreased up to 22%, mean LV GS up to −5.2%; RVESV enlarged up to 93 ml, RVEDV up to 131 ml, RV EF reduced up to 29%, RV GS up to −4.9%). Owing to progression of HF symptoms (development of ascites, hydropericardium and pulmonary hypertension to PAP mean 39 mmHg), a successful orthotropic cardiac transplantation was performed.
In the removed heart, the findings were non-specific with diffuse alteration in myocytes including variation in size, nuclear variation, and interstitial fibrosis.
DNA mutations were detected using a single-stranded conformational polymorphism method and direct sequencing of the amplified LMNA exons. We detected the mutation c.569G>C in the third exon of the gene.
DISCUSSION
A laminopathy was suspected in a patient with DCM and conduction defect (intermittent complete heart block), cardiac arrhythmias (slow AF, ventricular tachyarrhythmias), and skeletal muscle abnormality with an increased creatine kinase level. This diagnosis was confirmed by genetic testing with the analysis of the LMNA gene. We detected a novel mutation c.569G>C in the third exon of the gene which substituted arginine residue by the proline at codon 190 – p.Arg190Pro. The patient was a heterozygous carrier of this mutation. We consider that the nucleotide replacement c.569G>C (rs267607571) of the patient emerged de novo, since this mutation was not present in her parents, who did not have cardiovascular disease nor skeletal muscle involvement. The clinical and genetic status of this family is presented in Fig. 1c as the schematic diagram of the MOGE(S) system classification [5].
The codon 190, located in the rod-domain of lamin A/C, is the mutational hot spot. Several mutations were described in this position previously—p.Arg190Gln, p.Arg190Trp, p.Arg190fsX22, enclosed in the UND-LMNA mutation database [6]. Most cases were associated with DCM with conduction disease.
We think that the molecular basis of the disease is the breakdown of lamin A/C dimerization. Like all intermediate filaments, this protein has a strong tendency to formation of alpha-helical coiled coil (CC) by their rod-domains [7]. The proline intercalation, known as ‘helix breaker’, in codon 190 would prevent the CC formation (confirmed by Jpred3 and NetSurfP secondary structure prediction algorithms [8, 9]) and thus abolish the assembly of normal nuclear lamina. We hypothesize that such molecular changes in the lamin A/C structure can diminish muscle mechanical stability which is critical during muscle contraction.
Although it is not possible to exclude other genomic changes in our patient, it would be appropriate to consider that the disruption of the nuclear architecture secondary to the p.Arg190Pro mutation could lead to DCM.
REFERENCES
- 1.van Berlo JH, de Voogt WG, van der Kooi AJ, van Tintelen JP, Bonne G, Yaou RB, et al. Meta-analysis of clinical characteristics of 299 carriers of LMNA gene mutations: do lamin A/C mutations portend a high risk of sudden death? J Mol Med. 2005;83:79–83. doi: 10.1007/s00109-004-0589-1. [DOI] [PubMed] [Google Scholar]
- 2.Arbustini E, Pilotto A, Repetto A, Grasso M, Negri A, Diegoli M, et al. Autosomal dominant dilated cardiomyopathy with atrioventricular block: a lamin A/C defect-related disease. J Am Coll Cardiol. 2002;39:981–90. doi: 10.1016/s0735-1097(02)01724-2. [DOI] [PubMed] [Google Scholar]
- 3.Charron P, Arad M, Arbustini E, Basso C, Bilinska Z, Elliott P, et al. Genetic counselling and testing in cardiomyopathies: a position statement of the European Society of Cardiology Working Group on Myocardial and Pericardial Diseases. Eur Heart J. 2010;31:2715–26. doi: 10.1093/eurheartj/ehq271. [DOI] [PubMed] [Google Scholar]
- 4.Charron Ph, Arbustini E, Bonne G. What should the cardiologist know about lamin disease? Arrhythm Electrophysiol Rev. 2012;1:22–8. doi: 10.15420/aer.2012.1.22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Arbustini E, Narula N, Dec GW, Reddy KS, Greenberg B, Kushwaha S, et al. The MOGE(S) classification for a phenotype–genotype nomenclature of cardiomyopathy. J Am Coll Cardiol. 2013;62:2046–72. doi: 10.1016/j.jacc.2013.08.1644. [DOI] [PubMed] [Google Scholar]
- 6.UND-LMNA mutation database. http://www.umd.be/LMNA/ (5 February 2014, date last accessed)
- 7.Parry DA. Coiled coils in alpha-helix-containin proteins: analysis of the residue types within the heptad repeat and the use of these data in the prediction of coiled coils in other protein. Biosci Rep. 1982;2:1017–24. doi: 10.1007/BF01122170. [DOI] [PubMed] [Google Scholar]
- 8.Cole C, Barber JD, Barton GJ. The Jpred 3 secondary structure prediction server. Nucleic Acids Res. 2008;36:W197–201. doi: 10.1093/nar/gkn238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Petersen B, Petersen TN, Andersen P, Nielsen M, Lundegaard C. A generic method for assignment of reliability scores applied to solvent accessibility predictions. BMC Struc Biol. 2009;9:51. doi: 10.1186/1472-6807-9-51. [DOI] [PMC free article] [PubMed] [Google Scholar]