

Abstract
Electron cryo-microscopy has become a routine technique to determine structure of biochemically purified herpes simplex virus capsid particles. This chapter describes the procedures of specimen preparation by cryopreservation; low dose and low temperature imaging in an electron cryo-microscope; and data processing for reconstruction. This methodology has yielded subnanometer resolution structures of the icosahedral capsid shell where alpha helices and beta sheets of individual subunits can be recognized. A relaxation of the symmetry in the reconstruction steps allows us to resolve the DNA packaging protein located at one of the 12 vertices in the capsid.
Keywords: Herpes Simplex Virus, Virus Structure, Cryo-EM, Cryo-ET, Electron Cryo-microscopy, Frozen Hydrated, Vitrification, Symmetry and Symmetry-Free Reconstruction, Sub-Tomogram Averaging
1. Introduction
In this chapter we will discuss the preparation of frozen, hydrated herpes simplex virus type I (HSV-1) embedded in vitreous ice. This technique, which is typical of electron cryo-microscopy (cryo-EM), relies on achieving a frozen hydrated state, which preserves the sample in its solution environment. This sample preparation and imaging modality has been used to study the 3-dimensional (3D) structure of HSV-1 in great detail, resolving the organization of the infectious virion (1) (Figure 1A) and biochemically isolated capsid shell (2) (Figure 1B) as well as the structure of the machinery necessary for loading dsDNA into the virus (i.e. the portal) (3) (Figure 1C). Electron Cryo-Tomography (cryo-ET) has also been used to image infectious HSV-1 virions, revealing an unexpected tail-like feature in the pleomorphic tegument layer associated with the portal complex (4) (Figure 1D). The methods discussed within, while specifically tailored to HSV-1, are generally applicable to other specimens and require only moderate modifications to optimize for other biological systems.
Figure 1.
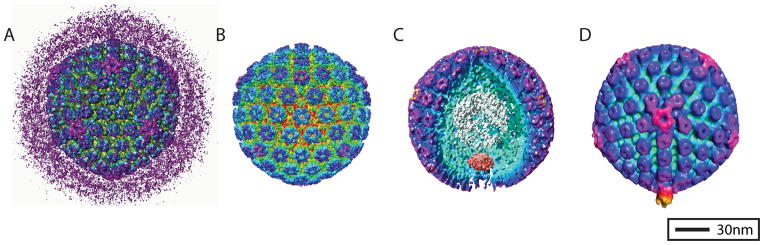
HSV-1 3D reconstructions. Each of these four reconstructions is radially colored to show surface features of the virus. One of the first icosahedral 3D reconstructions of HSV-1 was of the whole infectious virion (A) (1). The pleomorphic layer of tegument proteins that surround the HSV-1 capsid in the virion are readily visualized in the reconstruction. The first subnanometer resolution reconstruction of HSV-1 was of purified B-capsids (B) (2). The first asymmetric reconstruction of HSV-1 was of purified B-capsids (C) [EMD-5259](3). A portion of the capsid shell has been resected from the reconstruction to show the portal complex sitting beneath a unique vertex of the capsid shell. The first sub-tomogram average from cryo-ET of HSV-1 virions revealed a novel organization of protein in the pleomorphic tegument layer that resembled the tail of bacteriophage (D) [EMD-5453] (4).
2. Materials
There are many materials are commonly used for the purposes of cryo-EM and cryo-ET of icosahedral viruses. The items listed in the subsequent sections are available from many vendors and are not generally specialized to the field of cryo-EM.
2.1 Herpes Simplex Virus type I
Depending upon the focus of the study, the investigator must decide whether to purify capsids, whole virions, or work with unpurified sample. The specifics of virus purification are not detailed within this chapter, and have been well reviewed in the past (5). The preferred concentration of the virus sample is ~1010 particles/ml which enhances the data collection efficiency. While it is not a requirement to have a 99% pure particle sample, the presence of heterogeneity would increase the computational complexity in deriving the final 3D structure.
2.3 Electron Microscopy Grids
Electron microscopy grids, which are 3.05 mm in diameter, are the medium on which a sample is frozen prior to imaging in an electron cryo-microscope (Figure 2). The EM grid contains a metal meshwork on which a thin holey carbon film (also known as a support film) is placed. This film acts as a substrate for the sample and contains holes, in which the ice-embedded virus particles are suspended in random orientations. The physical characteristics of a grid (i.e. mesh size, hole size, support material) are typically optimized on a per-project basis. The mesh size is the number of bars per inch, and the hole size is the diameter of each hole in the support film (typically in μm). Grids are also available in a variety of materials (e.g. copper, molybdenum, gold, etc.), the choice of which material to choose depends on the project. Copper is most often the choice for single-particle cryo-EM sample preparation due to a lower cost per-grid. However, copper grids can be toxic to some specimens and do not permit cell cultures, necessitating alternative grid materials (e.g. gold) for these specific applications.
Figure 2.

Grid orientation for sample application. The two sides of Quantifoil™ TEM microscopy grids have a different luster, the side that glares in light is purely copper and the matte side is carbon coated. It is important that the sample is applied to the carbon side as it is more hydrophilic.
2.3 Sample Vitrification Apparatus
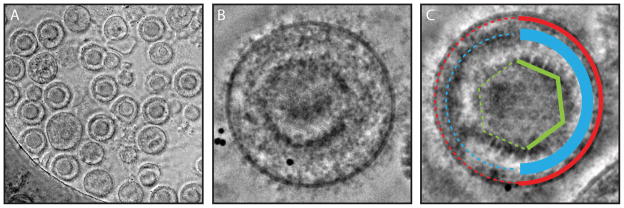
One of the most important steps in cryo-EM is vitrification of the sample via plunge freezing. The most common method for sample vitrification requires freezing the sample in a thin layer of ice on an electron microscopy grid (6, 7). Vitrification is a process whereby a sample is frozen at a rapid rate such that crystalline ice does not form. Typically, when frozen at a slow rate, water will form a hexagonal or cubic lattice, thus the sample may be damaged and not fully embedded in the ice matrix causing loss in the sample’s structural integrity (Figure 3). Since the formation of non-vitreous ice can damage the ultrastructural elements of macromolecules, samples preserved by vitrification, make it possible to resolve near-atomic features in the resulting 3D density map (for examples: (8–14)).
Figure 3.

Ice contamination. When the sample in suspension on a grid is not fully vitrified during freezing it is possible to see other forms of ice. Hexagonal ice formation will look like striations in the image (A) and a Fourier transform of the image will contain discrete ice peaks (B). Cubic ice formation will look like nodules in the image (C) and a Fourier transform of the image will contain an “ice ring” in the Fourier transform at a specific spatial frequency (D). N.B. The virus sample shown in panels A and C are of bacteriophage epsilon15 not HSV-1.
There are several commercially available semi-automated freezing devices from a variety of vendors; Vitrobot (FEI), EM-GP™ (Leica), Cp3™ (Gatan). The primary advantage of these devices is that by automating many of the steps of the grid freezing process there is a high level of consistency in the frozen grids they produce. Many features like mechanical blotting and a humidified sample chamber make it possible to tightly control the ice thickness. Furthermore, the grid-to-grid reproducibility achieved by the semi-automatic plunge freezing devices streamlines the process of optimizing grid-freezing conditions for each specimen.
2.4 Electron Cryo-microscope
The electron cryo-microscope is an extremely specialized tool that is available from just a few vendors worldwide, with most installations either from JEOL (www.jeol.co.jp/en/) or FEI (www.fei.com/). Apart from the specific companies that supply these pieces of equipment, there are many basic concepts that define their applicability for resolving the structure of viruses. Select features of these devices are outlined below.
Electron microscopes are typically defined by the accelerating voltage of the electron. It typically ranges from 80 to 300kV. Higher voltage microscopes are particularly useful for studying thicker specimens like the HSV-1 virion because of its higher penetrating power (15).
The two most common electron sources are filament (e.g LaB6 or Tungsten) and field emission gun (FEG). FEGs have the advantage of improved spatial and temporal coherence to preserve higher resolution signal even with highly defocused images. Nevertheless, filament electron guns are still used for high-resolution data collection and are a fraction of the cost of an FEG.
The specimen cryo-holder is what electron microscopy grids are loaded onto at liquid nitrogen temperature before being inserted into the microscope. To keep the specimen in a frozen-hydrated state while in the microscope, the cryo-specimen holder must be maintained close to or below liquid nitrogen temperature (−170 C), ensuring that the ice remains vitreous (7), and minimizes the damage from the electron beam (16).
An energy filter is an optional component of an electron microscope that enhances contrast in the recorded images (17, 18). Energy filters serve to reduce the background noise of images by reducing the number of inelastically scattered electrons that reach the detector, thus improving contrast.
A Zernike phase plate is a device placed in the focal plane of the objective lens of an electron microscope (19) that increases image contrast by selectively phase shifting only the electrons scattered by the sample (Figure 4). Phase plate technology has slowly made a niche in cryo-EM for visualizing low contrast macromolecular complexes (Figure 5)(3, 19, 20) and cells (21).
Figure 4.

Zernike phase contrast imaging. HSV capsids imaged under conventional cryo-EM conditions are characteristically low contrast (A). The same sample when imaged with Zernike phase contrast cryo-EM has markedly increased contrast (B).
Figure 5.
Zernike phase plate image of HSV-1 virions. Heterogeneity of the virion sample is visualized in an individual image (A). The numerous components of the virion (e.g. Red – Envelope, Blue – Tegument, Green - Capsid) are revealed in the high contrast phase plate image (B, C).
2.5 Electron Detection Device
The manner in which images from an electron microscope are recorded has a direct impact on throughput and quality of the resulting 3D reconstructions. The three most common media for electron detection are photographic film, charged coupled devices (CCDs), and direct detection devices (DDDs). The biggest differences between these three media is that CCDs and DDDs provide digital images that can be processed in real time, whereas film must be removed from the microscope, developed, and scanned before the images can be processed computationally. It is clear that the DDD is the superior recording media, based largely on the fact that the Detective Quantum Efficiency (DQE) for these devices is dramatically higher than for other media (22–24). Another advantage of these detectors is their high acquisition frame-rates (>30 frames per second), making it possible to align individual frames prior to integrating them into a single image, thus minimizing any specimen or stage movements (i.e. drift) that can occur during imaging.
2.6 Electron Micrograph Repository
A single imaging session on the microscope can contain as many as 500 digital images, and a single near atomic-resolution project can take a few days to weeks to complete data collection, depending on the quality of the specimen grids and particle concentration. This equates to terabytes of data per project, depending upon the detector used. Regardless of what detector is used to acquire these, the electron micrographs should be stored in a protected environment. Databases are available to store these images as well as archiving pertinent image metadata (i.e. sample preparation information, freezing conditions, lens parameters, etc.)(25, 26).
2.7 High-end Workstation
A final piece of hardware for 3D virus image reconstruction is a high-end graphics workstation to process, visualize, and manipulate the electron density volume. The visualization software packages mentioned in this review (e.g. Chimera (27), EMAN2 (28), Amira) have been developed to work with many different operating systems.
3. Methods
Every aspect of sample preparation and image acquisition can have a dramatic impact on the quality of the resulting 3D reconstruction. As data is pooled across multiple grid preparations and imaging sessions prior to data processing, variability in how each of the following steps is performed can affect these results.
3.1 Cryo-EM Virus Sample Preparation
For single particle virus reconstruction, it is highly desirable to have highly purified virus sample and a high enough concentration to permit sufficient particle numbers for each image frame. Ideally there should be enough particle separation to ensure that there are no overlapping particles in the projection images characteristic of transmission electron microscopy.
After purification of HSV-1 (Section 2.1) the virus pellet is stored in a buffer solution to keep it stable. For cryo-EM it is advisable to avoid detergents and other chemicals such as DMSO, sugars, glycerol, etc. Additionally, it is important to consider the temperature dependence of pH in these buffer solutions. As such, phosphate buffered solution is an ideal buffer solution as it has a relatively small change in pH across the nearly 200°C change in temperature experienced during freezing (29).
Depending upon the sample, purification typically yields far more sample than is needed for freezing. As only 2–3μL of sample is applied to each grid before freezing, it is likely that only a small portion of the purified virus will be used for a given reconstruction. Preservation of the purified sample can either be short- or long-term. For HSV-1, capsids can be stored at either 4°C or −81°C however the mature virion should always be stored at 4°C. It is important to note that immediately prior to freezing the sample on grids, the virus should be thawed on ice.
3.2 Grid Preparation
Once the desired grid material, hole size, and hole distribution has been chosen, the grids must be washed to remove any surface contamination prior to being used.
Cleaning grids: Remove grids from the storage box they are shipped in and place them one-by-one into a small vial of acetone, making sure they are submerged. Gently shake the vial periodically to make sure that grids do not stick to each other. Allow grids to soak in acetone for up to 24 hours, making sure that the acetone does not completely evaporate.
Washing grids: After the grids have soaked in acetone, wash the grids while they are still in the vial using micron-filtered distilled water. As you fill the vial with water, the grids will remain at the bottom of the vial, pour off the top layer of water and repeat at least twice.
Drying grids: Place two pieces of filter paper in the bottom of a petri dish and pour the contents of the vial onto the filter paper, washing the vial with micron-filtered distilled water to ensure all grids are out of the vial. Using tweezers, carefully pick up each grid and place them onto a separate dry piece of filter paper. Place the dry filter paper with the grids on it in a clean petri dish, cover the petri dish, and place under an incandescent lamp to dry.
3.3 Sample Vitrification
When freezing a sample it is important to plasma clean or glow discharge the grid prior to the application of the sample, ensuring that the sample adheres well to the grid. The process of freezing a grid is broken into three steps; sample application, sample blotting (one or two sided), and plunge freezing (videos of this process, specific to each device, are available from individual vendors). Please note that BSL-2 precautions should be taken while handling infectious HSV-1 virions.
3.4 Grid Transfer to Microscope
The process of transferring a frozen hydrated sample grid from storage into the electron microscope puts the sample at risk for contamination due to extreme differences in temperature between the sample and environment. Furthermore, any moderate change in grid temperature can cause regions of the frozen hydrated sample to undergo phase transitions from vitreous to crystalline ice thus damaging the specimen. Grid transfer stations are designed to facilitate specimen transfer from the grid storage button to the sample cryo-holder in a cryo-protected environment. These transfer stations are typically designed to accommodate a sample cryo-holder specific to the cryo-microscope.
3.5 Microscope Alignment
Beyond good sample preparation technique, accurate microscope alignment is a necessity for high-resolution cryo-EM. The alignment procedure includes aligning the gun, the beam tilt, the condenser and objective lens, the apertures, the specimen height and the astigmatism of the objective lens. The process of microscope alignment is performed on a top down basis, starting with the electron source and moving down the microscope column. High-resolution alignment of the electron microscope is best performed by an individual specifically trained for the microscope, however new automation software in some contemporary microscopes is making accurate alignment easier for the general user.
3.6 Data Collection for Single Particle Reconstruction
There are many factors that must be considered before embarking on imaging a virus for the purposes of high-resolution reconstruction. First and foremost is the structural question at hand and what resolution you need to address that question. While resolution may seem like a linear goal, the amount of data required to achieve a specific resolution is inversely proportional to the resolution desired (30). Accordingly, one should target their reconstruction to the resolution they desire, 20 Å for capsomere morphology, 12 Å for individual protein domains or components, 8 Å for long α-helices and large β-sheets, 4.5 Å for short α-helices and β-sheets and possibly a C-alpha backbone trace, and <4.0 Å for individual bulky side chains.
Dose optimization: Total accumulated electron dose is important because it is directly related to the signal-to-noise ratio (SNR) of the electron micrographs. The higher the SNR, the easier it is to accurately align and reconstruct a 3D volume from the raw data. Increasing dose has the cost of damaging the specimen, and more than 25 e−/Å2 has been shown to ablate high-resolution features for cryo-EM data (16). For single particle data collection of HSV-1 capsids and virions, the dose for a single image is typically set between 20 and 25 e−/Å2, enough to get high-resolution signal, but not enough to significantly damage the specimen.
Magnification selection: The magnification at which images are acquired should be chosen so that the target resolution is approximately 4 times the sampling rate. This process should also take into consideration the native pixel size and noise profile of the detector (22). For example, if an 8 Å resolution reconstruction is desired, the magnification used should correspond to a sampling of approximately 2 Å/pixel in the micrograph. Care must be taken when choosing magnification, as choosing a magnification higher than required, will unnecessarily increase the time need to obtain the reconstruction at the target resolution.
Defocus selection: Images are often defocused to increase contrast, however this method has the downside of reducing the high-resolution signals in an image (18). For HSV-1, data collection with a defocus range of 0.5 to 2.5 μm with 300 kV electrons is optimal for initial model generation and high-resolution 3D reconstruction.
Image acquisition: When recording images of frozen hydrated particles, it is important to find optimal areas for data collection (i.e. good particle concentration and thin vitreous ice). As a single grid can have thousands of potential areas for imaging it is important to search the grid without damaging the specimen with a low electron dose rate. Some microscopes are equipped with software which automates this complicated process to minimize beam damage (31–33).
3.7 Data Storage
Digital detection devices output standardized files for every image recorded by the device. For all digital images, it is imperative they are stored with multiple redundant backups to ensure that work is not lost due to hardware failures. EMEN2 is one database software that is publicly available for data archiving and dissemination (25, 26).
3.8 Data Pre-processing
Before single-particle images can be reconstructed into a 3D volume, the individual images must be preprocessed. Numerous programs are available to facilitate and automate these processes (28, 34) which have been well reviewed recently and provide a comprehensive background of both theory and application for virus reconstruction (35).
Screening for optimal images: Data acquisition for a single structural project can consist of hundreds if not thousands of cryo-EM images. Prior to processing this data for a reconstruction, it is important to exclude any images that may contain aberrations (e.g drift, vibration, astigmatism). As not every aberration is readily apparent from visual inspection of the raw image, it is important to inspect the power spectrum (i.e. Fourier Transform) of the particle images where these aberrations can be readily identified.
Boxing particles: The images that are aligned and reconstructed into a 3D density map are of single particles obtained from boxing electron micrographs. EMAN and EMAN2 are two such programs that are available to facilitate this process (28, 34). One of the advantages of cryo-EM is the ability to visualize and selectively process heterogeneous samples. It is important that when boxing particles from images, every effort is taken to selectively box particles of the same type. In the case of HSV-1, a biochemically purified capsid sample can still contain A-, B-, and C-capsids (Figure 6), however through the process of boxing, these particles can be digitally and visually separated prior to processing.
Image quality assessment: Images collected on an electron microscope are modulated by the contrast transfer function (CTF)(36, 37). The CTF pattern is indicative of the image quality and can be used to screen out poor quality images. Determining the parameters of the CTF is necessary for proper image reconstruction and is a well-established procedure in the cryo-EM field (18). The CTF for a set of particles can be automatically fitted using either EMAN or EMAN2 (28, 34).
Figure 6.

Zernike phase plate image of HSV-1 capsids. Three distinct capsids are produced during HSV-1 replication and numerous capsids can be seen in high magnification images (A). A-capsids are empty shells (B), B-capsids contain condensed scaffolding proteins (C), and C-capsids contain dsDNA which appears as a whorl like pattern within the capsid (D).
3.9 Image Reconstruction
The process of generating a 3D volume from a collection of 2D projection images is a computationally intensive process of particle alignment, CTF correction, refinement, and reconstruction. The complexity of the reconstruction depends on the assumed symmetry of the particles and the target resolution. Generally speaking, near-atomic resolution reconstructions can require hundreds of thousands of CPU hours to complete in some software (38). There are numerous software packages (e.g. SPIDER(39), EMAN (34), IMIRIS (40), EMAN2 (28), MPSA (41), FREALIGN (42), and jalign (43)) that are available to process this data, and their application for processing viruses has been well reviewed previously (35).
3.10 Single Particle Icosahedral Reconstruction
Icosahedral reconstructions assume a symmetrical arrangement of capsid proteins with 60 asymmetric units related to each other by 5, 3 and 2 fold symmetry operations. Each asymmetric unit is composed of multiple proteins (e.g. 16 VP5, 15 VP26, 10–11 VP23, 5–6 VP19C in HSV-1 capsid). Not all of the structural proteins in the HSV-1 virion are organized with icosahedral symmetry whereas the capsid proteins are largely icosahedrally arranged.
Initial model generation: The first step in a 3D reconstruction is generating an initial model from scratch to avoid model bias in the final reconstruction. The initial model should be produced from the raw data itself or just a smooth sphere to avoid any model bias in the refinement process.
Particle alignment: The specific principles behind single particle alignment have been thoroughly detailed previously (35). The two most common approaches for single particle alignment are projection matching and cross common-line correlation, both of which have been used for near-atomic resolution reconstructions (8–14). As the name suggests, projection matching processes individual single particle images and builds class averages from similarly oriented particles. Cross common-lines based particle alignment takes advantage of internal icosahedral symmetry within the particle to align individual images to a model (41, 44, 45). In either case, a model generated from a few particle images or a randomly generated sphere is used as an initial model for the particle alignment refinement. Projection matching is more computationally intensive than cross common-lines, and is independent of particle symmetry. Since HSV-1 capsid is a large icosahedral virus (60-fold symmetric), the cross common-lines approach for particle alignment is computationally efficient. During the particle refinement step, the images will be corrected for the CTF effects. There are many reconstruction software packages which can carry out all these steps such as SPIDER(39), EMAN (34), IMIRIS (40), EMAN2 (28), MPSA (41), FREALIGN (42), and jalign (43).
3D reconstruction: A 3D density map is computed from the aligned and CTF corrected particle images. Single particle reconstruction software packages(e.g. SPIDER(39), EMAN (34), IMIRIS (40), EMAN2 (28), MPSA (41), FREALIGN (42), and jalign (43)) each have their own implementation of algorithms for model generation and the implementation of these methods are described within the tutorials for each of the software packages..
3.11 Single Particle Reconstruction of Asymmetric Vertex Specific Proteins
Symmetry-free reconstruction is similar to section 3.10, with the exception that no assumptions are made about the arrangement of asymmetric units (i.e. capsid proteins) with respect to each other in the final reconstruction though the initial particle orientation is determined with the icosahedral particle alignment step as described above. These reconstructions are technically more challenging, but allow for the visualization of unique features in the capsid shell that are not icosahedrally present in all asymmetric units (i.e. only at one of the twelve vertices) (3, 4). The manner in which these two reconstruction techniques differ is described below.
Initial model generation: Identical to icosahedral reconstruction to find the particle orientation.
Symmetry-free particle alignment: When searching for a unique vertex in the icosahedral capsid, one must differentiate one of the 12 vertices from the other 11. Once this vertex has been identified, determining the correct rotational orientation of the protein component in this pseudo 5-fold environment must then be made (3, 4, 46).
3D reconstruction: Once the symmetry-free orientations have been determined, the model is reconstructed without imposing icosahedral symmetry on the final reconstruction.
3.12 3D Reconstruction Deposition
Once a 3D reconstruction of a virus has been produced, investigators are encouraged to submit the 3D model to the Electron Microscopy Data Base (http://www.emdatabank.org/). Once the manuscript pertaining to the 3D reconstruction has been published the model can be made available for dissemination and referenced through the unique EMD identifier (i.e. EMD-5259).
3.13 Data Interpretation
Once a 3D volume has been generated, the final step is analysis of the map. A key element to data interpretation is knowledge of the limits of map visualization and interpretability. In general, one of the best metrics by which a 3D model can be evaluated is the computed resolution of the map (as described below). To date, the highest resolution published structure of HSV-1 is of the B-capsid to 8.5 Å (2). A subnanometer resolution map of the capsid is shown in Figure 1B. At this resolution, it was possible to identify numerous secondary structural features in the major capsid protein, VP5, and its upper domain features match well with the crystal structure (47). The first symmetry free reconstruction of HSV-1 was at 24 Å and revealed the dsDNA packaging protein situated beneath one of the icosahedral vertices (Figure 1C)(3). A symmetry-free reconstruction of intact virions demonstrated novel portal vertex associated tegument extending from the portal vertex outwards from the capsid shell (Figure 1D)(4).
3D Density Visualization: Volume rendering of large macromolecular complexes is typically done with a handful of tools. There are many freely available software packages to facilitate this process, Chimera (27), Coot (48), and Pymol (http://www.pymol.org), in addition to commercially licensed solutions such as Amira (http://www.amiravis.com) and Avizo (http://www.vsg3d.com/avizo/).
Resolution determination: There are a few ways in which the resolution of a 3D density map can be evaluated. The first is visual inspection of the reconstruction for the appearance of long rod shaped densities (α-helices), which is an indication that the resolution of the map is subnanometer (i.e. < 10 Å). A more accurate quantitative method is the calculation of resolution from the Fourier Shell Correlation (FSC) (49, 50) between two independently determined density maps of the same sample from two data sets. A 0.143 criterion in the FSC plot can be used to estimate the resolution when the two independent data sets are combined (8, 51–53).
Map Segmentation: Interpreting the structure derived from a 3D reconstruction is a time consuming process that requires the macromolecular complex to be broken down into more manageable pieces. Understanding how these components fit together for the entirety of the structure is at the heart of understanding the macromolecular assembly. Depending upon the complexity of the data, this process can be very time consuming, so programs have been developed to automate certain aspects of this process (54–56).
For HSV-1, the capsid is comprised of over a 1000 copies of four viral proteins, VP5, VP19c, VP23, and VP26. Since little secondary structural information is known about the proteins that comprise the capsid shell, accurately segmenting out these components can be difficult and relies on high-resolution maps where the boundaries between protein subunits are clearly delineated.
3.14 Conclusion
Cryo-EM is unique in the fact that it allows for the direct visualization of virus structures in their hydrated state without the need for heavy metal stains. The success of cryo-EM for resolving the structure of HSV-1 capsid associated components is seen in the 3D reconstructions of the capsid and virion. Icosahedral reconstructions have been a mainstay in the cryo-EM field for the past 40 years, enabling reconstructions to reach the point of near-atomic resolutions such that individual amino acid side-chains can be visualized in the resulting 3D models. Recently, asymmetric cryo-EM reconstructions have for the first time identified the machinery responsible for packaging the HSV-1 genome in the context of the capsid shell (3). Beyond the single particle approach, advances have been made in cryo-ET, which have allowed for the identification of a novel tail-like capsid associated structure from HSV-1 virions (4, 57). These complementary imaging modalities have paved the way for a structural approach to gain insight into the life cycle of HSV-1.
Acknowledgments
This research was supported by grants from Robert Welch Foundation (Q1242) and National Institutes of Health (P41GM103832 and R56AI075208 to W.C.; T15LM007093 through the Gulf Coast Consortia and T32GM007330 through the MSTP to R.H.R.).
References
- 1.Zhou ZH, Chen DH, Jakana J, Rixon FJ, Chiu W. Visualization of tegument-capsid interactions and DNA in intact herpes simplex virus type 1 virions. J Virol. 1999;73:3210–3218. doi: 10.1128/jvi.73.4.3210-3218.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Zhou ZH, Dougherty M, Jakana J, He J, Rixon FJ, Chiu W. Seeing the herpesvirus capsid at 8.5 A. Science. 2000;288:877–880. doi: 10.1126/science.288.5467.877. [DOI] [PubMed] [Google Scholar]
- 3.Rochat RH, Liu X, Murata K, Nagayama K, Rixon FJ, Chiu W. Seeing the portal in herpes simplex virus type 1 B capsids. J Virol. 2011;85:1871–1874. doi: 10.1128/JVI.01663-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Schmid MF, Hecksel CW, Rochat RH, Bhella D, Chiu W, Rixon FJ. A tail-like assembly at the portal vertex in intact herpes simplex type-1 virions. PLoS Pathog. 2012;8:e1002961. doi: 10.1371/journal.ppat.1002961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Roberts AP, Abaitua F, O’Hare P, McNab D, Rixon FJ, Pasdeloup D. Differing roles of inner tegument proteins pUL36 and pUL37 during entry of herpes simplex virus type 1. J Virol. 2009;83:105–116. doi: 10.1128/JVI.01032-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Adrian M, Dubochet J, Lepault J, McDowall AW. Cryo-electron microscopy of viruses. Nature. 1984;308:32–36. doi: 10.1038/308032a0. [DOI] [PubMed] [Google Scholar]
- 7.Dubochet J, Adrian M, Chang JJ, Homo JC, Lepault J, McDowall AW, Schultz P. Cryo-electron microscopy of vitrified specimens. Q Rev Biophys. 1988;21:129–228. doi: 10.1017/s0033583500004297. [DOI] [PubMed] [Google Scholar]
- 8.Baker ML, Hryc CF, Zhang Q, Wu W, Jakana J, Haase-Pettingell C, Afonine PV, Adams PD, King JA, Jiang W, Chiu W. Validated near-atomic resolution structure of bacteriophage epsilon15 derived from cryo-EM and modeling. Proc Natl Acad Sci U S A. 2013;110:12301–12306. doi: 10.1073/pnas.1309947110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jiang W, Baker ML, Jakana J, Weigele PR, King J, Chiu W. Backbone structure of the infectious epsilon15 virus capsid revealed by electron cryomicroscopy. Nature. 2008;451:1130–1134. doi: 10.1038/nature06665. [DOI] [PubMed] [Google Scholar]
- 10.Ludtke SJ, Baker ML, Chen DH, Song JL, Chuang DT, Chiu W. De Novo backbone trace of GroEL from single particle electron cryomicroscopy. Structure. 2008;16:441–448. doi: 10.1016/j.str.2008.02.007. [DOI] [PubMed] [Google Scholar]
- 11.Yu X, Jin L, Zhou ZH. 3.88 A structure of cytoplasmic polyhedrosis virus by cryo-electron microscopy. Nature. 2008;453:415–419. doi: 10.1038/nature06893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhang J, Baker ML, Schroder GF, Douglas NR, Reissmann S, Jakana J, Dougherty M, Fu CJ, Levitt M, Ludtke SJ, Frydman J, Chiu W. Mechanism of folding chamber closure in a group II chaperonin. Nature. 2010;463:379–383. doi: 10.1038/nature08701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhang J, Ma B, DiMaio F, Douglas NR, Joachimiak LA, Baker D, Frydman J, Levitt M, Chiu W. Cryo-EM structure of a group II chaperonin in the prehydrolysis ATP-bound state leading to lid closure. Structure. 2011;19:633–639. doi: 10.1016/j.str.2011.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhang X, Settembre E, Xu C, Dormitzer PR, Bellamy R, Harrison SC, Grigorieff N. Near-atomic resolution using electron cryomicroscopy and single-particle reconstruction. Proc Natl Acad Sci U S A. 2008;105:1867–1872. doi: 10.1073/pnas.0711623105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhou ZH, Chiu W. Determination of icosahedral virus structures by electron cryomicroscopy at subnanometer resolution. Adv Protein Chem. 2003;64:93–124. doi: 10.1016/s0065-3233(03)01003-9. [DOI] [PubMed] [Google Scholar]
- 16.Bammes BE, Jakana J, Schmid MF, Chiu W. Radiation damage effects at four specimen temperatures from 4 to 100 K. J Struct Biol. 2010;169:331–341. doi: 10.1016/j.jsb.2009.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Langmore JP, Smith MF. Quantitative energy-filtered electron microscopy of biological molecules in ice. Ultramicroscopy. 1992;46:349–373. doi: 10.1016/0304-3991(92)90024-e. [DOI] [PubMed] [Google Scholar]
- 18.Rochat RH, Chiu W. In: Comprehensive Biophysics, (2012) Egelman EH, editor. Vol. 1. Academic Press; Oxford: pp. 311–340. [Google Scholar]
- 19.Danev R, Kanamaru S, Marko M, Nagayama K. Zernike phase contrast cryo-electron tomography. J Struct Biol. 2010;171:174–181. doi: 10.1016/j.jsb.2010.03.013. [DOI] [PubMed] [Google Scholar]
- 20.Murata K, Liu X, Danev R, Jakana J, Schmid MF, King J, Nagayama K, Chiu W. Zernike phase contrast cryo-electron microscopy and tomography for structure determination at nanometer and subnanometer resolutions. Structure. 2010;18:903–912. doi: 10.1016/j.str.2010.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dai W, Fu C, Raytcheva D, Flanagan J, Khant HA, Liu X, Rochat RH, Haase-Pettingell C, Piret J, Ludtke SJ, Nagayama K, Schmid MF, King JA, Chiu W. Visualizing virus assembly intermediates inside marine cyanobacteria. Nature. 2013;502:707–710. doi: 10.1038/nature12604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bammes BE, Rochat RH, Jakana J, Chen DH, Chiu W. Direct electron detection yields cryo-EM reconstructions at resolutions beyond 3/4 Nyquist frequency. J Struct Biol. 2012;177:589–601. doi: 10.1016/j.jsb.2012.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Li X, Mooney P, Zheng S, Booth CR, Braunfeld MB, Gubbens S, Agard DA, Cheng Y. Electron counting and beam-induced motion correction enable near-atomic-resolution single-particle cryo-EM. Nat Methods. 2013;10:584–590. doi: 10.1038/nmeth.2472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bai XC, Fernandez IS, McMullan G, Scheres SH. Ribosome structures to near-atomic resolution from thirty thousand cryo-EM particles. Elife. 2013;2:e00461. doi: 10.7554/eLife.00461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ludtke SJ, Nason L, Tu H, Peng L, Chiu W. Object oriented database and electronic notebook for transmission electron microscopy. Microscopy and Microanalysis. 2003;9:556–565. doi: 10.1017/S1431927603030575. [DOI] [PubMed] [Google Scholar]
- 26.Rees I, Langley E, Chiu W, Ludtke SJ. EMEN2: an object oriented database and electronic lab notebook. Microsc Microanal. 2013;19:1–10. doi: 10.1017/S1431927612014043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, Ferrin TE. UCSF Chimera--a visualization system for exploratory research and analysis. J Comput Chem. 2004;25:1605–1612. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- 28.Tang G, Peng L, Baldwin PR, Mann DS, Jiang W, Rees I, Ludtke SJ. EMAN2: an extensible image processing suite for electron microscopy. J Struct Biol. 2007;157:38–46. doi: 10.1016/j.jsb.2006.05.009. [DOI] [PubMed] [Google Scholar]
- 29.Good N, Winget G, Winter W, Connolly T, Izawa S, Singh R. Hydrogen ion buffers for biological research. Biochemistry. 1966;5:467–477. doi: 10.1021/bi00866a011. [DOI] [PubMed] [Google Scholar]
- 30.Crowther RA, Amos LA, Finch JT, DeRosier DJ, Klug A. Three dimensional reconstructions of spherical viruses by Fourier synthesis from electron micrographs. Nature. 1970;226:421–425. doi: 10.1038/226421a0. [DOI] [PubMed] [Google Scholar]
- 31.Suloway C, Pulokas J, Fellmann D, Cheng A, Guerra F, Quispe J, Stagg S, Potter CS, Carragher B. Automated molecular microscopy: the new Leginon system. J Struct Biol. 2005;151:41–60. doi: 10.1016/j.jsb.2005.03.010. [DOI] [PubMed] [Google Scholar]
- 32.Zhang J, Nakamura N, Shimizu Y, Liang N, Liu X, Jakana J, Marsh MP, Booth CR, Shinkawa T, Nakata M, Chiu W. JADAS: a customizable automated data acquisition system and its application to ice-embedded single particles. J Struct Biol. 2009;165:1–9. doi: 10.1016/j.jsb.2008.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mastronarde DN. Automated electron microscope tomography using robust prediction of specimen movements. J Struct Biol. 2005;152:36–51. doi: 10.1016/j.jsb.2005.07.007. [DOI] [PubMed] [Google Scholar]
- 34.Ludtke SJ, Baldwin PR, Chiu W. EMAN: semiautomated software for high-resolution single-particle reconstructions. J Struct Biol. 1999;128:82–97. doi: 10.1006/jsbi.1999.4174. [DOI] [PubMed] [Google Scholar]
- 35.Chang J, Liu X, Rochat RH, Baker ML, Chiu W. Reconstructing virus structures from nanometer to near-atomic resolutions with cryo-electron microscopy and tomography. Adv Exp Med Biol. 2012;726:49–90. doi: 10.1007/978-1-4614-0980-9_4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Erickson HP. The Fourier transform of an electron micrograph - First order and second order theory of image formation. Adv Opt Elec Microsc. 1973;5:163–199. [Google Scholar]
- 37.Thon F. In: Electron Microscopy in Material Sciences. Valdre U, editor. Academic Press, Inc; New York: 1971. pp. 571–625. [Google Scholar]
- 38.Chen DH, Baker ML, Hryc CF, DiMaio F, Jakana J, Wu W, Dougherty M, Haase-Pettingell C, Schmid MF, Jiang W, Baker D, King JA, Chiu W. Structural basis for scaffolding-mediated assembly and maturation of a dsDNA virus. Proc Natl Acad Sci U S A. 2011;108:1355–1360. doi: 10.1073/pnas.1015739108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Frank J, Radermacher M, Penczek P, Zhu J, Li Y, Ladjadj M, Leith A. SPIDER and WEB: processing and visualization of images in 3D electron microscopy and related fields. J Struct Biol. 1996;116:190–199. doi: 10.1006/jsbi.1996.0030. [DOI] [PubMed] [Google Scholar]
- 40.Liang Y, Ke EY, Zhou ZH. IMIRS: a high-resolution 3D reconstruction package integrated with a relational image database. J Struct Biol. 2002;137:292–304. doi: 10.1016/s1047-8477(02)00014-x. [DOI] [PubMed] [Google Scholar]
- 41.Liu X, Jiang W, Jakana J, Chiu W. Averaging tens to hundreds of icosahedral particle images to resolve protein secondary structure elements using a Multi-Path Simulated Annealing optimization algorithm. J Struct Biol. 2007;160:11–27. doi: 10.1016/j.jsb.2007.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Grigorieff N. FREALIGN: high-resolution refinement of single particle structures. J Struct Biol. 2007;157:117–125. doi: 10.1016/j.jsb.2006.05.004. [DOI] [PubMed] [Google Scholar]
- 43.Guo F, Jiang W. Humana Press; New York: 2013. in press. [Google Scholar]
- 44.Fuller SD, Butcher SJ, Cheng RH, Baker TS. Three-dimensional reconstruction of icosahedral particles--the uncommon line. J Struct Biol. 1996;116:48–55. doi: 10.1006/jsbi.1996.0009. [DOI] [PubMed] [Google Scholar]
- 45.Zhou ZH, Chiu W, Haskell K, Spears H, Jr, Jakana J, Rixon FJ, Scott LR. Refinement of herpesvirus B-capsid structure on parallel supercomputers. Biophys J. 1998;74:576–588. doi: 10.1016/S0006-3495(98)77816-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Liu X, Zhang Q, Murata K, Baker ML, Sullivan MB, Fu C, Dougherty MT, Schmid MF, Osburne MS, Chisholm SW, Chiu W. Structural changes in a marine podovirus associated with release of its genome into Prochlorococcus. Nat Struct Mol Biol. 2010;17:830–836. doi: 10.1038/nsmb.1823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bowman BR, Baker ML, Rixon FJ, Chiu W, Quiocho FA. Structure of the herpesvirus major capsid protein. EMBO J. 2003;22:757–765. doi: 10.1093/emboj/cdg086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Emsley P, Lohkamp B, Scott WG, Cowtan K. Features and development of Coot. Acta Crystallogr D Biol Crystallogr. 2010;66:486–501. doi: 10.1107/S0907444910007493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Harauz G, van Heel M. Exact filters for general geometry three dimensional reconstruction. Optik. 1986;73:146–156. [Google Scholar]
- 50.Saxton WO, Baumeister W. The correlation averaging of a regularly arranged bacterial cell envelope protein. J Microsc. 1982;127:127–138. doi: 10.1111/j.1365-2818.1982.tb00405.x. [DOI] [PubMed] [Google Scholar]
- 51.Liao L, Kuang SQ, Yuan Y, Gonzalez SM, O’Malley BW, Xu J. Molecular structure and biological function of the cancer-amplified nuclear receptor coactivator SRC-3/AIB1. J Steroid Biochem Mol Biol. 2002;83:3–14. doi: 10.1016/s0960-0760(02)00254-6. [DOI] [PubMed] [Google Scholar]
- 52.Rosenthal PB, Henderson R. Optimal determination of particle orientation, absolute hand, and contrast loss in single-particle electron cryomicroscopy. J Mol Biol. 2003;333:721–745. doi: 10.1016/j.jmb.2003.07.013. [DOI] [PubMed] [Google Scholar]
- 53.van Heel M, Schatz M. Fourier shell correlation threshold criteria. J Struct Biol. 2005;151:250–262. doi: 10.1016/j.jsb.2005.05.009. [DOI] [PubMed] [Google Scholar]
- 54.Frangakis AS, Hegerl R. Segmentation of two- and three-dimensional data from electron microscopy using eigenvector analysis. J Struct Biol. 2002;138:105–113. doi: 10.1016/s1047-8477(02)00032-1. [DOI] [PubMed] [Google Scholar]
- 55.Pintilie GD, Zhang J, Goddard TD, Chiu W, Gossard DC. Quantitative analysis of cryo-EM density map segmentation by watershed and scale-space filtering, and fitting of structures by alignment to regions. J Struct Biol. 2010;170:427–438. doi: 10.1016/j.jsb.2010.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Volkmann N. A novel three-dimensional variant of the watershed transform for segmentation of electron density maps. J Struct Biol. 2002;138:123–129. doi: 10.1016/s1047-8477(02)00009-6. [DOI] [PubMed] [Google Scholar]
- 57.Forster F, Reiner H. In: Methods in cell biology. 29. McIntosh RJ, editor. Vol. 79. Elsevier; 2007. pp. 741–767. [DOI] [PubMed] [Google Scholar]