

Abstract
Glucuronidation is a major metabolism process of detoxification for carcinogens, 4-(methylnitrosamino)-1-(3-pyridy)-1-butanone (NNK) and 1,2-dimethylhydrazine (DMH), of reactive oxygen species (ROS). However, intestinal E. coli β-glucuronidase (eβG) has been considered pivotal to colorectal carcinogenesis. Specific inhibition of eβG may prevent reactivating the glucuronide-carcinogen and protect the intestine from ROS-mediated carcinogenesis. In order to develop specific eβG inhibitors, we found that 59 candidate compounds obtained from the initial virtual screening had high inhibition specificity against eβG but not human βG. In particular, we found that compounds 7145 and 4041 with naphthalenylidene-benzenesulfonamide (NYBS) are highly effective and selective to inhibit eβG activity. Compound 4041 (IC50 = 2.8 μM) shows a higher inhibiting ability than compound 7145 (IC50 = 31.6 μM) against eβG. Furthermore, the molecular docking analysis indicates that compound 4041 has two hydrophobic contacts to residues L361 and I363 in the bacterial loop, but 7145 has one contact to L361. Only compound 4041 can bind to key residue (E413) at active site of eβG via hydrogen-bonding interactions. These novel NYBS-based eβG specific inhibitors may provide as novel candidate compounds, which specifically inhibit eβG to reduce eβG-based carcinogenesis and intestinal injury.
1. Introduction
Glucuronidation represents a major route of drug metabolism in the liver [1–3]. Many carcinogens and xenobiotics are detoxified by conjugation with a glucuronide acid to increase their water solubility, thus facilitating their excretion [4, 5]. For example, the 4-(methylnitrosamino)-1-(3-pyridy1)-1-butanone (NNK) and 1,2-dimethylhydrazine (DMH), which lead to reactive oxygen species- (ROS-) mediated DNA damage, mutagenesis, and carcinogenesis, can be metabolized and detoxified to glucuronide form by UDP-glucuronosyltransferases (UGTs) [6–8]. However, when the glucuronide conjugates enter the intestine through enterohepatic circulation, they can be hydrolyzed and their toxicity can be reversed by E. coli β-glucuronidase (eβG) [9–11], which would induce intestinal injury [5, 12–14] and colon carcinogenesis. In addition, chemotherapy-induced diarrhea (CID) and intestinal damage have been clinically demonstrated in several chemotherapeutic drugs such as CPT-11 [15–17], 5-FU [18], and oxaliplatin [19]. There is a specific βG inhibitor, glucaro-1,5-lactone, which has been shown to alleviate CPT-11-induced mucosal damage in the small intestine in vivo [5]. However, glucaro-1, 5-lactone of the current treatments against CID is limited and not effective, since it preferentially inhibits human βG (hβG) activity [20], which may induce mucopolysaccharidoses (MPS) [21, 22]. The current treatments against CID are limited and not effective. Therefore, it is crucial to develop an eβG specific inhibitor, which cannot affect hβG activity, as an effective treatment against carcinogenesis and CID.
The crystal structures of hβG and eβG have been reported [16, 17]. In addition, eβG has a unique “bacteria loop” (LGIGFEAGNKPKELYSE) [16] which is absent in hβG. Some known drugs such as Amoxapine [23, 24] and Loxapine [23] and eβG inhibitors [16] have been also demonstrated to interact with the residues of bacterial loop and active sites of eβG [16, 24]. Those reports indicate that the area around the unique loop and the active site is an important target for eβG inhibitor selection.
For the development of specific eβG inhibitors, we demonstrated a crystal structure of recombinant eβG (provided by Steve R. Roffler, Institute of Biomedical Sciences, Academia Sinica, Taipei, Taiwan) in complex with D-glucaro-1, 5-lactone which revealed that the inhibitor was bound at the residues (E413, E504) of active site. And, we further compared the active center between eβG and hβG though overleap. We obtained candidate compounds that selectively inhibit eβG via computational screening by DOCK 4.0 program [25, 26] and X-ray crystal structure of eβG. A chemical database (SPECS) containing ~300,000 commercially available compounds was computationally screened against a grid box enclosing the unique bacterial loop at eβG active site. To prove whether the candidate compounds can effectively inhibit eβG without affecting hβG activity, compounds were examined based on their specific inhibition for eβG versus hβG by in vitro βG activity-based assays. The binding motifs of eβG specific inhibitors were determined by molecular docking studies. The novel eβG specific inhibitor may provide a highly effective and selective agent to prevent eβG-based carcinogenesis and CID.
2. Materials and Methods
2.1. Expression and Purification of βG Protein
Plasmid pRESTB containing βG gene and a histidine tag at the N-terminus was constructed as described [27]. Recombinant βG (human and E. coli) was produced by isopropyl β-D-thiogalactopyranoside (IPTG) induction of BL21 (DE3) bacteria. βG was purified from bacterial supernatants by affinity chromatography on nickels Sepharose 6 Fast Flow (GE Healthcare). The column was washed by phosphate-buffered saline (PBS), with 50 mM imidazole, and βG was eluted by PBS with 250 mM imidazole. The purified βG was desalted on a Sephadex G-25 column equilibrated with PBS and stored at −80°C.
2.2. Virtual Screening of eβG Specific Inhibitors
The virtual screening was performed using the DOCK 4.0 program and the X-ray crystal structure of eβG (provide by Steve R. Roffler). The B-chain structure of protein, water molecules, and the cocrystallized inhibitor D-glucaro-1,5-lactone were removed. The remaining A-chain protein structure was used to prepare the target site for docking simulations. The active-site region of eβG was specified as the target site for ligand docking in virtual screening. Briefly, a molecular surface around the target site was generated with the MS program using a 1.4 Å probe radius and this surface was used to generate with the SPHGEN program 43 overlapping spheres to fill the target site. A grid box enclosing the target site was created for grid calculations with dimensions 19.3 × 22.4 × 15.6 Å. The force filled scoring grids were calculated with the GRID program using a distance-dependent dielectric constant of 4r, an energy cutoff distance of 10 Å, and a grid spacing of 0.3 Å. The database for virtual screening was the SPECS compound collection, which included ~300,000 commercially available compounds (downloaded from the ZINC database web site). The DOCK 4.0 program performs docking simulations using a distance-matching algorithm. The matching parameters used to run virtual screening were set as follows: distance tolerance = 0.5, distance minimum = 2.5, nodes maximum = 10, and nodes minimum = 4. The SPECS database was computationally screened against the active site of eβG using the force field scoring function based on interaction energy. Virtual screening was performed on a Silicon Graphics Octane workstation with dual 270 MHz MIPS R12000 processors.
For compound selection, the docking models of the 4724 top-ranked compounds (energy score values ≤−42.00 kcal/mol) were visually inspected using the software PyMOL. Together with consideration of chemical diversity, the selection of compounds was assisted by analysis of the docking models with respect to shape fitting and hydrogen-bonding and hydrophobic interactions. Finally, we selected 59 compounds for enzyme inhibition assays against E. coli and human βGs. The compounds for testing were purchased from the SPECS Company. The SPECS ID number and docking energy score for compounds are listed in the supporting information.
2.3. In Vitro βG-Activity Assay of eβG Specific Inhibitors
The candidate compounds were purchased from SPECS (The Netherlands). Each candidate was provided as a solid power and dissolved in 100% DMSO (Sigma-Aldrich) to 10 mM as stock. Candidates were screened for their inhibition specificity of eβG versus hβG, which were conducted at pH 7.3 or pH 5.4, in triplicate, respectively. 40 μL purified βG was treated with 10 μL compound solution at 37°C for 30 min and sequentially incubated with 50 μL of pNPG (Sigma-Aldrich) at 37°C for 30 min. Reactions were quenched with 5 μL of 2 N sodium hydroxide (Sigma-Aldrich). Each reaction consisted of 3.75 ng purified βG, 50 μM compound, and 5 mM pNPG in PBS containing 10% DMSO and 0.05% BSA (Sigma-Aldrich). βG-activities were measured by color development of pNP detected on a microplate reader at OD 405 nm. Results are displayed as percent of βG activity compared with the untreated control. For IC50 determination, compounds at various concentrations (100 μM to 0.001 μM) were added.
2.4. Molecular Docking Studies of eβG Specific Inhibitors
The crystal structure of eβG for the virtual screening was also utilized in the docking studies of compounds 7145 and 4041. Hydrogen atoms were added to the A-chain protein structure, and the resulting structure was used in the docking simulations. The 3D structures of compounds were built and optimized by energy minimization using the MM2 force field and a minimum RMS gradient of 0.05 in the software Chem3D 6.0 (CambridgeSoft Corp., Cambridge, MA). Docking simulations were performed using the GOLD 5.0 program [28] on an HP xw6600 workstation with Intel Xeon E5450/3.0 GHz Quadcores as the processors. The GOLD program utilizes a genetic algorithm (GA) to perform flexible ligand docking simulations. In the present study, for each of the 30 independent GA runs, a maximum number of 100000 GA operations was performed on a single population of 100 individuals. Operator weights for crossover, mutation, and migration were set to 95, 95, and 10, respectively. The GoldScore fitness function was applied for scoring the docking poses of compounds. The docking region was defined to encompass the active site of eβG. The best docking solution (with the highest GOLD fitness score) for a compound was chosen to represent the most favorable predicted binding mode to eβG.
3. Results
3.1. Comparison of the Active Site Structures of eβG and hβG
To identify the active site of eβG, recombinant full-length eβG was purified and shown to hydrolyze pNPG to PNP for detecting βG activity. The enzyme was crystallized in complex with an established inhibitor, D-glucaro-1, 5-lactone. Crystal structure of eβG (provided by Steve R. Roffler) in complex with D-glucaro-1, 5-lactone revealed that the inhibitor was bound at residues (E413 and E504) of the active site (Figure 1(a)). To compare the structures of the active centers between eβG and hβG (PDB ID 1BHG), βGs were analyzed by computer simulation technology. After superimposition, the crystallized structure of eβG is 45% similar to hβG. Moreover, there is a “bacterial loop” within eβG which is absent in hβG (Figure 1(b)). Similar results have been also shown in other reports [16]. This eβG unique loop of the active center is an ideal target site for screening compounds that can selectively inhibit eβG activity.
Figure 1.

The crystal structure of eβG and hβG. (a) The crystal structure of eβG bound with the inhibitor D-glucaro-1,5-lactone in the active site was used in the virtual screening. A docking box (red line) was defined to enclose the active site for virtual compound screening. (b) The eβG (green) and hβG (purple) were modeled by superimposing. The eβG contains a “bacterial loop” (yellow) not found in the hβG. The E413 and E504 are two catalytic residues in the active site of eβG.
3.2. In Silico Virtual Screening of eβG Inhibitor Candidates
To identify potential eβG inhibitors that can selectively block eβG activity, but not hβG, the virtual screening proceeded based on the different structures of the active center between eβG and hβG. The SPECS database (~300,000 commercially available compounds) was computationally screened against the “grid box” which contains the bacterial loop of eβG and active site using the DOCK program (version 4.0). Fifty-nine candidate compounds were acquired from the initial virtual screening which was designed to target the bacterial loop of eβG and its active site. The docking energy scores of 59 candidate compounds measured by the DOCK program are −43 to −55 kcal/mol (Table S1 in the supplementary material available online at http://dx.doi.org/10.1155/2014/740815).
3.3. Screening of eβG Inhibitor Candidates by In Vitro βG Activity Assay
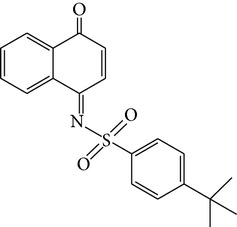
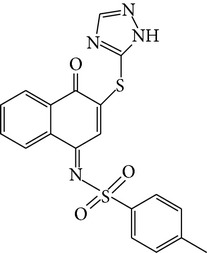
To prove whether these 59 candidate compounds can effectively inhibit eβG without affecting hβG activity, 50 μM compounds were examined for their specific inhibition for eβG versus hβG by in vitro βG-based activity assays, in which the conversion of pNPG to PNP was detected by measuring the increases in PNP absorbance at OD 405 nm. The result showed that all the 59 candidate compounds displayed selective inhibition against eβG activity. The inhibiting ability against eβG activity, especially, was >95% in 7 candidates of eβG specific inhibitors (Table S1). Based on these results, we concluded that the pocket site in the unique loop and active site of eβG are an ideal site to screen eβG specific inhibitors through virtual screening. We found that compound 7145 (4-tert-butyl-N-(4-oxo-1(4H)-naphthalenylidene-benzenesulfonamide) can inhibit >95% eβG activity and does not hamper hβG activity at 50 μM condition (Table S1). The result indicated that the derivatives of naphthalenylidene-benzenesulfonamide (NYBS) might effectively and specifically inhibit the eβG activity. Based on the NYBS structure, we performed the substructure search and then found compound 4041 (4-methyl-N-(4-oxo-3-(1H-1,2,4-triazol-5ylsulfanyl)-1(4H)-naphthalenylidene)benzenesulfonamide). In particular, compound 4041 has been shown to be a more potent eβG antagonist than compound 7145. Figure 2 and Table 1 show that while compound 4041 (IC50 = 2.8 μM) can selectively inhibit >80% eβG activity, at 10 μM, the inhibition of compound 7145 (IC50 = 31.6 μM) is 55% inhibition. Compared to D-saccharic acid 1,4-lactone (saccharolactone), which showed higher inhibition on hβG, our candidate compounds displayed specificity against eβG activity (Figure 2). Based on these results, we concluded that the derivatives of NYBS may provide a novel specific inhibitor to reduce eβG-based intestinal injury and CID.
Figure 2.
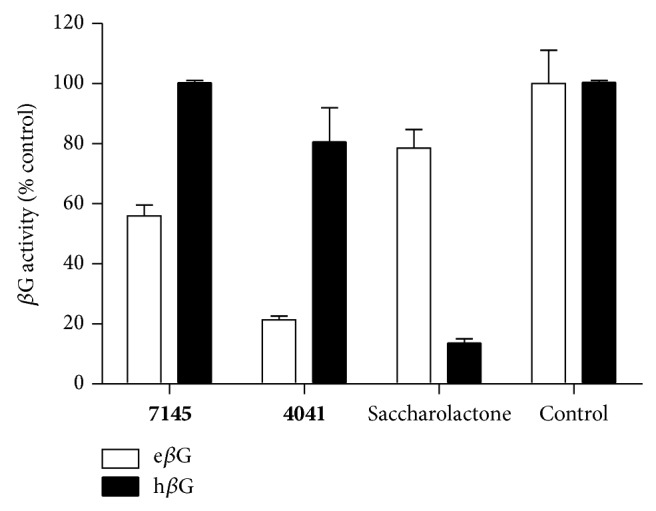
Specific inhibition of compounds 7145 and 4041 against eβG. Compound 7145 and compound 4041 acquired from ligand docking in virtual screening were evaluated based on their selective inhibition for recombinant eβG versus hβG. 10 μM of compound 7145, compound 4041, saccharolactone, and 10% DMSO (control) was incubated with purified eβG (□) and hβG (■), respectively. βG activity was determined by hydrolysis of the pNPG substrate. Error bars represent SD; N = 3.
Table 1.
The structure, IC50, and GOLD fitness scores of compound 7145 and compound 4041 docked into the active site of eβG.
| Compound | Structure | GOLD fitness scorea | IC50 (μM) |
|---|---|---|---|
| 7145 |
|
62.09 | 31.6 |
|
| |||
| 4041 |
|
64.91 | 2.8 |
aDocking simulations were performed using the GOLD 5.0 program.
3.4. Molecular Docking Studies of eβG Specific Inhibitors
To predict the binding modes of compounds 7145 and 4041 in the active site of eβG, we performed molecular docking studies using the GOLD 5.0 program. As depicted in Figure 3, the docking model revealed that compound 7145 forms four hydrogen bonds to eβG. Three of the hydrogen bonds, to residues Y468, Y472, and R562, arise from the SO2 group of compound 7145. The carbonyl group on the bicyclic 4-oxo-1(4H)-naphthalenylidene ring can form one hydrogen bond with H296. Compound 7145 makeshydrophobic interactions with the surrounding residues, including W549, F554, F164, V355, V446, M447, F448, Y468, and Y472. The residue L361 in the bacterial loop makes hydrophobic contact with compound 7145. Compound 7145 showed a GOLD fitness score of 62.09.
Figure 3.
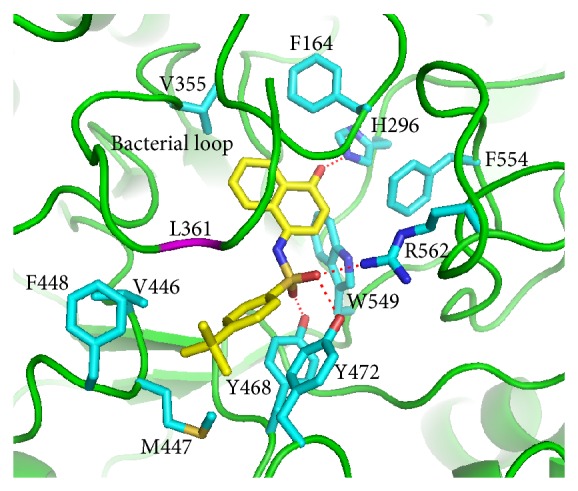
Binding model of compound 7145. Predicted binding mode of compound 7145 in the active site of eβG from the docking study. Compound 7145 (yellow) and some amino acid residues (cyan) interacting with the inhibitor are shown as stick structures. The red dashed lines indicate hydrogen-bonding interactions. The residue L361 (purple) in the bacterial loop makes hydrophobic contact with compound 7145.
The docked orientation of compound 4041 is considerably different from that of compound 7145 (Figure 3). For compound 4041, the bicyclic 4-oxo-1(4H)-naphthalenylidene ring points towards M447 and makes a π-π stacking interaction with Y472. In contrast, the bicyclic ring of compound 7145 is oriented in the opposite direction to M447 and located very close to the experimental binding position of the inhibitor D-glucaro-1,5-lactone. Compound 4041 is hydrogen bonded to residues Y472 and R562 through the SO2 group and to E413 through the 1,2,4-triazole moiety. Compound 4041 makes hydrophobic interactions with the surrounding residues, including V446, M447, Y472, and L561. The residues L361 and I363 in the bacterial loop make hydrophobic contact with compound 4041 (Figure 4). Compound 4041 has a GOLD fitness score of 64.91 higher than that of compound 7145. Figure 5 shows an overlay of the docking pose of compound 4041 with the bound orientation of an eβG-specific inhibitor [16] observed in the cocrystal structure of eβG. In comparison to the cocrystallized inhibitor, compound 4041 has similar binding interactions with eβG, including hydrogen bonding to the catalytic residue E413 and close contact with the bacterial loop. Based on these results, we suggest that binding to the active site and the bacterial loop of eβG may provide selective abilities of inhibitors against eβG.
Figure 4.
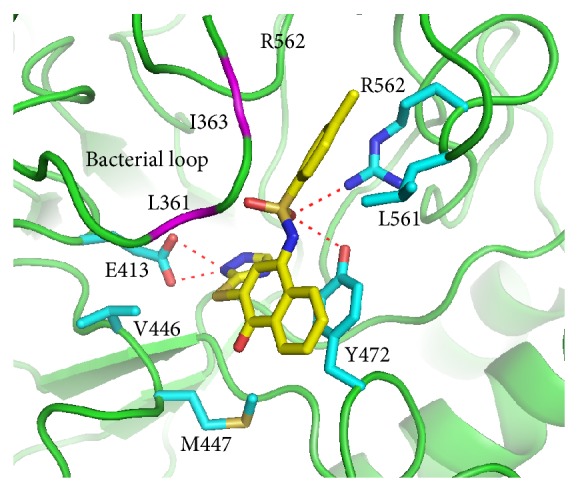
Binding model of compound 4041. Predicted binding mode of compound 4041 in the active site of eβG from the docking study. Compound 4041 (yellow) and some amino acid residues (cyan) interacting with the inhibitor are shown as stick structures. The red dashed lines indicate hydrogen-bonding interactions. The residues L361 and I363 (purple) in the bacterial loop make hydrophobic contact with compound 4041.
Figure 5.
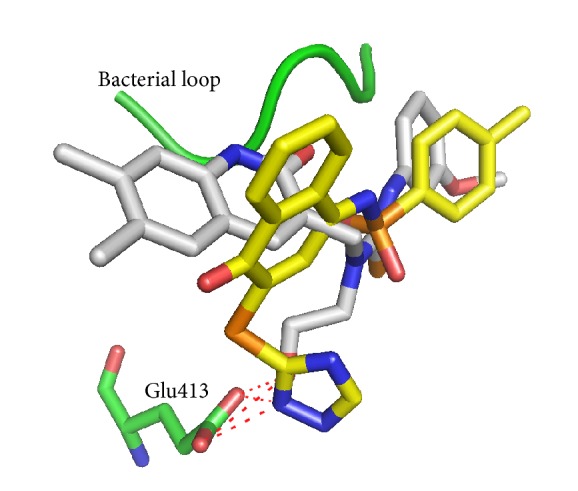
Binding model of compound 4041 in the crystal structure. Overlay of the docking pose of compound 4041 (yellow) with the bound orientation of an eβG-specific inhibitor (gray) observed in the cocrystal structure of eβG.
4. Discussion
In this study, we have obtained potent and selective eβG inhibitors from in silico virtually screening and further confirmed their inhibition specificity by in vitro βG activity-based assay. All the 59 candidate compounds from the initial screening showed high effective and selective inhibition against eβG. We identified the two most promising compounds, compound 7145 and its derivate compound 4041, showing IC50 values of 31.6 μM and 2.8 μM, respectively. Importantly, compound 4041 with naphthalenylidene-benzenesulfonamide displayed inhibition selectivity against eβG by binding to the active site at E413 and the unique loop of eβG at L361 and I363.
High-throughput screening (HTS) allows researchers to screen millions of compounds for lead identification in drug discovery. However, this method is limited by the size of compound library. Generally, a compound library is quite costly, and the screening process is time-consuming; thus, the limitations have become more apparent. Hence, virtual screening has become an important tool to access novel drugs for lead indentation [29]. The hit rate of virtual screening can reach up to 2–24% which is much higher than HTS with 0.01–0.001% [30]. In our study, we obtained 59 potential eβG inhibitors via virtual screening of a library which consisted of ~300,000 compounds. All candidate compounds showed specific inhibition against eβG, but not hβG, and met the criteria as virtual screening. The structure-based virtual screening can select compounds with no range limitation and narrow down the candidates for further evaluation, which saves both money and time.
hβG is a lysosomal enzyme of normal tissues, and quite low levels of hβG are found in serum [31, 32]. In contrast, eβG is mainly found in the intestine. Both hβG and eβG catalyze hydrolysis of β-D-glucuronic acid residues from the nonreducing end of glycosaminoglycans [33, 34], but the enzyme has a unique acidic optimum pH. While hβG displays maximal catalytic activity at pH 4–4.5 [32, 35], eβG exhibits optimal activity at neutral pH. Inhibiting hβG may cause MPS [21, 22], a lysosomal storage disease that can affect appearance, physical abilities, organ and system functioning, and, in most cases, mental development. It is crucial to screen compounds that can only block eβG activity but not affect hβG.
A unique loop structure was found in eβG which lacked hβG after superimposition of two βGs, which provides a target site to screen compounds that can distinguish the two βGs [16]. We found 59 candidate compounds which can selectively inhibit eβG activity through molecular docking against the grid box enclosing the bacterial loop and active site of eβG. Some known drugs, such as Amoxapine and Loxapine, have been demonstrated to interact with the residues of bacterial loop and active site of eβG and inhibit variant bacterial βG activity [24]. Wallace and colleagues indicated that the key residues of bacterial loop are L361 and F365 [16], indicating that we can develop the eβG specific inhibitor by targeting the unique loop of eβG. In this report, compound 4041 can bind to E413 (key residue in active site of eβG) through the 1,2,4-triazole moiety but not show in the compound 7145. Furthermore, compound 4041 has twohydrophobic contacts to residues L361 and I363 in the bacterial loop. But, compound 7145 shows one hydrophobic contact withresidue L361. In eβG activity assay, compound 4041 (IC50 = 2.8 μM) also shows a higher inhibiting ability than compound 7145 (IC50 = 31.6 μM). We concluded that the inhibiting ability of eβG has positive correlation with the interacting quantities to the active site and the unique loop of eβG.
eβG inhibitors can be developed as a chemotherapy adjuvant to reduce CID [16, 24]. CID is a main side effect that occurs in up to 50–80% of patients depending on chemotherapy regimen [36]. There are several studies indicating that inhibiting βG activity can reduce CID and intestinal injury [37]. Inhibition of intestinal βG by antibiotics could reduce CPT-11-induced diarrhea in vivo. But, antibiotics will kill all native gut floras, including probiotics within the digestive tract, which is not recommended for chemotherapeutic patients. Moreover, the eβG has also been considered to play a pivotal role in the development of colon carcinogenesis. For example, the DMH and NNK (ROS based carcinogen) have been report that their glucuronide metabolite may be re-toxic by eβG and induce intestinal damage and colon tumor in vivo [6–8, 12–14]. eβG specific inhibitors may act as colon cancer chemoprevention agents by reducing the generation of xenobiotics from glucuronide metabolites. Thus, the specific eβG inhibitor can be applied in nutrient supplement for cancer prevention.
5. Conclusions
In conclusion, we have identified that two compounds, compound 7145 and compound 4041, can selectively inhibit eβG activity without disrupting hβG activity by binding to the active site and the unique loop within eβG. Because of their high specificity and efficacy against eβG, they have great potential to be developed as a chemotherapy adjuvant for antidiarrhea treatment and cancer chemoprevention agent. Moreover, we proved that inhibitors for the desire enzymes can be selected from virtual screening based on the structure docking showing a high hit rate, which may provide a fast and inexpensive approach for new drug discovery.
Supplementary Material
Fifty-nine candidate compounds were acquired from the initial virtually screening which was designed to target the bacterial loop of eβG and its active site. The docking energy scores of 59 candidate compounds measured by the DOCK program are -43 to -55 kcal/mol. (Table S1) The candidate compounds were purchased from SPECS (Zoetermeer, The Netherlands). Each candidate was rovided as a solid power and dissolved in 100% DMSO (Sigma-Aldrich, MO, USA) to 10 mM as stock. Candidates were screening for their inhibition specificity of eβG verse hβG, which were conducted at pH 7.3 or pH 5.4 in triplicate, respectively. 40 µL purified βG was treated with 10 µL compound solution at 37 °C for 30 min, and sequentially incubated with 50 µL of pNPG (Sigma-Aldrich) at 37 °C for 30 min. Reactions were quenched with 5 µL of 2 N sodium hydroxide (Sigma-Aldrich). Each reaction consisted of 3.75 ng purified βG, 50 µM compound, and 5 mM pNPG in PBS containing 10% DMSO and 0.05% BSA (Sigma-Aldrich). βG-activities were measured by color development of pNP detected on a microplate reader at OD 405 nm. Results are displayed as percent of βG activity compared with the untreated control. The result showed that all the 59 candidate compounds displayed selective inhibition against eβG activity. Especially, the inhibiting ability against eβG activity was >95% in 7 candidates of eβG specific inhibitors (Table S1).
Acknowledgments
This work was supported by grants from the National Research Program for Biopharmaceuticals, Ministry of Science and Technology, Taipei, Taiwan (MOST 103-2325-B-037-007, MOST 103-2325-B-041-001, NSC 101-2320-B-041-001-MY2, and NSC 102-2320-B-038-043-MY2), the Ministry of Health and Welfare, Taiwan (MOHW103-TD-B-111-05), the National Health Research Institutes, Taiwan (NHRI-EX103-10238SC), the China Medical University, Taichung, Taiwan (CMU99-N1-19-1 and CMU99-N1-19-2), 103NSYSU-KMU Joint Research Project (NSYSUKMU103 I-003), Comprehensive Cancer Center of Taipei Medical University/Health and Welfare Surcharge of Tobacco Products (MOHW103-TD-B-111-01), and the Grant of Biosignature in Colorectal Cancers, Academia Sinica, Taiwan. This study is also supported partially by Kaohsiung Medical University “Aim for the Top 500 Universities Grant” (Grants nos. KMU-TP103C00, KMU-TP103C01, KMU-TP103C11, KMU-TP103H10, and KMU-DT103005).
Conflict of Interests
The authors declare that there is no conflict of interests regarding the publication of this paper.
Authors' Contribution
Ta-Chun Cheng and Kuo-Hsiang Chuang contributed equally.
References
- 1.Nelson G. M., Swank A. E., Brooks L. R., Bailey K. C., George S. E. Metabolism, microflora effects, and genotoxicity in haloacetic acid-treated cultures of rat cecal microbiota. Toxicological Sciences. 2001;60(2):232–241. doi: 10.1093/toxsci/60.2.232. [DOI] [PubMed] [Google Scholar]
- 2.Rowland I. The influence of the gut microflora on food toxicity. Proceedings of the Nutrition Society. 1981;40(1):67–74. doi: 10.1079/PNS19810011. [DOI] [PubMed] [Google Scholar]
- 3.Humblot C., Murkovic M., Rigottier-Gois L., et al. β-glucuronidase in human intestinal microbiota is necessary for the colonic genotoxicity of the food-borne carcinogen 2-amino-3-methylimidazo[4,5-f]quinoline in rats. Carcinogenesis. 2007;28(11):2419–2425. doi: 10.1093/carcin/bgm170. [DOI] [PubMed] [Google Scholar]
- 4.Rowland A., Miners J. O., Mackenzie P. I. The UDP-glucuronosyltransferases: their role in drug metabolism and detoxification. International Journal of Biochemistry and Cell Biology. 2013;45(6):1121–1132. doi: 10.1016/j.biocel.2013.02.019. [DOI] [PubMed] [Google Scholar]
- 5.Takasuna K., Hagiwara T., Watanabe K., et al. Optimal antidiarrhea treatment for antitumor agent irinotecan hydrochloride (CPT-11)-induced delayed diarrhea. Cancer Chemotherapy and Pharmacology. 2006;58(4):494–503. doi: 10.1007/s00280-006-0187-8. [DOI] [PubMed] [Google Scholar]
- 6.Chihara T., Shimpo K., Kaneko T., et al. Inhibition of 1, 2-dimethylhydrazine-induced mucin-depleted foci and O6-methylguanine DNA adducts in the rat colorectum by boiled garlic powder. Asian Pacific Journal of Cancer Prevention. 2010;11(5):1301–1304. [PubMed] [Google Scholar]
- 7.Kim P. M., Wells P. G. Genoprotection by UDP-glucuronosyltransferases in peroxidase-dependent, reactive oxygen species-mediated micronucleus initiation by the carcinogens 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone and benzo[a]pyrene. Cancer Research. 1996;56(7):1526–1532. [PubMed] [Google Scholar]
- 8.Kim M., Miyamoto S., Sugie S., et al. A tobacco-specific carcinogen, NNK, enhances AOM/DSS-induced colon carcinogenesis in male A/J mice. In Vivo. 2008;22(5):557–564. [PubMed] [Google Scholar]
- 9.Kaderlik K. R., Minchin R. F., Mulder G. J., et al. Metabolic activation pathway for the formation of DNA adducts of the carcinogen 2-amino-1-methyl-6-phenylimidazo[4,5-b]pyridine (PhIP) in rat extrahepatic tissues. Carcinogenesis. 1994;15(8):1703–1709. doi: 10.1093/carcin/15.8.1703. [DOI] [PubMed] [Google Scholar]
- 10.Nalini N., Manju V., Menon V. P. Effect of coconut cake on the bacterial enzyme activity in 1,2-dimethyl hydrazine induced colon cancer. Clinica Chimica Acta. 2004;342(1-2):203–210. doi: 10.1016/j.cccn.2004.01.001. [DOI] [PubMed] [Google Scholar]
- 11.Nalini N., Sabitha K., Viswanathan P., Menon V. P. Influence of spices on the bacterial (enzyme) activity in experimental colon cancer. Journal of Ethnopharmacology. 1998;62(1):15–24. doi: 10.1016/S0378-8741(98)00007-5. [DOI] [PubMed] [Google Scholar]
- 12.Likhachev A., Anisimov V., Paranova L., Pozharisski K. Effect of exogenous β-glucuronidase on the carcinogenicity of 1,2-dimethylhydrazine in rats: evidence that carcinogenic intermediates form conjugates and act through their subsequent enzymatic release. Carcinogenesis. 1985;6(5):679–681. doi: 10.1093/carcin/6.5.679. [DOI] [PubMed] [Google Scholar]
- 13.Newaz S. N., Fang W. F., Strobel H. W. Metabolism of the carcinogen 1,2-dimethylhydrazine by isolated human colon microsomes and human colon tumor cells in culture. Cancer. 1983;52(5):794–798. doi: 10.1002/1097-0142(19830901)52:5<794::AID-CNCR2820520507>3.0.CO;2-G. [DOI] [PubMed] [Google Scholar]
- 14.Wolter S., Frank N. Metabolism of 1,2-dimethylhydrazine in isolated perfused rat liver. Chemico-Biological Interactions. 1982;42(3):335–344. doi: 10.1016/0009-2797(82)90077-1. [DOI] [PubMed] [Google Scholar]
- 15.Kobayashi K. Chemotherapy-induced diarrhea. Gan To Kagaku Ryoho. 2003;30(6):765–771. [PubMed] [Google Scholar]
- 16.Wallace B. D., Wang H., Lane K. T., et al. Alleviating cancer drug toxicity by inhibiting a bacterial enzyme. Science. 2010;330(6005):831–835. doi: 10.1126/science.1191175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Roberts A. B., Wallace B. D., Venkatesh M. K., Mani S., Redinbo M. R. Molecular insights into microbial β-glucuronidase inhibition to abrogate CPT-11 toxicity. Molecular Pharmacology. 2013;84(2):208–217. doi: 10.1124/mol.113.085852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cascinu S., Bichisao E., Amadori D., et al. High-dose loperamide in the treatment of 5-fluorouracil-induced diarrhea in colorectal cancer patients. Supportive Care in Cancer. 2000;8(1):65–67. doi: 10.1007/s005209900085. [DOI] [PubMed] [Google Scholar]
- 19.Dranitsaris G., Maroun J., Shah A. Severe chemotherapy-induced diarrhea in patients with colorectal cancer: a cost of illness analysis. Supportive Care in Cancer. 2005;13(5):318–324. doi: 10.1007/s00520-004-0738-7. [DOI] [PubMed] [Google Scholar]
- 20.Fittkau M., Voigt W., Holzhausen H.-J., Schmoll H.-J. Saccharic acid 1.4-lactone protects against CPT-11-induced mucosa damage in rats. Journal of Cancer Research and Clinical Oncology. 2004;130(7):388–394. doi: 10.1007/s00432-004-0557-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Constantopoulos G., Rees S., Cragg B. G., Barranger J. A., Brady R. O. Experimental animal model for mucopolysaccharidosis: suramin-induced glycosaminoglycan and sphingolipid accumulation in the rat. Proceedings of the National Academy of Sciences of the United States of America. 1980;77(6):3700–3704. doi: 10.1073/pnas.77.6.3700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gillett P. M., Schreiber R. A., Jevon G. P., et al. Mucopolysaccharidosis type VII (Sly syndrome) presenting as neonatal cholestasis with hepatosplenomegaly. Journal of Pediatric Gastroenterology and Nutrition. 2001;33(2):216–220. doi: 10.1097/00005176-200108000-00025. [DOI] [PubMed] [Google Scholar]
- 23.Ahmad S., Hughes M. A., Yeh L.-A., Scott J. E. Potential repurposing of known drugs as potent bacterial β-glucuronidase inhibitors. Journal of Biomolecular Screening. 2012;17(7):957–965. doi: 10.1177/1087057112444927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kong R., Liu T., Zhu X., et al. Old drug new use—amoxapine and its metabolites as potent bacterial β-glucuronidase inhibitors for alleviating cancer drug toxicity. Clinical Cancer Research. 2014;20(13):3521–3530. doi: 10.1158/1078-0432.CCR-14-0395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kuntz I. D. Structure-based strategies for drug design and discovery. Science. 1992;257(5073):1078–1082. doi: 10.1126/science.257.5073.1078. [DOI] [PubMed] [Google Scholar]
- 26.Meng E. C., Shoichet B. K., Kuntz I. D. Automated docking with grid-based energy evaluation. Journal of Computational Chemistry. 1992;13(4):505–524. [Google Scholar]
- 27.Cheng T.-L., Chen B.-M., Chan L.-Y., Wu P.-Y., Chern J.-W., Roffler S. R. Poly(ethylene glycol) modification of β-glucuronidase-antibody conjugates for solid-tumor therapy by targeted activation of glucuronide prodrugs. Cancer Immunology Immunotherapy. 1997;44(6):305–315. doi: 10.1007/s002620050387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Jones G., Willett P., Glen R. C., Leach A. R., Taylor R. Development and validation of a genetic algorithm for flexible docking. Journal of Molecular Biology. 1997;267(3):727–748. doi: 10.1006/jmbi.1996.0897. [DOI] [PubMed] [Google Scholar]
- 29.Cohen N. C., Blaney J. M., Humblet C., Gund P., Barry D. C. Molecular modeling software and methods for medicinal chemistry. Journal of Medicinal Chemistry. 1990;33(3):883–894. doi: 10.1021/jm00165a001. [DOI] [PubMed] [Google Scholar]
- 30.Maggio E. T., Ramnarayan K. Recent developments in computational proteomics. Drug Discovery Today. 2001;6(19):996–1004. doi: 10.1016/S1359-6446(01)02003-7. [DOI] [PubMed] [Google Scholar]
- 31.Su Y.-C., Chuang K.-H., Wang Y.-M., et al. Gene expression imaging by enzymatic catalysis of a fluorescent probe via membrane-anchored β-glucuronidase. Gene Therapy. 2007;14(7):565–574. doi: 10.1038/sj.gt.3302896. [DOI] [PubMed] [Google Scholar]
- 32.Leu Y.-L., Roffler S. R., Chern J.-W. Design and synthesis of water-soluble glucuronide derivatives of camptothecin for cancer prodrug monotherapy and antibody-directed enzyme prodrug therapy (ADEPT) Journal of Medicinal Chemistry. 1999;42(18):3623–3628. doi: 10.1021/jm990124q. [DOI] [PubMed] [Google Scholar]
- 33.Wong A. W., He S., Grubb J. H., Sly W. S., Withers S. G. Identification of Glu-540 as the catalytic nucleophile of human β- glucuronidase using electrospray mass spectrometry. The Journal of Biological Chemistry. 1998;273(51):34057–34062. doi: 10.1074/jbc.273.51.34057. [DOI] [PubMed] [Google Scholar]
- 34.Legigan T., Clarhaut J., Renoux B., et al. Synthesis and antitumor efficacy of a β-glucuronidase-responsive albumin-binding prodrug of doxorubicin. Journal of Medicinal Chemistry. 2012;55(9):4516–4520. doi: 10.1021/jm300348r. [DOI] [PubMed] [Google Scholar]
- 35.Cheng T.-C., Roffler S. R., Tzou S.-C., et al. An activity-based near-infrared glucuronide trapping probe for imaging β-glucuronidase expression in deep tissues. Journal of the American Chemical Society. 2012;134(6):3103–3110. doi: 10.1021/ja209335z. [DOI] [PubMed] [Google Scholar]
- 36.Stein A., Voigt W., Jordan K. Chemotherapy-induced diarrhea: pathophysiology, frequency and guideline-based management. Therapeutic Advances in Medical Oncology. 2010;2(1):51–63. doi: 10.1177/1758834009355164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kim D.-H., Jin Y.-H. Intestinal bacterial β-glucuronidase activity of patients with colon cancer. Archives of Pharmacal Research. 2001;24(6):564–567. doi: 10.1007/BF02975166. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fifty-nine candidate compounds were acquired from the initial virtually screening which was designed to target the bacterial loop of eβG and its active site. The docking energy scores of 59 candidate compounds measured by the DOCK program are -43 to -55 kcal/mol. (Table S1) The candidate compounds were purchased from SPECS (Zoetermeer, The Netherlands). Each candidate was rovided as a solid power and dissolved in 100% DMSO (Sigma-Aldrich, MO, USA) to 10 mM as stock. Candidates were screening for their inhibition specificity of eβG verse hβG, which were conducted at pH 7.3 or pH 5.4 in triplicate, respectively. 40 µL purified βG was treated with 10 µL compound solution at 37 °C for 30 min, and sequentially incubated with 50 µL of pNPG (Sigma-Aldrich) at 37 °C for 30 min. Reactions were quenched with 5 µL of 2 N sodium hydroxide (Sigma-Aldrich). Each reaction consisted of 3.75 ng purified βG, 50 µM compound, and 5 mM pNPG in PBS containing 10% DMSO and 0.05% BSA (Sigma-Aldrich). βG-activities were measured by color development of pNP detected on a microplate reader at OD 405 nm. Results are displayed as percent of βG activity compared with the untreated control. The result showed that all the 59 candidate compounds displayed selective inhibition against eβG activity. Especially, the inhibiting ability against eβG activity was >95% in 7 candidates of eβG specific inhibitors (Table S1).