

Abstract
CD38 is a multifunctional enzyme that catalyzes the formation of the endogenous Ca2+-mobilizing messengers cyclic ADP-ribose (cADPR) and nicotinic acid adenosine dinucleotide phosphate (NAADP) for the activation of ryanodine receptors (RyRs) of sarcoplasmic reticulum and NAADP-sensitive Ca2+ release channels in endolysosomes, respectively. It plays important roles in systemic vascular functions, but there is little information on CD38 in pulmonary arterial smooth muscle cells (PASMCs). Earlier studies suggested a redox-sensing role of CD38 in hypoxic pulmonary vasoconstriction. This study sought to characterize its roles in angiotensin II (Ang II)–induced Ca2+ release (AICR) in PASMCs. Examination of CD38 expression in various rat arteries found high levels of CD38 mRNA and protein in pulmonary arteries. The Ang II–elicited Ca2+ response consisted of extracellular Ca2+ influx and intracellular Ca2+ release in PASMCs. AICR activated in the absence of extracellular Ca2+ was reduced by pharmacological or siRNA inhibition of CD38, by the cADPR antagonist 8-bromo-cADPR or ryanodine, and by the NAADP antagonist Ned-19 or disruption of endolysosomal Ca2+ stores with the vacuolar H+-ATPase inhibitor bafilomycin A1. Suppression of AICR by the inhibitions of cADPR- and NAADP-dependent pathways were nonadditive, indicating interdependence of RyR- and NAADP-gated Ca2+ release. Furthermore, AICR was inhibited by the protein kinase C inhibitor staurosporine, the nonspecific NADPH oxidase (NOX) inhibitors apocynin and diphenyleneiodonium, the NOX2-specific inhibitor gp91ds-tat, and the scavenger of reactive oxygen species (ROS) tempol. These results provide the first evidence that Ang II activates CD38-dependent Ca2+ release via the NOX2-ROS pathway in PASMCs.
Keywords: CD38, cyclic ADP-ribose, nicotinic acid adenosine dinucleotide phosphate, angiotensin II, NADPH oxidase
Clinical Relevance
Ca2+ signaling in pulmonary arterial smooth muscle cells (PASMCs) is important in many different physiological processes, including contraction, proliferation, migration, and gene transcription. CD38 is a master Ca2+ regulator that catalyzes the formation of the endogenous Ca2+-mobilizing messengers cyclic adenosine diphosphate ribose and nicotinic acid adenosine dinucleotide phosphate (NAADP) for the activation of ryanodine receptors of sarcoplasmic reticulum and NAADP-sensitive Ca2+ release channels in endolysosomes, respectively. However, its physiological roles in PASMCs have not been examined systematically. This paper provides the first evidence that CD38 is a major player in vasoactive agonist–induced vascular response.
CD38 is a 45-kD transmembrane glycoprotein that is ubiquitously distributed in mammalian tissues, including inflammatory cells, brain tissue, pancreas, cardiac muscles, and airway and vascular smooth muscle cells (VSMCs) (1). It is a multifunctional enzyme that serves as an ADP–ribosyl cyclase that synthesizes cyclic ADP-ribose (cADPR) from β-NAD+ and as a catalase that produces nicotinic acid adenosine diphosphate (NAADP) from β-NADP+ through a base-exchange reaction. cADPR and NAADP can be hydrolyzed also by CD38 to form ADP-ribose and ADP-ribose phosphate, respectively. cADPR is the endogenous ligand of ryanodine receptors (RyRs) (2). It binds to the FK506-binding protein, an accessory protein that stabilizes RyRs, causing its dissociation from RyRs to initiate Ca2+ release from sarcoplasmic reticulum (SR) (3). NAADP, the most potent Ca2+ mobilizing messenger to date, triggers Ca2+ release from endolysosomes (4). Recent studies have identified the two-pore channels (TPC1 and TPC2) as the NAADP-activated Ca2+ release channels (5, 6). It has been suggested that NAADP binds directly to TPCs or to an accessory protein of the TPC complex to activate endolysosomal Ca2+ release (7).
CD38 expression has been reported in some systemic arteries, including aorta and the coronary and renal arteries (8–10). Endogenous vasoconstrictors, such as norepinephrine, endothelin-1 (ET-1), and angiotensin II (Ang II), have been shown to activate the cyclase and catalase activities of CD38, contributing to the elevation of [Ca2+]i and vasoconstriction (11–16). In general, these vasoconstrictory agonists of G-protein–coupled receptors activate phospholipase C (PLC) to synthesize inositol trisphosphate (IP3) and diacyglycerol from phosphatidylinositol bisphosphate. IP3 activates IP3 receptors to release Ca2+ from SR in VSMCs. Evidence from other studies suggests that Ang II and ET-1 can also activate NADPH oxidases (NOXs) to increase the production of reactive oxygen species (ROS) in VSMCs (17). Several studies showed that ROS production is associated with CD38 activation (18–20). These observations suggest that CD38 may play an important role in the regulation of agonist-induced vascular responses.
CD38 expression and its activity have been reported in rat pulmonary arteries (PAs) and pulmonary arterial smooth muscle cells (PASMCs) (21, 22). Previous studies from our and other laboratories have identified the expression of RyR and TPC subtypes and have characterized Ca2+ release signals from RyR-gated Ca2+ stores (23–25) and NAADP-gated acidic endo-lysosomes in rat PASMCs (13, 21). Hence, all the components of the CD38-dependent Ca2+ pathways are operational in PASMCs. CD38 of PASMCs was originally proposed as a redox sensor for hypoxic pulmonary vasoconstriction (HPV) (26). It has been suggested that a reduction of β-NAD+:β-NADH ratio during hypoxia stimulates cyclase activity and inhibits hydrolase activity of CD38, leading to the accumulation of cADPR to activate RyR-gated Ca2+ release for the sustained HPV (22, 27). This hypothesis has yet to be verified because a recent study failed to observe the contribution of CD38 to HPV in isolated rat PAs without precontracted with a vasoactive agonist (28). In contrast, CD38 may play a significant role in agonist-induced Ca2+ release in PASMCs. We have previously shown that the integrin peptide ligand Gly-Arg-Gly-Asp-Ser-Pro (GRGDSP) stimulates the production of cADPR and activates Ca2+ release from both the RyR-gated and the endo/lysosomal Ca2+ stores (21). Furthermore, ET-1 elicits Ca2+ response in PASMCs, partially through NAADP-gated Ca2+ release and cross-activation of RyRs by Ca2+-induced Ca2+ release (13, 29). Apart from the abovementioned studies, there is no related information on any other vasoactive agonist and on the mechanism underlying agonist-induced activation of CD38 in PASMCs. In this study, we sought to examine the CD38-dependent mechanism in the regulation of pulmonary vascular reactivity by investigating Ang II–induced release (AICR). We compared the expression of CD38 in PAs and systemic arteries, determined the contribution of CD38-dependent Ca2+ pathways in AICR, and examined the signaling mechanism of Ang II–induced activation of CD38 in PASMCs. Our results show that Ang II activates CD38-dependent Ca2+ release via a protein kinase C (PKC)-NOX2-ROS pathway in PASMCs.
Materials and Methods
PASMC Preparation
All animal procedures are conformed to the Laboratory Animals Care and Use guidelines published by the National Institutes of Health and approved by the Johns Hopkins University Animal Care and Use Committee. PASMCs were isolated from intralobar PAs (300–800 μm) of male Wistar rats (150–200 g) and transiently cultured (18–20 h) as described (13).
Western Blotting
CD38 protein in PAs and PASMCs were determined following a standard protocol (13). Primary antibodies of CD38 (1:500 dilution) (SC-7049; Santa Cruz) and actin (1:5,000 dilution) (SC-1615; Santa Cruz) and horseradish peroxidase–coupled secondary antibody (1:5,000 dilution) (SC-2020; Santa Cruz) were used. A CD38-specific blocking peptide (SC-7049P) was premixed with CD38 antibody (5-fold excess) and incubated overnight at 4°C before use. After CD38 detection, membrane was stripped and reprobed for actin determination. The CD38 value was normalized with actin of each sample to correct for sample variability.
Quantitative RT-PCR
Total RNA from arteries and PASMCs were extracted, and first-strand cDNA was prepared as described (13). Quantitative RT-PCR (qRT-PCR) was performed using primers for 18S rRNA and CD38 of the following sequences: 18S rRNA, forward 5′-CGGCTACCACATCCAAGGAA-3′ and reverse 5′-AGCTGGAATTACCGCGGC-3′ (X01117.1, position 452–639) and CD38, forward 5′-TGGAGCAAGTCCAAACACCTGGC-3′ and reverse 5′- CTGGGGTCTCCACACCACCTGA-3′ (NM_013127.1, position 382–500). The qRT-PCR reaction consisted of an initial step at 95°C for 5 minutes followed by 40 cycles at 95°C for 15 seconds, 60°C for 30 seconds, and 72°C for 1 minute and was performed using the iQ5 system (Bio-Rad, Hercules, CA). Absolute copy number was calculated using standard curves generated from purified PCR products of known copy number. Data were normalized by the copy number of 18S rRNA in each sample to compensate for sample variability.
Calcium Imaging
PASMCs were loaded with fluo-3 AM, and [Ca2+]i was determined using fluorescence microscopy as described (13). [Ca2+]i was calculated using the equation: [Ca2+]i = [KD(F – Fbg)]/(Fmax – F), where F is the fluorescence intensity, Fbg is the background fluorescence, and Fmax is the maximum fluorescence. The KD value of fluo-3 is 1.1 μM. Fmax was determined in situ using the Ca2+ ionophone 4-Bromo-A23187 (Calbiochem, La Jolla, CA) and 10 mM Ca2+. Fbg was measured in an area devoid of cells after Mn2+ quenching.
siRNA Knockdown of CD38
Isolated PASMCs were rested in 0.5% FBS–containing HAM’s F-12 media (Mediatech, Herndon, VA) overnight and then cultured in 5% FBS-SmGM (Lonza, Walkersville, MD) for 6 days with two cell passages. Small interfering RNA (siRNA) for CD38 was purchased from Origene (Rockville, MD) (SR509476A, sequence: 5′-ACCAUACCAUGUAACAAGACUCUCT-3′) along with the scrambled control sequence. PASMCs were transfected with 100 nM siRNA or scramble control by electroporation using an Amaxa Nucleofactor (Lonza) and immediately seeded on coverslips in 5% FBS-SmGM. After 24 hours, medium was changed to serum-free SmGM for overnight starvation. [Ca2+]i measurement and Western blot were performed within 48 hours after the siRNA transfection.
Statistical Analysis
Data are shown as mean ± SEM. Statistical significance (P < 0.05) was assessed by unpaired Student’s t tests or ANOVA with Holm-Sidak method or Newman-Keuls post hoc analyses if applicable.
Results
Expression Profile of CD38 in Vascular Smooth Muscle
To determine CD38 protein expression in pulmonary and systemic arteries, the specificity of the antibody was first verified using a specific blocking peptide. CD38 was detected as a single band around 45 kD in the resolved protein samples of PA, renal artery (RA), and cerebral artery (CA) (Figure 1A). The signals were completely blocked by the blocking peptide, whereas the nonspecific signals were unaffected. The molecular size of CD38 detected in CA samples was slightly smaller compared with those in PA and RA, presumably due to differences in post-translational modification of glycosylation and phosphorylation. CD38 protein expression in different types of arteries, including aorta, PA, mesenteric artery (MA), femoral artery (FA), tail artery (TA), RA, and CA, as well as isolated PASMCs were examined (Figure 1B). Clear signals of CD38 were detected in PA, RA, CA, and PASMCs compared with the weaker signals in aorta, MA, FA, and TA. Semiquantitative comparison using β-actin for normalization showed the relative abundance of CD38 protein is in the order of CA > RA = PA > MA > aorta = FA = TA (Figure 1C). qRT-PCR showed that the expression profile of CD38 transcript in different types of arteries was similar to that of CD38 protein, although a higher expression level was seen in the aorta (Figure 1D). These results indicate that CD38 protein is differentially expressed in different types of arteries with clear expression in PA and PASMCs.
Figure 1.

CD38 expression in rat pulmonary and systemic arteries. (A) Verification of the specificity of CD38 antibody in samples of pulmonary artery (PA), renal artery (RA), and cerebral artery (CA) with (right panel) and without (left panel) pretreatment of the antibody with a blocking peptide. (B) An immunoblot of CD38 from samples of aorta, PA, mesenteric (MA), RA, femoral (FA), tail (TA), CA, and pulmonary arterial smooth muscle cells (PASMCs). (C) Averaged normalized CD38 protein level in various arteries using β-actin for normalization (n = 5). (D) Quantitative real-time RT-PCR analysis of CD38 mRNA expression in different vascular smooth muscle tissues. The data are normalized with 18S RNA (n = 5). a.u., arbitrary units.
AICR in PASMCs Was Inhibited by Nicotinamide
CD38 contributes to agonist-induced Ca2+ mobilization in several types of cells. Here, we examine the involvement of CD38 in Ang II–induced Ca2+ mobilization in PASMCs. Ang II–induced Ca2+ response was elicited in the presence extracellular Ca2+ or 100 seconds after external solution was switched to Ca2+-free (with 1 mM EGTA) solution (Figures 2A and 2B). Ang II at concentrations of 10 nM to 1 μM elicited a concentration-dependent increase in [Ca2+]i, which raised rapidly to the peak and returned to the baseline within 50 to 100 seconds. The changes in [Ca2+]i (∆[Ca2+]i) induced by 10 nM, 100 nM, and 1 μM Ang II were 88.5 ± 32.6 (n = 6), 405.6 ± 51.3 (n = 7), and 741.4 ± 122.5 nM (n = 6), respectively, in the presence of 2 mM Ca2+ and 69.6 ± 26 (n = 6), 165.5 ± 68 (n = 7), and 418.6 ± 126.4 nM (n = 6), respectively, in the absence of extracellular Ca2+ (Figure 2C). Ca2+ responses activated by 100 nM Ang II were significantly reduced after removal of extracellular Ca2+ (P = 0.016). These results show that Ca2+ response elicited by Ang II in PASMCs consisted of Ca2+ release and Ca2+ influx.
Figure 2.

Nicotinamide (NA) inhibited angiotension II (Ang II)–induced Ca2+ release in rat PASMCs. (A and B) Average Ca2+ transient (Δ[Ca2+]i) activated by different concentrations of Ang II (10 nM, 100 nM, and 1 μM) in the presence (2 mM Ca2+) or absence (containing 1 mM EGTA) of extracellular Ca2+. (C) The average increase (Δ peak, right panel) in [Ca2+]i induced by different concentrations of Ang II (n = 6–7 experiments in each group; P < 0.05). *Significant difference between the groups. (D) Averaged traces of Ang II–induced Ca2+ transients elicited 100 seconds after removal of Ca2+ in the absence or presence of different concentrations (2, 5, 10, and 20 mM) of the CD38 inhibitor NA. (E) Averaged peak change in [Ca2+]i in the various groups (n = 8–10 experiments in each group). *Significant differences when compared with control (5 mM, P = 0.007; 10 mM, P < 0.001; and 20 mM, P < 0.001).
To examine whether Ang II–induced Ca2+ release (AICR) is dependent on CD38, PASMCs were incubated for 20 minutes with the CD38 inhibitor nicotinamide (NA) before application of Ang II (100 nM) under Ca2+-free conditions. NA at concentrations between 2 and 20 mM had no significant effect on the basal [Ca2+]i in PASMCs but caused concentration-dependent inhibition of AICR (Figures 2D and 2E). Δ[Ca2+]i induced by Ang II was 469.9 ± 60.2 nM (n = 10) in the absence of NA and was 375 ± 65.8 (n = 8; P = N.S.), 268.9 ± 49.3 (n = 8; P = 0.007), 221.7 ± 31.6 (n = 9; P < 0.001), and 152.9 ± 28.6 nM (n = 10; P < 0.001) in the presence of 2, 5, 10, and 20 mM NA, respectively. Inhibition of AICR by NA suggests that CD38-dependent Ca2+ pathways contribute to the Ang II–mediated Ca2+ response in PASMCs.
Knockdown of CD38 Decreased AICR in PASMCs
To further verify the contribution of CD38 to Ang II–induced Ca2+ response in PASMCs, AICR was examined in PASMCs transfected with a CD38-specific siRNA or a control sequence. The efficacy of CD38 siRNA treatment was determined by Western blot (Figures 3A and 3B). CD38 protein level was reduced by 69.05 ± 5.35% in CD38 siRNA–transfected PASMCs compared with that in scrambled sequence transfected cells (P = 0.006; n = 5). The reduction of CD38 protein expression in the siRNA-transfected PASMCs was associated with attenuated AICR (Figures 3C and 3D). Peak Δ[Ca2+]i values induced by Ang II (100 nM) were 1,028 ± 114.5 nM in control cells (n = 10) and 552.6 ± 103.1 nM in CD38 siRNA (n = 8) (P = 0.003). NA (20 mM) inhibition of AICR, which was clearly observed in the scrambled sequence transfected cells (637.8 ± 119.2 nM [n = 8]; P = 0.012), was absent in the CD38 siRNA–transfected PASMCs (574.6 ± 71.9 nM [n = 8]). Accordingly, the percent reduction in peak Δ[Ca2+]i caused by 20 mM NA was 37.96 ± 11.59% in the control cells and −3.97 ± 13.02% in CD38 siRNA–transfected PASMCs. These results provide the direct evidence that AICR is mediated, in part, through CD38 activation in PASMCs.
Figure 3.

Effect of small interfering RNA (siRNA)-mediated suppression of CD38 expression on Ang II–induced Ca2+ release (AICR) in PASMCs. (A) An immunoblot of CD38 from samples of PASMCs transfected with a CD38-targeting siRNA and a control scrambled sequence. (B) The mean CD38 protein levels from the two groups after normalization with β-actin (n = 5). *Significant decrease in expression compared with the scrambled control (P = 0.006). (C) The mean traces of AICR in the control scrambled sequence and CD38 siRNA–transfected PASMCs in the absence or presence of 20 mM NA. (D) The mean peak Δ[Ca2+]i in the control scrambled sequence and CD38 siRNA–transfected PASMCs in the absence or presence of 20 mM NA (n = 8 for each group). *Significant difference from the control group in the absence of NA. n.s., no significant difference between the groups indicated.
AICR in PASMCs Is Mediated in Part by the cADPR-RyR and NAADP-Endolysosomal Pathways
The cyclase activity of CD38 generates cADPR, which causes Ca2+ release via RyRs of SR in various types of cells, including VSMCs. To examine the participation of the cADPR-RyRs pathway in PASMCs, we determined the effects of the specific cADPR antagonist 8-bromo-cADPR and ryanodine on the AICR in PASMCs. Preincubation of PASMCs with various concentrations of 8-bromo-cADPR (0.1–10 μM) for 20 minutes had no effect on the basal [Ca2+]i but caused significant reductions in the peak of AICR (Figures 4A and 4B). An apparent maximal inhibition of 40 to 50% of the control response was reached at 1 μM 8-bromo-cADPR. Moreover, inhibition of RyRs with 50 μM ryanodine reduced the peak AICR from 299.5 ± 57.3 nM to 125.4 ± 21.1 nM (n = 9; P = 0.034) (Figures 4C and 4D). These data suggest that AICR is mediated in part through activation of RyR by cADPR in PASMCs.
Figure 4.

Effects of inhibition of cyclic ADP-ribose (cADPR)-dependent pathway on AICR in PASMCs. (A) The mean traces of AICR in the absence or presence of 1 and 10 μM cADPR antagonist 8-Br-cADPR. (B) The mean peak Δ[Ca2+]i elicited by Ang II in the presence of various concentrations of 8-Br-cADPR (n = 6–9 experiments in each group). (C) Ang II–induced changes in [Ca2+]i in the absence or presence of 50 μM ryanodine. (D) The mean peak Δ[Ca2+]i elicited by Ang II with or without preincubation with ryanodine (n = 9 experiments). *P < 0.05 from control.
In addition to the cADPR-RyRs pathway, CD38 protein also mobilizes Ca2+ through production of NAADP, which activates two-pore channels (TPCs) of the endo/lysosomes (5). To evaluate the contribution of the NAADP–lysosomal Ca2+ pathways, we examine the AICR in the presence of the specific NAADP antagonist Ned-19. Our previous study showed that 1 μM Ned-19 significantly inhibited intracellular Ca2+ release induced by membrane-permeable NAADP-AM in PASMCs without the side effects on RyRs and IP3Rs-mediated Ca2+ release (13). Preincubation of PASMCs with 1 μM Ned-19 for 20 minutes significantly reduced the peak Ca2+ response induced by 100 nM Ang II from 186.9 ± 44 nM to 67.6 ± 14.8 nM (n = 9; P = 0.021) (Figure 5A). Furthermore, bafilomycin A1 (3 μM), which blocks the vacuolar H+-ATPase and disrupts the lysosomal proton gradient for Ca2+ uptake (30, 31), significantly suppressed AICR (control: 299.5 ± 57.3 nM [n = 9]; bafilomycin A1: 125.4 ± 21.1 nM [n = 8]; P = 0.014) (Figures 5C and 5D). These results suggest that NAADP-mediated Ca2+ release from endo/lysosomal stores also contributes significantly to the Ang II–induced Ca2+ response in PASMCs.
Figure 5.
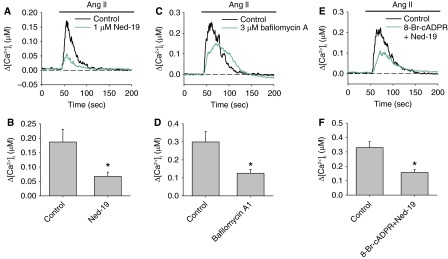
Effect of inhibition of nicotinic acid adenosine diphosphate (NAADP)-dependent pathway on AICR in PASMCs. (A) The mean traces of Ang II–induced changes in [Ca2+]i in the absence or presence of the NAADP antagonist Ned-19 (1 μM). (B) The mean peak changes in [Ca2+]i in each group (n = 9 experiments; P = 0.021). (C and D) AICR in PASMCs with or without preincubation with the vacuolar H+-ATPase blocker bafilomycin A1 (3 μM) (n = 8–9 experiments; P = 0.014). (E and F) AICR in PASMCs with or without pretreatment with 8-Br-cADPR (10 μM) and Ned-19 (1 μM) (n = 10–12 experiments; P = 0.002). *Significantly different from control.
CD38-dependent cADPR-RyRs and NAADP-lysosome pathways may operate independently or interdependently to facilitate Ang II–induced Ca2+ mobilization. Our previous study provided evidence that Ca2+ release from NAADP-sensitive stores is amplified by cross-activation of Ca2+ release from RyR-gated Ca2+ stores in PASMCs (13). This possibility was examined by inhibiting both pathways with 10 μM 8-Br-cADPR and 1 μM Ned-19. The combined inhibitory effect of 8-Br-cADPR and Ned-19 (peak Δ[Ca2+]i: control = 330.4 ± 40.8 nM [n = 12]; 8-Br-cADPR+Ned-19 = 157.7 ± 19.5 nM [n = 10]; P = 0.002) was similar to the inhibition caused by 8-Br-cADPR or Ned-19 alone, indicating that the effects of two inhibitors are nonadditive (Figures 5E and 5F). These results suggest that the two Ca2+ release pathways operate interdependently during AICR in PASMCs.
Ang II–Induced CD38 Activation Is Mediated by NOX-Dependent Pathways in PASMCs
To elucidate the signal transduction mechanism of AICR, we examined whether Ang II–induced CD38 activation is mediated by the PKC-NOX-ROS pathway. Preincubation of PASMCs with the PKC inhibitor staurosporine (stau) (10 nM) for 20 minutes significantly reduced the peak ACIR induced by 100 nM Ang II from 332.9 ± 30.9 nM (n = 8) to 204.7 ± 34.3 nM (P = 0.019 [n = 9]) (Figures 6A and 6B). Further application of 20 mM NA to inhibit CD38 did not cause additional reduction in the AICR (stau+NA: 163.7 ± 37.2 nM [n = 10]; P = 0.432). These results suggest that activation of PKC is required for Ang II–induced CD38 activation.
Figure 6.

Protein kinase C–NADPH oxidase–reactive oxygen species (PKC-NOX-ROS)-dependent activation of CD38 in PASMCs. (A and B) Mean traces and mean peak Δ[Ca2+]i showing the effect of PKC inhibition with staurosporine (Stau) (10 nM) on AICR in the absence or presence of NA (20 mM) in PASMCs (n = 8–10 experiments in each group). (C and D) Mean traces and mean peak Δ[Ca2+]i showing the effect of the NOX inhibitor apocynin (30 μM) on AICR in the absence or presence of NA (n = 5–6 experiments in each group). (E and F) Mean traces and mean peak Δ[Ca2+]i showing the effect of apocynin (30 μM) on AICR in the absence or presence of 8-Br-cADPR (10 μM) and Ned-19 (1 μM) (n = 8–9 experiments in each group). (G and H) The effect of the NOX inhibitor diphenyleneiodonium (DPI) (30 μM) on AICR in the absence or presence of NA (n = 8–10 experiments in each groups). (I and J) The effect of the ROS scavenger tempol (100 μM) on AICR in the absence or presence of NA (n = 9 experiments in each group). *Significant inhibition of AICR compared with control. There was no additional significant inhibition of AICR in cells treated with NA compared with staurosporine, apocynin, DPI, or tempol alone.
To determine the contribution of NOX in Ang II–induced CD38 activation, AICR was measured in the absence or presence of the NOX inhibitor apocynin. Peak Ca2+ response elicited by Ang II was significantly reduced by 30 μM apocycnin (control: 304 ± 56.9 nM [n = 5]; apocynin: 96.7 ± 25.1 nM [n = 6]; P < 0.001). Inhibition of CD38 by NA in the presence of apocynin did not lead to an additional decrease in AICR (apocynin+NA: 111.4 ± 18.1 nM [n = 6]) (Figures 6C and 6D). Similarly, inhibition of CD38-dependent Ca2+ pathways with 8-Br-cADPR and Ned-19 had no further effect on AICR in the presence apocynin (apocynin: 136.6 ± 29.6 nM [n = 8]; apocynin+8-Br-cADPR+Ned-19: 150.1 ± 67.6 nM [n = 9]) (Figures 6E and 6F). This suggests that NOX is required for Ang II activation of the cADPR and NAADP pathways. Moreover, AICR was significantly reduced by another NOX inhibitor, diphenyleneiodonium (DPI) (30 μM) (control: 333 ± 30.9 nM [n = 8]; DPI: 205.7 ± 34.3 nM [n = 9]; P = 0.019). NA did not caused an additional decrease in AICR after inhibition of NOX by DPI (DPI+NA: 164 ± 37.2 nM [n = 10]) (Figures 6G and GH). These results clearly suggest that NOX mediates Ang II–induced CD38 activation in PASMCs.
AICR was examined in the presence of the superoxide scavenger tempol. Preincubation of PASMCs with 100 μM tempol caused significant reduction of AICR (control: 227.6 ± 33.9 nM [n = 8]; tempol: 120.5 ± 18.4 nM [n = 9]; P = 0.007), and addition of NA did not cause further reduction in the Ca2+ response (tempol+NA: 92.7 ± 22.4 nM [n = 9]) (Figures 6I and 6J). These data indicate that Ang II–induced activation of CD38 is mediated by ROS.
Ang II–Induced Activation of CD38 Is Specifically Mediated by NOX2
Ang II is known to activate NOX1 and NOX2 in other cell types (32, 33). To further examine the specific NOX involvement in Ang II–mediated CD38 activation, we tested AICR in the presence of the NOX1-specific antagonist ML171 and the NOX2-specific inhibitory peptide gp91ds-tat. Exposure to 2 μM ML171 did not suppress AICR (control: 326 ± 51.3 nM [n = 6]; ML171: 315 ± 61.4 nM [n = 6]) in PASMCs (Figures 7A and 7B). In contrast, pretreatment of PASMCs with 10 μM gp91 ds-tat caused significant reduction of AICR (control: 364.6 ± 38.7 nM [n = 13]; gp91 ds-tat: 252.4 ± 24.9 nM [n = 13]; P = 0.045) (Figures 7C and 7D). Furthermore, there was no significant difference between the AICR activated in gp91ds-tat–treated and gp91ds-tat+NA–treated cells (206.5 ± 44.7 nM [n = 8]). These results suggest that Ang II–induced CD38-dependent Ca2+ release is mainly mediated by the activation of NOX2. Our results suggest that Ang II activates CD38 through PKC-dependent activation of ROS production from NOX2.
Figure 7.
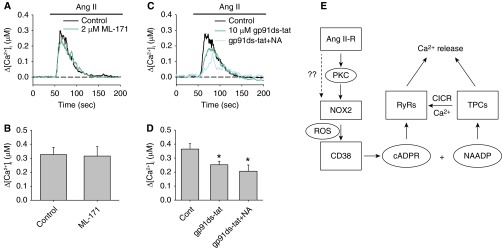
Effects of NOX1 and NOX2 specific antagonist on AICR in PASMCs. (A and B) The effect of the NOX1-specific inhibitor ML171 (30 μM) on AICR in PASMCs. (C and D) The effects of the NOX2-specific peptide inhibitor gp91 ds-tat (10 μM) on AICR in PASMCs in the absence or presence of NA (n = 8–13 experiments in each group). (E) A schematic diagram showing the proposed signaling pathways for AICR. *Significant inhibition of AICR compared with control. There was no significant different between gp91 ds-tat and gp91 ds-tat+NA–treated cells. CICR, Ca2+-induced Ca2+ release; NAADP, nicotinic acid adenosine dinucleotide phosphate; RyR, ryanodine receptor; TPC, two-pore channel.
Discussion
CD38, through its multifunctional enzymatic activities, generates cADPR and NAADP to mobilize intracellular Ca2+ in many different cell types (1). It contributes to agonist-induced vasoconstriction of systemic arteries (16, 34, 35), but its functions in pulmonary vasculature are unclear. In the present study, we found high expression levels of CD38 protein and mRNA in PA smooth muscle compared with other vascular smooth muscles. Ang II evokes intracellular Ca2+ release in part through CD38-dependent cADPR and NAADP pathways in PASMCs. Furthermore, Ang II stimulates CD38 through activation of NOX2. This study hence provides clear evidence that CD38 is a potent regulator of Ca2+ homeostasis in pulmonary vasculature and establishes a signaling pathway for AICR in PASMCs.
CD38 expression has been reported in systemic and PAs (8, 21, 22, 35). We found that CD38 protein level varies greatly among different vascular smooth muscles, with the highest level in CA, followed by PA and RA; the expression levels in aorta, MA, FA, and TA are low. This result suggests that the contribution of CD38 to the physiological functions of various vasculatures may be different. Vascular functions of CD38 have not been examined in cerebral arteries despite the abundance of expression, but they are well characterized in renal arteries and arterioles (10, 11, 14, 34, 36). It has been shown that CD38-dependent pathways modulate cytosolic [Ca2+]i and contractile functions in renal afferent arteries and arterioles and can account for 50% of the contractile response to Ang II, ET-1, and norepinephrine (10). cADPR-mediated contraction has also been reported in mesenteric arteries (35). The comparable expression of CD38 protein in PA and RA suggests that CD38 may play similarly important role in pulmonary vasculatures.
The contribution of CD38-dependent pathways in Ca2+ signaling is clearly evident in PASMCs. Removal of extracellular Ca2+ revealed that approximately 50% of the Ang II–induced Ca2+ response is mediated by intracellular Ca2+ release, suggesting that extracellular Ca2+ influx and intracellular Ca2+ release are important for Ang II–induced Ca2+ mobilization in PASMCs. When focusing on the Ca2+ release processes, we found that pharmacological inhibition of CD38 with NA or suppression of CD38 expression with siRNA caused significant inhibition of AICR in PASMCs. NA has been widely used for the inhibition of CD38. The IC50 of NA for CD38 inhibition is approximately 1.5 mM in sea urchin (37) and 3.1 mM in rat abdominal aorta (8), as determined by the cyclase assay. NA at 2 to 10 mM also caused concentration-dependent inhibition of CD38 cyclase activity in homogenate of coronary artery (38) and effectively inhibited agonist-induced vasoconstriction in systemic arteries and the Ca2+ response in arterial myocytes (16, 34, 35). The concentration-dependent inhibition of AICR by NA in PASMCs is therefore consistent with CD38 inhibition reported in systemic vascular smooth muscle. Moreover, siRNA knockdown of CD38 caused similar inhibition of AICR compared with NA, and NA failed to cause further reduction of AICR in CD38 siRNA–transfected cells. These results suggest that CD38 plays a significant role in the agonist-induced Ca2+ release in PASMCs and that NA does not cause major side effect on Ca2+ release independent of CD38 under our experimental conditions.
CD38 activation mobilizes intracellular Ca2+ through cADPR-dependent Ca2+ release from RyR-gated stores and NAADP-dependent Ca2+ release from the acidic endo-lysosomes. Previous work by our and other laboratories have identified all three RyR subtypes in PASMCs, with RyR2 being the most abundant subtype (23, 39). RyR1 and RyR2 are associated mainly with SR in the subsarcolemmal region, whereas RyR3 is localized predominantly in the perinuclear SR (23, 40). Mild activation of RyRs triggers discernible local Ca2+ events or Ca2+ sparks, and strong activation can lead to regenerative global Ca2+ release in PASMCs (23–25). NAADP-mediated Ca2+ release has also been described in PASMCs (5). We have recently identified the expression of NAADP-sensitive TPC1 and TPC2 in PASMCs and have characterized the NAADP-mediated global and local Ca2+ events using the membrane-permeable NAADP-AM (13, 40). The NAADP-mediated Ca2+ release in PASMCs is initiated from bafilomycin-sensitive acidic endo-lysosomes and is blocked by the specific NAADP antagonist Ned-19, which does not affect the voltage-dependent Ca2+ entry, store-operated Ca2+ entry, and RyR-gated and IP3-gated Ca2+ release in PASMCs (13, 41). These studies hence show that all the effectors of the CD38-dependent cADPR and NAADP pathways are operational in PASMCs.
In this study, antagonism of cADPR with the 8-bromo-cADPR or inhibition of RyRs with ryanodine effectively suppressed AICR. Inhibition of NAADP with Ned-19 (41) or depleting endo/lysosomal Ca2+ stores by disrupting H+ gradient for H+-Ca2+ exchange with the vacuolar H+-ATPase inhibitor bafilomycin A1 also reduced AICR. These results suggest that Ang II stimulates both the cyclase and catalase activities of CD38 to generate cADPR and NAADP, respectively, for eliciting the Ca2+ response. The cADPR- and NAADP-dependent mechanisms operate interdependently during AICR. This is reflected in the similar suppression of AICR by coinhibition of cADPR and NAADP pathways, compared with the inhibition of either the cADPR or the NAADP pathway alone. The interaction between the two Ca2+ pathways is consistent with others and our previous findings of cross-activation of RyRs by NAADP-mediated Ca2+ release in PASMCs (13, 29, 42). NAADP channels are spatially or functional coupled with RyRs through Ca2+-induced Ca2+ release (13, 29, 40), and cADPR may further sensitize RyRs for the cross-activation under physiological stimulation. The remaining AICR in PASMCs after coinhibition of cADPR and NAADP is likely mediated by IP3R-gated Ca2+ release through the well-recognized PLC-IP3 pathway.
There is substantial evidence suggesting that Ang II enhances ROS production through activation of NOX, contributing to Ca2+ response in VSMCs (17). Several studies have shown that CD38 activity is regulated by redox state and oxidative stress (19, 20, 22, 27, 43). However, the mechanism of Ang II–induced CD38 activation in PASMCs has not been established. We found that Ang II activates CD38-dependent Ca2+ release in PASMCs via ROS generation by NOX. This is based on the findings that AICR was inhibited significantly by the common NOX inhibitors apocynin and DPI and by the ROS scavenger tempol. Inhibition of CD38 with NA, or inhibition of the cADPR and NAADP pathways, in the presence of the NOX inhibitors or ROS scavenger did not further suppress AICR. These observations are consistent with reports in renal afferent arterioles and cardiac myocytes that Ang II stimulates NOX to generate ROS, leading to enhanced ADP–ribosyl cyclase activity (36, 44).
There are seven NOX homologs (NOX1–5 and DUOX1–2) with different subunit compositions, physiological functions, and activation mechanisms (45). Multiple isoforms of NOX, including NOX1, NOX2, and NOX4, are expressed in systemic arteries (17, 45) and PAs (46, 47). Ang II–induced activation of NOX1 appears to be important in systemic VSMCs of large arteries, whereas NOX2 may be more important in resistance arteries (32, 33). Ang II activates NOX2 by PKC-dependent phosphorylation of the cytosolic subunit p47phox to facilitate the assembly of the active oxidase complex (17). NOX1 and NOX2 activation is also critically modulated by the small GTP-binding protein Rac1 (17, 48, 49). It has been shown in coronary arteries that ET-1 stimulates Rac1-NOX1–dependent ROS production to activate CD38 (49). In this study, preincubation of PASMCs with the specific NOX1 inhibitor ML171 had no significant inhibitory effect on AICR. In contrast, the NOX2-specific peptide inhibitor gp91ds-tat caused a significant decrease in AICR. Furthermore, inhibition of CD38 by NA in the presence of gp91 ds-tat did not cause further reduction in the Ang II–induced response. This suggests that Ang II–induced activation of CD38 in PASMCs is mediated specifically by NOX2. Moreover, suppression of AICR with the PKC inhibitor staurosporine is consistent with Ang II–induced CD38 activation through the PKC-NOX2-ROS pathway to elicit Ca2+ release in PASMCs. However, Ang II activation of NOX2 may require other signaling pathways, such as Rac1 (17, 48), and PKC may also be modulated by the CD38 signaling pathways downstream of NOX2. CD38 of PASMCs can be activated through mechanisms unrelated to the PKC-NOX-ROS pathway. For example, activation of integrins with the GRGDSP peptide can elicit a CD38-dependent Ca2+ response in PASMCs (21). Because integrins are not G-protein–coupled receptors and because ligation of integrins activates complex signaling pathways, including focal adhesion kinase, integrin-linked kinase, receptor tyrosine kinase, and Src (50), it is likely that integrins activate CD38 through mechanisms different from Ang II.
In conclusion, we have characterized systematically the mechanism of Ca2+ release induced by Ang II and have demonstrated that it is in part mediated by CD38 activation through NOX2-dependent ROS production, leading to synergistic Ca2+ release from cADRP- and NAADP-gated Ca2+ stores of PASMCs. A schematic diagram illustrating the proposed mechanism for Ang II–induced activation of CD38-dependent Ca2+ release is shown in Figure 7E. These findings provide the evidence to support an important role of CD38 for Ca2+ signaling in pulmonary vasculature.
Footnotes
This work was supported by National Institutes of Health grants R01 HL071835 and R01 HL075134 (J.S.K.S.) and an American Heart Association Grant-in-Aid Award (J.S.K.S.).
Author Contributions: S.L., O.P., Y.J., and X.-R.Y. performed the experiments. S.L. analyzed the data. S.L. and J.S.K.S. interpreted the results of the experiments. S.L. drafted the manuscript. J.S.K.S. edited and revised the manuscript. J.S.K.S. participated in conception and design of the research.
Originally Published in Press as DOI: 10.1165/rcmb.2014-0141OC on July 31, 2014
Author disclosures are available with the text of this article at www.atsjournals.org.
References
- 1.Malavasi F, Deaglio S, Funaro A, Ferrero E, Horenstein AL, Ortolan E, Vaisitti T, Aydin S. Evolution and function of the ADP ribosyl cyclase/CD38 gene family in physiology and pathology. Physiol Rev. 2008;88:841–886. doi: 10.1152/physrev.00035.2007. [DOI] [PubMed] [Google Scholar]
- 2.Galione A, Churchill GC. Cyclic ADP ribose as a calcium-mobilizing messenger. Sci STKE. 2000;2000:pe1. doi: 10.1126/stke.2000.41.pe1. [DOI] [PubMed] [Google Scholar]
- 3.Noguchi N, Takasawa S, Nata K, Tohgo A, Kato I, Ikehata F, Yonekura H, Okamoto H. Cyclic ADP-ribose binds to FK506-binding protein 12.6 to release Ca2+ from islet microsomes. J Biol Chem. 1997;272:3133–3136. doi: 10.1074/jbc.272.6.3133. [DOI] [PubMed] [Google Scholar]
- 4.Lee HC, Aarhus R. A derivative of NADP mobilizes calcium stores insensitive to inositol trisphosphate and cyclic ADP-ribose. J Biol Chem. 1995;270:2152–2157. doi: 10.1074/jbc.270.5.2152. [DOI] [PubMed] [Google Scholar]
- 5.Calcraft PJ, Ruas M, Pan Z, Cheng X, Arredouani A, Hao X, Tang J, Rietdorf K, Teboul L, Chuang KT, et al. NAADP mobilizes calcium from acidic organelles through two-pore channels. Nature. 2009;459:596–600. doi: 10.1038/nature08030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Brailoiu E, Churamani D, Cai X, Schrlau MG, Brailoiu GC, Gao X, Hooper R, Boulware MJ, Dun NJ, Marchant JS, et al. Essential requirement for two-pore channel 1 in NAADP-mediated calcium signaling. J Cell Biol. 2009;186:201–209. doi: 10.1083/jcb.200904073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lin-Moshier Y, Walseth TF, Churamani D, Davidson SM, Slama JT, Hooper R, Brailoiu E, Patel S, Marchant JS. Photoaffinity labeling of nicotinic acid adenine dinucleotide phosphate (NAADP) targets in mammalian cells. J Biol Chem. 2012;287:2296–2307. doi: 10.1074/jbc.M111.305813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.de Toledo FG, Cheng J, Liang M, Chini EN, Dousa TP. ADP-Ribosyl cyclase in rat vascular smooth muscle cells: properties and regulation. Circ Res. 2000;86:1153–1159. doi: 10.1161/01.res.86.11.1153. [DOI] [PubMed] [Google Scholar]
- 9.Jia SJ, Jin S, Zhang F, Yi F, Dewey WL, Li PL. Formation and function of ceramide-enriched membrane platforms with CD38 during M1-receptor stimulation in bovine coronary arterial myocytes. Am J Physiol Heart Circ Physiol. 2008;295:H1743–H1752. doi: 10.1152/ajpheart.00617.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Thai TL, Arendshorst WJ. Mice lacking the ADP ribosyl cyclase CD38 exhibit attenuated renal vasoconstriction to angiotensin II, endothelin-1, and norepinephrine. Am J Physiol Renal Physiol. 2009;297:F169–F176. doi: 10.1152/ajprenal.00079.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Thai TL, Churchill GC, Arendshorst WJ. NAADP receptors mediate calcium signaling stimulated by endothelin-1 and norepinephrine in renal afferent arterioles. Am J Physiol Renal Physiol. 2009;297:F510–F516. doi: 10.1152/ajprenal.00116.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kim SY, Gul R, Rah SY, Kim SH, Park SK, Im MJ, Kwon HJ, Kim UH. Molecular mechanism of ADP-ribosyl cyclase activation in angiotensin II signaling in murine mesangial cells. Am J Physiol Renal Physiol. 2008;294:F982–F989. doi: 10.1152/ajprenal.00483.2007. [DOI] [PubMed] [Google Scholar]
- 13.Jiang YL, Lin AH, Xia Y, Lee S, Paudel O, Sun H, Yang XR, Ran P, Sham JS. Nicotinic acid adenine dinucleotide phosphate (NAADP) activates global and heterogeneous local Ca2+ signals from NAADP- and ryanodine receptor-gated Ca2+ stores in pulmonary arterial myocytes. J Biol Chem. 2013;288:10381–10394. doi: 10.1074/jbc.M112.423053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Thai TL, Arendshorst WJ. ADP-ribosyl cyclase and ryanodine receptors mediate endothelin ETA and ETB receptor-induced renal vasoconstriction in vivo. Am J Physiol Renal Physiol. 2008;295:F360–F368. doi: 10.1152/ajprenal.00512.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gul R, Kim SY, Park KH, Kim BJ, Kim SJ, Im MJ, Kim UH. A novel signaling pathway of ADP-ribosyl cyclase activation by angiotensin II in adult rat cardiomyocytes. Am J Physiol Heart Circ Physiol. 2008;295:H77–H88. doi: 10.1152/ajpheart.01355.2007. [DOI] [PubMed] [Google Scholar]
- 16.Fellner SK, Arendshorst WJ. Angiotensin II Ca2+ signaling in rat afferent arterioles: stimulation of cyclic ADP ribose and IP3 pathways. Am J Physiol Renal Physiol. 2005;288:F785–F791. doi: 10.1152/ajprenal.00372.2004. [DOI] [PubMed] [Google Scholar]
- 17.Garrido AM, Griendling KK. NADPH oxidases and angiotensin II receptor signaling. Mol Cell Endocrinol. 2009;302:148–158. doi: 10.1016/j.mce.2008.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ge Y, Jiang W, Gan L, Wang L, Sun C, Ni P, Liu Y, Wu S, Gu L, Zheng W, et al. Mouse embryonic fibroblasts from CD38 knockout mice are resistant to oxidative stresses through inhibition of reactive oxygen species production and Ca(2+) overload. Biochem Biophys Res Commun. 2010;399:167–172. doi: 10.1016/j.bbrc.2010.07.040. [DOI] [PubMed] [Google Scholar]
- 19.Zhang AY, Yi F, Teggatz EG, Zou AP, Li PL. Enhanced production and action of cyclic ADP-ribose during oxidative stress in small bovine coronary arterial smooth muscle. Microvasc Res. 2004;67:159–167. doi: 10.1016/j.mvr.2003.11.001. [DOI] [PubMed] [Google Scholar]
- 20.Kumasaka S, Shoji H, Okabe E. Novel mechanisms involved in superoxide anion radical-triggered Ca2+ release from cardiac sarcoplasmic reticulum linked to cyclic ADP-ribose stimulation. Antioxid Redox Signal. 1999;1:55–69. doi: 10.1089/ars.1999.1.1-55. [DOI] [PubMed] [Google Scholar]
- 21.Umesh A, Thompson MA, Chini EN, Yip KP, Sham JS. Integrin ligands mobilize Ca2+ from ryanodine receptor-gated stores and lysosome-related acidic organelles in pulmonary arterial smooth muscle cells. J Biol Chem. 2006;281:34312–34323. doi: 10.1074/jbc.M606765200. [DOI] [PubMed] [Google Scholar]
- 22.Wilson HL, Dipp M, Thomas JM, Lad C, Galione A, Evans AM. Adp-ribosyl cyclase and cyclic ADP-ribose hydrolase act as a redox sensor. a primary role for cyclic ADP-ribose in hypoxic pulmonary vasoconstriction. J Biol Chem. 2001;276:11180–11188. doi: 10.1074/jbc.M004849200. [DOI] [PubMed] [Google Scholar]
- 23.Yang XR, Lin MJ, Yip KP, Jeyakumar LH, Fleischer S, Leung GP, Sham JS. Multiple ryanodine receptor subtypes and heterogeneous ryanodine receptor-gated Ca2+ stores in pulmonary arterial smooth muscle cells. Am J Physiol Lung Cell Mol Physiol. 2005;289:L338–L348. doi: 10.1152/ajplung.00328.2004. [DOI] [PubMed] [Google Scholar]
- 24.Zhang WM, Yip KP, Lin MJ, Shimoda LA, Li WH, Sham JS. ET-1 activates Ca2+ sparks in PASMC: local Ca2+ signaling between inositol trisphosphate and ryanodine receptors. Am J Physiol Lung Cell Mol Physiol. 2003;285:L680–L690. doi: 10.1152/ajplung.00067.2003. [DOI] [PubMed] [Google Scholar]
- 25.Remillard CV, Zhang WM, Shimoda LA, Sham JS. Physiological properties and functions of Ca(2+) sparks in rat intrapulmonary arterial smooth muscle cells. Am J Physiol Lung Cell Mol Physiol. 2002;283:L433–L444. doi: 10.1152/ajplung.00468.2001. [DOI] [PubMed] [Google Scholar]
- 26.Evans AM, Dipp M. Hypoxic pulmonary vasoconstriction: cyclic adenosine diphosphate-ribose, smooth muscle Ca(2+) stores and the endothelium. Respir Physiol Neurobiol. 2002;132:3–15. doi: 10.1016/s1569-9048(02)00046-0. [DOI] [PubMed] [Google Scholar]
- 27.Dipp M, Evans AM. Cyclic ADP-ribose is the primary trigger for hypoxic pulmonary vasoconstriction in the rat lung in situ. Circ Res. 2001;89:77–83. doi: 10.1161/hh1301.093616. [DOI] [PubMed] [Google Scholar]
- 28.Connolly MJ, Prieto-Lloret J, Becker S, Ward JP, Aaronson PI. Hypoxic pulmonary vasoconstriction in the absence of pretone: essential role for intracellular Ca2+ release. J Physiol. 2013;591:4473–4498. doi: 10.1113/jphysiol.2013.253682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kinnear NP, Boittin FX, Thomas JM, Galione A, Evans AM. Lysosome-sarcoplasmic reticulum junctions: a trigger zone for calcium signaling by nicotinic acid adenine dinucleotide phosphate and endothelin-1. J Biol Chem. 2004;279:54319–54326. doi: 10.1074/jbc.M406132200. [DOI] [PubMed] [Google Scholar]
- 30.Crider BP, Xie XS, Stone DK. Bafilomycin inhibits proton flow through the H+ channel of vacuolar proton pumps. J Biol Chem. 1994;269:17379–17381. [PubMed] [Google Scholar]
- 31.Christensen KA, Myers JT, Swanson JA. pH-dependent regulation of lysosomal calcium in macrophages. J Cell Sci. 2002;115:599–607. doi: 10.1242/jcs.115.3.599. [DOI] [PubMed] [Google Scholar]
- 32.Lassègue B, Sorescu D, Szöcs K, Yin Q, Akers M, Zhang Y, Grant SL, Lambeth JD, Griendling KK. Novel gp91(phox) homologues in vascular smooth muscle cells : nox1 mediates angiotensin II-induced superoxide formation and redox-sensitive signaling pathways. Circ Res. 2001;88:888–894. doi: 10.1161/hh0901.090299. [DOI] [PubMed] [Google Scholar]
- 33.Touyz RM, Chen X, Tabet F, Yao G, He G, Quinn MT, Pagano PJ, Schiffrin EL. Expression of a functionally active gp91phox-containing neutrophil-type NAD(P)H oxidase in smooth muscle cells from human resistance arteries: regulation by angiotensin II. Circ Res. 2002;90:1205–1213. doi: 10.1161/01.res.0000020404.01971.2f. [DOI] [PubMed] [Google Scholar]
- 34.Fellner SK, Arendshorst W. Endothelin-A and -B receptors, superoxide, and Ca2+ signaling in afferent arterioles. Am J Physiol Renal Physiol. 2007;292:F175–F184. doi: 10.1152/ajprenal.00050.2006. [DOI] [PubMed] [Google Scholar]
- 35.Giulumian AD, Meszaros LG, Fuchs LC. Endothelin-1-induced contraction of mesenteric small arteries is mediated by ryanodine receptor Ca2+ channels and cyclic ADP-ribose. J Cardiovasc Pharmacol. 2000;36:758–763. doi: 10.1097/00005344-200012000-00011. [DOI] [PubMed] [Google Scholar]
- 36.Fellner SK, Arendshorst WJ. Angiotensin II, reactive oxygen species, and Ca2+ signaling in afferent arterioles. Am J Physiol Renal Physiol. 2005;289:F1012–F1019. doi: 10.1152/ajprenal.00144.2005. [DOI] [PubMed] [Google Scholar]
- 37.Sethi JK, Empson RM, Galione A. Nicotinamide inhibits cyclic ADP-ribose-mediated calcium signalling in sea urchin eggs. Biochem J. 1996;319:613–617. doi: 10.1042/bj3190613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Geiger J, Zou AP, Campbell WB, Li PL. Inhibition of cADP-ribose formation produces vasodilation in bovine coronary arteries. Hypertension. 2000;35:397–402. doi: 10.1161/01.hyp.35.1.397. [DOI] [PubMed] [Google Scholar]
- 39.Zheng YM, Wang QS, Liu QH, Rathore R, Yadav V, Wang YX. Heterogeneous gene expression and functional activity of ryanodine receptors in resistance and conduit pulmonary as well as mesenteric artery smooth muscle cells. J Vasc Res. 2008;45:469–479. doi: 10.1159/000127438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kinnear NP, Wyatt CN, Clark JH, Calcraft PJ, Fleischer S, Jeyakumar LH, Nixon GF, Evans AM. Lysosomes co-localize with ryanodine receptor subtype 3 to form a trigger zone for calcium signalling by NAADP in rat pulmonary arterial smooth muscle. Cell Calcium. 2008;44:190–201. doi: 10.1016/j.ceca.2007.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Naylor E, Arredouani A, Vasudevan SR, Lewis AM, Parkesh R, Mizote A, Rosen D, Thomas JM, Izumi M, Ganesan A, et al. Identification of a chemical probe for NAADP by virtual screening. Nat Chem Biol. 2009;5:220–226. doi: 10.1038/nchembio.150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Boittin FX, Galione A, Evans AM. Nicotinic acid adenine dinucleotide phosphate mediates Ca2+ signals and contraction in arterial smooth muscle via a two-pool mechanism. Circ Res. 2002;91:1168–1175. doi: 10.1161/01.res.0000047507.22487.85. [DOI] [PubMed] [Google Scholar]
- 43.Okabe E, Tsujimoto Y, Kobayashi Y. Calmodulin and cyclic ADP-ribose interaction in Ca2+ signaling related to cardiac sarcoplasmic reticulum: superoxide anion radical-triggered Ca2+ release. Antioxid Redox Signal. 2000;2:47–54. doi: 10.1089/ars.2000.2.1-47. [DOI] [PubMed] [Google Scholar]
- 44.Gul R, Shawl AI, Kim SH, Kim UH. Cooperative interaction between reactive oxygen species and Ca2+ signals contributes to angiotensin II-induced hypertrophy in adult rat cardiomyocytes. Am J Physiol Heart Circ Physiol. 2012;302:H901–H909. doi: 10.1152/ajpheart.00250.2011. [DOI] [PubMed] [Google Scholar]
- 45.Rodiño-Janeiro BK, Paradela-Dobarro B, Castiñeiras-Landeira MI, Raposeiras-Roubín S, González-Juanatey JR, Alvarez E. Current status of NADPH oxidase research in cardiovascular pharmacology. Vasc Health Risk Manag. 2013;9:401–428. doi: 10.2147/VHRM.S33053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Liu JQ, Zelko IN, Erbynn EM, Sham JS, Folz RJ. Hypoxic pulmonary hypertension: role of superoxide and NADPH oxidase (gp91phox) Am J Physiol Lung Cell Mol Physiol. 2006;290:L2–L10. doi: 10.1152/ajplung.00135.2005. [DOI] [PubMed] [Google Scholar]
- 47.Mittal M, Roth M, König P, Hofmann S, Dony E, Goyal P, Selbitz AC, Schermuly RT, Ghofrani HA, Kwapiszewska G, et al. Hypoxia-dependent regulation of nonphagocytic NADPH oxidase subunit NOX4 in the pulmonary vasculature. Circ Res. 2007;101:258–267. doi: 10.1161/CIRCRESAHA.107.148015. [DOI] [PubMed] [Google Scholar]
- 48.Hordijk PL. Regulation of NADPH oxidases: the role of Rac proteins. Circ Res. 2006;98:453–462. doi: 10.1161/01.RES.0000204727.46710.5e. [DOI] [PubMed] [Google Scholar]
- 49.Xu M, Li XX, Ritter JK, Abais JM, Zhang Y, Li PL. Contribution of NADPH oxidase to membrane CD38 internalization and activation in coronary arterial myocytes. PLoS One. 2013;8:e71212. doi: 10.1371/journal.pone.0071212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lal H, Verma SK, Foster DM, Golden HB, Reneau JC, Watson LE, Singh H, Dostal DE. Integrins and proximal signaling mechanisms in cardiovascular disease. Front Biosci (Landmark Ed) 2009;14:2307–2334. doi: 10.2741/3381. [DOI] [PubMed] [Google Scholar]