

Abstract
Asbestos causes asbestosis and malignancies by mechanisms that are not fully established. Alveolar epithelial cell (AEC) injury and repair are crucial determinants of the fibrogenic potential of noxious agents such as asbestos. We previously showed that mitochondrial reactive oxygen species mediate asbestos-induced AEC intrinsic apoptosis and that mitochondrial human 8-oxoguanine-DNA glycosylase 1 (OGG1), a DNA repair enzyme, prevents oxidant-induced AEC apoptosis. We reasoned that OGG1 deficiency augments asbestos-induced pulmonary fibrosis. Compared with intratracheal instillation of PBS (50 μl) or titanium dioxide (100 μg/50 μl), crocidolite or Libby amphibole asbestos (100 μg/50 μl) each augmented pulmonary fibrosis in wild-type C57BL/6J (WT) mice after 3 weeks as assessed by histology, fibrosis score, lung collagen via Sircol, and type 1 collagen expression; these effects persisted at 2 months. Compared with WT mice, Ogg1 homozygous knockout (Ogg1−/−) mice exhibit increased pulmonary fibrosis after crocidolite exposure and apoptosis in cells at the bronchoalveolar duct junctions as assessed via cleaved caspase-3 immunostaining. AEC involvement was verified by colocalization studies using surfactant protein C. Asbestos increased endoplasmic reticulum stress in the lungs of WT and Ogg1−/− mice. Compared with WT, alveolar type 2 cells isolated from Ogg1−/− mice have increased mtDNA damage, reduced mitochondrial aconitase expression, and increased P53 and cleaved caspase-9 expression, and these changes were enhanced 3 weeks after crocidolite exposure. These findings suggest an important role for AEC mtDNA integrity maintained by OGG1 in the pathogenesis of pulmonary fibrosis that may represent a novel therapeutic target.
Keywords: asbestosis, pulmonary fibrosis, mitochondria, 8-oxyguanosine DNA glycosylase
Clinical Relevance
We show that asbestos-induced pulmonary fibrosis in mice deficient in 8-oxoguanine-DNA glycosylase 1 (OGG1) is augmented in comparison to their wild-type counterparts due to increased mitochondrial (mt) DNA damage, decreased alveolar epithelial cell (AEC) mitochondrial aconitase-2 (ACO-2) levels, and increased AEC apoptosis. We reason that the prevention of asbestos-induced AEC mtDNA damage and apoptosis by mitochondrial OGG1 and ACO-2 may be an innovative target for the molecular events underlying asbestosis and possibly other degenerative diseases.
Asbestos-related lung diseases remain a common public health problem worldwide. Exposure to asbestos fibers has been epidemiologically linked to pulmonary fibrosis (asbestosis), pleural abnormalities, and malignancies such as bronchogenic carcinoma and mesothelioma (1–3). Although much is known about the mechanisms by which asbestos fibers cause pulmonary toxicity, the detailed molecular pathways are not fully understood. The long latency period (15–40 yr) between asbestos exposure and lung disease presents an opportunity to prevent the progression from exposure to disease, but there are no proven treatment regimens. Ineffective repair of damaged alveolar epithelial cells (AECs) and AEC apoptosis are implicated in mediating pulmonary fibrosis in humans with idiopathic pulmonary fibrosis (IPF) and in animal models of fibrotic lung disease, including asbestosis (for reviews, see References 4–7). Genetic approaches targeting apoptosis of alveolar epithelial type 2 (AT2) cells in mice and humans demonstrate an important role for AECs in mediating pulmonary fibrosis (8, 9). Asbestos fibers are internalized by AECs and inflammatory cells soon after exposure, resulting in the production of iron-derived free reactive oxygen species (ROS), DNA damage, and apoptosis (1, 3, 7).
Oxidative stress causes a multiplicity of DNA base adducts, the most abundant of which is 8-oxo-7,8-dihydroxyguanine (8-oxoG). In replicating cells, 8-oxoG can pair with adenine instead of cytosine, resulting in transversion mutations, which are implicated in ageing, neurodegenerative diseases, and cancer (3, 10). Inefficient repair of mitochondrial DNA (mtDNA) damage and mutations lead to mitochondrial dysfunction, mitochondrial ROS production, intrinsic apoptosis, and inflammatory signaling, which may be important in neoplastic transformation and pulmonary fibrosis (6, 11). In lung mesothelial cells, mtDNA is severalfold more sensitive to crocidolite asbestos–induced DNA damage than nuclear DNA (12). Our group previously reported that oxidative stress caused by asbestos fibers (exogenous) or H2O2 (endogenous) induces AEC mitochondrial ROS production, mtDNA damage, mitochondrial dysfunction, P53 activation, and intrinsic apoptosis (13–16). In mitochondria, the base excision repair (BER) pathway is primarily responsible for 8-oxoG mtDNA damage repair, ensuring long-term cell survival (17). Human 8-oxyguanine DNA glycosylase 1 (hOGG1) is a bifunctional BER protein that recognizes and removes 8-oxoG in the DNA located in the mitochondria and in the nucleus (18). Homozygous OGG1 knockout (Ogg1−/−) mice have substantial accumulations of 8-oxoG in the mtDNA and, to a lesser degree, the nuclear DNA but are otherwise phenotypically normal under regular breeding conditions (19–22). We showed that overexpression of a wild-type (WT) mitochondria-targeted human 8-oxoguanine-DNA glycosylase 1 (mt-hOGG1 WT) and a mitochondria-targeted long α/β 317–323 hOGG1 mutant incapable of DNA repair (mt-hOGG1 MUT) prevents oxidant (amosite asbestos or H2O2)-induced intrinsic AEC apoptosis, despite high levels of mitochondrial ROS stress, by maintaining mtDNA integrity in part because of a novel function of OGG1 chaperoning mitochondrial aconitase (ACO-2) from oxidative degradation (13, 16). Collectively, these findings suggest an important role for OGG1 in mitigating AEC mtDNA damage and apoptosis in the setting of oxidative stress as occurs after asbestos exposure.
In this study, we hypothesized that mice deficient in OGG1 are more prone to pulmonary fibrosis after asbestos exposure than their WT (C57BL/6J) counterparts due in part to increased AEC mtDNA damage and apoptosis. We show that, as compared with a single intratracheal instillation of PBS or titanium dioxide (TiO2), crocidolite or Libby amphibole asbestos fibers increased pulmonary fibrosis in WT mice as expected and that Ogg1−/− mice exhibit increased fibrosis. Furthermore, crocidolite asbestos augmented apoptosis in cells at the bronchoalveolar duct (BAD) junctions as assessed via cleaved caspase-3 (CC-3) immunostaining, and AEC involvement was verified by colocalization with surfactant protein C (SFTPC). Compared with AT2 cells isolated from WT mice, AT2 cells from Ogg1−/− mice have increased mtDNA damage, reduced ACO-2 expression, and increased P53 expression at baseline, and these changes were enhanced after crocidolite exposure for 3 weeks. Taken together, these findings suggest a crucial role for AEC OGG1 maintenance of mtDNA in preventing AEC apoptosis in the pathogenesis of pulmonary fibrosis.
Materials and Methods
Mice
Male, 8- to 10-week-old C57BL/6J mice (Jackson Labs, Bar Harbor, ME) and Ogg1−/− mice (backcrossed 10 generations on a C57BL/6J background; details are provided in the online supplement) were used for all experiments described herein. All the animal studies were approved by the Institutional Animal Use and Care Committees (IACUC) at Northwestern University and the Jesse Brown VA Medical Center. We confirmed that all of our Ogg1−/− mice were Ogg1 knockouts based upon PCR on genomic DNA obtained from tail samples of Ogg1−/− and WT mice to identify an 800-bp product of the Ogg1 gene as compared with the 377-bp product in WT mice, indicating that the Ogg1 gene is disrupted as expected (Figures 1A and 1B) (23). We also showed that the OGG1 protein (37 kD) present in AT2 cell homogenates from WT mice was absent from Ogg1−/− knockout mice (Figure 1A). The experimental protocol is shown in Figure 1C. Briefly, WT or Ogg1−/− mice were instilled intratracheally (details are provided in the online supplement) with control (PBS [50 μl] or TiO2 [100 μg in 50 μl]) or amphibole asbestos fibers (see below; 100 μg in 50 μl), and the lungs were harvested after 3 weeks or 2 months for various end-points, including (1) type-1 collagen by Western blotting, (2) Sircol collagen assay, (3) CC-3 and SFTPC immunohistochemistry (IHC), and (4) fibrosis score, as described below.
Figure 1.
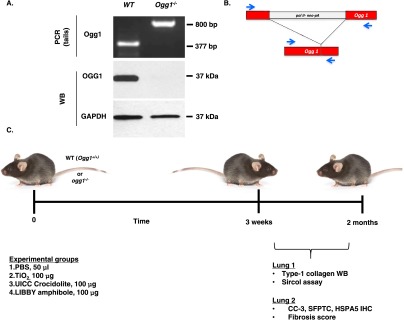
Phenotypic validation of 8-oxoguanine DNA glycosylase knockout (Ogg1−/−) mice and experimental design. (A) Genomic PCR of wild-type (WT) and Ogg1−/− tail DNA: 800 bp Ogg1−/− and 377 bp Ogg1+/+ (WT) amplification product. AT2 cells from WT and Ogg1−/− mice verifying lack of OGG1 protein in Ogg1−/− knockout mice. (B) Representation of Ogg1−/− gene knockout. Modified from Sakumi and colleagues (23). (C) Experimental timeline and scheme. Mouse image is from The Jackson Laboratory 2014, mouse model ID 664, C57BL/6J (used with permission). CC-3, cleaved caspase-3; HSPA5, heat shock protein 5; SFTPC, surfactant protein C.
Reagents
Union International Centre le Cancer (UICC) reference standard crocidolite asbestos fibers and Libby amphibole fibers were provided by Andy Ghio (US Environmental Protection Agency) (asbestos fiber characterization is provided in the online supplement) and handled for intratracheal instillation into mice as described in the online supplement.
Cell Culture
Primary mouse AT2 cells were isolated from the lungs of WT or Ogg1−/− mice as previously described (14, 24). Primary isolated AT2 cells were plated in 6-well plates (1 × 106 cells/well) and grown to confluence over 24 hours before harvesting protein extracts for Western blotting and, in separate experiments, obtaining nuclear DNA and mtDNA for a PCR-based DNA damage assay (see below).
Lung Protein Extraction and Western Blotting
Lung tissue lysates were collected for lung homogenization in cell lysis buffer (Cell Signaling, Danvers, MA), and immunobloting was performed as described previously (13, 25) using specific antibodies directed against OGG1 (Novus, Denver, CO), antiaconitase 2 antibody (Abcam, Cambridge, UK), anti–CC-3 (Cell Signaling), CC-9 (Cell Signaling), Pro-SFTPC (EMD Millipore Billerica, MA), type-1 collagen (Southern Biotech, Birmingham, AL), P53 (Santa Cruz Biotech, Santa Cruz, CA), and COXIV (Cell Signaling).
Fibrosis Scores and Lung Collagen Determination
Fibrosis scores in WT and Ogg1−/− mice were determined from Masson’s trichrome–stained specimens by an investigator (A.Y.Y.) blinded to the code according to the Pathology Standards for Asbestosis, as we have previously described (26). Collagen levels were determined by a Sircol assay and Western blotting for type-1 collagen. Further details are provided in the online supplement.
Semiquantitative Analysis of CC-3, SFTPC, and Heat Shock Protein 5 IHC
In each lung, the number of CC-3–positive AECs were counted per alveolar branch point and expressed as number of positive cells per 220 AECs ± SEM from two or more animals in each group. For CC-3–SFTPC colocalization studies, all CC-3–positive cells in fibrotic lesions or at the BAD were counted and assessed for SFTPC status on the corresponding area of a serial SFTPC IHC slide. The fraction of CC-3–positive cells that is also SFTPC positive is expressed as a percentage of approximately 200 cells assessed for each condition on serial sections as previously described (33). CC-3–heat shock protein 5 (HSPA5) colocalization studies were conducted on corresponding areas of serial CC-3 and HSPA5 IHC slides. The fraction of HSPA5-positive cells and the fraction of CC-3 cells that were also HSPA5 positive were calculated and expressed as described above for the CC-3–SFTPC colocalization studies.
PCR-Based Mitochondrial mtDNA Damage Assay
Genomic DNA, including nuclear and mtDNA, was assessed by quantitative PCR as described elsewhere (16, 31). Mitochondrial small fragment fluorescence values were used for normalization of the large fragment values. The number of mitochondrial lesions was calculated by the following equation
Statistical Analysis
Data were expressed as the means ± SEM (n = 6 unless otherwise stated) and analyzed as detailed in the online supplement.
Results
Asbestos-Induced Pulmonary Fibrosis Is Augmented in Ogg1−/− Mice as Compared with WT Mice
To confirm that asbestos induces pulmonary fibrosis in C57BL/6J WT mice, we compared intratracheally instilled PBS (Control, vehicle; 50 μl), TiO2 (Control particulate; 100 μg in 50 μl PBS), and amphibole asbestos fibers (UICC crocidolite or Libby amphibole fibers; 100 μg in 50 μl PBS) after a 3-week or 2-month exposure period. As expected, crocidolite asbestos induced significant destruction of the normal lung architecture that was most prominent at the BAD junctions. Compared with our negative controls (PBS or TiO2), crocidolite asbestos augmented lung fibrosis in WT mice after a 3-week exposure period as assessed by lung histology (Figure 2A), Masson’s trichrome staining of collagen (Figure 3A), lung tissue fibrosis score (0 ± 0 versus 0 ± 0 versus 5.0 ± 0.3, respectively; P < 0.05) (Figure 4A), and lung collagen levels as assessed by a Sircol assay (0.31 ± 0.05 versus 0.24 ± 0.02 versus 0.59 ± 0.03 mg/ml, respectively; P < 0.05 by ANOVA) (Figure 4B) and Western blotting of type-1 collagen seen as a 129- to 131-kD doublet (Figure 4C). Compared with crocidolite asbestos, mice exposed to Libby amphibole fibers for 3 weeks induced worse lung fibrosis as assessed by the fibrosis score (4 ± 0.4 versus 7.5 ± 0.5, respectively; P < 0.05), Sircol (0.48 ± 0.03 versus 0.74 ± 0.06 mg/ml, respectively; P < 0.05), and type-1 collagen expression by Western blotting (see Figures E1 and E2 in the online supplement). The lung fibrotic changes after crocidolite exposure noted at 3 weeks persisted at 2 months (Figure E3).
Figure 2.

Compared with WT mice, asbestos-induced pulmonary fibrosis at 3 weeks is more severe in Ogg1−/− mice. Three weeks after intratracheal instillation with PBS, TiO2, or crocidolite asbestos, serial mouse lung sections were stained with hematoxylin and eosin. WT (A) and Ogg1−/− (B) mice after treatment (upper row, scale bar = 0.1 mm; lower row, scale bar = 0.05 mm). Square on upper row shows enlargement in lower row.
Figure 3.

Compared with WT mice, pulmonary fibrosis after asbestos exposure at 3 weeks is more severe in Ogg1−/− mice (continued). Mice were treated with PBS, TiO2, or crocidolite asbestos as described in Figure 2. Serial lung sections were subject to Masson’s trichrome stain. WT (A) and Ogg1−/− (B) mice after treatment (upper row, scale bar = 0.1 mm; lower row, scale bar = 0.05 mm). Square on upper row shows enlargement in lower row. Collagen fibers are stained blue.
Figure 4.

Lung fibrosis scores and collagen levels are increased in Ogg1−/− versus WT mice exposed to TiO2 or crocidolite asbestos. WT and Ogg1−/− mice were treated with TiO2 or crocidolite asbestos. Three weeks after intratracheal instillation, serial lung sections were scored for fibrosis or whole lungs subject to Sircol assay or type-1 collagen Western blotting. (A) Lung fibrosis scores. *P < 0.05, PBS or TiO2; †P < 0.05, WT versus Ogg1−/−; n WT + PBS = 6, n WT + TiO2 = 9, n WT + CROC = 10, n Ogg1−/− + PBS = 6, n Ogg1−/− + TiO2 = 8, n Ogg1−/− + CROC = 10. (B) Collagen levels assessed via Sircol assay. *P < 0.05, PBS or TiO2; †P < 0.05, WT versus Ogg1−/−; n B6 + PBS = 6, n WT + TiO2 = 9, n WT + CROC = 10, n Ogg1−/− + PBS = 6, Ogg1−/− + TiO2 = 8, Ogg1−/− + CROC = 10. Fibrosis score = [(Severity 0–4) × (Extent 1–3)]. (C) Western blot of type-I collagen aftere exposure to crocidolite asbestos for 21 days. The 129- to 131-kD doublet is marked with double arrows.
Having established that pulmonary fibrosis is consistently evident at 3 weeks after asbestos exposure in our murine model, we determined if Ogg1−/− mice are more susceptible to asbestosis as compared with WT mice 3 weeks after intratracheal instillation of crocidolite asbestos. Similar to WT mice after intratracheal instillation of PBS or TiO2, Ogg1−/− mice have normal-appearing lung architecture (Figures 2A and 2B, left and center panels). Compared with WT mice, we also detected negligible increases in lung collagen deposition in Ogg1−/− mice after intratracheal instillation of PBS or TiO2 as assessed by Masson’s trichrome staining (Figures 3A and 3B, left and center panels), the lung fibrosis score (PBS: 0 ± 0 versus 0 ± 0, respectively; TiO2: 0 ± 0 versus 0 ± 0, respectively) (Figure 4A), and lung collagen deposition assessed by the Sircol assay (PBS: 0.31 ± 0.05 versus 0.31 ± 0.06 mg/ml, respectively; TiO2: 0.24 ± 0.02 versus 0.36 ± 0.03 mg/ml, respectively) (Figure 4B) or type-1 collagen expression (Figure 4C). Compared with our negative controls (PBS or TiO2), Ogg1−/− mice exposed to intratracheally instilled crocidolite asbestos for 3 weeks induced destruction of the normal lung architecture that was most prominent at the BAD junctions (Figure 2, right panel) and augmented collagen deposition (Figure 3, right panel), which is characteristic of fibrotic lung injury. As compared with WT mice, Ogg1−/− mice exhibited more fibrosis after crocidolite exposure as assessed by lung tissue fibrosis score (5.0 ± 0.3 versus 7.0 ± 0.5, respectively; P < 0.05) (Figure 4A) and lung collagen levels as assessed by a Sircol assay (0.59 ± 0.03 versus 0.86 ± 0.02 mg/ml, respectively; P < 0.05) (Figure 4B) and Western blotting of type-I collagen (Figure 4C). Taken together, these findings show that asbestosis is significantly augmented in Ogg1−/− mice as compared with WT mice 3 weeks after a single intratracheal instillation of crocidolite asbestos.
Crocidolite Asbestos-Induced AEC Apoptosis in Cells at the BAD Junction at 3 Weeks Is Augmented in Ogg1−/− Mice
Ineffective repair of damaged AEC and apoptosis is implicated in the pathobiology of pulmonary fibrosis in humans and animals (4–8). Asbestos-induced fibrotic lesions and apoptotic AECs are typically seen initially at the BAD junctions where fiber deposition is most evident (32). To determine if apoptotic cells can be found in fibrotic lung lesions in our model, we investigated the presence of CC-3 in cells present at the BAD junctions using IHC and semiquantitative analysis of WT and Ogg1−/− mouse lung sections 3 weeks after a single intratracheal instillation of PBS, TiO2, or crocidolite asbestos. Unlike our negative controls, in which CC-3–positive cells were < 1% of 220 cells assessed at the BAD junctions, we observed a significant increase in CC-3–positive cells in our crocidolite-treated WT and Ogg1−/− mice (Figures 5A and 5B). Using semiquantitative analysis, Ogg1−/− mice exposed to crocidolite asbestos had more CC-3–positive cells than WT (15.8 ± 0.8% versus 11.8 ± 0.7% CC-3–positive cells per 220 cells at the BAD junctions, respectively; P < 0.05).
Figure 5.

Asbestos-induced CC-3 activation in cells at the bronchoalveolar duct (BAD) junction is augmented in Ogg1−/− versus WT mice and colocalization of CC-3–positive BAD cells and AECs (SFTPC positive) in asbestos-exposed mice. Lungs from WT and Ogg1−/− mice were harvested 3 weeks after intratracheal instillation of PBS, TiO2 or crocidolite asbestos, subject to CC-3 or SFTPC immunohistochemistry (IHC) on serial sections. (A) Anti–CC-3 IHC in WT mice (top row) or Ogg1−/− mice (bottom row). Scale bar = 0.05 mm. (B) Semiquantitative analysis of 220 cells at the BAD junctions. *P < 0.05, PBS or TiO2; †P < 0.05, WT versus Ogg1−/−. n WT + PBS = 3, n WT + TiO2 = 3, n WT + CROC = 3, n Ogg1−/− + PBS = 3, n Ogg1−/− + TiO2 = 3, n Ogg1−/− + CROC = 3. (C) IHC on serial sections for CC-3 and SFTPC. Two regions are shown. Red arrows mark double-positive cells; yellow arrows mark CC-3–positive, SFTPC-negative cells. Square on upper row shows enlargement in lower row. Scale bar = 0.08 mm.
To determine whether AT2 cells are apoptotic in our Ogg1−/− mice, we performed colocalization IHC studies using CC-3 (a marker of apoptosis) and SFTPC (a marker of AT2 cells) (33) on serial sections of crocidolite-exposed Ogg1−/− mouse lungs. Figure 5C shows two regions of serial CC-3–SFTPC IHC staining demonstrating apoptotic AECs (positive for CC-3 and SFTPC; red arrows) as well as non-AECs (positive for CC-3 and negative for SFTPC; yellow arrows). Semiquantitative analysis of approximately 200 cells showed that AT2 cells accounted for nearly 40% of the apoptotic cells. These findings show that AT2 cell apoptosis occurs in the BAD junctions of asbestos-exposed Ogg1−/− mouse lungs and that non-AECs also undergo apoptosis.
As summarized recently (5, 7), mounting evidence convincingly shows that endoplasmic reticulum (ER) stress occurs in AECs undergoing apoptosis in subjects with IPF. Diverse fibrotic stimuli, including bleomycin, viruses, and misfolded mutant surfactant proteins, can induce an ER stress response that results in AEC apoptosis (9, 27–29). Because we recently showed that asbestos fibers can induce AEC ER stress that results in intrinsic apoptosis in vitro (30), we determined whether ER stress was present in WT and Ogg1−/− mouse lungs 3 weeks after a single intratracheal instillation of TiO2 or crocidolite asbestos. We used IHC to assess HSPA5, a protein involved in modulating ER stress, and whether HSPA5 colocalized with apoptotic (+CC-3 staining) cells present at the BAD junctions. TiO2 induced negligible HSPA5 expression in the lungs of WT and Ogg1−/− mice (Figure E4). Similar to apoptosis (Figure 5B), TiO2-induced HSPA5 expression occurred in < 1% of cells at the BAD junction in WT and Ogg1−/− mice. In contrast to TiO2, crocidolite-exposed mice demonstrated ER stress in apoptotic cells (positive for CC-3 and HSPA5; yellow arrows) and demonstrated ER stress in cells not undergoing apoptosis (negative for CC-3 and positive for HSPA5). Semiquantitative analysis of approximately 200 cells at the BAD junctions in WT and Ogg1−/− mice revealed that crocidolite asbestos augmented HSPA5 activation in apoptotic cells (12.9 and 16.1%, respectively) and in nonapoptotic cells (84.0 and 92.0%, respectively). These findings show that asbestos fibers induce ER stress in cells at the BAD junctions, including apoptotic cells, in WT and Ogg1−/− mice.
mtDNA Damage Is Augmented in AECs Isolated from Lungs of Ogg1−/− Mice versus WT
As reviewed elsewhere (6, 7), accumulating evidence implicates a direct association between mtDNA damage and intrinsic apoptosis in many cell types. We recently showed that AEC mtDNA damage is an important determinant of asbestos-induced apoptosis and that OGG1 and ACO-2 modulate mtDNA damage (16). However, the role of mtDNA damage in mediating AEC apoptosis in vivo is unclear. To investigate whether mtDNA damage occurs in AT2 cells, we exposed WT and Ogg1−/− mice to intratracheally instilled crocidolite or TiO2 for 3 weeks and then isolated primary AT2 cells for assessment of mitochondria and nuclear DNA damage using a quantitative PCR–based measurement (16, 31). We detected negligible nuclear DNA damage as assessed by reductions in nuclear DNA copy number or number of lesions per 10 kB after exposure to TiO2 or crocidolite asbestos (Figure 6). In contrast, reduced mtDNA copy number and increased mtDNA lesions were evident in WT mice exposed to crocidolite and in Ogg1−/− mice exposed to TiO2 or crocidolite (Figure 6). AT2 cells from WT mice had significant mtDNA damage after crocidolite exposure as compared with TiO2 exposure (0.056 ± 0.007 versus 0.0 ± 0 lesions/10 kb, respectively]; P < 0.05 [n = 4]) (Figure 6). AT2 cells from Ogg1−/− mice exposed to TiO2 exhibited mtDNA damage, and AT2 cell mtDNA damage was augmented further in the crocidolite asbestos–exposed Ogg1−/− mice (0.073 ± 0.011 and 0.105 ± 0.005 lesions/10 kb, respectively; P < 0.05 [n = 4]). These data show that the loss of OGG1 augments AT2 cell mtDNA damage under basal conditions and that crocidolite asbestos augments mtDNA damage.
Figure 6.

Mitochondrial but not nuclear DNA damage is increased in AT2 cells isolated from Ogg1−/− mice after TiO2 exposure (negative control) and augmented further after asbestos exposure. WT and Ogg1−/− mice were given a single intratracheal instillation of TiO2 or crocidolite asbestos. Three weeks after exposure, AT2 cells were isolated from lungs and used for a fluorescent PCR-based nuclear DNA or mtDNA damage assay. (A) Nuclear and mitochondrial DNA damage, relative fluorescence ratio. (B) Nuclear and mitochondrial DNA relative ratio converted to ratio DNA lesions/10 kb DNA: mitochondrial small fragment fluorescence values were used for normalization of the large fragment values (31). The number of mitochondrial lesions was calculated as described in the Materials and Methods section. *P < 0.05 versus WT TiO2. †P < 0.05 versus WT CROC. §P < 0.05 versus Ogg1−/− TiO2.
AT2 Cells from Ogg1−/− Mice Have Reduced ACO-2 Expression and Increased P53 and CC-9 Expression
We previously showed a novel role for mtOGG1 in chaperoning ACO-2 from oxidative degradation in vitro that accounted for the protective effects of mtOGG1 against oxidant-induced AEC mtDNA damage and intrinsic apoptosis (13, 16). We have also reported that P53 modulates asbestos-induced AEC mitochondria-regulated apoptosis and that OGG1 and ACO-2 can regulate this response (15, 16). There is evidence linking P53, OGG1, and ACO-2 expression, but the molecular mechanisms involved and whether asbestos-exposed AT2 cells behave similarly in vivo are unclear (7, 8). Crocidolite asbestos reduced ACO-2 levels by > 50% as compared with TiO2 in AT2 cells from WT mice (0.44 ± 0.03 versus 1.0 ± 0 relative density units, respectively; P < 0.05 [n = 3]) (Figure 7A). As compared with AT2 cells from WT mice, ACO-2 protein expression was reduced in AT2 cells from Ogg1−/− mice after exposure to TiO2 (1.0 ± 0 versus 0.39 ± 0.11 relative density units, respectively; P < 0.05 [n = 3]) or crocidolite (1.0 ± 0 versus 0.30 ± 0.08 relative density units, respectively; P < 0.05 [n = 3]). Although ACO-2 levels in AT2 cells from Ogg1−/− mice exposed to TiO2 or crocidolite were lower than from AT2 cells from WT mice after crocidolite exposure, these differences did not reach statistical significance. AT2 cells isolated from WT mice 3 weeks after a single intratracheal instillation of crocidolite asbestos had increased P53 expression as compared with TiO2 (1.55 ± 0.16 versus 1.0 ± 0 relative density units, respectively; P < 0.05 [n = 4]) and marked activation of cleaved caspase-9 (CC-9) (7.24 ± 0.03 versus 1.0 ± 0 relative density units, respectively; P < 0.05 [n = 3]) (Figures 7B and 7C). AT2 cells isolated from Ogg1−/− mice after a single intratracheal instillation of crocidolite asbestos had increased P53 expression as compared with TiO2 (3.90 ± 0.59 versus 1.92 ± 0.48 relative density units, respectively; P < 0.05 [n = 3]) and when compared with AT2 cells from WT mice exposed to crocidolite (3.90 ± 0.0.59 versus 1.55 ± 0 relative density units, respectively; P < 0.05 [n = 3]). Compared with AT2 cells from WT mice, CC-9 activation was augmented in AT2 cells from Ogg1−/− mice after exposure to TiO2 or crocidolite (1.0 ± 0 versus 7.17 ± 0.011 versus 8.35 ± 0.82, respectively; P > 0.05 [n = 3]), although the difference between TiO2 and crocidolite in Ogg1−/− mice did not reach statistical significance. Collectively, these data demonstrate that the loss of OGG1 reduces AT2 cell ACO-2 levels in vivo under basal conditions after TiO2 exposure as compared with AT2 cells from WT mice and that OGG1 deficiency augments asbestos-induced CC-9 and P53 expression 3 weeks after crocidolite asbestos exposure.
Figure 7.

Asbestos decreases AT2 cell mitochondrial ACO-2 expression levels and augments P53 and intrinsic apoptosis. AT2 cells were isolated from the lungs of WT and Ogg1−/− mice 3 weeks after exposure to TiO2 or crocidolite asbestos as described in Figure 6 and then assessed for ACO-2, P53, and CC-9 protein expression. (A) Representative Western blot and a densitometric quantification of ACO-2 expression from mitochondrial fractions of AT2 cells from three experiments. *P < 0.01 versus WT + TiO2. †P < 0.001 versus WT + TiO2 (n = 3). (B) Representative P53 and CC-9 protein expression from AT2 cell extracts. (C) Densitometric analysis of P53 (n = 4) and CC-9 (n = 3) protein levels. *P < 0.05 versus WT + TiO2. †P < 0.05 versus WT + CROC. §P < 0.05 versus Ogg1−/− + TiO2. (D) Hypothetical model (see text for details).
Discussion
The major findings of this study are that, as compared with WT mice, Ogg1−/− mice that are deficient in 8-oxo-G oxidative DNA damage repair have increased pulmonary fibrosis and AEC apoptosis after crocidolite asbestos exposure. Furthermore, compared with TiO2, crocidolite asbestos fibers induce greater levels of mtDNA damage in primary isolated AT2 cells from WT mice, and mtDNA damage is greater than nuclear DNA damage. Finally, as compared with AT2 cells from WT mice, AT2 cells from Ogg1−/− mice have decreased ACO-2 levels and increased mtDNA damage at baseline, and these deleterious effects are augmented after crocidolite asbestos exposure, accompanied by increased P53 expression and intrinsic apoptosis. Collectively, these findings suggest that OGG1 deficiency augments AEC mtDNA damage and apoptosis in vivo that appear crucial for the development of pulmonary fibrosis after asbestos exposure.
An important finding of this study is that, as compared with WT mice, pulmonary fibrosis in Ogg1−/− mice is worse 3 weeks after a single intratracheal instillation of crocidolite asbestos as assessed by histology (Figures 2 and 3) and quantified by the lung fibrosis score (Figure 4A) and biochemical measures (Sircol [Figure 4B] and type 1 collagen levels [Figure 4C]). These changes persist 2 months after asbestos instillation (Figure E3). The finding of a relationship between depletion of OGG1, an 8-oxo-G DNA BER enzyme, and pulmonary fibrosis after asbestos exposure is novel. Our data showing that the lack of OGG1 promotes asbestos-induced pulmonary fibrosis add to the growing list of studies showing OGG1’s potential protective role against diverse diseases, including various cancers (e.g., colorectal [34)] lung [35], and otolaryngeal [36]), cardiac fibrosis (37), Barrett’s esophagus (38), type 2 diabetes mellitus (39), obesity/metabolic syndrome (40), and neurodegenerative diseases such as Parkinson’s disease (41) and Huntington’s disease (42). Our findings in the lungs of Ogg1−/− mice are also in accord with the enhanced age-related nigrostriatal pathway loss and motor defects noted in Ogg1−/− mice as compared with their WT counterparts (43). Taken together, these findings implicate an important role of OGG1 in DNA repair in the prevention of diverse diseases, including pulmonary fibrosis. In this study, we used a single intratracheal instillation dose of crocidolite asbestos (100 μg) that is well established in causing murine lung fibrosis. A limitation of the present study is the use of a single dose of asbestos. Future studies assessing the effects of low-dose amphibole asbestos fibers over extended exposure periods will be of interest.
Although the detailed molecular mechanisms by which OGG1 knockout mice develop increased pulmonary fibrosis after asbestos exposure requires additional study, we explored several possibilities. First, we determined if apoptotic AECs could be detected in fibrotic lung lesions given the crucial role of AEC apoptosis in the pathogenesis of pulmonary fibrosis and that mitochondrial OGG1 overexpression can attenuate asbestos-induced AEC apoptosis in vitro (1, 2, 5–9, 13, 16). As compared with controls (PBS and TiO2), crocidolite asbestos augmented apoptotic cells at the BAD junctions over 25-fold in WT mice (Figures 5A and 5B: 0.3 versus 0.45 versus 11.8% CC-3–positive cells, respectively), and asbestos-induced apoptosis at the BAD junctions was further increased in Ogg1−/− mice (15.8%). Colocalization IHC studies confirmed that approximately 40% of the apoptotic cells at the BAD junctions were of AT2 lineage (Figure 5C). Further, unlike TiO2, crocidolite asbestos induced marked ER stress (HSPA5) in cells at the BAD junctions, including apoptotic cells, in WT and Ogg1−/− mouse lungs (Figure E4). Our findings implicating AEC apoptosis in augmenting pulmonary fibrosis after asbestos exposure in the Ogg1−/− mice are in accord with an accumulating body of evidence, reviewed in detail elsewhere (1, 2, 5–9), that ineffective repair of damaged AECs and AEC apoptosis are key events underlying pulmonary fibrosis in humans with IPF and in animal models of fibrotic lung disease, including asbestosis. Our findings are concordant with a recent study in humans with Hermansky-Pudlak syndrome showing that the alveolar epithelium determines susceptibility to lung fibrosis (44). An important role for the alveolar epithelium is further supported by the finding that targeted damage of AT2 cells appears necessary for triggering murine pulmonary fibrosis and that alveolar deposition of AT2 cells, but not alveolar macrophages, attenuates bleomycin-related fibrosis (8, 45). Moreover, our findings of an ER stress response in apoptotic cells in fibrotic lungs are in accord with numerous groups showing ER stress in apoptotic AECs in vitro and in vivo (27–30). Our data do not exclude a role for other cells, such as inflammatory cells/macrophages, in modulating the fibrotic response noted in our model. Emerging evidence suggests that human induced pluripotent stem cells and Clara cells may be important for AT2 cell differentiation after lung injury (46). Although the causal role of AECs and/or other cell types in mediating pulmonary fibrosis after asbestos exposure is unclear from the present study, AEC-targeted OGG1 over- and underexpression studies would better inform our understanding of the role of the alveolar epithelium.
A second mechanism that we explored in this study is whether OGG1 deficiency promotes mtDNA damage in AT2 cells. We found that crocidolite asbestos fibers induce greater levels of mtDNA in primary isolated AT2 cells from WT mice 3 weeks after exposure and that mtDNA damage is greater than nuclear DNA damage (Figure 6). Compared with WT mice, AT2 cells from Ogg1−/− mice had greater mtDNA damage 3 weeks after TiO2 exposure and even greater crocidolite-induced AT2 cell mtDNA damage (Figure 6). Our findings are in accord with studies showing that oxidative mtDNA damage is more abundant and long lasting than nuclear DNA damage presumably because of the proximity of mtDNA to the electron transport chain and because mtDNA lacks histones, which are protective against ROS (47, 48). Our in vivo studies reported herein also concur with in vitro studies by others as well as our group showing that persistent mtDNA damage triggers intrinsic apoptosis, including AECs exposed to asbestos fibers in vitro (16, 49). The present study suggesting that asbestos-induced AEC mtDNA damage triggers pulmonary fibrosis parallels work by others showing that mtDNA damage is implicated in the pathophysiology of diverse conditions, such as atherosclerosis, cardiac fibrosis/heart failure, diaphragmatic dysfunction from mechanical ventilation, and cancer (34–37, 50–52). Mitochondria-targeted OGG1 attenuates ventilator-induced lung injury in mice in part by decreasing the levels of mtDNA damage in the lungs (53). Thus, our data showing asbestos-induced AEC mtDNA damage in the setting of pulmonary fibrosis add to the accumulating evidence that implicates ongoing mtDNA damage in the development of a wide array of diseases.
A third mechanism investigated herein to account for the increased susceptibility of Ogg1−/− mice to asbestos-induced pulmonary fibrosis is whether AEC ACO-2 levels are reduced. ACO-2 is a mitochondrial tricarboxylic acid cycle enzyme that can act as a biosensor for oxidative stress and preserves mtDNA, independent of its tricarboxylic acid activity in yeast (54, 55). Compared with primary isolated AT2 cells from WT mice, AT2 cells from the Ogg1−/− mice have decreased ACO-2 levels 3 weeks after a single intratracheal instillation of TiO2 (Figure 7). A single intratracheal instillation of crocidolite asbestos reduced ACO-2 expression in AT2 cells from WT mice, and AT2 cell ACO-2 levels were further reduced in Ogg1 −/− mice, although this latter difference did not reach statistical significance (Figure 7A). Moreover, alterations in AT2 cell ACO-2 expression occur in association with increased P53 expression and intrinsic apoptosis (increased CC-9 levels). These in vivo findings parallel recent in vitro work by our group demonstrating that mt-hOGG1 and ACO-2 act in concert to preserve AEC mtDNA integrity in the setting of oxidative stress (asbestos or H2O2), thereby preventing mitochondrial dysfunction, P53 mitochondrial expression, and intrinsic apoptosis (13, 16). Using immunoprecipitation techniques, we previously reported a novel chaperone function of ACO-2 by mt-hOGG1–WT and an mt-hOGG1–Mut incapable of mtDNA repair (13). Also, OGG1 and ACO-2 over- and under-gene expression of studies showed that both are important for preserving AEC mtDNA integrity and mitigating intrinsic apoptosis (13, 16). Collectively, these findings suggest that OGG1/ACO-2 may be novel therapeutic targets important for ameliorating oxidant-induced mtDNA damage, which, if left inadequately repaired, can result in AEC apoptosis and pulmonary fibrosis.
Our findings of P53 activation in WT and Ogg1−/− mice exposed to asbestos are consistent with a P53 DNA damage response (Figure 7). These findings are in accord with recent work from our group showing that overexpression of mt-hOGG1 WT, mt-hOGG1 Mut, and ACO-2 in AECs attenuate the P53 DNA damage response (16). Our data are also in agreement with studies showing that P53 activation is required for oxidant-induced apoptosis in hOGG1-deficient human fibroblasts (56) and that P53 can sensitize HepG2 cells to oxidative stress by reducing mtDNA (57). Accumulating studies suggest a key association between P53, OGG1, and ACO-2, including (1) P53 regulates OGG1 gene transcription in colon and renal epithelial cells (56), (2) P53-deficient cells have reduced OGG1 protein expression and activity (56), and (3) P53 can reduce ACO-2 gene expression and activity (58, 59). Mitochondrial P53 can, under certain conditions, promote cell survival or death by promoting mtDNA repair (60–62). Furthermore, P53 levels are detected in lung cancers of patients exposed to asbestos and tobacco (63), and P53 point mutations are present in the lung epithelium of asbestos-exposed individuals (64). Whole-genome gene profiling studies in lung epithelial and mesothelial cells confirm that P53 activation and a DNA damage response occurs that may play a crucial role in the regulation of tumor suppression and the control of cell death or survival in the setting of asbestos exposure (65, 66). Collectively, these findings support an important role for P53 in the profibrotic lung response after asbestos exposure, which has implications for our understanding of the malignant potential of asbestos fibers. However, determining the precise molecular mechanisms by which P53, OGG1, and ACO-2 coordinately regulate mtDNA integrity in AECs and how this affects cell fate requires further study.
In summary, we show that asbestos-induced pulmonary fibrosis is augmented in Ogg1−/− mice as compared with their WT counterparts. Moreover, pulmonary fibrosis after asbestos exposure in WT mice is associated with an increased number of apoptotic cells at the BAD junctions, an ER stress response, decreased AT2 cell ACO-2 levels, and increased mtDNA damage, P53 expression, and CC-9 expression, all of which are augmented in Ogg1−/− mice. Our findings implicate a novel role for mitochondrial OGG1 and ACO-2 in preserving AEC mtDNA integrity in the setting of oxidative stress resulting from asbestos exposure that induces an AEC P53 DNA damage response and intrinsic apoptosis and pulmonary fibrosis. A hypothetical model summarizing the key events studied herein is shown in Figure 7D. We reason that the prevention of asbestos-induced AEC mtDNA damage and apoptosis by mitochondrial OGG1 chaperoning of ACO-2 may be an innovative target for the molecular events underlying asbestos-induced toxicity as occurs in the setting of pulmonary fibrosis and possibly other degenerative diseases.
Footnotes
This work was supported by National Institutes of Health grants RO1ES020357 (D.W.K.), R01HL071643 (K.R.), and NCICA0605531 and by a VA Merit award (D.W.K.).
This article has an online supplement, which is accessible from this issue's table of contents at www.atsjournals.org
Originally Published in Press as DOI: 10.1165/rcmb.2014-0038OC on June 11, 2014
Author disclosures are available with the text of this article at www.atsjournals.org.
References
- 1.Mossman BT, Lippmann M, Hesterberg TW, Kelsey KT, Barchowsky A, Bonner JC. Pulmonary endpoints (lung carcinomas and asbestosis) following inhalation exposure to asbestos. J Toxicol Environ Health. 2011;14:76–121. doi: 10.1080/10937404.2011.556047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kamp DW. Asbestos-induced lung diseases: an update. Transl Res. 2009;153:143–152. doi: 10.1016/j.trsl.2009.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Huang SX, Jaurand MC, Kamp DW, Whysner J, Hei TK. Role of mutagenicity in asbestos fiber-induced carcinogenicity and other diseases. J Toxicol Environ Health B Crit Rev. 2011;14:179–245. doi: 10.1080/10937404.2011.556051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.King TE, Jr, Pardo A, Selman M. Idiopathic pulmonary fibrosis. Lancet. 2011;378:1949–1961. doi: 10.1016/S0140-6736(11)60052-4. [DOI] [PubMed] [Google Scholar]
- 5.Noble PW, Barkauskas CE, Jiang D. Pulmonary fibrosis: patterns and perpetrators. J Clin Invest. 2012;122:2756–2762. doi: 10.1172/JCI60323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cheresh P, Kim SJ, Tulasiram S, Kamp DW. Oxidative stress and pulmonary fibrosis. Biochim Biophys Acta. 2013;1832:1028–1040. doi: 10.1016/j.bbadis.2012.11.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Liu G, Cheresh P, Kamp DW. Molecular basis of asbestos-induced lung disease. Annu Rev Pathol. 2013;8:161–187. doi: 10.1146/annurev-pathol-020712-163942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sisson TH, Mendez M, Choi K, Subbotina N, Courey A, Cunningham A, Dave A, Engelhardt JF, Liu X, White ES, et al. Targeted injury of type II alveolar epithelial cells induces pulmonary fibrosis. Am J Respir Crit Care Med. 2010;181:254–263. doi: 10.1164/rccm.200810-1615OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Uhal BD, Nguyen H. The Witschi Hypothesis revisited after 35 years: genetic proof from SFTPC BRICHOS domain mutations. Am J Physiol Lung Cell Mol Physiol. 2013;305:L906–L911. doi: 10.1152/ajplung.00246.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gredilla R, Bohr VA, Stevnsner T. Mitochondrial DNA repair and association with aging: an update. Exp Gerontol. 2010;45:478–488. doi: 10.1016/j.exger.2010.01.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kamp DW, Shacter E, Weitzman SA. Chronic inflammation and cancer: the role of the mitochondria. Oncology (Williston Park) 2011;25:400–410, 413. [PubMed] [Google Scholar]
- 12.Shukla A, Jung M, Stern M, Fukagawa NK, Taatjes DJ, Sawyer D, Van Houten B, Mossman BT. Asbestos induces mitochondrial DNA damage and dysfunction linked to the development of apoptosis. Am J Physiol Lung Cell Mol Physiol. 2003;285:L1018–L1025. doi: 10.1152/ajplung.00038.2003. [DOI] [PubMed] [Google Scholar]
- 13.Panduri V, Liu G, Surapureddi S, Kondapalli J, Soberanes S, de Souza-Pinto NC, Bohr VA, Budinger GR, Schumacker PT, Weitzman SA, et al. Role of mitochondrial hOGG1 and aconitase in oxidant-induced lung epithelial cell apoptosis. Free Radic Biol Med. 2009;47:750–759. doi: 10.1016/j.freeradbiomed.2009.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Panduri V, Weitzman SA, Chandel N, Kamp DW. The mitochondria-regulated death pathway mediates asbestos-induced alveolar epithelial cell apoptosis. Am J Respir Cell Mol Biol. 2003;28:241–248. doi: 10.1165/rcmb.4903. [DOI] [PubMed] [Google Scholar]
- 15.Panduri V, Surapureddi S, Soberanes S, Weitzman SA, Chandel N, Kamp DW. P53 mediates amosite asbestos-induced alveolar epithelial cell mitochondria-regulated apoptosis. Am J Respir Cell Mol Biol. 2006;34:443–452. doi: 10.1165/rcmb.2005-0352OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kim SJ, Cheresh P, Williams D, Cheng Y, Ridge K, Schumacker PT, Weitzman S, Bohr VA, Kamp DW. Mitochondria-targeted Ogg1 and aconitase-2 prevent oxidant-induced mitochondrial DNA damage in alveolar epithelial cells. J Biol Chem. 2014;289:6165–6176. doi: 10.1074/jbc.M113.515130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Karahalil B, Hogue BA, de Souza-Pinto NC, Bohr VA. Base excision repair capacity in mitochondria and nuclei: tissue-specific variations. FASEB J. 2002;16:1895–1902. doi: 10.1096/fj.02-0463com. [DOI] [PubMed] [Google Scholar]
- 18.Bjoras M, Luna L, Johnsen B, Hoff E, Haug T, Rognes T, Seeberg E. Opposite base-dependent reactions of a human base excision repair enzyme on DNA containing 7,8-dihydro-8-oxoguanine and abasic sites. EMBO J. 1997;16:6314–6322. doi: 10.1093/emboj/16.20.6314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bohr VA, Stevnsner T, de Souza-Pinto NC. Mitochondrial DNA repair of oxidative damage in mammalian cells. Gene. 2002;286:127–134. doi: 10.1016/s0378-1119(01)00813-7. [DOI] [PubMed] [Google Scholar]
- 20.Klungland A, Rosewell I, Hollenbach S, Larsen E, Daly G, Epe B, Seeberg E, Lindahl T, Barnes DE. Accumulation of premutagenic DNA lesions in mice defective in removal of oxidative base damage. Proc Natl Acad Sci USA. 1999;96:13300–13305. doi: 10.1073/pnas.96.23.13300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Arai T, Kelly VP, Minowa O, Noda T, Nishimura S. High accumulation of oxidative DNA damage, 8-hydroxyguanine, in Mmh/Ogg1 deficient mice by chronic oxidative stress. Carcinogenesis. 2002;23:2005–2010. doi: 10.1093/carcin/23.12.2005. [DOI] [PubMed] [Google Scholar]
- 22.de Souza-Pinto NC, Eide L, Hogue BA, Thybo T, Stevnsner T, Seeberg E, Klungland A, Bohr VA. Repair of 8-oxodeoxyguanosine lesions in mitochondrial dna depends on the oxoguanine dna glycosylase (OGG1) gene and 8-oxoguanine accumulates in the mitochondrial dna of OGG1-defective mice. Cancer Res. 2001;61:5378–5381. [PubMed] [Google Scholar]
- 23.Sakumi K, Tominaga Y, Furuichi M, Xu P, Tsuzuki T, Sekiguchi M, Nakabeppu Y. Ogg1 knockout-associated lung tumorigenesis and its suppression by Mth1 gene disruption. Cancer Res. 2003;63:902–905. [PubMed] [Google Scholar]
- 24.Panduri V, Weitzman SA, Chandel NS, Kamp DW. Mitochondrial-derived free radicals mediate asbestos-induced alveolar epithelial cell apoptosis. Am J Physiol Lung Cell Mol Physiol. 2004;286:L1220–L1227. doi: 10.1152/ajplung.00371.2003. [DOI] [PubMed] [Google Scholar]
- 25.Soberanes S, Panduri V, Mutlu GM, Ghio A, Bundinger GR, Kamp DW. P53 mediates particulate matter-induced alveolar epithelial cell mitochondria-regulated apoptosis. Am J Respir Crit Care Med. 2006;174:1229–1238. doi: 10.1164/rccm.200602-203OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kamp DW, Israbian VA, Yeldandi AV, Panos RJ, Graceffa P, Weitzman SA. Phytic acid, an iron chelator, attenuates pulmonary inflammation and fibrosis in rats after intratracheal instillation of asbestos. Toxicol Pathol. 1995;23:689–695. doi: 10.1177/019262339502300606. [DOI] [PubMed] [Google Scholar]
- 27.Lawson WE, Crossno PF, Polosukhin VV, Roldan J, Cheng DS, Lane KB, Blackwell TR, Xu C, Markin C, Lare LB, et al. Endoplasmic reticulum stress in alveolar epithelial cells is prominent in IPF: association with altered surfactant protein processing and herpesvirus infection. Am J Physiol Lung Cell Mol Physiol. 2008;286:L1119–L1126. doi: 10.1152/ajplung.00382.2007. [DOI] [PubMed] [Google Scholar]
- 28.Korfei M, Ruppert C, Mahavadi P, Henneke I, Markart P, Koch M, Lang G, Fink L, Bohle R-M, Seeger W, et al. Epithelial endoplasmic reticulum stress and apoptosis in sporadic ideopathic pulmonary fibrosis. Am J Respir Crit Care Med. 2008;178:838–846. doi: 10.1164/rccm.200802-313OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lawson WE, Cheng DS, Degryse AL, Tanjore H, Polosukhin VV, Xu XC, Newcomb DC, Jones BR, Roldan J, Lane KB, et al. Endoplasmic reticulum stress enhances fibrotic remodeling in the lungs. Proc Natl Sci USA. 2011;108:10562–10567. doi: 10.1073/pnas.1107559108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kamp DW, Liu G, Cheresh P, Kim SJ, Mueller A, Lam AP, Trejo H, Williams D, Tulasiram S, Baker M, et al. Asbestos-induced alveolar epithelial cell apoptosis: the role of endoplasmic reticulum stress response. Am J Respir Cell Mol Biol. 2013;9:892–901. doi: 10.1165/rcmb.2013-0053OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Santos JH, Mandavilli BS, Van Houten B. Measuring oxidative mtDNA damage and repair using quantitative PCR. Methods Mol Biol. 2002;197:159–176. doi: 10.1385/1-59259-284-8:159. [DOI] [PubMed] [Google Scholar]
- 32.Chang LY, Overby LH, Brody AR, Crapo JD. Progressive lung cell reactions and extracellular matrix production after a brief exposure to asbestos. Am J Pathol. 1988;131:156–170. [PMC free article] [PubMed] [Google Scholar]
- 33.Phelps DS, Floros J. Localization of pulmonary surfactant proteins using immunohistochemistry and tissue in situ hybridization. Exp Lung Res. 1991;17:985–995. doi: 10.3109/01902149109064330. [DOI] [PubMed] [Google Scholar]
- 34.Przybylowska K, Kabzinski J, Sygut A, Dziki L, Dziki A, Majsterek I. An association selected polymorphisms of XRCC1, OGG1 and MUTYH gene and the level of efficiency oxidative DNA damage repair with a risk of colorectal cancer. Mutat Res. 2013;745–746:6–15. doi: 10.1016/j.mrfmmm.2013.04.002. [DOI] [PubMed] [Google Scholar]
- 35.Duan WX, Hua RX, Yi W, Shen LJ, Jin ZX, Zhao YH, Yi DH, Chen WS, Yu SQ. The association between OGG1 Ser326Cys polymorphism and lung cancer susceptibility: a meta-analysis of 27 studies. PLoS ONE. 2012;7:e35970. doi: 10.1371/journal.pone.0035970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Elahi A, Zheng Z, Park J, Eyring K, McCaffrey T, Lazarus P. The human OGG1 DNA repair enzyme and its association with orolaryngeal cancer risk. Carcinogenesis. 2002;23:1229–1234. doi: 10.1093/carcin/23.7.1229. [DOI] [PubMed] [Google Scholar]
- 37.Wang J, Wang Q, Watson LJ, Jones SP, Epstein PN. Cardiac overexpression of 8-oxoguanine DNA glycosylase 1 protects mitochondrial DNA and reduces cardiac fibrosis following transaortic constriction. Am J Physiol Heart Circ Physiol. 2011;301:H2073–H2080. doi: 10.1152/ajpheart.00157.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Cardin F, Piciocchi M, Tieppo C, Maddalo G, Zaninotto G, Mesicoli C, Rugge M, Farinati F.Oxidative DNA damage in Barrett mucosa: correlation with telomeric dysfunction and P53 mutation Ann Surg OncolIn press) [DOI] [PubMed] [Google Scholar]
- 39.Daimon M, Oizumi T, Toriyama S, Karasawa S, Jimbu Y, Wada K, Kameda W, Susa S, Muramatsu M, Kubota I, et al. Association of the Ser326Cys polymorphism in the OGG1 gene with type 2 DM. Biochem Biophys Res Commun. 2009;386:26–29. doi: 10.1016/j.bbrc.2009.05.119. [DOI] [PubMed] [Google Scholar]
- 40.Sampath H, Vartanian V, Rollins MR, Sakumi K, Nakabeppu Y, Lloyd RS. 8-Oxoguanine DNA glycosylase (OGG1) deficiency increases susceptibility to obesity and metabolic dysfunction. PLoS ONE. 2012;7:e51697. doi: 10.1371/journal.pone.0051697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Fukae J, Mizuno Y, Hattori N. Mitochondrial dysfunction in Parkinson's disease. Mitochondrion. 2007;7:58–62. doi: 10.1016/j.mito.2006.12.002. [DOI] [PubMed] [Google Scholar]
- 42.Berger F, Vaslin L, Belin L, Asselain B, Forlani S, Humbert S, Durr A, Hall J. The impact of single-nucleotide polymorphisms (SNPs) in OGG1 and XPC on the age at onset of Huntington disease. Mutat Res. 2013;755:115–119. doi: 10.1016/j.mrgentox.2013.04.020. [DOI] [PubMed] [Google Scholar]
- 43.Cardozo-Pelaez F, Sanchez-Contreras M, Nevin AB. Ogg1 null mice exhibit age-associated loss of the nigrostriatal pathway and increased sensitivity to MPTP. Neurochem Int. 2012;61:721–730. doi: 10.1016/j.neuint.2012.06.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Young LR, Gulleman PM, Bridges JP, Weaver TE, Deutsch GH, Blackwell TS, McCormack FX. The alveolar epithelium determines susceptibility to lung fibrosis in Hermansky-Pudlak syndrome. Am J Respir Crit Care Med. 2012;186:1014–1024. doi: 10.1164/rccm.201207-1206OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Serrano-Mollar A, Nacher M, Gay-Jordi G, Closa D, Xaubet A, Bulbena O. Intratracheal transplantation of alveolar type II cells reverses bleomycin-induced lung fibrosis. Am J Respir Crit Care Med. 2007;176:1261–1268. doi: 10.1164/rccm.200610-1491OC. [DOI] [PubMed] [Google Scholar]
- 46.Zheng D, Limmon GV, Yin L, Leung NH, Yu H, Chow VT, Chen J. A cellular pathway involved in Clara cell to alveolar type II cell differentiation after severe lung injury. PLoS ONE. 2013;8:e71028. doi: 10.1371/journal.pone.0071028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Yakes FM, Van Houten B. Mitochondrial DNA damage is more extensive and persists longer than nuclear DNA damage in human cells following oxidative stress. Proc Natl Acad Sci USA. 1997;94:514–519. doi: 10.1073/pnas.94.2.514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Van Houten B, Woshner V, Santos JH. Role of mitochondrial DNA in toxic responses to oxidative stress. DNA Repair (Amst) 2006;5:145–152. doi: 10.1016/j.dnarep.2005.03.002. [DOI] [PubMed] [Google Scholar]
- 49.Santos JH, Hunakova L, Chen Y, Bortner C, Van Houten B. Cell sorting experiments link persistent mitochondrial DNA damage with loss of mitochondrial membrane potential and apoptotic cell death. J Biol Chem. 2003;278:1728–1734. doi: 10.1074/jbc.M208752200. [DOI] [PubMed] [Google Scholar]
- 50.Tsutsui H. Oxidative stress in heart failure: the role of mitochondria. Intern Med. 2001;40:1177–1182. doi: 10.2169/internalmedicine.40.1177. [DOI] [PubMed] [Google Scholar]
- 51.Ding Z, Liu S, Wang X, Khaidakov M, Dai Y, Mehta JL. Oxidant stress in mitochondrial DNA damage, autophagy and inflammation in atherosclerosis. Sci Rep. 2013;3:1077. doi: 10.1038/srep01077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Picard M, Jung B, Liang F, Azuelos I, Hussain S, Goldberg P, Godin R, Danialou G, Chaturvedi R, Rygiel K, et al. Mitochondrial dysfunction and lipid accumulation in the human diaphragm during mechanical ventilation. Am J Respir Crit Care Med. 2012;186:1140–1149. doi: 10.1164/rccm.201206-0982OC. [DOI] [PubMed] [Google Scholar]
- 53.Hashizume M, Mouner M, Chouteau JM, Gorodnya OM, Ruchko MV, Potter BJ, Wilson GL, Gillespie MN, Parker JC. Mitochondrial-targeted DNA repair enzyme 8-oxoguanine DNA glycosylase 1 protects against ventilator-induced lung injury in intact mice. Am J Physiol Lung Cell Mol Physiol. 2013;304:L287–L297. doi: 10.1152/ajplung.00071.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Bulteau AL, Ikeda-Saito M, Szweda LI. Redox-dependent modulation of aconitase activity in intact mitochondria. Biochemistry. 2003;42:14846–14855. doi: 10.1021/bi0353979. [DOI] [PubMed] [Google Scholar]
- 55.Chen XJ, Wang X, Kaufman BA, Butow RA. Aconitase couples metabolic regulation to mitochondrial DNA maintenance. Science. 2005;307:714–717. doi: 10.1126/science.1106391. [DOI] [PubMed] [Google Scholar]
- 56.Youn CK, Song PI, Kim MH, Kim JS, Hyun JW, Choi SJ, Yoon SP, Chung MH, Chang IY, You HJ. Human 8-oxoguanine DNA glycosylase suppresses the oxidative stress induced apoptosis through a P53-mediated signaling pathway in human fibroblasts. Mol Cancer Res. 2007;5:1083–1098. doi: 10.1158/1541-7786.MCR-06-0432. [DOI] [PubMed] [Google Scholar]
- 57.Koczor CA, Torres RA, Fields EJ, Boyd A, Lewis W. Mitochondrial matrix P53 sensitizes cells to oxidative stress. Mitochondrion. 2013;13:277–281. doi: 10.1016/j.mito.2013.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Roepke M, Diestel A, Bajbouj K, Walluscheck D, Schonfeld P, Roessner A, Schneider-Stock R, Gali-Muhtasib H. Lack of P53 augments thymoquinone-induced apoptosis and caspase activation in human osteosarcoma cells. Cancer Biol Ther. 2007;6:160–169. doi: 10.4161/cbt.6.2.3575. [DOI] [PubMed] [Google Scholar]
- 59.Tsui KH, Feng TH, Lin YF, Chang PL, Juang HH. P53 downregulates the gene expression of mitochondrial aconitase in human prostate carcinoma cells. Prostate. 2011;71:62–70. doi: 10.1002/pros.21222. [DOI] [PubMed] [Google Scholar]
- 60.Vaseva AV, Marchenko ND, Ji K, Tsirka SE, Holzmann S, Moll UM. P53 opens the mitochondrial permeability transition pore to trigger necrosis. Cell. 2012;149:1536–1548. doi: 10.1016/j.cell.2012.05.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Nantajit D, Fan M, Duru N, Wen Y, Reed JC, Li JJ. Cyclin B1/Cdk1 phosphorylation of mitochondrial P53 induces anti-apoptotic response. PLoS ONE. 2010;5:e12341. doi: 10.1371/journal.pone.0012341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Essmann F, Pohlmann S, Gillissen B, Daniel PT, Schulze-Osthoff K, Janicke RU. Irradiation-induced translocation of P53 to mitochondria in the absence of apoptosis. J Biol Chem. 2005;280:37169–37177. doi: 10.1074/jbc.M502052200. [DOI] [PubMed] [Google Scholar]
- 63.Nuorva K, Makitaro R, Huhti E, Kamel D, Vahakangas K, Bloigu R, Soini Y, Paakko P. P53 protein accumulation in lung carcinomas of patients exposed to asbestos and tobacco smoke. Am J Respir Crit Care Med. 1994;150:528–533. doi: 10.1164/ajrccm.150.2.8049841. [DOI] [PubMed] [Google Scholar]
- 64.Husgafvel-Pursiainen K, Kannio A, Oksa P, Suitiala T, Koskinen H, Partanen R, Hemminki K, Smith S, Rosenstock-Leibu R, Brandt-Rauf PW. Mutations, tissue accumulations, and serum levels of P53 in patients with occupational cancers from asbestos and silica exposure. Environ Mol Mutagen. 1997;30:224–230. [PubMed] [Google Scholar]
- 65.Nymark P, Lindholm PM, Korpela MV, Lahti L, Ruosaari S, Kaski S, Hollmen J, Anttila S, Kinnula VL, Knuutila S. Gene expression profiles in asbestos-exposed epithelial and mesothelial lung cell lines. BMC Genomics. 2007;8:62. doi: 10.1186/1471-2164-8-62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Hevel JM, Olson-Buelow LC, Ganesan B, Stevens JR, Hardman JP, Aust AE. Novel functional view of the crocidolite asbestos-treated A549 human lung epithelial transcriptome reveals an intricate network of pathways with opposing functions. BMC Genomics. 2008;9:376. doi: 10.1186/1471-2164-9-376. [DOI] [PMC free article] [PubMed] [Google Scholar]