

Abstract
Reversible changes in lung microstructure accompany lung inflammation, although alterations in tissue micromechanics and their impact on inflammation remain unknown. This study investigated changes in extracellular matrix (ECM) remodeling and tissue stiffness in a model of LPS-induced inflammation and examined the role of lipoxin analog 15-epi-lipoxin A4 (eLXA4) in the reduction of stiffness-dependent exacerbation of the inflammatory process. Atomic force microscopy measurements of live lung slices were used to directly measure local tissue stiffness changes induced by intratracheal injection of LPS. Effects of LPS on ECM properties and inflammatory response were evaluated in an animal model of LPS-induced lung injury, live lung tissue slices, and pulmonary endothelial cell (EC) culture. In vivo, LPS increased perivascular stiffness in lung slices monitored by atomic force microscopy and stimulated expression of ECM proteins fibronectin, collagen I, and ECM crosslinker enzyme, lysyl oxidase. Increased stiffness and ECM remodeling escalated LPS-induced VCAM1 and ICAM1 expression and IL-8 production by lung ECs. Stiffness-dependent exacerbation of inflammatory signaling was confirmed in pulmonary ECs grown on substrates with high and low stiffness. eLXA4 inhibited LPS-increased stiffness in lung cross sections, attenuated stiffness-dependent enhancement of EC inflammatory activation, and restored lung compliance in vivo. This study shows that increased local vascular stiffness exacerbates lung inflammation. Attenuation of local stiffening of lung vasculature represents a novel mechanism of lipoxin antiinflammatory action.
Keywords: endothelium, lung inflammation, vascular leak, stiffness, lipoxin
Clinical Relevance
Lipoxin post-treatment attenuates lung injury induced by bacterial pathogens. This study demonstrates a novel mechanism of the beneficial effects by lipoxin on acute lung injury recovery. Lipoxin abrogates the positive feedback escalation of lung inflammation and organ dysfunction via attenuation of extracellular matrix remodeling and perivascular tissue stiffening.
Acute respiratory distress syndrome remains a serious condition, with an overall mortality rate of 30 to 40% (1, 2) and limited selection of therapeutic options. Thus, further investigation into the mechanisms of acute respiratory distress syndrome remains a top priority for the cardiopulmonary field. The onset of lung injury is characterized by increased vascular permeability and expression of adhesive surface molecules (ICAM-1, VCAM-1, and E-selectin) by activated lung vascular endothelium, which promotes leukocyte adhesion, recruitment to the lung, and inflammatory cytokine production (3–5).
Transition from the acute phase of lung inflammation to lung recovery is accompanied by change in the spectrum of lipid mediators present in the lung. Lipoxin A4 (LXA4) belongs to a group of lipid mediators generated later in the course of acute lung injury (ALI) that suppress the inflammation phase and promote the resolution phase (reviewed in References 6 and 7). The LXA4 aspirin-triggered analog, 15-epi-LXA4 (eLXA4), shares antiinflammatory properties with native lipoxin, such as stimulation of Escherichia coli phagocytosis and inactivation of E. coli–induced cytokine production (8). In vivo induction of eLXA4 by statins markedly decreases acute lung inflammation and stimulates the resolution phase (9).
The molecular mechanisms of LXA4 antiinflammatory action are currently under investigation and include modulation of redox signaling (10), presqualene diphosphate turnover (11), inhibition of VEGF receptor-2–activated signaling, phospholipase C-γ, ERK1/2, and Akt kinases (12, 13). A recent study by Carlo and colleagues demonstrates inhibition of agonist-induced assembly of NADPH oxidase complex by eLXA4 and inhibition of superoxide anion generation by human neutrophils (14).
The decrease in lung compliance observed in patients and in animal models of lung dysfunction reflects significant alterations of lung tissue mechanical properties. Increased substrate stiffness contributes to endothelial barrier disruption induced by inflammatory mediators (15). Bacterial pathogens and bacteria-derived inflammatory compounds stimulate production of extracellular matrix (ECM) proteins by lung cells (16, 17) and trigger fibrin and ECM deposition in the human inflamed lungs (18, 19), which may increase lung tissue stiffness. However, the changes in micromechanical environment of lung vasculature and their contribution to the lung inflammatory process remain unclear. Although tissue remodeling and increased ECM deposition has been observed in the course of ALI, its effects on the changes in micromechanical environment of the lung vasculature and the potential contribution to the lung inflammatory process remain unknown.
In this study, we used models of ALI in vivo precision-cut live lung slices and pulmonary endothelial cells (ECs) to test the effect of eLXA4 on perivascular mechanical properties, modulation of ECM synthesis, and endothelial inflammatory activation induced by bacterial LPS. We tested the hypothesis that eLXA4 suppresses the LPS-induced ECM production and ECM stiffening mediated by activated vascular endothelium and that this effect of eLXA4 on lung vascular mechanical microenvironment contributes to the attenuation of endothelial inflammatory activation and lung injury.
Materials and Methods
Further details are provided in the online supplement.
Cell Culture
Human pulmonary artery ECs were obtained from Lonza (Allendale, NJ).
Preparation of Polyacrylamide Substrates for Cell Culture
Polyacrylamide substrates were prepared on glass coverslips with an acrylamide/bis-acrylamide ratio to obtain gels with shear elastic moduli of 1.5, 2.8, and 40 kPa. Collagen was covalently attached to the top surface of the polyacrylamide hydrogel as described elsewhere (20, 21).
Measurement of IL-8
IL-8 levels were determined in human pulmonary artery EC–conditioned media by ELISA (ELISA MAX Deluxe set; Biolegend, San Diego, CA) following the manufacturer’s protocol.
Immunofluorescence Staining
Endothelial monolayers were subjected to immunofluorescence staining as described (22). Slides were analyzed using a Nikon video imaging system (Nikon Instech Co., Tokyo, Japan). Images were processed with Image J software (National Institutes of Health, Washington, DC) and Adobe Photoshop 7.0 (Adobe Systems, San Jose, CA) software.
Western Blot
Protein extracts were separated by SDS-PAGE and transferred to polyvinylidene difluoride membranes, and the membranes were incubated with specific antibodies of interest.
Quantitative RT-PCR
Analysis of human and mouse fibronectin, collagen type I A1, and lysyl oxidase (LOX) mRNA expression was performed by quantitative real-time RT-PCR. Gene expression fold changes were calculated according to the ΔΔCt method (23). Procedure details are provided in the online supplement.
LOX Activity Assay
LOX activity in conditioned medium was measured using a LOX activity assay (AAT Bioquest, Sunnyvale, CA) according to the manufacturer’s instructions.
Human Lung Slices
Human donor lungs that could not be transplanted were obtained from deceased donors through the Gift of Hope/Regional Organ Bank of Illinois and were stored at 4°C for up to 2 days before use. Precision-cut lung slices were obtained as previously described (24).
Atomic Force Microscope Analysis of Vascular Stiffness in Live Lung Slices
Atomic force microscope (AFM) measurements were performed on fresh, fully hydrated lung tissue sections. Procedure details are provided in the online supplement.
Animal Studies
All experimental protocols involving the use of animals were approved by the University of Chicago Institutional Animal Care and Use Committee for the humane treatment of experimental animals. C57BL/6J mice were randomized to concurrently receive sterile saline solution or eLXA4 (100 μg/kg) by injection in the external jugular vein 5 hours after LPS challenge (0.83 mg/kg; E. coli O55:B5). After 48 hours of LPS challenge, animals were killed by exsanguination under anesthesia. Measurements of cell count, protein concentration, myeloperoxidase activity, Evans blue extravasation, and histological assessment of lung injury were conducted as described (25, 26).
Statistical Analysis
Results are expressed as means ± SD of three to eight independent experiments. Stimulated samples were compared with controls by unpaired Student’s t test. For multiple-group comparisons, one-way ANOVA and Tukey’s post hoc multiple-comparison test were used. P < 0.05 was considered statistically significant.
Results
eLXA4 Attenuates LPS-Induced Lung Inflammation, Vascular Leak, and Neutrophil Infiltration
Intratracheal instillation of LPS for 48 hours significantly increased protein content, increased total and neutrophil cell count in the BAL samples, and elevated myeloperoxidase activity in the lung tissue homogenates (Figures 1A–1D). A single intravenous injection of eLXA4 5 hours after intratracheal LPS instillation substantially attenuated all parameters of LPS-induced lung dysfunction, whereas administration of the lipoxin receptor inhibitor WRW4 diminished the protective effects of eLXA4.
Figure 1.

Effect of 15-epi-lipoxin A4 (eLXA4) on LPS-induced lung inflammation and barrier dysfunction. C57BL/6J mice were challenged with vehicle (Veh) or LPS (0.63 mg/kg, intratracheally) with or without eLXA4 post-treatment (100 μg/kg, intravenously, 5 h after LPS administration) with or without FPR2/ALX receptor inhibitor (FPR2i, 100 μg/kg). Control animals were treated with sterile saline solution. (A–C) Protein concentration (A), total cell count (B), and PMN cell count (C) were determined in bronchoalveolar lavage (BAL) fluid collected 48 hours after treatments. (D) Myeloperoxidase (MPO) activity was measured in lung homogenates as described in Materials and Methods. Data are expressed as mean ± SD. *P < 0.05 as compared with LPS treatment (n = 5). (E) Evans blue dye (30 ml/kg, intravenously) was injected 2 hours before termination of the experiment. Lung vascular permeability was assessed by Evans blue accumulation in the lung tissue. The quantitative analysis of Evans blue–labeled albumin extravasation was performed by spectrophotometric analysis of Evans blue extracted from the lung tissue samples. *P < 0.05 (n = 5). (F) Histological analysis of lung tissue by hematoxilin and eosin staining (original magnification: ×40).
LPS-induced lung vascular leak was detected by Evans blue dye accumulation in the lung parenchyma. Images of original lung preparations depicting Evans blue extravasation are shown in Figure 1E (upper panel). Post-treatment with eLXA4 decreased the Evans blue accumulation in the lungs of LPS-exposed mice. Quantitative analysis of Evans blue–labeled albumin extravasation further confirmed these results (Figure 1E, lower panel). Morphological analysis of lung injury by hematoxylin and eosin staining was used as an additional parameter of neutrophilic infiltration and inflammation and showed marked attenuation of LPS-induced lung inflammation by eLXA4 (Figure 1F).
eLXA4 Attenuates LPS Effects on Lung Stiffness
We measured transthoracic static compliance, which is an index of lung edema caused by ALI. Pressure–volume curves were generated in saline-treated mice, in LPS-treated mice, and in mice treated with LPS followed by eLXA4 post-treatment 5 hours after LPS. Lung volume in experimental animals was measured beginning at ≥ 30 cm H2O, which corresponded to total lung capacity. Composite changes in transthoracic static compliance were derived from the linear portion of the pressure–volume curves in each group. Transthoracic static compliance was measured at 10 cm H2O (Figure 2A). Static compliance decreased by −0.16 ± 0.02 cm H2O after LPS treatment. This reduction in transthoracic static compliance caused by LPS was attenuated to −0.05 ± 0.012 cm H2O in mice post-treated with eLXA4 (P < 0.01 versus LPS-treated mice) (Figure 2A).
Figure 2.

Effect of eLXA4 on LPS-induced changes in the lung compliance and perivascular stiffness. C57BL/6J mice were challenged with vehicle or LPS (0.83 mg/kg, intratracheally) with or without eLXA4 post-treatment (100 μg/kg, intravenously, 5 h after LPS administration). Control animals were treated with sterile saline solution. (A) Measurements of lung compliance were performed using the Flexivent system as described in Materials and Methods. eLXA4 reduced transthoracic static compliance caused by intratracheal administration of LPS. Measurements are means ± SE at 10 cm H2O for each group. *P < 0.05 compared with LPS-treated mice (n = 3). (B) Representative bright field microscopy images of precision-cut live lung slices before the atomic force microscope (AFM) measurements. AW, airway. The vessel cross-section areas selected for AFM probe application are shown by arrows. (C) Elastic modulus measurements in precision-cut lung slices from control and LPS-treated mice were performed using the AFM approach described in Materials and Methods. Cantilever spring constant, k = 63 pN/nm (determined by thermal resonance method). Typical recordings are presented. Forces were measured in a PBS environment using the same probe and identical parameters for all measurements. Shown are representative force curves for each condition. The plot represents, for each of the three conditions, measurements from at least five different vessels on at least two different tissue sections. Indenter depth was 500 nm or less, for which over 500 stable nanoindentations were taken for each condition. (D) Statistical analysis of elastic modulus measurements. The filled square is the mean, the line above or below is the median, the box represents the 25th to 75th percentile of data, and the vertical line shows the data range. Origin8 Data Analysis software was used for this analysis.
Using precision-cut lung slices, we directly evaluated local changes in lung vascular viscoelastic properties. Cross-sections of blood capillaries were identified by bright field microscopy of live lung slices, and the AFM cantilever was aimed at these regions shown in Figure 2B. Peak force was reached during 50-μm indentations. Indenter depth was 500 nm or less, for which over 500 stable nanoindentations were taken for each condition. The raw AFM force data from precision-cut lung slices obtained from control mice and from LPS-treated and LPS + eLXA4–treated mice are shown in Figure 2C. The AFM-determined elastic modulus of the microvessel walls from the lungs of LPS-treated mice was dramatically increased (42.12 ± 15.142 kPa versus 3.293 ± 1.177 kPa in control; P < 0.001), whereas eLXA4 post-treatment returned the elastic modulus of the vessel wall to the levels in the untreated mouse lung (3.959 ± 2.028 kPa) (Figure 2D).
eLXA4 Attenuates LPS-Increased Synthesis of ECM Proteins and Expression of LOX, ICAM1, and VCAM1
The AFM studies and lung compliance measurements demonstrated LPS-induced lung stiffening. Because increased ECM production contributes to this effect, we performed comprehensive analysis of lung tissue in vivo and ex vivo and of pulmonary ECs. Intratracheal injection of LPS increased expression of the ECM component fibronectin, which was detected in the lung tissue samples (Figure 3A, left panel). This increase was accompanied by increased expression of endothelial surface adhesion molecules VCAM1 and ICAM1, indicating endothelial inflammatory activation (Figure 3A, right panel). We next analyzed these parameters in the cultured precision-cut human lung slices exposed to LPS with and without eLXA4 post-treatment. LPS increased protein expression of fibronectin and ICAM1 (Figure 3B). LPS also increased expression of LOX, the enzyme involved in ECM crosslinking leading to tissue stiffening (27). These changes were attenuated in the LPS-challenged lung slices post-treated with eLXA4. Treatment with lipoxin receptor inhibitor WRW4 before eLXA4 abolished its protective effect. Finally, we measured ICAM1, VCAM1, fibronectin, and LOX expression in human pulmonary EC cultures challenged with LPS for 48 hours with or without 5 hours post-treatment with eLXA4. Similarly to lung tissue, LPS-induced up-regulation of ECM and cell adhesion proteins was blunted by eLXA4 in the cultured cells (Figure 3C). These data also demonstrate involvement of lung vascular endothelium in ECM remodeling.
Figure 3.

Effect of eLXA4 on LPS-induced expression of fibronectin (FN), lysyl oxidase (LOX), ICAM1, and VCAM1. (A) C57BL/6J mice were challenged with vehicle or LPS with or without eLXA4 post-treatment. (B and C) Human lung slices (B) and human pulmonary artery ECs (HPAECs) (C) were stimulated with LPS (200 nM) followed by eLXA4 post-treatment (100 nM, 5 h after LPS). After 48 hours, expression of FN, LOX, ICAM1, and VCAM1 was determined by Western blot analysis with specific antibodies. Equal protein loading was confirmed by membrane reprobing with β-actin antibody. Shown are representative results of four independent experiments.
The effects of LPS on the expression of fibronectin, LOX, and the other major ECM protein, collagen-1A, were further verified by quantitative RT-PCR. Consistent with Western blot results, LPS significantly increased fibronectin, LOX, and collagen-1A mRNA levels in the mouse lungs and cultured pulmonary ECs (Figure 4). This effect was attenuated by administration of eLXA4 5 hours after LPS challenge.
Figure 4.

Effect of eLXA4 on LPS-induced mRNA expression of fibronectin, collagen-1A, and LOX. (A) C57BL/6J mice were challenged with vehicle or LPS with or without eLXA4 post-treatment (5 h after LPS administration). (B) HPAECs were stimulated with LPS (200 ng/ml) in culture followed by eLXA4 post-treatment (100 nM, 5 h after LPS). Analysis of FN, collagen 1A (Col1A), and LOX mRNA levels in lung and cell samples was performed by RT-PCR as described in Materials and Methods. *P < 0.05 as compared with LPS alone (n = 6).
eLXA4 Attenuates LPS-Induced Activation of LOX and ECM Deposition by Pulmonary ECs
Precision-cut human lung slices were cultured for 48 hours with or without LPS. eLXA4 was added to selected slices 5 hours after LPS administration. Similarly, human pulmonary EC cultures were exposed to LPS with or without eLXA4 post-treatment. LOX enzymatic activity was measured in conditioned medium. Increased LOX activity was observed in LPS-treated lung slices and EC cultures and was attenuated by eLXA4 post-treatment (Figures 5A and 5B). In control experiments, cotreatment with the LOX inhibitor β-aminopropionitrile attenuated LPS-induced LOX activation. Pharmacological inhibitor of lipoxin FPR2 receptor abolished the protective effects of eLXA4 against LPS-induced LOX activation.
Figure 5.

Effect of eLXA4 on LPS-induced LOX enzymatic activation and fibronectin deposition. (A and B) LOX enzymatic activity in preconditioned medium of human lung slices (A) and –HPAECs (B) treated with LPS, LPS + LOX inhibitor β-aminopropionitrile, LPS + eLXA4, or LPS + FPRi + eLXA4. *P < 0.05 versus LPS alone. (C) Pulmonary ECs were treated with LPS alone or LPS followed by post-treatment with eLXA4 (5 h after LPS). Actin cytoskeleton staining of control and treated EC monolayers was performed using Texas Red phalloidin (top panel). Fibronectin deposition was visualized on coverslips after cell detachment by staining with fibronectin antibody (middle panel). ICAM1 expression was examined by cell staining with ICAM1 antibody (bottom panel).
LPS-induced endothelial activation was reflected by an increased number of actin stress fibers and formation of paracellular gaps in the LPS-stimulated EC monolayers (Figure 5C, top panel). LPS-induced ECM deposition was further evaluated using immunofluorescence staining with fibronectin antibody. Fibronectin bundles deposited by cultured ECs were visualized by fibronectin-specific antibody (Figure 5C, middle panel) after detachment of cell monolayers using EGTA treatment as described in Materials and Methods. LPS markedly increased overall antifibronectin immunoreactivity and caused the appearance of thicker fibronectin bundles. These LPS effects were attenuated by EC post-treatment with eLXA4. These results show that extracellular deposition of fibronectin by cultured ECs was stimulated by LPS and attenuated by eLXA4. Immunofluorescence detection of ICAM1 showed that LPS induced ICAM1 expression by pulmonary ECs, which was abolished by eLXA4 cotreatment (Figure 5C, bottom panel).
Increased Matrix Deposition by LPS-Treated Pulmonary ECs Enhances TNF-α–Induced ICAM1 Expression and IL-8 Production
These studies tested whether increased matrix deposition by LPS-stimulated ECs affects cell response to proinflammatory cytokines. In these experiments, control or LPS-challenged pulmonary ECs were cultured for 3 days on compliant polyacrylamide substrates (2.8 kPa) to allow for ECM deposition. Then, cells were detached using a 15-minute incubation with sterile EGTA, the deposited matrix was additionally washed with sterile PBS, and fresh pulmonary ECs were seeded on predeposited matrix. After cell attachment and spreading, freshly seeded EC cultures were stimulated with TNF-α for 4 hours. Cells reseeded on matrix produced by LPS-stimulated ECs responded to TNF-α by increased ICAM1 and VCAM1 expression (Figure 6A). Increased ECM deposition by initial EC culture challenged with LPS is reflected by increased fibronectin immunoreactivity in the total lysates, as compared with β-actin normalization control (Figure 6A, bottom panels). ECs seeded on the matrix deposited by LPS-stimulated cells also showed increased IL-8 expression in response to TNF-α (Figure 6B).
Figure 6.

Extracellular matrix (ECM) deposition by LPS-treated ECs exacerbates TNF-α–induced activation of naive ECs. Human pulmonary ECs were cultured on polyacrylamide hydrogels (2.8 kPa) for 3 days with or without LPS (200 ng/ml). After gentle cell detachment and washing cycles, naive ECs were plated on deposited ECM and stimulated with TNF-α (2 ng/ml, 4 h). (A) ICAM1, VCAM1, and FN protein levels in total SDS samples collected from each well were analyzed by Western blot. (B) IL-8 production in response to TNF-α (2 ng/ml, 4 h) by naive ECs plated on ECM deposited by control and LPS-stimulated cells was evaluated by ELISA assay. *P < 0.05 (n = 5). (C) ECs were cultured for 3 days with or without LPS or with LPS and LOX inhibitor β-aminopropionitrile (LOXi; 100 μM). TNF-α–induced ICAM1 expression by naive ECs plated on deposited ECM was evaluated by Western blot of total lysates. Equal protein loading was confirmed by membrane reprobing with β-actin antibody. Shown are representative results of four independent experiments.
In the next series of experiments, primary seeded ECs were grown in the presence of LPS with or without the LOX irreversible inhibitor β-aminopropionitrile. After 3 days of culture, the cells were detached as described above, and fresh pulmonary ECs were reseeded onto predeposited matrix and stimulated with TNF-α. The LOX inhibitor attenuated enhancement of TNF-α–induced ICAM1 expression by predeposited matrix (Figure 6C). These results indicate that LPS induces activation of LOX in addition to stimulation of ECM expression by pulmonary ECs, suggesting the importance of LOX-mediated ECM crosslinking leading to ECM stiffening, which is likely required for full inflammatory activation of TNF-α–treated fresh cells grown on this preformed matrix.
Increased Matrix Stiffness Augments EC Inflammatory Response and Paracellular Gap Formation Caused by TNF-α and LPS
Results of experiments described above prompted us to test the effects of substrate stiffness on EC responses to inflammatory agonists directly. EC monolayers grown on soft (1.5 kPa) and stiff (40 kPa) polyacrylamide gels were stimulated with TNF-α or LPS for 4 hours, and ICAM1 and VCAM1 expression and IL-8 production were evaluated. Cells grown on stiff substrates responded to TNF-α and LPS by higher levels of ICAM1 and VCAM1 expression and IL-8 production by stimulated ECs (Figures 7A and 7B).
Figure 7.
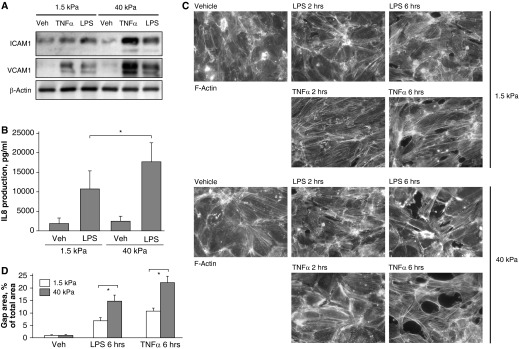
Effect of substrate stiffness on TNF-α– and LPS-induced activation of ICAM1 and VCAM1 expression, IL-8 production, and actin cytoskeleton remodeling. Pulmonary ECs were grown on polyacrylamide gels of different stiffness (1.5 and 40 kPa) and treated with TNF-α (2 ng/ml) or LPS (200 ng/ml) for 4 hours. (A) Expression of ICAM1 and VCAM1 was determined by Western blot analysis. Equal protein loading was confirmed by membrane reprobing with β-actin antibody. (B) IL-8 production in response to LPS was evaluated by ELISA assay. *P < 0.05 (n = 6). (C) F-actin was visualized after 2 and 6 hours by immunofluorescence staining with Texas Red–conjugated phalloidin. (D) Quantitative analysis of paracellular gap formation in control and treated HPAECs. Data are expressed as mean ± SD of four independent experiments. *P < 0.05 (n = 3).
A more pronounced inflammatory response by cells grown on a stiffer substrate was also associated with a higher level of barrier disruption in EC monolayers upon LPS and TNF-α stimulation, which was monitored by F-actin immunofluorescence staining with Texas Red phalloidin (Figure 7C) and quantitative analysis of paracellular gap formation (Figure 7D).
Discussion
This study demonstrates for the first time the local changes in perivascular stiffness, which developed in inflamed lungs. We performed over 30 independent AFM measurements in different areas of the microvessel tissue cross-section for each experimental condition. The remarkably reproducible differences between local tissue stiffness in the lung slices from control, LPS-treated, and LPS + LXA4–treated animals reflect local mechanical properties of microvascular tissue defined by deposited matrix and/or swollen capillary walls in LPS-treated mouse lungs. This report is also the first demonstration of inflammation enhancement by increased local tissue stiffness. LPS-induced increases in lung perivascular stiffness from LPS-treated mice determined in the AFM study were accompanied by increased fibronectin and collagen-1 expression and LOX activity in three models: the lungs of LPS-treated mice, LPS-challenged human lung slices ex vivo, and human pulmonary ECs. These data indicate that LPS-activated vascular cells produce ECM, which alters local mechanical microenvironment in the inflamed microvasculature. Although it was not investigated in this study, other vascular cells (smooth muscle cells and fibroblasts) may also participate in the LPS-induced vascular remodeling in addition to vascular endothelium. LOX activation stimulates crosslinking of collagen fibers, leading to the formation of more rigid ECM scaffolds (28, 29). Increased LOX activity was observed in tumors and fibrotic tissues and was associated with increased stiffness of ECM (27, 30).
The direct effects of substrate stiffness on EC inflammatory activation were measured using polyacrylamide hydrogels, revealing enhancement of TNF-α–induced ICAM1 and VCAM1 expression and IL-8 production by ECs grown on stiff substrates. LPS-induced ECM production and deposition by cultured ECs observed in this study as well as activation of LOX suggests increased production of matrix with increased stiffness. This possibility was addressed by analysis of inflammatory responses of naive ECs reseeded on ECM deposited by LPS-stimulated human pulmonary ECs. Our results showed that (1) ECM formed by LPS-challenged ECs exacerbated ICAM1 and IL-8 production by naive ECs in response to TNF-α and (2) this exacerbation effect was significantly attenuated by ECM deposition in the presence of LOX inhibitor. Because these experiments were performed on hydrogels of physiologically relevant stiffness (31), the observed effects of LOX inhibition strongly suggest the role of endogenous ECM stiffening in the potentiation of EC inflammation.
The mechanisms of stiffness-dependent regulation of inflammation remain unclear. The magnitude of RhoA GTPase–mediated signaling activation is dependent on substrate stiffness and may determine the path of stem cell differentiation (32), myofibroblast differentiation (33), or endothelial permeability response to agonists (15). On the other hand, Rho signaling is essential for full activation of LPS-induced inflammatory signaling cascades (34–36). This study shows the enhancement of EC inflammatory activation by LPS-induced ECM production and LOX-dependent ECM crosslinking leading to ECM stiffening. Because increased ECM stiffness enhances agonist-induced activation of RhoA signaling, which in turn modulates the inflammatory response to LPS, we speculate that stiffness-dependent enhancement of RhoA may be a plausible mechanism of stiffness-dependent augmentation of vascular leak and inflammation in the model of lung dysfunction induced by LPS or other inflammatory stimuli. This intriguing question requires further investigation.
The second major finding of this study is attenuation of LPS-induced lung tissue stiffening and inflammation by LXA4. Such protective effects of intravenous LXA4 injection in the animal ALI model was achieved even 5 hours after LPS challenge. LXA4 abrogated LPS-induced activation of ECM protein expression as well as expression and activation of LOX. These data suggest the novel role for LXA4 in down-regulation of lung tissue remodeling and restoration of lung vascular mechanical microenvironment. We also cannot exclude the direct effects of LXA4 on LPS-induced cytokine production and inflammatory adhesion molecule expression. However, enhanced inflammatory activation of ECs grown on stiff substrate and the inhibitory effects of LXA4 on ECM production and lung perivascular stiffness support the mechanism of inflammatory signaling modulation by LXA4 via change in lung tissue micromechanics.
In summary, these results demonstrate a novel modality of substrate stiffness in the modulation of the EC inflammatory response. LPS or other inflammatory interventions activate inflammatory cascades in ECs but also stimulate deposition of ECM and activation of LOX activity, leading to local tissue stiffening. In turn, increased ECM stiffness further exacerbates inflammatory activation of lung vascular endothelium, reflected by increased expression of ICAM1 and VCAM1 and production of IL-8. This feedback mechanism leads to escalation of vascular inflammation and ALI but may be intercepted by administration of LXA4. Thus, LXA4 exhibits multifaceted modalities in suppressing inflammation and promoting lung repair, including a novel mechanism for control of vascular endothelial inflammation by changes in lung micromechanics. We speculate that disrupted control of inflammation and ECM micromechanics normally performed by LXA4 and other proresolving lipid mediators may lead to the development of fibroproliferative acute respiratory distress syndrome, which is associated with poor prognosis and high mortality.
Acknowledgments
Acknowledgments
The authors thank Tera Lavoie and Julian Solway for assistance with preparations of precision-cut lung slices and valuable advice on lung tissue culturing and Jacob Riehm for excellent technical assistance in mounting the tissue sections for the AFM analysis.
Footnotes
This work was supported by National Institutes of Health grants HL89257, HL107920, HL76259, and NS067247 and by an American Asthma Foundation Early Excellence Award.
This article has an online supplement, which is accessible from this issue's table of contents at www.atsjournals.org
Originally Published in Press as DOI: 10.1165/rcmb.2013-0468OC on July 3, 2014
Author disclosures are available with the text of this article at www.atsjournals.org.
References
- 1.Ricard JD, Dreyfuss D, Saumon G. Ventilator-induced lung injury. Eur Respir J Suppl. 2003;42:2s–9s. doi: 10.1183/09031936.03.00420103. [DOI] [PubMed] [Google Scholar]
- 2.Matthay MA, Zimmerman GA, Esmon C, Bhattacharya J, Coller B, Doerschuk CM, Floros J, Gimbrone MA, Jr, Hoffman E, Hubmayr RD, et al. Future research directions in acute lung injury: summary of a national heart, lung, and blood institute working group. Am J Respir Crit Care Med. 2003;167:1027–1035. doi: 10.1164/rccm.200208-966WS. [DOI] [PubMed] [Google Scholar]
- 3.Frank JA, Matthay MA. Science review: mechanisms of ventilator-induced injury. Crit Care. 2003;7:233–241. doi: 10.1186/cc1829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lionetti V, Recchia FA, Ranieri VM. Overview of ventilator-induced lung injury mechanisms. Curr Opin Crit Care. 2005;11:82–86. doi: 10.1097/00075198-200502000-00013. [DOI] [PubMed] [Google Scholar]
- 5.Maniatis NA, Orfanos SE. The endothelium in acute lung injury/acute respiratory distress syndrome. Curr Opin Crit Care. 2008;14:22–30. doi: 10.1097/MCC.0b013e3282f269b9. [DOI] [PubMed] [Google Scholar]
- 6.Serhan CN, Chiang N. Endogenous pro-resolving and anti-inflammatory lipid mediators: a new pharmacologic genus. Br J Pharmacol. 2008;153:S200–S215. doi: 10.1038/sj.bjp.0707489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Haworth O, Levy BD. Endogenous lipid mediators in the resolution of airway inflammation. Eur Respir J. 2007;30:980–992. doi: 10.1183/09031936.00005807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Prescott D, McKay DM. Aspirin-triggered lipoxin enhances macrophage phagocytosis of bacteria while inhibiting inflammatory cytokine production. Am J Physiol Gastrointest Liver Physiol. 2011;301:G487–G497. doi: 10.1152/ajpgi.00042.2011. [DOI] [PubMed] [Google Scholar]
- 9.Planaguma A, Pfeffer MA, Rubin G, Croze R, Uddin M, Serhan CN, Levy BD. Lovastatin decreases acute mucosal inflammation via 15-epi-lipoxin a4. Mucosal Immunol. 2010;3:270–279. doi: 10.1038/mi.2009.141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Nascimento-Silva V, Arruda MA, Barja-Fidalgo C, Fierro IM. Aspirin-triggered lipoxin a4 blocks reactive oxygen species generation in endothelial cells: a novel antioxidative mechanism. Thromb Haemost. 2007;97:88–98. [PubMed] [Google Scholar]
- 11.Bonnans C, Levy BD. Lipid mediators as agonists for the resolution of acute lung inflammation and injury. Am J Respir Cell Mol Biol. 2007;36:201–205. doi: 10.1165/rcmb.2006-0269TR. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Baker N, O'Meara SJ, Scannell M, Maderna P, Godson C. Lipoxin a4: anti-inflammatory and anti-angiogenic impact on endothelial cells. J Immunol. 2009;182:3819–3826. doi: 10.4049/jimmunol.0803175. [DOI] [PubMed] [Google Scholar]
- 13.El Kebir D, Jozsef L, Pan W, Wang L, Petasis NA, Serhan CN, Filep JG. 15-epi-lipoxin a4 inhibits myeloperoxidase signaling and enhances resolution of acute lung injury. Am J Respir Crit Care Med. 2009;180:311–319. doi: 10.1164/rccm.200810-1601OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Carlo T, Kalwa H, Levy BD. 15-epi-lipoxin a4 inhibits human neutrophil superoxide anion generation by regulating polyisoprenyl diphosphate phosphatase 1. FASEB J. 2013;27:2733–2741. doi: 10.1096/fj.12-223982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Birukova AA, Tian X, Cokic I, Beckham Y, Gardel ML, Birukov KG. Endothelial barrier disruption and recovery is controlled by substrate stiffness. Microvasc Res. 2013;87:50–57. doi: 10.1016/j.mvr.2012.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hagiwara S, Iwasaka H, Matsumoto S, Noguchi T, Yoshioka H. Coexpression of hsp47 gene and type i and type iii collagen genes in lps-induced pulmonary fibrosis in rats. Lung. 2007;185:31–37. doi: 10.1007/s00408-006-0085-1. [DOI] [PubMed] [Google Scholar]
- 17.Chao MC, Garcia CS, de Oliveira MB, Santos RS, Lucas IH, Silva PL, Vieira-Abreu A, de Castro-Faria-Neto HC, Parra-Cuentas ER, Capelozzi VL, et al. Degree of endothelium injury promotes fibroelastogenesis in experimental acute lung injury. Respir Physiol Neurobiol. 2010;173:179–188. doi: 10.1016/j.resp.2010.08.005. [DOI] [PubMed] [Google Scholar]
- 18.Wygrecka M, Jablonska E, Guenther A, Preissner KT, Markart P. Current view on alveolar coagulation and fibrinolysis in acute inflammatory and chronic interstitial lung diseases. Thromb Haemost. 2008;99:494–501. doi: 10.1160/TH07-11-0666. [DOI] [PubMed] [Google Scholar]
- 19.Burnham EL, Janssen WJ, Riches DW, Moss M, Downey GP. The fibroproliferative response in acute respiratory distress syndrome: mechanisms and clinical significance. Eur Respir J. 2014;43:276–285. doi: 10.1183/09031936.00196412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Aratyn-Schaus Y, Oakes PW, Stricker J, Winter SP, Gardel ML. Preparation of complaint matrices for quantifying cellular contraction. J Vis Exp. 2010;46:2173. doi: 10.3791/2173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yeung T, Georges PC, Flanagan LA, Marg B, Ortiz M, Funaki M, Zahir N, Ming W, Weaver V, Janmey PA. Effects of substrate stiffness on cell morphology, cytoskeletal structure, and adhesion. Cell Motil Cytoskeleton. 2005;60:24–34. doi: 10.1002/cm.20041. [DOI] [PubMed] [Google Scholar]
- 22.Birukova AA, Fu P, Xing J, Yakubov B, Cokic I, Birukov KG. Mechanotransduction by gef-h1 as a novel mechanism of ventilator-induced vascular endothelial permeability. Am J Physiol Lung Cell Mol Physiol. 2010;298:L837–L848. doi: 10.1152/ajplung.00263.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Pfaffl MW. A new mathematical model for relative quantification in real-time rt-pcr. Nucleic Acids Res. 2001;29:e45. doi: 10.1093/nar/29.9.e45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lavoie TL, Krishnan R, Siegel HR, Maston ED, Fredberg JJ, Solway J, Dowell ML. Dilatation of the constricted human airway by tidal expansion of lung parenchyma. Am J Respir Crit Care Med. 2012;186:225–232. doi: 10.1164/rccm.201202-0368OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Birukova AA, Fu P, Chatchavalvanich S, Burdette D, Oskolkova O, Bochkov VN, Birukov KG. Polar head groups are important for barrier protective effects of oxidized phospholipids on pulmonary endothelium. Am J Physiol Lung Cell Mol Physiol. 2007;292:L924–L935. doi: 10.1152/ajplung.00395.2006. [DOI] [PubMed] [Google Scholar]
- 26.Fu P, Birukova AA, Xing J, Sammani S, Murley JS, Garcia JG, Grdina DJ, Birukov KG. Amifostine reduces lung vascular permeability via suppression of inflammatory signalling. Eur Respir J. 2009;33:612–624. doi: 10.1183/09031936.00014808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Levental KR, Yu H, Kass L, Lakins JN, Egeblad M, Erler JT, Fong SF, Csiszar K, Giaccia A, Weninger W, et al. Matrix crosslinking forces tumor progression by enhancing integrin signaling. Cell. 2009;139:891–906. doi: 10.1016/j.cell.2009.10.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kagan HM, Li W. Lysyl oxidase: properties, specificity, and biological roles inside and outside of the cell. J Cell Biochem. 2003;88:660–672. doi: 10.1002/jcb.10413. [DOI] [PubMed] [Google Scholar]
- 29.Karsdal MA, Nielsen MJ, Sand JM, Henriksen K, Genovese F, Bay-Jensen AC, Smith V, Adamkewicz JI, Christiansen C, Leeming DJ. Extracellular matrix remodeling: the common denominator in connective tissue diseases. Possibilities for evaluation and current understanding of the matrix as more than a passive architecture, but a key player in tissue failure. Assay Drug Dev Technol. 2013;11:70–92. doi: 10.1089/adt.2012.474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Adam O, Theobald K, Lavall D, Grube M, Kroemer HK, Ameling S, Schafers HJ, Bohm M, Laufs U. Increased lysyl oxidase expression and collagen cross-linking during atrial fibrillation. J Mol Cell Cardiol. 2011;50:678–685. doi: 10.1016/j.yjmcc.2010.12.019. [DOI] [PubMed] [Google Scholar]
- 31.Stroka KM, Aranda-Espinoza H. Endothelial cell substrate stiffness influences neutrophil transmigration via myosin light chain kinase-dependent cell contraction. Blood. 2011;118:1632–1640. doi: 10.1182/blood-2010-11-321125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Engler AJ, Sen S, Sweeney HL, Discher DE. Matrix elasticity directs stem cell lineage specification. Cell. 2006;126:677–689. doi: 10.1016/j.cell.2006.06.044. [DOI] [PubMed] [Google Scholar]
- 33.Huang X, Yang N, Fiore VF, Barker TH, Sun Y, Morris SW, Ding Q, Thannickal VJ, Zhou Y. Matrix stiffness-induced myofibroblast differentiation is mediated by intrinsic mechanotransduction. Am J Respir Cell Mol Biol. 2012;47:340–348. doi: 10.1165/rcmb.2012-0050OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Xing J, Birukova AA. Anp attenuates inflammatory signaling and rho pathway of lung endothelial permeability induced by lps and tnfalpha. Microvasc Res. 2010;79:26–62. doi: 10.1016/j.mvr.2009.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.He F, Peng J, Deng XL, Yang LF, Wu LW, Zhang CL, Yin F. Rhoa and nf-kappab are involved in lipopolysaccharide-induced brain microvascular cell line hyperpermeability. Neuroscience. 2011;188:35–47. doi: 10.1016/j.neuroscience.2011.04.025. [DOI] [PubMed] [Google Scholar]
- 36.Guo F, Zhou Z, Dou Y, Tang J, Gao C, Huan J. Gef-h1/rhoa signalling pathway mediates lipopolysaccharide-induced intercellular adhesion molecular-1 expression in endothelial cells via activation of p38 and nf-kappab. Cytokine. 2012;57:417–428. doi: 10.1016/j.cyto.2011.12.009. [DOI] [PubMed] [Google Scholar]