

Abstract
Bacterial pneumonia is a common public health problem associated with significant mortality, morbidity, and cost. Neutrophils are usually the earliest leukocytes to respond to bacteria in the lungs. Neutrophils rapidly sequester in the pulmonary microvasculature and migrate into the lung parenchyma and alveolar spaces, where they perform numerous effector functions for host defense. Previous studies showed that migrated neutrophils produce IFN-γ early during pneumonia induced by Streptococcus pneumoniae and that early production of IFN-γ regulates bacterial clearance. IFN-γ production by neutrophils requires Rac2, Hck/Lyn/Fgr Src family tyrosine kinases, and NADPH oxidase. Our current studies examined the mechanisms that regulate IFN-γ production by lung neutrophils during acute S. pneumoniae pneumonia in mice and its function. We demonstrate that IFN-γ production by neutrophils is a tightly regulated process that does not require IL-12. The adaptor molecule MyD88 is critical for IFN-γ production by neutrophils. The guanine nucleotide exchange factor CalDAG-GEFI modulates IFN-γ production. The CD11/CD18 complex, CD44, Toll-like receptors 2 and 4, TRIF, and Nrf2 are not required for IFN-γ production by neutrophils. The recently described neutrophil–dendritic cell hybrid cell, identified by its expression of Ly6G and CD11c, is present at low numbers in pneumonic lungs and is not a source of IFN-γ. IFN-γ produced by neutrophils early during acute S. pneumoniae pneumonia induces transcription of target genes in the lungs, which are critical for host defense. These studies underline the complexity of the neutrophil responses during pneumonia in the acute inflammatory response and in subsequent resolution or initiation of immune responses.
Keywords: pneumonia, neutrophils, IFN-γ, S. pneumoniae, mice
Clinical Relevance
This research adds to our knowledge base of pneumonia and how host defense occurs and to our understanding of neutrophils and their function during inflammation. This research affects clinical medicine by suggesting that targeting neutrophil function, particularly the production of IFN-γ, will modulate host defense, resolution of inflammation, and the initiation of the innate and specific immune processes.
Bacterial pneumonia is a common public health problem with considerable morbidity and mortality (1). It is one of the most common precursors to the development of acute lung injury and acute respiratory distress syndrome. Neutrophils are usually the earliest leukocyte to respond to bacteria in the lungs. Neutrophils rapidly sequester in the pulmonary microvasculature and migrate into the lung parenchyma and alveolar spaces where they perform numerous effector functions.
Recent studies have shown that neutrophils can no longer be considered simply carriers of the machinery to produce superoxide and other reactive oxygen species (ROS) and proteases but rather that they are very capable of transcribing and translating proteins in a regulated manner (2). The mediators that they produce depend on the inflammatory stimulus and the lung environment. For example, our recent studies have shown that migrated neutrophils produce IFN-γ during pneumonia induced by Streptococcus pneumoniae and Staphylococcus aureus, but not in pneumonias caused by Escherichia coli or Pseudomonas aeruginosa, when assessed by flow cytometry, mass spectroscopy, or ELISA (3). At this time early in the evolution of pneumonias, 98% of the IFN-γ–expressing cells are neutrophils (3). This IFN-γ is important in host defense because at 24 hours after intratracheal instillation of S. pneumoniae bacterial clearance was impaired in mice deficient in IFN-γ (3). Our studies showed that one mechanism through which IFN-γ regulates bacterial clearance is by regulating the production of neutrophil extracellular traps (3).
Very little is understood about the pathways through which IFN-γ is induced in neutrophils. The cytokine IL-12 is produced by dendritic cells, neutrophils, and monocytes/macrophages and induces inflammatory processes, including IFN-γ production by lymphocytes and natural killer (NK) cells (4). Several studies have shown that IL-12 can also induce IFN-γ production by macrophages and dendritic cells (5–7). A critical mediator of IL-12 signaling is signal transducer and activator of transcription (STAT) 4, which is phosphorylated upon IL-12 binding to its receptor (8, 9). Phosphorylated STAT4 then translocates to the nucleus and binds to target genes to regulate their transcription. Our initial studies tested the hypothesis that IL-12 is required for IFN-γ production by neutrophils through a STAT4-dependent pathway. Because the studies presented in this manuscript show no role for IL-12 in IFN-γ production by neutrophils, other hypotheses were pursued.
Our previous studies provided clues about important pathways for neutrophil production of IFN-γ. Three nonreceptor Src family kinases expressed on neutrophils (Hck, Fgr, and Lyn), the small GTPase Rac2, and the gp91phox (Nox2) component of the phagocyte NADPH oxidase are required for IFN-γ production by neutrophils during S. pneumoniae pneumonia (3). A signaling pathway in which the Src family kinases activate Rac2 and leads to assembly and activation of the NADPH oxidase has been described (10). The NADPH oxidase complex catalyzes the production of superoxide from oxygen, which leads to a cascade of ROS and potentially to oxidative stress (11). This stress activates the transcription factor Nrf2, which switches on cytoprotective pathways by regulating transcription of antioxidant and detoxifying genes (12, 13). Recent reports have also demonstrated a requirement for Nrf2 in regulating the innate immune response during infection or injury (14–20). Whether the observed role of NADPH oxidase in IFN-γ production is due to Nrf2 target genes regulating IFN-γ is an important question we address. Furthermore, three members of the Src family kinases (Fgr, Hck, and Lyn), Rac2, and NADPH oxidase are downstream of signaling induced by the CD11/CD18 leukocyte adhesion complex (10, 21–24). The glycoprotein CD44, a major receptor for hyaluronan and other extracellular matrix components, is also important for activating β2 integrins (25). The studies described herein also seek to determine whether CD11/CD18 and CD44 are required for the production of IFN-γ by neutrophils.
Toll-like receptors (TLRs) initiate signaling cascades and induce production of mediators in response to pathogens. Of particular interest for this study are TLR2, which recognizes whole S. pneumoniae and peptidoglycan from the cell wall of gram-positive organisms (26, 27), and TLR4, which is best known for recognizing LPS but also capable of recognizing other ligands, including the pneumococcal pore-forming toxin pneumolysin (28). TLR2 also appears to contribute to the host response to purified pneumolysin in vivo (29). The initial signaling immediately downstream of TLRs is mediated by the TIR-domain–containing adaptors MyD88 and TRIF (30). MyD88 mediates signaling downstream of all TLRs except TLR3 and in some cases TLR4, whereas TRIF mediates MyD88-independent signaling downstream of TLR4 and TLR3 (31–33). MyD88 regulates neutrophil recruitment to the lung during bacterial pneumonia (34, 35). TRIF is critical for host responses in pneumonias caused by the gram-negative organisms E. coli (36), Klebsiella pneumoniae (37), and P. aeruginosa (38).
Our studies thus pursued questions about the mechanisms through which IFN-γ expression in neutrophils is regulated during S. pneumoniae pneumonia. Because these studies show that IL-12 is not a regulator of IFN-γ expression, whereas nonreceptor Src tyrosine kinases, Rac2, and NADPH oxidase are required for IFN-γ production (3), subsequent studies tested hypotheses about what is downstream and upstream of these signaling molecules. These hypotheses tested the role of the oxidant stress–activated transcription factor Nrf2 in regulating IFN-γ production and of NADPH oxidase–generated ROS in regulating mitogen-activated protein kinase (MAPK). A role for the CD11/CD18 adhesion complex (β2 leukocyte integrins) was postulated because the complex is upstream of these signaling molecules and because CD11b/CD18 is also a complement receptor (CR3); a role for CD44 was also postulated. These studies also tested the hypothesis that TLR2 and TLR4 and the TLR downstream adaptor molecules MyD88 and TRIF are important regulatory molecules in IFN-γ production by neutrophils.
Materials and Methods
Further details are provided in the online supplement.
Mice
Adult C57BL/6 wild-type (WT) and mutant mice backcrossed for ≥10 generations were purchased or obtained from collaborators. Age- and gender-matched mice were studied at 6 to 8 weeks of age. All animal studies were performed in compliance with the United States Department of Health and Human Services Guide for the Care and Use of Laboratory Animals. Animal studies were reviewed and approved by our Institutional Animal Care and Use Committee.
Bacterial Pneumonia
Pneumonia was induced by intratracheal instillation of S. pneumoniae (serotype 19, ATCC 49619) suspended in PBS into the left lung at a dose of 2.3 μl/g mouse body weight (details are provided in the online supplement).
Bone marrow (BM) chimeric mice were generated as described in the online supplement.
Isolation of single lung cells, immunostaining to identify neutrophils and intracellular IFN-γ, and flow cytometry data analysis were performed as previously described (3) and are detailed in the online supplement. Isolation of neutrophils from single cell suspensions of mouse lungs was performed using biotin–anti-Ly6G and magnetic beads coated with antibiotin (MACS beads; Miltenyi Biotec, San Diego, CA).
Gene expression profiling using Affymetrix arrays is described in the online supplement.
Results
Neutrophils, but Not Neutrophil–Dendritic Cell Hybrids, Produce IFN-γ during S. pneumoniae Pneumonia
S. pneumoniae caused a large increase in the number of neutrophils in the lung (Figure 1). Consistent with our previously published work (3), neutrophils in the pneumonic lung produced IFN-γ during pneumonia induced by S. pneumoniae (Figure 1). The median fluorescence intensity (MFI) of IFN-γ staining was significantly higher in neutrophils from mice given S. pneumoniae compared with that of neutrophils from mice given PBS, whether comparing all neutrophils (20 ± 2.3 versus 2.1 ± 0.3, respectively) or neutrophils expressing IFN-γ (31 ± 1.1 versus 16 ± 0.5, respectively).
Figure 1.
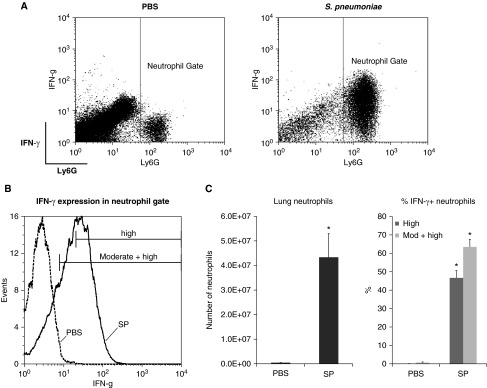
Lung neutrophils produce IFN-γ during Streptococcus pneumoniae (SP) pneumonia. (A) Dot plots show Ly6G and IFN-γ expression of lung cells from wild-type (WT) mice given PBS or SP. Cells were gated to exclude debris and doublets. Neutrophils were identified by high expression of Ly6G. (B) Representative histogram shows IFN-γ expression of neutrophils from the lungs of WT mice given PBS or SP. The percentage of high IFN-γ expressors was determined by setting the threshold of staining above that seen with the isotype-matched antibody control, effectively excluding cells with dim or moderate staining. The percentage of cells expressing moderate or high IFN-γ was determined by creating gates based on scatter and staining characteristics and setting the staining threshold with the negative staining controls at ≤2%. (C) The total number of neutrophils in the left lung and the percentage of IFN-γ+ neutrophils in the lung in WT mice at 24 hours after intratracheal instillation of PBS or SP. Data are expressed as mean ± SEM (n = 4). High expression: the percentage of neutrophils with high expression, as calculated using analysis 1, as described in Materials and Methods. High + moderate: the percentage of neutrophils that expressed either moderate or high levels of IFN-γ, as calculated using analysis 2. *P < 0.05 compared with PBS instillation.
Our published data show that T cells, NK cells, and macrophages are not producing IFN-γ at 24 hours (3). In fact, > 98% of the IFN-γ–expressing cells in lung digests are neutrophils, as marked by high expression levels of Ly6G. Recent reports have indicated that CD11c-expressing neutrophils arise during inflammation and may be important in bacterial clearance and antigen presentation by functioning as neutrophil–dendritic cell hybrids (39, 40). The number of lung cells coexpressing CD11c and Ly6G was greater in WT mice with S. pneumoniae pneumonia compared with those given PBS (4.0 ± 0.73 × 104 versus 0.13 ± 0.04 × 104 [n = 4]; P < 0.05 using t test). However, CD11c+ Ly6G+ cells constituted a minuscule proportion of cells in the lungs of S. pneumoniae and PBS-treated mice (0.08 ± 0.02% versus 0.01 ± 0.0%, respectively) and a very low proportion of the IFN-γ+ cells expressed CD11c in mice with S. pneumoniae pneumonia (0.4 ± 0.3%). These data indicate that CD11c+ neutrophils are not contributing to IFN-γ production during pneumonia.
IL-12 Is Not Required for IFN-γ Production by Lung Neutrophils
IL-12a–deficient mice lack the unique p35 subunit of IL-12 and express decreased levels of IFN-γ during infection with Listeria monocytogenes (41). The number of neutrophils that migrated into the lungs was similar in WT and IL-12–deficient (Il12a−/−) mice during 24-hour S. pneumoniae pneumonia. The percentage of Ly6G-negative cells that expressed IFN-γ was similarly low in WT and IL-12–deficient mice (1.4 ± 0.3% versus 1.1 ± 0.3%, respectively). In WT mice, over half the lung neutrophils expressed IFN-γ (Figure 2). Most importantly, the percentage of IFN-γ+ neutrophils was similar in WT and IL-12a–deficient mice when gating was based on high expression (analysis 1) or on moderate and high expression (analysis 2) (Figure 2A). The IFN-γ MFI and the distribution of intensity of IFN-γ staining was also not different between genotypes (Table 1). These data indicate that IL-12 is not required for IFN-γ production by neutrophils.
Figure 2.

IL-12 is not required for IFN-γ production by neutrophils. (A) The total number of neutrophils in the left lung and the percentage of IFN-γ+ neutrophils in the lung were similar in WT and IL-12a–deficient (Il12a−/−) mice at 24 hours after intratracheal instillation of SP. Data are expressed as mean ± SEM; n = 7 (WT) and n = 8 (Il12a−/−). *P < 0.05 compared with WT mice. (B) STAT4 is expressed by neutrophils but is not phosphorylated in neutrophils during SP pneumonia. The phosphorylated form of STAT4 (pSTAT4) was not detected in neutrophils isolated from lungs of two WT mice 6 hours after intratracheal instillation of PBS or SP. PBS- and SP-treated neutrophils expressed total STAT4. pSTAT4 was detected in isolated WT splenocytes incubated in the presence of media (Med), SP, or phorbol ester (PMA). (C) STAT4 in lung neutrophils is not immunoprecipitated by an antibody that recognizes phosphorylated tyrosine residues (PY20). Neutrophils were isolated from the lungs of WT mice 6 hours after intratracheal administration of PBS or SP. The neutrophils were lysed, and immunoprecipitation with the antiphosphotyrosine antibody was performed using standard procedures. IP, immunoprecipitated fraction; S, supernatant (nonimmunoprecipitated fraction). The positive control was obtained from splenocytes treated with PMA for 4 hours.
Table 1.
The Level of IFN-γ Expression by Neutrophils From Mice with Targeted Mutations in the Indicated Molecules Compared with Wild-Type Mice*
| Molecule Deficient in Deficient Mice | MFI of All Neutrophils |
MFI of IFN-γ+ Neutrophils |
||
|---|---|---|---|---|
| Deficient | WT | Deficient | WT | |
| IL-12a | 15 ± 1.5 | 16 ± 1.4 | 32 ± 1.3 | 33 ± 2.1 |
| Nrf2 | 19 ± 1.8 | 19 ± 1.7 | 30 ± 2.5 | 29 ± 2.2 |
| gp91phox | 2.3 ± 0.7* | 19 ± 1.7 | 13 ± 1.2† | 29 ± 2.2 |
| CD11a | 14 ± 2.0 | 20 ± 1.3 | 43 ± 2.0 | 43 ± 0.7 |
| CD11b | 24 ± 4.1 | 20 ± 1.3 | 44 ± 4.6 | 43 ± 0.7 |
| CD18 (Chimeric mice) | 15 ± 2.0 | 16 ± 2.4 | 27 ± 1.0 | 29 ± 1.6 |
| CalDAG GEFI | 16 ± 2.3 (P = 0.05) | 9.9 ± 0.6 | 31 ± 2.2 | 25 ± 1.0 |
| CD44 | 8.9 ± 1.2 | 8.0 ± 1.0 | 31 ± 1.0 | 33 ± 1.6 |
| TLR2/4 | 11 ± 1.9 | 13 ± 1.8 | 31 ± 1.5 | 32 ± 2.1 |
| TLR2 | 11 ± 2.9 | 13 ± 1.8 | 32 ± 0.2 | 32 ± 2.1 |
| TLR4 | 14 ± 1.6 | 13 ± 1.8 | 34 ± 1.7 | 32 ± 2.1 |
Definition of abbreviations: MFI, median fluorescence intensity; TLR, Toll-like receptor; WT, wild type.
The data show the MFI of lung neutrophils and of IFN-γ–expressing lung neutrophils in age- and gender-matched mutant and WT mice during 24-h Streptococcus pneumoniae pneumonia. MFI was estimated as the median channel value for all events that meet the gating criteria after subtracting the signal of the respective isotype control. Values are mean ± SEM.
P ≤ 0.05 compared with WT (t test or ANOVA with Scheffe post hoc test as appropriate).
IL-12 activates the transcription factor STAT4, which plays a critical role in IFN-γ production by lymphocytes and NK cells (8, 9). STAT4 is expressed in neutrophils from the lungs of mice given PBS or S. pneumoniae (Figure 2B). However, the phosphorylated form of STAT4 was not detected in these lung neutrophils using an antibody specific for STAT4 phosphorylated at tyrosine 693 or after immunoprecipitation of tyrosine-phosphorylated proteins with an antiphosphotyrosine antibody (Figure 2C). Taken together, these studies suggest that IL-12 and phosphorylation of STAT4 are not required for IFN-γ expression by neutrophils during S. pneumoniae pneumonia.
The Leukocyte Adhesion Complex CD11/CD18 Does Not Contribute to IFN-γ Production
Our previous study demonstrated that production of IFN-γ by neutrophils during S. pneumoniae pneumonia required Rac2, the Src family kinases Hck/Fgr/Lyn, and the gp91phox component of NADPH oxidase (3). Studies in the literature have demonstrated that these molecules are downstream of leukocyte integrin engagement in outside-in signaling pathways that modulate neutrophil adhesion, motility, and activation (21, 22, 24, 42). To determine the role of adhesion molecules in IFN-γ production by neutrophils, mice deficient in leukocyte β2 integrins (the CD11/CD18 adhesion complex) were studied.
The leukocyte integrin CD11/CD18 expressed on neutrophils mediates adhesion of neutrophils to endothelial cells and to bacteria. CD11a/CD18 (LFA-1) and CD11b/CD18 (Mac1 or CR3) can bind ICAM-1 expressed on endothelial and other cells. CD11b/CD18 can also bind to the complement fragment iC3b-coating bacteria, extracellular matrix components, or other cells.
The role of the CD11a/CD18 and CD11b/CD18 in initiating IFN-γ expression was studied using mice deficient in CD11a or CD11b. The total number of lung neutrophils in the pneumonic lung and the percentage of lung neutrophils that expressed both high and moderate levels of intracellular IFN-γ were not different among CD11a-deficient (Itgal−/−), CD11b-deficient (Itgam−/−), and WT mice (Table 2). The intensity of IFN-γ staining tended to be less in neutrophils from CD11a-deficient mice, but this trend was not statistically significant (Table 1). These data indicate that CD11a/CD18 and CD11b/CD18 are not required for IFN-γ production by neutrophils.
Table 2.
The CD11/CD18 Adhesion Complex and CD44 Do Not Contribute to IFN-γ Production*
| Molecule Deficient in Deficient Mice | Neutrophils in Pneumonic Lung (× 106) (n) |
Neutrophils Expressing IFN-γ (%) |
Neutrophils Expressing Moderate or High Levels of IFN-γ (%) |
|||
|---|---|---|---|---|---|---|
| Deficient | WT | Deficient | WT | Deficient | WT | |
| CD11a | 64 ± 5.9 | 48 ± 4.3 | 16 ± 1.3 | 18 ± 0.9 | 36 ± 2.2 | 45 ± 3.0 |
| CD11b | 44 ± 6.5 | 48 ± 4.3 | 19 ± 4.3 | 18 ± 0.9 | 44 ± 2.7 | 45 ± 3.0 |
| CD18 (Chimeric mice) | 49 ± 6.1† | 0.38 ± 0.10 | 21 ± 1.8 | 23 ± 1.8 | 56 ± 4.5 | 58 ± 5.1 |
| CD44 | 21 ± 5.1 | 17 ± 5.1 | 25 ± 1.5 | 25 ± 2.9 | 42 ± 3.2 | 41 ± 3.8 |
Definition of abbreviation: WT, wild type.
The number of lung neutrophils in the pneumonic lung and the percentage that express IFN-γ in mutant and age- and sex-matched WT mice during 24-h Streptococcus pneumoniae pneumonia are shown. Values are mean ± SE.
P ≤ 0.05 compared with WT (t test or ANOVA with Scheffe post hoc test as appropriate). Data are from at least two experiments.
A direct comparison of WT and CD18-deficient neutrophils was made by reconstituting lethally irradiated WT mice with a mixture of WT and CD18-deficient BM cells. The total number of lung neutrophils in chimeric mice was similar to the number of lung neutrophils in WT mice in the study addressing the roles of CD11a and CD11b (Table 2). The proportion of neutrophils that are CD18 deficient is much higher in the lung than in the blood (99.5 ± 0.2% versus 42 ± 4.8%, respectively), suggesting that CD18-deficient neutrophils are retained and may preferentially migrate in the lungs. The proportion of WT and CD18-deficient lung neutrophils that made IFN-γ was similar (Table 2), as was the level of IFN-γ expression. Taken together, these data indicate that CD11/CD18 adhesion complex is not required for IFN-γ production by neutrophils. Thus, Rac2, the Src kinases Hck/Fgr/Lyn, and NADPH oxidase mediate IFN-γ production through pathways that do not require ligation of β2 integrins on the neutrophil surface.
Signaling through CalDAG-GEFI Suppresses IFN-γ Production
CalDAG-GEFI is a guanine nucleotide exchange factor regulated by calcium. It acts as an activator of β2 integrins in neutrophils and other integrins in platelets by activating Rap1. The proportion of neutrophils expressing IFN-γ was significantly greater in CalDAG-GEFI–deficient (Rasgrp2−/−) mice compared with WT mice (Figure 3), and the intensity of IFN-γ staining tended to be greater (Table 1). Thus, signaling by CalDAG-GEFI appears to inhibit rather than induce IFN-γ expression by neutrophils.
Figure 3.

Deficiency of CalDAG-GEFI augments IFN-γ production by neutrophils. The percentage of lung neutrophils expressing IFN-γ is significantly higher in CalDAG-GEFI–deficient (Rasgrp2−/−) mice compared with WT mice. Data are mean ± SEM from four separate experiments; n = 10 (WT) or n = 8 (Rasgrp2−/−). *P < 0.05 compared with WT mice (t test).
CD44 Does Not Contribute to IFN-γ Production
CD44 is a major receptor for hyaluronan and binds collagen and E-selectin. Several reports have indicated that CD44 ligation can up-regulate the expression of ICAM-1 and modulate integrin-dependent adhesion (43–45) as well as activate β2 integrins on macrophages (25). However, the proportion of IFN-γ–producing neutrophils in CD44-deficient mice and the intensity of IFN-γ expression were not different compared with those in WT mice, indicating that CD44 is not required for IFN-γ production by neutrophils (Tables 1 and 2).
MyD88 but Not TRIF Regulate IFN-γ Production
Neutrophil numbers in the bronchoalveolar lavage fluid (BALF) were significantly less in MyD88-deficient (Myd88−/−) mice compared with WT mice (Table 3), consistent with the previously described role of MyD88 in regulating neutrophil recruitment during pulmonary inflammation (34, 35). The percentage of IFN-γ+ neutrophils and the intensity of IFN-γ staining in neutrophils in the BALF and lung tissue after lavage were significantly less in Myd88−/− mice, indicating a role for MyD88 in mediating IFN-γ production by neutrophils (Table 3). The percentages of IFN-γ+ neutrophils and the intensity of IFN-γ staining of neutrophils in the BALF and lung tissue after lavage were similar in WT and TRIF-deficient (Ticam1−/−) mice (Table 3), indicating that TRIF is not required for IFN-γ production by neutrophils during S. pneumoniae pneumonia. The total number of neutrophils in the BALF was significantly higher in the Ticam1−/− mice; consequently, the number of IFN-γ+ cells in the BALF was significantly higher in Ticam1−/− compared with WT mice.
Table 3.
MyD88 but Not TRIF Modulates Expression of IFN-γ in Lung Neutrophils, Particularly Those in the Bronchoalveolar Lavage Fluid*
| Lung Neutrophils | Study Comparing WT and Myd88−/− Mice |
Study Comparing WT and Ticam1−/− Mice |
||
|---|---|---|---|---|
| Myd88−/− | WT | Ticam1−/− | WT | |
| Total neutrophils in BALF ×106 | 1.0 ± 0.1† | 3.2 ± 0.4 | 5.4 ± 0.3† | 3.9 ± 0.2 |
| % IFN-γ+ neutrophils in BALF | ||||
| % high | 13 ± 1.6† | 27 ± 3.2 | 26 ± 2.2 | 24 ± 2.2 |
| % high + moderate | 28 ± 3.4† | 51 ± 4.6 | 51 ± 7.6 | 51 ± 6.4 |
| Total neutrophils in lung digest after BAL ×106 | 19 ± 2.0 | 21 ± 4.0 | 22 ± 1.3 | 23 ± 3.0 |
| % IFN-γ+ neutrophils in lung tissue after BALF | ||||
| % high | 16 ± 2.6† | 30 ± 2.4 | 29 ± 1.9 | 22 ± 3.3 |
| % high + moderate | 22 ± 4.1‡ | 36 ± 5.6 | 56 ± 3.6 | 50 ± 6.6 |
| MFI of all neutrophils | ||||
| BALF | 5.9 ± 0.9† | 16 ± 2.4 | 11 ± 1.0 | 11 ± 0.6 |
| Lung digest | 5.0 ± 1.0† | 14 ± 1.2 | 11 ± 1.0 | 9.0 ± 1.1 |
| MFI of IFN-γ+ neutrophils | ||||
| BALF | 30 ± 1.4 | 33 ± 3.0 | 24 ± 3.2 | 23 ± 2.2 |
| Lung digest | 39 ± 2.3 | 48 ± 7.1 | 23 ± 1.9 | 20 ± 1.4 |
Definition of abbreviations: BAL, bronchoalveolar lavage; BALF, bronchoalveolar lavage fluid; MFI, median fluorescence intensity; WT, wild type.
The role of MyD88 and TRIF was examined in neutrophils in BALF and lung tissue after lavage. Flow cytometry was performed to detect intracellular IFN-γ levels in neutrophils (Ly6G+) in the BALF and in the lung tissue after lavage of Streptococcus pneumoniae–infected mice. Isotype-matched control antibody was used for assessing the level of background staining. The mutant genotypes were compared with gender- and age-matched WT mice. Data are expressed as the mean ± SEM (n = 5–7).
P ≤ 0.05 compared with WT using two-tailed t tests.
P = 0.07 compared with WT (t test).
TLR2 and TLR4 Do Not Contribute to IFN-γ Production
The role of TLR2 and TLR4 in IFN-γ production by neutrophils in S. pneumoniae pneumonia was examined using mice singly deficient in TLR2 (Tlr2−/−) or TLR4 (Tlr4−/−) or mice lacking both TLR2 and TLR4 (Tlr2/4−/−). Mice singly deficient in TLR2 or TLR4 were examined coincidently with WT and Tlr2/4−/− mice. There was no significant difference in the proportion of IFN-γ+ neutrophils or in the total number of neutrophils in the lung when comparing all four genotypes (Table 4).
Table 4.
Signaling Initiated Through Toll-Like Receptors 2 and 4 Does Not Modulate Expression of IFN-γ in Lung Neutrophils*
| Molecule Deficient in Deficient Mice | Neutrophils in Pneumonic Lung (n) |
Neutrophils Expressing IFN-γ (%) |
Neutrophils Expressing Moderate or High Levels of IFN-γ (%) |
|||
|---|---|---|---|---|---|---|
| Deficient | WT | Deficient | WT | Deficient | WT | |
| TLR2/4 | 23 ± 1.1 | 29 ± 3.2 | 20 ± 3.7 | 24 ± 3.0 | 38 ± 7.3 | 44 ± 4.5 |
| TLR2 | 24 ± 1.7 | 29 ± 3.2 | 20 ± 5.2 | 24 ± 3.0 | 37 ± 8.6 | 44 ± 4.5 |
| TLR4 | 31 ± 5.0 | 29 ± 3.2 | 26 ± 3.7 | 24 ± 3.0 | 46 ± 6.0 | 44 ± 4.5 |
Definition of abbreviations: TLR, Toll-like receptor; WT, wild type.
TLR2 and TLR4 are not required for IFN-γ production by neutrophils. No significant difference in the total number of neutrophils and the percentage of IFN-γ+ neutrophils in the pneumonic lung in coincidently studied WT and mice lacking TLR2 (Tlr2−/−), TLR4 (Tlr4−/−), or both TLR2 and TLR4 (Tlr2/4−/−). Data are mean ± SEM (n = 4–5 per genotype).
To examine the role of TLR2 and TLR4 in neutrophil recruitment to the lungs and IFN-γ production, neutrophils in the BALF and neutrophils remaining in the lung tissue after lavage were assessed. No significant differences were observed between WT and Tlr2/4−/− mice in the number of neutrophils, in the proportion that expressed IFN-γ, or in the intensity of IFN-γ staining in the BALF and lung tissue after lavage (Table 5). These studies suggest that TLR2 and TLR4 are not required for neutrophil recruitment to the lungs or IFN-γ production by neutrophils at 24 hours during S. pneumoniae pneumonia.
Table 5.
Toll-Like Receptors 2 and 4 Are Not Required for IFN-γ Expression by Neutrophils in the Airspace and Lung Tissue*
| Lung Neutrophils | Tlr2/4−/− | WT |
|---|---|---|
| Total neutrophils in BALF ×106 | 3.5 ± 0.4 | 3.0 ± 0.7 |
| % IFN-γ+ neutrophils in BALF | ||
| % high | 35 ± 10 | 37 ± 9.2 |
| % high + moderate | 50 ± 5.4 | 54 ± 2.7 |
| Total neutrophils in lung digest after BAL ×106 | 38 ± 7.4 | 26 ± 5.2 |
| % IFN-γ+ neutrophils in lung tissue after BALF | ||
| % high | 35 ± 3.8 | 44 ± 7.6 |
| % high + moderate | 51 ± 7.9 | 56 ± 10.4 |
| MFI of all neutrophils | ||
| BALF | 8.7 ± 2.1 | 9.6 ± 1.5 |
| Lung digest | 8.8 ± 2.3 | 11 ± 3.2 |
| MFI of IFN-γ+ neutrophils | ||
| BALF | 17 ± 1.9 | 17 ± 1.9 |
| Lung digest | 18 ± 3.2 | 17 ± 2.9 |
Definition of abbreviations: BAL, bronchoalveolar lavage; BALF, bronchoalveolar lavage fluid; MFI, median fluorescence intensity; TLR, Toll-like receptor; WT, wild type.
IFN-γ expression was determined in neutrophils (Ly6G+) in the BALF and in the lung tissue after lavage of Streptococcus pneumoniae–infected WT mice and mice lacking both TLR2 and TLR4 (Tlr2/4−/−). No significant differences were found using t test to compare genotypes (n = 4 from two separate experiments).
NADPH Oxidase–Generated Oxidants Are Required for IFN-γ Production, but Nrf2 and Its Gene Products Are Not
Our previous work showed that the gp91phox (Nox2, gene name Cybb) component of the phagocyte NADPH oxidase is required for IFN-γ production by neutrophils (3). In neutrophils, NADPH oxidase is the main source of superoxide. Free radicals such as superoxide can cause oxidative stress, which in turn activates the cytoprotective transcription factor Nrf2. To determine whether Nrf2 is required for the production of IFN-γ by neutrophils, Nrf2-deficient (Nfe2l2−/−) mice were studied concurrently with WT and Nox2-deficient (Cybb−/−) mice. The percentage of lung neutrophils that expressed intracellular IFN-γ was similar in WT mice and Nrf2-deficient mice (Figure 4) whether high expressors or high + moderate expressors are compared. The level of expression was also not different in these two genotypes when compared using the MFI or the distribution of IFN-γ staining (Table 1). In contrast, neutrophils from Nox2-deficient mice expressed almost no IFN-γ (Figure 4), and the MFI was similarly low and not different from isotype controls (Table 1). The number of neutrophils in the lungs was not significantly different among WT, Nrf2-deficient, and Nox2-deficient mice (Figure 4). These data indicate that Nrf2 and Nrf2-dependent target genes are not required for IFN-γ production by neutrophils. NADPH oxidase may regulate IFN-γ production by modulating other oxidant-sensitive transcription factors and pathways.
Figure 4.

NADPH oxidase is required for IFN-γ production, but the redox-sensitive transcription factor Nrf2 is not. The total number of neutrophils and the percentage of IFN-γ+ neutrophils in the pneumonic lung in WT, Nrf2-deficient (Nfe2l2−/−), and Nox2 (gp91phox)-deficient (Cybb−/−) mice. Data are mean ± SEM (n = 3–5). *P ≤ 0.05 compared with the other groups (ANOVA with Tukey HSD post hoc test). Similar results were obtained in a separate study.
To rule out the possibility that the defect in IFN-γ production observed in Nox2-deficient mice is due to differences in cell death, membrane integrity and apoptosis were assessed in lung neutrophils from WT and Nox2-deficient mice. Membrane integrity of lung neutrophils was similarly high in WT and Nox2-deficient mice, as assessed with a fixable viability dye (94 ± 1% versus 95 ± 0.2%, respectively [n = 4]). The percentage of viable neutrophils was similar between IFN-γ expressors and nonexpressors in both genotypes. Cell viability as determined by 7-AAD exclusion was slightly less in WT compared with Nox2-deficient mice (90 ± 0.4% versus 92 ± 0.6%, respectively; P < 0.05, t test). The proportion of neutrophils that bound Annexin V on their surface but excluded 7-AAD (consistent with membrane changes that occur early during apoptosis) was not significantly different between WT and Nox2-deficient mice (26 ± 3.8% versus 34 ± 2.7%, respectively). Thus, differences in neutrophil viability are unlikely to be the reason for the lack of IFN-γ production in the absence of Nox2.
MAPK activity depends on phosphorylation of a few critical residues, which is regulated by kinases and phosphatases. ROS modulate the activity of MAPKs by inhibiting phosphatases, resulting in sustained MAPK activation (46, 47). The levels of phosphorylated and total MAPKs were determined in neutrophils isolated from the lungs of WT and Nox2-deficient mice given S. pneumoniae. There was no significant difference between WT and Nox2-deficient neutrophils in the levels of phosphorylated or total p38 and extracellular signal-regulated kinases (ERK) MAPK or in the level of total c-Jun N-terminal kinase (JNK) (Figure 5). However, phosphorylated JNK was significantly greater in Nox2-deficient neutrophils compared with WT neutrophils by approximately 30% (Figure 5). Thus, ROS appear to be regulating JNK activity during S. pneumoniae pneumonia by limiting the amount of JNK in its phosphorylated active state.
Figure 5.

Deficiency of Nox2 results in augmented phosphorylation of c-Jun N-terminal kinase (JNK) and no change in phosphorylation of extracellular signal-regulated kinases (ERKs) or p38 mitogen-activated protein kinases (MAPKs) in neutrophils. The total amount of each MAPK was similar in the two genotypes. Representative immunoblots showing protein levels of phosphorylated and total MAPKs in lung neutrophils from WT and Nox2-deficient (Cybb−/−) mice during S. pneumoniae pneumonia. Graphs show densitometric measurements in arbitrary units (AU) (mean ± SEM; n = 4–5 mice per genotype repeated in two or three separate experiments). *P ≤ 0.05 compared with WT (t test).
IFN-γ Produced Early during Acute S. pneumoniae Pneumonia Regulates Gene Transcription in the Lungs
To determine the effect of neutrophil IFN-γ on host defense, Ifng−/− mice were lethally irradiated and reconstituted with either WT or Ifng−/− BM. They were studied after 3 to 4 weeks, when virtually all neutrophils are donor derived. Gene expression in lungs from these mice was profiled at 24 hours after intratracheal administration of PBS (control) or after a low or high dose of S. pneumoniae. In Ifng−/− mice given PBS, no genes were differentially expressed in mice reconstituted with WT compared with Ifng−/− BM, using standard filtering criteria (FDR-adjusted P ≤ 0.05; fold change ≥ 2). Administration of a high dose of S. pneumoniae resulted in differential expression of 3,013 and 2,667 transcripts in mice reconstituted with WT or Ifng−/− cells, respectively, and administration of a low dose of S. pneumoniae induced 2,577 and 2,436 transcripts in mice reconstituted with WT or Ifng−/− BM cells, respectively (Table 6). Transcripts that were uniquely changed by S. pneumoniae in Ifng−/− mice reconstituted with WT BM compared with Ifng−/− mice reconstituted with Ifng−/− BM were identified (Table 6). However, a number of these unique transcripts were close to the statistical cutoff of 2.0-fold change, so that the ratio of the fold change in WT BM compared with the fold change in Ifng−/− BM was often close to 1. When this ratio was calculated for all genes counted in Table 6, far fewer had a ratio greater than 1.5 (Table 7). There were 22 genes in both low- and high-dose pneumonias that showed a ratio of over 1.5 (i.e., were 50% greater in WT BM compared with Ifng−/− BM), and these transcripts are listed in Table E1 in the online supplement. Eight of these genes are known to be IFN-γ regulated (bold font).
Table 6.
The Number of Up- or Down-Regulated Transcripts That Were Differentially Expressed During High- or Low-Dose Streptococcus pneumoniae Pneumonia Compared with PBS Controls in Ifng−/− Mice That Were Reconstituted with Wild-Type Bone Marrow or IFN-γ–Deficient Bone Marrow or Were Expressed in Both (Common)*
| Transcripts | Up-Regulated ≥ 2-Fold | Down-Regulated ≥ 2-Fold | Total |
|---|---|---|---|
| High SP versus PBS | |||
| Common | 1,101 | 1,214 | 2,315 |
| Unique to WT BM | 334 | 364 | 698 |
| Unique to Ifng−/− BM | 141 | 211 | 352 |
| Low SP versus PBS | |||
| Common | 1,041 | 1,060 | 2,101 |
| Unique to WT BM | 188 | 288 | 476 |
| Unique to Ifng−/− BM | 175 | 160 | 335 |
Definition of abbreviations: BM, bone marrow; SP, Streptococcus pneumoniae; WT, wild type.
The hematopoietic system of Ifng−/− mice was reconstituted with WT or Ifng−/− BM cells. Transcripts were considered to be differentially expressed after passing filtering criteria (FDR-adjusted P ≤ 0.05; fold change ≥ 2 compared with PBS). n = 4 mice in each group.
Table 7.
The Number of Genes in Table 6 That Also Met the Additional Criterion of the Ratio of the Fold Change in Streptococcus pneumoniae Compared with PBS > 1.5
| Transcripts | ≥2-Fold Up and ≥1.5× Ratio | ≥2-Fold Down and ≥1.5× Ratio | Total |
|---|---|---|---|
| High SP versus PBS | |||
| Common | 131 | 39 | 170 |
| Unique to WT BM | 103 | 57 | 160 |
| Unique to Ifng−/− BM | 22 | 22 | 44 |
| Low SP versus PBS | |||
| Common | 75 | 34 | 109 |
| Unique to WT BM | 50 | 30 | 80 |
| Unique to Ifng−/− BM | 20 | 6 | 26 |
Definition of abbreviations: BM, bone marrow; SP, S. pneumoniae; WT, wild type.
IFN-γ mRNA was barely detectable in all mice given PBS. In the mice reconstituted with WT BM, IFN-γ expression was up-regulated 10-fold and 9.6-fold during high- and low-dose pneumonia, respectively. The Ifng−/− mice have a stop codon in exon 2 of the Ifng gene. However, variable-length mRNA is produced that is recognized by the probes that target the Ifng gene. Expression of the mutant Ifng mRNA in mice reconstituted with Ifng−/− BM was up-regulated 1.8-fold and 2.1-fold at high- and low-dose pneumonia, respectively, and these changes were significantly less compared with those observed in mice with WT BM (adjusted P < 0.001, ANOVA). This mRNA appears unstable because less was produced, but a stop codon in exon 2 would not be expected to affect the promoter region’s function.
There were 22 transcripts whose expression was at least 2-fold greater in WT BM-reconstituted mice compared with Ifng−/− BM-reconstituted mice during low- and high-dose pneumonia (Table 8). Except for Ciita, Pydc3, and GM22079, the expression of the remaining 19 transcripts was increased during high-dose S. pneumoniae pneumonia compared with PBS in mice reconstituted with Ifng−/− BM, indicating that the effect of IFN-γ is partial. Ciita expression was down-regulated during pneumonia in mice reconstituted with Ifng−/− BM cells, and Pydc3 and GM22079 were unchanged, suggesting that IFN-γ is required for their up-regulation. Of the 22 differentially expressed transcripts, 18 have been previously identified in the literature as IFN-γ inducible genes, as curated in NCBI Gene and the Interferome database (48) (Table 8, bold font). The expression of an additional 16 genes increased 1.5- to 2.0-fold in WT BM-reconstituted mice compared with Ifng−/− BM-reconstituted mice during low- and high-dose S. pneumoniae pneumonia, and 10 of these are known to be regulated by IFN-γ (Table E2).
Table 8.
Transcripts That Are Significantly Differentially Expressed 2-Fold or Greater in Ifng−/− Mice Reconstituted with Wild-Type Bone Marrow Compared with Ifng−/− Bone Marrow–Reconstituted in Both High-Dose and Low-Dose Streptococcus pneumoniae Pneumonia
| Gene Symbol* | Description | High-Dose SP |
Low-Dose SP |
||||
|---|---|---|---|---|---|---|---|
| SP/PBS Fold Change in WT BM Mice | SP/PBS Fold Change in Ifng−/− BM Mice | Fold Change in WT BM Mice with SP Compared with Ifng−/− BM Mice with SP | SP/PBS Fold Change in WT BM Mice | SP/PBS Fold Change in Ifng−/− BM Mice | Fold Change in WT BM Mice with SP Compared with Ifng−/− BM Mice with SP | ||
| Apol10b | Apolipoprotein L 10B [Source:MGI Symbol;Acc:MGI:3043522] | 3.7 | 1.5 | 3.3 | 3.7 | 1.2 | 4.0 |
| Apol6 | Apolipoprotein L 6 [Source:MGI Symbol;Acc:MGI:1919189] | 4.1 | 1.6 | 2.8 | 5.1 | 1.3 | 4.2 |
| Batf2 | Basic leucine zipper transcription factor, ATF-like 2 [Source:MGI Symbol;Acc:MGI:192173 | 8.3 | 2.9 | 2.6 | 7.9 | 2.1 | 3.4 |
| BC023105 | cDNA sequence BC023105 [Source:MGI Symbol;Acc:MGI:2384767] | 8.9 | 3.8 | 2.9 | 10.0 | 4.3 | 3.0 |
| Ciita | Class II transactivator [Source:MGI Symbol;Acc:MGI:108445] | 1.4 | −1.9 | 2.4 | 2.1 | −1.4 | 2.5 |
| Cxcl9 | Chemokine (C-X-C motif) ligand 9 [Source:MGI Symbol;Acc:MGI:1352449] | 28.9 | 21.6 | 3.3 | 32.1 | 18.2 | 4.3 |
| Fam26f | Family with sequence similarity 26, member F [Source:MGI Symbol;Acc:MGI:2443082] | 12.9 | 3.2 | 3.7 | 10.9 | 2.2 | 4.6 |
| Gbp10 | Guanylate-binding protein 10 [Source:MGI Symbol;Acc:MGI:4359647] | 7.4 | 3.1 | 3.1 | 8.2 | 2.8 | 3.8 |
| Gbp11 | Guanylate binding protein 11 [Source:MGI Symbol;Acc:MGI:3646307] | 10.0 | 4.2 | 3.0 | 13.0 | 3.0 | 5.4 |
| Gbp4 | Guanylate binding protein 4 [Source:MGI Symbol;Acc:MGI:97072] | 4.1 | 2.1 | 3.1 | 4.8 | 1.9 | 4.2 |
| Gm12185 | Predicted gene 12,185 [Source:MGI Symbol;Acc:MGI:3652173] | 5.7 | 2.5 | 2.5 | 4.4 | 2.0 | 2.4 |
| Gm12250 | Predicted gene 12,250 [Source:MGI Symbol;Acc:MGI:3649299] | 20.5 | 12.8 | 2.2 | 18.8 | 11.1 | 2.3 |
| Gm22079 | Predicted gene, 22,079 [Source:MGI Symbol;Acc:MGI:5451856] | 3.1 | 1.1 | 2.9 | 3.7 | 1.4 | 2.6 |
| Gm4841 | Predicted gene 4,841 [Source:MGI Symbol;Acc:MGI:3643814] | 10.0 | 2.8 | 3.8 | 14.4 | 3.4 | 4.4 |
| Ido1 | Indoleamine 2,3-dioxygenase 1 [Source:MGI Symbol;Acc:MGI:96416] | 10.5 | 1.6 | 7.6 | 12.9 | 1.5 | 10.2 |
| Igtp | IFN-γ–induced GTPase [Source:MGI Symbol;Acc:MGI:107729] | 7.3 | 4.5 | 2.1 | 7.7 | 4.3 | 2.3 |
| Irf1 | IFN regulatory factor 1 [Source:MGI Symbol;Acc:MGI:96590] | 4.1 | 2.0 | 2.2 | 4.0 | 1.6 | 2.8 |
| Pydc3 | Pyrin domain containing 3 | 6.0 | −1.0 | 7.4 | 3.8 | −1.0 | 4.7 |
| Slamf8 | SLAM family member 8 [Source:MGI Symbol;Acc:MGI:1921998] | 6.7 | 1.7 | 3.2 | 9.7 | 2.6 | 3.0 |
| Socs1 | Suppressor of cytokine signaling 1 [Source:MGI Symbol;Acc:MGI:1354910] | 13.4 | 6.4 | 2.3 | 11.1 | 5.0 | 2.4 |
| Tgtp1 | T cell–specific GTPase 1 [Source:MGI Symbol;Acc:MGI:98734] | 10.9 | 2.0 | 5.7 | 11.5 | 2.1 | 5.7 |
| Tgtp2 | T cell–specific GTPase 2 [Source:MGI Symbol;Acc:MGI:3710083] | 13.0 | 7.4 | 2.8 | 13.9 | 5.9 | 3.7 |
Definition of abbreviations: BM, bone marrow; SP, S. pneumonia; WT, wild type.
Genes that have been previously described in the literature as IFN-γ regulated are shown in bold.
Gene Set Enrichment Analysis using gene ontology (GO) sets showed that genes involved in IFN-γ signaling and other immune response pathways were highly statistically enriched in the lungs of mice reconstituted with WT cells compared with those reconstituted with Ifng−/− cells (Table 9). The IFN-γ–inducible genes that accounted for the enrichment score in two GO sets focused on IFN-γ–induced responses (GO:0034341 and GO:0071346) (Table 9) are shown in Table E3. Nine of the genes in Table E2 were also identified in Table 8 or Table E1 (Ciita, Gbp10, Gbp4, Gbp5, Irf1, Irgm2, Nlrc5, Socs1, and Tgtp1).
Table 9.
Gene Set Enrichment Analysis–Identified Gene Ontology Biological Pathways Showing Highly Significant Enrichment in Wild-Type Bone Marrow–Reconstituted Mice with Streptococcus pneumoniae Compared with Ifng−/− Bone Marrow–Reconstituted Mice with S. pneumoniae (FDR ≤ 0.0001)*
| Gene Ontology Set ID | Functional Term |
|---|---|
| GO:0001562 | Response to protozoan |
| GO:0001819 | Positive regulation of cytokine production |
| GO:0042832 | Defense response to protozoan |
| GO:0035456 | Response to IFN-β |
| GO:0034341 | Response to IFN-γ |
| GO:0045087 | Innate immune response |
| GO:0031341 | Regulation of cell killing |
| GO:0071346 | Cellular response to IFN-γ |
| GO:0045428 | Regulation of nitric oxide biosynthetic process |
| GO:0001906 | Cell killing |
| GO:0031343 | Positive regulation of cell killing |
| GO:0046209 | Nitric oxide metabolic process |
| GO:0001909 | Leukocyte-mediated cytotoxicity |
| GO:0045429 | Positive regulation of nitric oxide biosynthetic process |
| GO:0045088 | Regulation of innate immune response |
| GO:0006809 | Nitric oxide biosynthetic process |
| GO:0035458 | Cellular response to IFN-β |
| GO:0002697 | Regulation of immune effector process |
| GO:0032732 | Positive regulation of interleukin-1 production |
| GO:0006955 | Immune response |
These pathways were also significantly enriched in mice with high-dose S. pneumoniae compared with PBS regardless of the donor bone marrow genotype.
STAT1 is downstream of the IFN-γ receptor and mediates most effects of IFN-γ. STAT1 is activated during S. pneumoniae pneumonia, as demonstrated by GSEA showing statistically significant enrichment of genes involved in tyrosine phosphorylation of STAT1 protein (GO:0042508 and GO:0042510). Furthermore, STAT1 binding sites are present upstream to the transcription start site of all genes identified in Tables 8, E2, and E3, many of which have been experimentally confirmed (49, 50).
Taken together, these data indicate that neutrophil-derived IFN-γ regulates gene expression in the lung during S. pneumoniae pneumonia and thus shapes the host response to bacteria in the lungs, the resolution of inflammation, and the initiation of the immune response.
Discussion
Our previous studies demonstrated that neutrophils recruited to the lungs in response to S. pneumoniae produce IFN-γ and that this IFN-γ appears to contribute to host defense (3). The studies presented here sought to determine the signaling pathways through which this IFN-γ is produced by neutrophils and the mechanisms through which this IFN-γ affects host defense and innate immunity.
A growing body of work has shown that neutrophils are capable of producing cytokines and other mediators during inflammation (2). IFN-γ production by neutrophils in the lung has been documented in response to Nocardia asteroides (51) or Saccharopolyspora rectivirgula (52). Neutrophils also make IFN-γ during inflammation in other tissues, including in the spleen during infection with Salmonella (53) or L. monocytogenes (54), during early graft loss of pancreatic islet transplants (55), in response to Francisella tularensis live vaccine strain (56), and in the kidney early in renal ischemia–reperfusion injury (57). These reports suggest that IFN-γ production by neutrophils early during infection or injury may be more widespread than generally appreciated.
This rapidly produced IFN-γ appears to be almost entirely from neutrophils. At 24 hours, neutrophils account for >98% of the IFN-γ production, and none is produced by T cells, NK cells, macrophages, or neutrophil–dendritic cell hybrids. IL-12 is not required for IFN-γ production by neutrophils. IL-12–independent IFN-γ production has been suggested to occur even in lymphocytes. For example, lymphocytes from IL-12a-deficient mice are still able to produce low levels of IFN-γ (41, 58). IL-12–independent IFN-γ production could occur through IL-15 and/or IL-18. IL-18 is produced by macrophages and dendritic and other cells, and it can induce IFN-γ production by T lymphocytes and NK cells in combination with IL-12 or other factors in the absence of IL-12 (59–61). Neutrophils from the lungs of mice exposed to S. rectivirgula produced IFN-γ when cultured in vitro with IL-12 and IL-18 or with IL-18 and IL-15 but not when cultured with any cytokine alone (62). Purified human peripheral blood neutrophils cultured with IL-12 produced a small amount of IFN-γ, and IFN-γ levels were augmented by the addition of TNF (63). Whether IL-18, IL-15, TNF, or other cytokines induce IL-12–independent IFN-γ production by neutrophils in response to S. pneumoniae in vivo has not been determined. However, S. pneumoniae does induce a small but significant increase in IL-15 and IL-18 mRNAs in the pneumonic lung.
Consistent with the finding that IL-12 is not required for IFN-γ production by neutrophils, STAT4 phosphorylation was not detected in lung neutrophils. In T lymphocytes and NK cells, the T-box transcription factors T-bet and Eomes regulate IFN-γ production (64, 65). T-bet has been implicated in IFN-γ production by T helper cells independent of IL-12 (66). However, T-bet is expressed at almost undetectable levels, and Eomes is not significantly expressed in lung neutrophils from mice given PBS or S. pneumoniae (unpublished data). These findings suggest that IFN-γ gene transcription in neutrophils may be regulated through pathways distinct from those in other cell types.
Our data indicate that CalDAG-GEFI inhibits IFN-γ expression. In neutrophils, CalDAG-GEFI is critical for integrin activation by activating the small GTPase Rap1 downstream of G protein–coupled receptor signaling (67) or rolling on E-selectin (68). CalDAG-GEFI can mediate neutrophil chemotaxis independent of integrins by regulating the actin cytoskeleton (69). The role of Rap1 in inducing IFN-γ is likely to be complex due to its many functions in neutrophils, endothelial cells, and other cells (70). Whether CalDAG-GEFI regulates NADPH oxidase or any of the other signaling molecules required for IFN-γ production by neutrophils apart from its role in Rap1 or integrin activation has not been demonstrated to our knowledge.
MyD88 was important in neutrophil recruitment and IFN-γ expression, but neither TLR2 nor TLR4 was required. These results suggest that redundant pathways for recognizing S. pneumoniae exist and that other TLRs and/or pattern recognition receptors upstream of MyD88 are sufficient to drive IFN-γ production. The defect in IFN-γ production in MyD88-deficient mice is not as large as that observed in Nox2-deficient mice, suggesting that other pattern recognition receptors that do not signal through MyD88 may be important in the recognition of and response to S. pneumoniae. Other pattern recognition receptors have been shown to play important roles in host defense against this organism, including TLR9 (71, 72), NLRP3 (73, 74), NOD2 (75), and DAI (76). Pore formation by pneumolysin may enable bacterial components to enter host cells, thereby facilitating recognition of bacterial components by intracellular pathogen recognition receptors. These studies do not address whether MyD88 in neutrophils or in other cell types, such as alveolar and bronchial epithelial cells and alveolar macrophages, is controlling IFN-γ production by neutrophils.
NADPH oxidase is required for IFN-γ production by neutrophils but not Nrf2, a transcription factor that is activated by oxidative stress. These results suggest that NADPH oxidase may regulate IFN-γ production by modulating activation of other oxidant-sensitive transcription factors, such as AP-1 and NF-κB, and/or by regulating phosphatase activity. In other cell types, oxidants have been shown to inhibit phosphatases, but our data show that JNK phosphorylation is greater in Nox2-deficient neutrophils. In contrast, phosphorylation of p38 MAPK and ERK is similar in WT and Nox2-deficient neutrophils. Whether the observed increase in JNK phosphorylation is the mechanism through which IFN-γ production is inhibited in Nox2-deficient mice remains to be determined. Neutrophils may have evolved complex mechanisms to regulate oxidant-responsive pathways as befits a cell that can generate large amounts of oxidants rapidly.
IFN-γ is important for host defense through its ability to activate neutrophils and other innate immune cells. For example, IFN-γ was required for timely clearance of S. pneumoniae and for the formation of neutrophil extracellular traps (3). Mice deficient in IFN-γ have decrements in the production of iNOS, also important for bactericidal activity. Moreover, this IFN-γ may shape the subsequent response by lymphocytes that migrate into the site of infection or injury after neutrophils. For example, Cxcl11 expression at 24 hours of pneumonia requires IFN-γ production (unpublished data). Cxcl11 is the ligand for Cxcr3, which is expressed on CD4+ Th1 and CD8+ cytotoxic lymphocytes, and Cxcl11 regulates migration of Th1 T cells into sites of inflammation (77).
The mechanisms through which the IFN-γ produced within 24 hours after instillation of S. pneumoniae contributes to host defense was evaluated by gene profiling of lungs from Ifng−/− mice with either Ifng−/− or WT BM. S. pneumoniae induces robust gene expression in the lungs, and a significant proportion of these induced genes are regulated by IFN-γ. Many of the genes identified in our analysis as IFN-γ–regulated genes are known IFN-γ targets that are critical players in immune response (including Ciita, Cxcl9, Ido1, Irf1, and Socs1). Regulation of expression of certain genes by IFN-γ may be direct, as in transcriptional regulation of target genes through the IFN-γ receptor and downstream STAT1 signaling, or indirect, for example by regulating recruitment of certain cell types. Thus, by producing IFN-γ, neutrophils can drive many processes in the host response to bacteria in the lungs.
In summary, these studies show that, at 24 hours after instillation of S. pneumoniae, neutrophils, but not neutrophil-dendritic cell hybrids, produced IFN-γ. No role for IL-12 was found for this process in neutrophils, in striking contrast to its critical role in IFN-γ production by T lymphocytes and NK cells (Figure 6). This IL-12–independent IFN-γ production is nearly abolished in Nox2-deficient mice, suggesting a role for oxidants in IFN-γ expression (3). However, Nrf2, a major transcription factor activated by oxidative stress, is not required for IFN-γ production. These NADPH oxidase–generated oxidants may be acting through limiting JNK phosphorylation. Building on our previous work showing that Src family tyrosine kinases, Rac2, and NADPH oxidase are required for IFN-γ production, our studies tested the role of the CD11/CD18 adhesion complex and CD44, which network with these signaling molecules. The data show that CD11/CD18 adhesion complex and CD44 are not required. The pathogen recognition receptors TLR2 and TLR4, either singly or in combination, are also not required, but the TLR adaptor molecule MyD88 is required for full expression of IFN-γ by neutrophils. Deficiency of TRIF, the other TLR adaptor protein, induced an increase in neutrophils and thus an increase in the total number of IFN-γ+ neutrophils, but TRIF is not required in IFN-γ production itself. Thus, the intracellular signaling pathway(s) that leads to IFN-γ transcription includes MyD88, Src family tyrosine kinases, Rac2, and NADPH oxidase–generated ROS and is negatively regulated by CalDAG-GEFI. Neutrophils are the only cells in the lung making IFN-γ during 24-hour S. pneumoniae pneumonia, and this IFN-γ is critical for the expression of many genes known to regulate macrophage activation, immune cell recruitment, leukocyte activation, cell killing, and other aspects of innate and specific immunity. These studies underline the complexity and the importance of the neutrophil responses in host defense in the acute inflammatory response and in subsequent resolution or initiation of innate and specific immune responses. IFN-γ production by neutrophils during pneumonia induced by S. pneumoniae is a tightly regulated process that likely involves interactions between multiple signaling pathways. Understanding these mechanisms may have important implications for treatment of infections and inflammatory diseases.
Figure 6.

Postulated pathway regulating IFN-γ production by neutrophils. Recognition of S. pneumoniae or its components by pattern recognition receptors (PRRs) results in activation of the adaptor molecule MyD88, which we postulate leads to activation of Src family kinases (SFK) Lyn/Fgr/Hck, the small GTPase Rac2, and NADPH oxidase. Production of oxidants by NADPH oxidase leads to production of IFN-γ through a process that does not require the oxidant-sensitive transcription factor Nrf2. IL-12 is not required for IFN-γ production by neutrophils. The adaptor molecule TRIF and the leukocyte adhesion complex CD11/CD18 (including complement receptor 3) are also not required. The small GTPase CalDAG-GEFI inhibits neutrophil IFN-γ production. Neutrophil-derived IFN-γ is critical for the expression of many genes known to regulate macrophage activation, immune cell recruitment, leukocyte activation, cell killing, and other aspects of innate and specific immunity. Dashed arrows indicate likely to include intervening steps. ROS, reactive oxygen species; X, not required.
Acknowledgments
Acknowledgments
The authors thank Elizabeth A. Duncan, Eric Pearlman, Casey Weaver, Michael J. Vernon, and Wanda K. O’Neal for their expert advice and many helpful discussions.
Footnotes
This work was supported by National Institutes of Health grants AI067798 (W.J.B.), HL 048160 (C.M.D.), and HL 052466 (C.M.D.). Flow cytometry was performed at the UNC Flow Cytometry Core Facility, which is supported in part by an NCI Center Core Support Grant P30CA016086 to the UNC Lineberger Comprehensive Cancer Center.
Author Contributions: Conception and design: J.C.G., M.Y., H.D., M.C.D., and C.M.D. Acquisition, analysis, and interpretation: J.C.G., M.Y., J.R.M., H.D., W.J.B., W.B., M.C.D., and C.M.D. Drafting the manuscript for important intellectual content: J.C.G., M.Y., H.D., W.B., M.C.D., and C.M.D.
This article has an online supplement, which is accessible from this issue’s table of contents at www.atsjournals.org
Originally Published in Press as DOI: 10.1165/rcmb.2013-0316OC on August 6, 2014
Author disclosures are available with the text of this article at www.atsjournals.org.
References
- 1.Mizgerd JP. Respiratory infection and the impact of pulmonary immunity on lung health and disease. Am J Respir Crit Care Med. 2012;186:824–829. doi: 10.1164/rccm.201206-1063PP. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Mantovani A, Cassatella MA, Costantini C, Jaillon S. Neutrophils in the activation and regulation of innate and adaptive immunity. Nat Rev Immunol. 2011;11:519–531. doi: 10.1038/nri3024. [DOI] [PubMed] [Google Scholar]
- 3.Yamada M, Gomez JC, Chugh PE, Lowell CA, Dinauer MC, Dittmer DP, Doerschuk CM. Interferon-γ production by neutrophils during bacterial pneumonia in mice. Am J Respir Crit Care Med. 2011;183:1391–1401. doi: 10.1164/rccm.201004-0592OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Trinchieri G. Interleukin-12 and the regulation of innate resistance and adaptive immunity. Nat Rev Immunol. 2003;3:133–146. doi: 10.1038/nri1001. [DOI] [PubMed] [Google Scholar]
- 5.Munder M, Mallo M, Eichmann K, Modolell M. Murine macrophages secrete interferon gamma upon combined stimulation with interleukin (IL)-12 and IL-18: a novel pathway of autocrine macrophage activation. J Exp Med. 1998;187:2103–2108. doi: 10.1084/jem.187.12.2103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Fukao T, Matsuda S, Koyasu S. Synergistic effects of IL-4 and IL-18 on IL-12-dependent IFN-gamma production by dendritic cells. J Immunol. 2000;164:64–71. doi: 10.4049/jimmunol.164.1.64. [DOI] [PubMed] [Google Scholar]
- 7.Ohteki T, Fukao T, Suzue K, Maki C, Ito M, Nakamura M, Koyasu S. Interleukin 12-dependent interferon gamma production by CD8alpha+ lymphoid dendritic cells. J Exp Med. 1999;189:1981–1986. doi: 10.1084/jem.189.12.1981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bacon CM, Petricoin EF, III, Ortaldo JR, Rees RC, Larner AC, Johnston JA, O’Shea JJ. Interleukin 12 induces tyrosine phosphorylation and activation of STAT4 in human lymphocytes. Proc Natl Acad Sci USA. 1995;92:7307–7311. doi: 10.1073/pnas.92.16.7307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Thierfelder WE, van Deursen JM, Yamamoto K, Tripp RA, Sarawar SR, Carson RT, Sangster MY, Vignali DA, Doherty PC, Grosveld GC, et al. Requirement for Stat4 in interleukin-12-mediated responses of natural killer and T cells. Nature. 1996;382:171–174. doi: 10.1038/382171a0. [DOI] [PubMed] [Google Scholar]
- 10.Li S, Yamauchi A, Marchal CC, Molitoris JK, Quilliam LA, Dinauer MC. Chemoattractant-stimulated Rac activation in wild-type and Rac2-deficient murine neutrophils: preferential activation of Rac2 and Rac2 gene dosage effect on neutrophil functions. J Immunol. 2002;169:5043–5051. doi: 10.4049/jimmunol.169.9.5043. [DOI] [PubMed] [Google Scholar]
- 11.Bedard K, Krause KH. The NOX family of ROS-generating NADPH oxidases: physiology and pathophysiology. Physiol Rev. 2007;87:245–313. doi: 10.1152/physrev.00044.2005. [DOI] [PubMed] [Google Scholar]
- 12.Cho HY, Reddy SP, Kleeberger SR. Nrf2 defends the lung from oxidative stress. Antioxid Redox Signal. 2006;8:76–87. doi: 10.1089/ars.2006.8.76. [DOI] [PubMed] [Google Scholar]
- 13.Kobayashi M, Yamamoto M. Nrf2-Keap1 regulation of cellular defense mechanisms against electrophiles and reactive oxygen species. Adv Enzyme Regul. 2006;46:113–140. doi: 10.1016/j.advenzreg.2006.01.007. [DOI] [PubMed] [Google Scholar]
- 14.Thimmulappa RK, Lee H, Rangasamy T, Reddy SP, Yamamoto M, Kensler TW, Biswal S. Nrf2 is a critical regulator of the innate immune response and survival during experimental sepsis. J Clin Invest. 2006;116:984–995. doi: 10.1172/JCI25790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Reddy NM, Suryanarayana V, Kalvakolanu DV, Yamamoto M, Kensler TW, Hassoun PM, Kleeberger SR, Reddy SP. Innate immunity against bacterial infection following hyperoxia exposure is impaired in NRF2-deficient mice. J Immunol. 2009;183:4601–4608. doi: 10.4049/jimmunol.0901754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cho HY, Imani F, Miller-DeGraff L, Walters D, Melendi GA, Yamamoto M, Polack FP, Kleeberger SR. Antiviral activity of Nrf2 in a murine model of respiratory syncytial virus disease. Am J Respir Crit Care Med. 2009;179:138–150. doi: 10.1164/rccm.200804-535OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kong X, Thimmulappa R, Craciun F, Harvey C, Singh A, Kombairaju P, Reddy SP, Remick D, Biswal S. Enhancing Nrf2 pathway by disruption of Keap1 in myeloid leukocytes protects against sepsis. Am J Respir Crit Care Med. 2011;184:928–938. doi: 10.1164/rccm.201102-0271OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Harvey CJ, Thimmulappa RK, Sethi S, Kong X, Yarmus L, Brown RH, Feller-Kopman D, Wise R, Biswal S. Targeting Nrf2 signaling improves bacterial clearance by alveolar macrophages in patients with COPD and in a mouse model. Sci Transl Med. 2011;3:78ra32. doi: 10.1126/scitranslmed.3002042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.MacGarvey NC, Suliman HB, Bartz RR, Fu P, Withers CM, Welty-Wolf KE, Piantadosi CA. Activation of mitochondrial biogenesis by heme oxygenase-1-mediated NF-E2-related factor-2 induction rescues mice from lethal Staphylococcus aureus sepsis. Am J Respir Crit Care Med. 2012;185:851–861. doi: 10.1164/rccm.201106-1152OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Piantadosi CA, Withers CM, Bartz RR, MacGarvey NC, Fu P, Sweeney TE, Welty-Wolf KE, Suliman HB. Heme oxygenase-1 couples activation of mitochondrial biogenesis to anti-inflammatory cytokine expression. J Biol Chem. 2011;286:16374–16385. doi: 10.1074/jbc.M110.207738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Berton G, Mócsai A, Lowell CA. Src and Syk kinases: key regulators of phagocytic cell activation. Trends Immunol. 2005;26:208–214. doi: 10.1016/j.it.2005.02.002. [DOI] [PubMed] [Google Scholar]
- 22.Dib K, Melander F, Axelsson L, Dagher MC, Aspenström P, Andersson T. Down-regulation of Rac activity during beta 2 integrin-mediated adhesion of human neutrophils. J Biol Chem. 2003;278:24181–24188. doi: 10.1074/jbc.M302300200. [DOI] [PubMed] [Google Scholar]
- 23.Giagulli C, Ottoboni L, Caveggion E, Rossi B, Lowell C, Constantin G, Laudanna C, Berton G. The Src family kinases Hck and Fgr are dispensable for inside-out, chemoattractant-induced signaling regulating beta 2 integrin affinity and valency in neutrophils, but are required for beta 2 integrin-mediated outside-in signaling involved in sustained adhesion. J Immunol. 2006;177:604–611. doi: 10.4049/jimmunol.177.1.604. [DOI] [PubMed] [Google Scholar]
- 24.Zhao T, Benard V, Bohl BP, Bokoch GM. The molecular basis for adhesion-mediated suppression of reactive oxygen species generation by human neutrophils. J Clin Invest. 2003;112:1732–1740. doi: 10.1172/JCI19108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Vachon E, Martin R, Kwok V, Cherepanov V, Chow CW, Doerschuk CM, Plumb J, Grinstein S, Downey GP. CD44-mediated phagocytosis induces inside-out activation of complement receptor-3 in murine macrophages. Blood. 2007;110:4492–4502. doi: 10.1182/blood-2007-02-076539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lien E, Sellati TJ, Yoshimura A, Flo TH, Rawadi G, Finberg RW, Carroll JD, Espevik T, Ingalls RR, Radolf JD, et al. Toll-like receptor 2 functions as a pattern recognition receptor for diverse bacterial products. J Biol Chem. 1999;274:33419–33425. doi: 10.1074/jbc.274.47.33419. [DOI] [PubMed] [Google Scholar]
- 27.Yoshimura A, Lien E, Ingalls RR, Tuomanen E, Dziarski R, Golenbock D. Cutting edge: recognition of Gram-positive bacterial cell wall components by the innate immune system occurs via Toll-like receptor 2. J Immunol. 1999;163:1–5. [PubMed] [Google Scholar]
- 28.Malley R, Henneke P, Morse SC, Cieslewicz MJ, Lipsitch M, Thompson CM, Kurt-Jones E, Paton JC, Wessels MR, Golenbock DT. Recognition of pneumolysin by Toll-like receptor 4 confers resistance to pneumococcal infection. Proc Natl Acad Sci USA. 2003;100:1966–1971. doi: 10.1073/pnas.0435928100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Dessing MC, Hirst RA, de Vos AF, van der Poll T. Role of Toll-like receptors 2 and 4 in pulmonary inflammation and injury induced by pneumolysin in mice. PLoS One. 2009;4:e7993. doi: 10.1371/journal.pone.0007993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ve T, Gay NJ, Mansell A, Kobe B, Kellie S. Adaptors in toll-like receptor signaling and their potential as therapeutic targets. Curr Drug Targets. 2012;13:1360–1374. doi: 10.2174/138945012803530260. [DOI] [PubMed] [Google Scholar]
- 31.Oshiumi H, Matsumoto M, Funami K, Akazawa T, Seya T. TICAM-1, an adaptor molecule that participates in Toll-like receptor 3-mediated interferon-beta induction. Nat Immunol. 2003;4:161–167. doi: 10.1038/ni886. [DOI] [PubMed] [Google Scholar]
- 32.Yamamoto M, Sato S, Mori K, Hoshino K, Takeuchi O, Takeda K, Akira S. Cutting edge: a novel Toll/IL-1 receptor domain-containing adapter that preferentially activates the IFN-beta promoter in the Toll-like receptor signaling. J Immunol. 2002;169:6668–6672. doi: 10.4049/jimmunol.169.12.6668. [DOI] [PubMed] [Google Scholar]
- 33.Yamamoto M, Sato S, Hemmi H, Hoshino K, Kaisho T, Sanjo H, Takeuchi O, Sugiyama M, Okabe M, Takeda K, et al. Role of adaptor TRIF in the MyD88-independent toll-like receptor signaling pathway. Science. 2003;301:640–643. doi: 10.1126/science.1087262. [DOI] [PubMed] [Google Scholar]
- 34.Skerrett SJ, Liggitt HD, Hajjar AM, Wilson CB. Cutting edge: myeloid differentiation factor 88 is essential for pulmonary host defense against Pseudomonas aeruginosa but not Staphylococcus aureus. J Immunol. 2004;172:3377–3381. doi: 10.4049/jimmunol.172.6.3377. [DOI] [PubMed] [Google Scholar]
- 35.Balamayooran G, Batra S, Fessler MB, Happel KI, Jeyaseelan S. Mechanisms of neutrophil accumulation in the lungs against bacteria. Am J Respir Cell Mol Biol. 2010;43:5–16. doi: 10.1165/rcmb.2009-0047TR. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Jeyaseelan S, Young SK, Fessler MB, Liu Y, Malcolm KC, Yamamoto M, Akira S, Worthen GS. Toll/IL-1 receptor domain-containing adaptor inducing IFN-beta (TRIF)-mediated signaling contributes to innate immune responses in the lung during Escherichia coli pneumonia. J Immunol. 2007;178:3153–3160. doi: 10.4049/jimmunol.178.5.3153. [DOI] [PubMed] [Google Scholar]
- 37.Cai S, Batra S, Shen L, Wakamatsu N, Jeyaseelan S. Both TRIF- and MyD88-dependent signaling contribute to host defense against pulmonary Klebsiella infection. J Immunol. 2009;183:6629–6638. doi: 10.4049/jimmunol.0901033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Power MR, Li B, Yamamoto M, Akira S, Lin TJ. A role of Toll-IL-1 receptor domain-containing adaptor-inducing IFN-beta in the host response to Pseudomonas aeruginosa lung infection in mice. J Immunol. 2007;178:3170–3176. doi: 10.4049/jimmunol.178.5.3170. [DOI] [PubMed] [Google Scholar]
- 39.Geng S, Matsushima H, Okamoto T, Yao Y, Lu R, Page K, Blumenthal RM, Ward NL, Miyazaki T, Takashima A. Emergence, origin, and function of neutrophil-dendritic cell hybrids in experimentally induced inflammatory lesions in mice. Blood. 2013;121:1690–1700. doi: 10.1182/blood-2012-07-445197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Matsushima H, Geng S, Lu R, Okamoto T, Yao Y, Mayuzumi N, Kotol PF, Chojnacki BJ, Miyazaki T, Gallo RL, et al. Neutrophil differentiation into a unique hybrid population exhibiting dual phenotype and functionality of neutrophils and dendritic cells. Blood. 2013;121:1677–1689. doi: 10.1182/blood-2012-07-445189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Mattner F, Magram J, Ferrante J, Launois P, Di Padova K, Behin R, Gately MK, Louis JA, Alber G. Genetically resistant mice lacking interleukin-12 are susceptible to infection with Leishmania major and mount a polarized Th2 cell response. Eur J Immunol. 1996;26:1553–1559. doi: 10.1002/eji.1830260722. [DOI] [PubMed] [Google Scholar]
- 42.Abram CL, Lowell CA. The ins and outs of leukocyte integrin signaling. Annu Rev Immunol. 2009;27:339–362. doi: 10.1146/annurev.immunol.021908.132554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Oertli B, Beck-Schimmer B, Fan X, Wüthrich RP. Mechanisms of hyaluronan-induced up-regulation of ICAM-1 and VCAM-1 expression by murine kidney tubular epithelial cells: hyaluronan triggers cell adhesion molecule expression through a mechanism involving activation of nuclear factor-kappa B and activating protein-1. J Immunol. 1998;161:3431–3437. [PubMed] [Google Scholar]
- 44.Fujii Y, Fujii K, Nakano K, Tanaka Y. Crosslinking of CD44 on human osteoblastic cells upregulates ICAM-1 and VCAM-1. FEBS Lett. 2003;539:45–50. doi: 10.1016/s0014-5793(03)00182-0. [DOI] [PubMed] [Google Scholar]
- 45.Fujisaki T, Tanaka Y, Fujii K, Mine S, Saito K, Yamada S, Yamashita U, Irimura T, Eto S. CD44 stimulation induces integrin-mediated adhesion of colon cancer cell lines to endothelial cells by up-regulation of integrins and c-Met and activation of integrins. Cancer Res. 1999;59:4427–4434. [PubMed] [Google Scholar]
- 46.Kamata H, Honda S, Maeda S, Chang L, Hirata H, Karin M. Reactive oxygen species promote TNFalpha-induced death and sustained JNK activation by inhibiting MAP kinase phosphatases. Cell. 2005;120:649–661. doi: 10.1016/j.cell.2004.12.041. [DOI] [PubMed] [Google Scholar]
- 47.Kim HS, Ullevig SL, Zamora D, Lee CF, Asmis R. Redox regulation of MAPK phosphatase 1 controls monocyte migration and macrophage recruitment. Proc Natl Acad Sci USA. 2012;109:E2803–E2812. doi: 10.1073/pnas.1212596109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Rusinova I, Forster S, Yu S, Kannan A, Masse M, Cumming H, Chapman R, Hertzog PJ. Interferome v2.0: an updated database of annotated interferon-regulated genes. Nucleic Acids Res. 2013;41:D1040–D1046. doi: 10.1093/nar/gks1215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Marinescu VD, Kohane IS, Riva A. MAPPER: a search engine for the computational identification of putative transcription factor binding sites in multiple genomes. BMC Bioinformatics. 2005;6:79. doi: 10.1186/1471-2105-6-79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Marinescu VD, Kohane IS, Riva A. The MAPPER database: a multi-genome catalog of putative transcription factor binding sites. Nucleic Acids Res. 2005;33:D91–D97. doi: 10.1093/nar/gki103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ellis TN, Beaman BL. Murine polymorphonuclear neutrophils produce interferon-gamma in response to pulmonary infection with Nocardia asteroides. J Leukoc Biol. 2002;72:373–381. [PubMed] [Google Scholar]
- 52.Nance S, Cross R, Yi AK, Fitzpatrick EA. IFN-gamma production by innate immune cells is sufficient for development of hypersensitivity pneumonitis. Eur J Immunol. 2005;35:1928–1938. doi: 10.1002/eji.200425762. [DOI] [PubMed] [Google Scholar]
- 53.Kirby AC, Yrlid U, Wick MJ. The innate immune response differs in primary and secondary Salmonella infection. J Immunol. 2002;169:4450–4459. doi: 10.4049/jimmunol.169.8.4450. [DOI] [PubMed] [Google Scholar]
- 54.Yin J, Ferguson TA. Identification of an IFN-gamma-producing neutrophil early in the response to Listeria monocytogenes. J Immunol. 2009;182:7069–7073. doi: 10.4049/jimmunol.0802410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Yasunami Y, Kojo S, Kitamura H, Toyofuku A, Satoh M, Nakano M, Nabeyama K, Nakamura Y, Matsuoka N, Ikeda S, et al. Valpha14 NK T cell-triggered IFN-gamma production by Gr-1+CD11b+ cells mediates early graft loss of syngeneic transplanted islets. J Exp Med. 2005;202:913–918. doi: 10.1084/jem.20050448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.De Pascalis R, Taylor BC, Elkins KL. Diverse myeloid and lymphoid cell subpopulations produce gamma interferon during early innate immune responses to Francisella tularensis live vaccine strain. Infect Immun. 2008;76:4311–4321. doi: 10.1128/IAI.00514-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Li L, Huang L, Sung SS, Lobo PI, Brown MG, Gregg RK, Engelhard VH, Okusa MD. NKT cell activation mediates neutrophil IFN-gamma production and renal ischemia-reperfusion injury. J Immunol. 2007;178:5899–5911. doi: 10.4049/jimmunol.178.9.5899. [DOI] [PubMed] [Google Scholar]
- 58.Brombacher F, Dorfmüller A, Magram J, Dai WJ, Köhler G, Wunderlin A, Palmer-Lehmann K, Gately MK, Alber G. IL-12 is dispensable for innate and adaptive immunity against low doses of Listeria monocytogenes. Int Immunol. 1999;11:325–332. doi: 10.1093/intimm/11.3.325. [DOI] [PubMed] [Google Scholar]
- 59.Freudenberg MA, Merlin T, Kalis C, Chvatchko Y, Stübig H, Galanos C. Cutting edge: a murine, IL-12-independent pathway of IFN-gamma induction by gram-negative bacteria based on STAT4 activation by Type I IFN and IL-18 signaling. J Immunol. 2002;169:1665–1668. doi: 10.4049/jimmunol.169.4.1665. [DOI] [PubMed] [Google Scholar]
- 60.Robinson D, Shibuya K, Mui A, Zonin F, Murphy E, Sana T, Hartley SB, Menon S, Kastelein R, Bazan F, et al. IGIF does not drive Th1 development but synergizes with IL-12 for interferon-gamma production and activates IRAK and NFkappaB. Immunity. 1997;7:571–581. doi: 10.1016/s1074-7613(00)80378-7. [DOI] [PubMed] [Google Scholar]
- 61.Müller U, Köhler G, Mossmann H, Schaub GA, Alber G, Di Santo JP, Brombacher F, Hölscher C. IL-12-independent IFN-gamma production by T cells in experimental Chagas’ disease is mediated by IL-18. J Immunol. 2001;167:3346–3353. doi: 10.4049/jimmunol.167.6.3346. [DOI] [PubMed] [Google Scholar]
- 62.Abdelsamed HA, Desai M, Nance SC, Fitzpatrick EA. T-bet controls severity of hypersensitivity pneumonitis. J Inflamm (Lond) 2011;8:15. doi: 10.1186/1476-9255-8-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Yeaman GR, Collins JE, Currie JK, Guyre PM, Wira CR, Fanger MW. IFN-gamma is produced by polymorphonuclear neutrophils in human uterine endometrium and by cultured peripheral blood polymorphonuclear neutrophils. J Immunol. 1998;160:5145–5153. [PubMed] [Google Scholar]
- 64.Balasubramani A, Mukasa R, Hatton RD, Weaver CT. Regulation of the Ifng locus in the context of T-lineage specification and plasticity. Immunol Rev. 2010;238:216–232. doi: 10.1111/j.1600-065X.2010.00961.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Gill S, Vasey AE, De Souza A, Baker J, Smith AT, Kohrt HE, Florek M, Gibbs KD, Jr, Tate K, Ritchie DS, et al. Rapid development of exhaustion and down-regulation of eomesodermin limit the antitumor activity of adoptively transferred murine natural killer cells. Blood. 2012;119:5758–5768. doi: 10.1182/blood-2012-03-415364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Mullen AC, High FA, Hutchins AS, Lee HW, Villarino AV, Livingston DM, Kung AL, Cereb N, Yao TP, Yang SY, et al. Role of T-bet in commitment of TH1 cells before IL-12-dependent selection. Science. 2001;292:1907–1910. doi: 10.1126/science.1059835. [DOI] [PubMed] [Google Scholar]
- 67.Bergmeier W, Goerge T, Wang HW, Crittenden JR, Baldwin AC, Cifuni SM, Housman DE, Graybiel AM, Wagner DD. Mice lacking the signaling molecule CalDAG-GEFI represent a model for leukocyte adhesion deficiency type III. J Clin Invest. 2007;117:1699–1707. doi: 10.1172/JCI30575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Stadtmann A, Brinkhaus L, Mueller H, Rossaint J, Bolomini-Vittori M, Bergmeier W, Van Aken H, Wagner DD, Laudanna C, Ley K, et al. Rap1a activation by CalDAG-GEFI and p38 MAPK is involved in E-selectin-dependent slow leukocyte rolling. Eur J Immunol. 2011;41:2074–2085. doi: 10.1002/eji.201041196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Carbo C, Duerschmied D, Goerge T, Hattori H, Sakai J, Cifuni SM, White GC, II, Chrzanowska-Wodnicka M, Luo HR, Wagner DD. Integrin-independent role of CalDAG-GEFI in neutrophil chemotaxis. J Leukoc Biol. 2010;88:313–319. doi: 10.1189/jlb.0110049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Frische EW, Zwartkruis FJ.Rap1, a mercenary among the Ras-like GTPases Dev Biol 20103401–9 [DOI] [PubMed] [Google Scholar]
- 71.Lee KS, Scanga CA, Bachelder EM, Chen Q, Snapper CM. TLR2 synergizes with both TLR4 and TLR9 for induction of the MyD88-dependent splenic cytokine and chemokine response to Streptococcus pneumoniae. Cell Immunol. 2007;245:103–110. doi: 10.1016/j.cellimm.2007.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Albiger B, Dahlberg S, Sandgren A, Wartha F, Beiter K, Katsuragi H, Akira S, Normark S, Henriques-Normark B. Toll-like receptor 9 acts at an early stage in host defence against pneumococcal infection. Cell Microbiol. 2007;9:633–644. doi: 10.1111/j.1462-5822.2006.00814.x. [DOI] [PubMed] [Google Scholar]
- 73.Witzenrath M, Pache F, Lorenz D, Koppe U, Gutbier B, Tabeling C, Reppe K, Meixenberger K, Dorhoi A, Ma J, et al. The NLRP3 inflammasome is differentially activated by pneumolysin variants and contributes to host defense in pneumococcal pneumonia. J Immunol. 2011;187:434–440. doi: 10.4049/jimmunol.1003143. [DOI] [PubMed] [Google Scholar]
- 74.McNeela EA, Burke A, Neill DR, Baxter C, Fernandes VE, Ferreira D, Smeaton S, El-Rachkidy R, McLoughlin RM, Mori A, et al. Pneumolysin activates the NLRP3 inflammasome and promotes proinflammatory cytokines independently of TLR4. PLoS Pathog. 2010;6:e1001191. doi: 10.1371/journal.ppat.1001191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Davis KM, Nakamura S, Weiser JN. Nod2 sensing of lysozyme-digested peptidoglycan promotes macrophage recruitment and clearance of S. pneumoniae colonization in mice. J Clin Invest. 2011;121:3666–3676. doi: 10.1172/JCI57761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Parker D, Martin FJ, Soong G, Harfenist BS, Aguilar JL, Ratner AJ, Fitzgerald KA, Schindler C, Prince A. Streptococcus pneumoniae DNA initiates type I interferon signaling in the respiratory tract. MBio. 2011;2:e00016–e11. doi: 10.1128/mBio.00016-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Groom JR, Luster AD. CXCR3 ligands: redundant, collaborative and antagonistic functions. Immunol Cell Biol. 2011;89:207–215. doi: 10.1038/icb.2010.158. [DOI] [PMC free article] [PubMed] [Google Scholar]