

Abstract
Cystic fibrosis transmembrane conductance regulator gene (CFTR) expression in human airway epithelial cells involves the recruitment of distal cis-regulatory elements, which are associated with airway-selective DNase hypersensitive sites at −44 kb and −35 kb from the gene. The −35-kb site encompasses an enhancer that is regulated by the immune mediators interferon regulatory factor 1 and 2 and by nuclear factor Y. Here we investigate the −44-kb element, which also has enhancer activity in vitro in airway epithelial cells but is inactive in intestinal epithelial cells. This site contains an antioxidant response element (ARE) that plays a critical role in its function in airway cell lines and primary human bronchial epithelial cells. The natural antioxidant sulforaphane (SFN) induces nuclear translocation of nuclear factor, erythroid 2-like 2 (Nrf2), a transcription factor that regulates genes with AREs in their promoters, many of which are involved in response to injury. Under normal conditions, the −44-kb ARE is occupied by the repressor BTB and CNC homology 1, basic leucine zipper transcription factor (Bach1), and v-Maf avian musculoaponeurotic fibrosarcoma oncogene homolog K (MafK) heterodimers. After 2 hours of SFN treatment, Nrf2 displaces these repressive factors and activates CFTR expression. Site-directed mutagenesis shows that both the ARE and an adjacent NF-κB binding site are required for activation of the –44-kb element in airway epithelial cells. Moreover, this element is functionally linked to the −35-kb enhancer in modulating CFTR expression in response to environmental stresses in the airway.
Keywords: CFTR gene expression, oxidative stress, antioxidant response element, airway epithelium
Clinical Relevance
Understanding the mechanisms of regulation of CFTR expression in the airway epithelium is important for the development of new therapies for cystic fibrosis.
Reprogramming of gene expression is pivotal to the maintenance of cellular homeostasis in response to environmental stress. Multiple molecular mechanisms contribute to this homeostasis, which is required for cell survival. Under conditions of oxidative stress, antioxidant response elements (AREs) are among the critical cis-regulatory elements that respond to modulate gene expression. AREs recruit nuclear factor, erythroid 2-like 2 (Nrf2), an important regulator of antioxidant response pathways, forming heterodimers with v-Maf avian musculoaponeurotic fibrosarcoma oncogene homolog K (MafK) proteins to activate the expression of antioxidant enzymes. In the absence of oxidative stress, AREs are occupied by a repressive BTB and CNC homology 1, basic leucine zipper transcription factor (Bach1)/MafK heterodimer (1–3).
The Nrf2 antioxidant response signaling can be induced by the naturally occurring antioxidant compound sulforaphane (SFN) (4–7). Triggered by SFN treatment, inactive Nrf2 dissociates from its repressor kelch-like ECH-associated protein 1 (Keap1) in the cytoplasm and translocates into the nucleus. Here it can regulate the transcription of target genes by recruitment to AREs (8, 9).
The cystic fibrosis transmembrane conductance regulator (CFTR) gene encodes a chloride ion channel, which is critical for regulating transepithelial anion secretion under normal conditions and in response to oxidative stress. In previous work, oxidative stress was shown to suppress the CFTR mRNA levels in Calu3 lung adenocarcinoma cells (10), although this appeared to result from reduced CFTR mRNA stability rather than from transcriptional repression of the gene. Other researchers also identified a conserved ARE in the CFTR promoter, which was reported to recruit Nrf2 and, in conjunction with the transcription factor YinYang 1 (YY1), to repress the CFTR promoter in human bronchial epithelial Beas2b cells (11). However, the CFTR promoter has many characteristics of a “house-keeping” gene promoter and lacks tissue-specific and temporal regulatory elements (12–14). These critical elements are located within introns of the CFTR gene and in flanking intergenic regions (15). In intestinal epithelial cells, cis-regulatory elements in the middle of the locus (in introns 10 and 11) include a strong enhancer that is brought to the gene promoter by a looping mechanism along with a weaker enhancer in the first intron (16). This 3D structure is stabilized by CCCTC-binding factor (CTCF), the cohesin complex, and a transcriptional network, including the pioneer factors forkhead box A1/A2 (FOXA1/A2), hepatocyte nuclear factor 1 (HNF1), and caudal type homeobox 2 (CDX2) (17–19). These sites do not apparently contribute to CFTR expression in the airway epithelium, where the important cis-regulatory elements are located outside the gene, associated with DNase hypersensitive sites (DHS) at −44 kb and −35 kb upstream of the CFTR translation start site (20, 21). The −35 kb DHS encompasses an airway-selective enhancer that is regulated by the immune mediators interferon regulatory factor 1 and 2 (IRF1/2) and by nuclear factor Y (NF-Y) (22).
Here we examine the airway-selective cis-regulatory element at DHS at −44 kb (DHS-44kb) to determine its function and mechanism of activation. We show that this sequence contains an ARE involved in the Nrf2-activated antioxidant signaling pathway. Exposure to oxidative stress initially activates CFTR expression, although, consistent with previous data, repression of CFTR expression occurs under prolonged conditions of oxidative stress. We show the importance of Bach1/MafK and Nrf2/MafK competition for occupancy at this element. Moreover, we show that the −44 kb DHS element is functionally linked to the −35 kb DHS (DHS-35kb), consistent with the invariant coexistence of these two sites. Our results suggest that the distal ARE is involved in regulation of CFTR expression in human airway epithelial cells under conditions of environmental stress.
Materials and Methods
Cell Culture and Exposure to Chemicals
Human bronchial epithelial (HBE) cells were cultured in BEGM (Lonza, Walkersville, MD). 16HBE14o- (23) and Caco2 cells (24) were grown in Dulbecco’s modified Eagle medium with 10% serum. Serum-starved (> 12 h) cells were treated with 10 μM SFN (Sigma-Aldrich, St. Louis, MO) in serum-free medium for 0, 2, 4, 6 hours or for 4 hours with 200 ng/ml LPS (L9134; Sigma-Aldrich) in PBS or PBS alone before harvest.
Plasmids and Reporter Assays
Sequences encompassing DHS-44kb (hg 19, chr7:117075400–117076000) and a 279-bp subfragment (hg19, chr7:117,075,558–117,075,836) were amplified using Pfu DNA polymerase (Stratagene, La Jolla, CA) and inserted into the enhancer site of the pGL3B 245 (CFTR basal promoter) luciferase reporter vector (25). Mutants were generated using the QuikChange Lightning Multi Site-Directed Mutagenesis Kit (20215; Agilent Technologies, Santa Clara, CA). Primers for PCR and mutagenesis are shown in Tables E1 and E2 in the online supplement.
pGL3B luciferase constructs were transiently cotransfected with a modified pRL Renilla luciferase control vector (Promega, Madison, WI) into 16HBE14o- cells with Lipofectin (Life Technologies, Carlsbad, CA). Firefly and Renilla (normalizer) luciferase activities were measured 48 hours after transfection (26).
Electrophoretic Mobility Gel Shift Assay
Electrophoretic mobility gel shift assay (EMSA) reactions using subfragments of the 279-bp DHS-44kb element were done by standard protocols (27). Probes and competitor sequences are shown in Table E3. Antibodies specific for NF-κB p65 (sc-372x; Santa Cruz Biotech, Santa Cruz, Ca), Bach1 (gift of Dr. K. Igarashi) (3), Nrf2 (sc-722x), and GR (sc-1003x) were used for supershift assays.
Chromatin Immunoprecipitation
Chromatin immunoprecipitation (ChiP) was performed by standard protocols (20) with 0.37% (for histone modifications) or 1% (for transcription factors) formaldehyde crosslinking. Antibodies were specific for H3K27Ac (ab4729; Abcam, Cambridge, UK), MafK (ab50322; Abcam), Bach1 (sc-14700x), Nrf2 (sc-722x), NF-κB p65 (sc-372x), normal rabbit IgG (Millipore 12–370; EMD Millipore, Billerica, MA), or normal goat IgG (sc-2028). Enrichments were calculated by quantitative PCR (qPCR) relative to input (for histone modifications) or IgG (for transcription factors). qPCR primers are shown in Table E4.
cDNA-Encoded Overexpression and Transient Small Interfering RNA–Mediated Depletion of Factors
Bach1 (28) and Nrf2 (36971; Addgene, Cambridge, MA) cDNAs were transfected into 16HBE14o- cells as described above and harvested after 48 hours. To examine the influence of each factor on the cis-acting element alone, cDNA clones were cotransfected with the reporter gene constructs described earlier.
Small interfering RNA (siRNA) specific for nuclear transcription factor Y, subunit A (NF-YA) (sc-2994) and relevant negative controls (sc-37007) were reverse-transfected into 16HBE14o- cells by Lipofectamine RNAiMAX (Life Technologies, Grand Island, NY), and cells were harvested after 72 hours.
Quantitative RT-PCR
Total RNA was extracted using TRIzol reagent (Life Technologies). cDNA synthesis was performed by TaqMan reverse-transcription reactions (Roche Applied Science). CFTR expression was measured using a TaqMan assay described previously (27, 29). Relative gene expression levels were calculated after normalization to 18S rRNA. Primer sequences are shown in Table E5.
Western Blot Analysis
Cells were lysed for cytoplasmic or nuclear extract as described previously (22). Western blots were probed with antibodies specific for Nrf2 (sc-722x), Bach1 (3), histone 3 (ab10799; Abcam, Cambridge, MA), and β-tubulin (T4026; Sigma-Aldrich).
Results
DHS-44kb Encompasses a Cell-Type Selective cis-Regulatory Element that Binds Multiple Factors
The cis-acting regulatory element associated with a DHS-44kb 5′ to the CFTR translation start site was first identified using DNase-chip (20, 21). It was seen in primary human bronchial and tracheal epithelial cells (NHBE and human tracheal epithelial [HTE]) and human airway cell lines 16HBE14o- and Calu3. DHS-44kb is always present with DHS-35kb, a distal enhancer regulating airway CFTR expression (22). Using open chromatin mapping by DNase-seq in HTE cells (30), the core of DHS-44kb was localized to 600 bp at hg 19, chr7:117075400–117076000 (Figure 1D). This region was marked by enrichment of H3K27Ac in 16HBE14o- cells, consistent with DHS-44kb encompassing an active enhancer element (Figure 1A).
Figure 1.

DNase hypersensitive site at −44 kb (DHS-44kb) encompasses a cell type–selective cis-regulatory element that binds multiple factors. (A–C) Cell type–selective enhancer properties of the DHS-44kb element. (A) H3K27Ac chromatin immunoprecipitation (ChIP) in16HBE14o- cells shows modest enrichment at DHS-44kb. Quantitative PCR analysis of chromatin from 16HBE14o- cells immunoprecipitated with an antibody specific to H3K27Ac. The primer sets shown (Table E4) amplify the promoter region and specific DHS regions. Data from a single representative ChIP experiment are shown as % input (n = 2). Error bars represent SEM. (B and C) Enhancer activity of DHS-44kb in luciferase reporter gene assays in 16HBE14o- and Caco2 cells. Cells were transfected with pGL3B luciferase reporter constructs driven by CFTR basal promoter (pGL3B 245) or with DHS-44kb fragments: 0.6 kb DHS and 279-bp subfragment, cloned into the enhancer site of the vector (F and R denote forward or reverse orientation). Luciferase data are shown relative to the CFTR basal promoter vector (= 1). Error bars represent SEM (n = 6). *P < 0.05 using unpaired t tests. (D) Scale diagram to show the location of oligonucleotides 44-I, 44-II, and 44-III, and 44-a, 44-b, and 44-c, used in electrophoretic mobility gel shift assays (EMSAs) and the conserved sequences at DHS-44 kb. (E) EMSAs identified transcription factor binding sites at DHS-44(279). 16HBE14o- nuclear extracts were incubated with 32P-labeled probes (Table E3), and DNA–protein complex was observed with oligonucleotides 44-I, 44-II, 44-a, and 44-c, which were competed by 50× excess of unlabeled corresponding probes but not nonspecific oligonucleotides (Table E3).
To confirm that DHS-44kb contained an enhancer of CFTR expression, the 0.6-kb DHS fragment (hg 19, chr7:117075400–117076000) was PCR amplified and inserted into the enhancer site of the pGL3B 245 vector. Luciferase reporter gene expression in this vector is driven by a 787-bp CFTR basal promoter fragment (25). The constructs were transfected into 16HBE14o- cells, and after 48 hours firefly luciferase activity was measured relative to Renilla luciferase. The 0.6-kb DHS-44kb sequence showed 24.1-fold (forward orientation) or 31.5-fold (reverse orientation) enhancer activity in comparison to the promoter activity of pGL3 245 alone (Figure 1B). When the same plasmids were transfected into Caco2 colon carcinoma cells, only marginal enhancer activity was measured (2.4-fold in forward orientations and 4.7-fold in reverse). In concurrent transfections, the intestinal-specific enhancer at intron 11 showed 30-fold enhancer activity in Caco2 cells (Figure 1C). These data suggest that the enhancer activity of the DHS-44kb element is specific to airway epithelial cells.
Next, using sequence conservation data (31) from Encode (32), a 279-bp highly conserved region was identified in the center of the 600-bp DHS-44 kb (hg19, chr7:117,075,558–117,075,836) (Figure 1D). This 279-bp fragment [DHS-44(279)] was cloned into the enhancer site of the pGL3B 245 vector, and its activity was compared with the 600-bp region. DHS-44 (279) showed 81.4-fold enhancement of pGL3 245 promoter activity in the forward orientation. This enhancement was 3-fold greater than the 600-bp sequence (Figure 1B), suggesting it contained the core functional element of DHS-44 kb. (The reverse orientation was not stable in the pGL3 245 vector.)
To identify the transcription factors driving the DHS-44(279) enhancer, the sequence was evaluated in segments 44-I, 44-II, and 44-III, where 44-I and 44-III correspond to highly conserved sequences (Figure 1D) (33). EMSAs were performed with 32P-labeled, double-stranded DNA probes for 44-I, 44-II, and 44-III (Table E3) and nuclear extracts from 16HBE14o- cells. Specific DNA–protein complexes were generated with 44-I and 44-II but not 44-III (Figure 1E), and these were effectively competed with 50-fold molar excess of the corresponding unlabeled probe but not an irrelevant oligonucleotide sequence (Figure 1E; see Table E3). These results suggest that critical transcription factors at DHS-44 interact with sequences in fragments 44-I and 44-II.
Further analysis of the 44-I and 44-II regions used raw data from our DNase-seq analysis of primary HTE (30) and bronchial (NHBE) cells (Harris Laboratory, 2011, unpublished data). These data enable the visualization of in vivo footprints (the canyon region between two neighboring DNase-seq peaks) (34) where accessibility to DNase I is restricted by bound transcription factors. In vivo footprints within the DHS-44(279) region (44-a, 44-b, and 44-c) (Figure 1D) were assayed by EMSA with 16HBE14o- nuclear extract (Figures 1E and 2B). Specific DNA–protein complexes were formed with 44-a, which is within 44-I; 44-b (Figure 2B), which is within 44-II; and 44-c, which overlaps 44-II and 44-III (Figures 1D and 1E). The unlabeled 44-a and 44-b also effectively competed the complexes formed by the larger fragments 44-I (Figure E1A) and 44-II (Figure 1E), suggesting that 44-a and 44-b encompass the critical transcription factor binding sites in the region.
Figure 2.

NF-κB (p65) binds to a NF-κB motif in DHS-44(279) in vitro but not in vivo. (A) Identification of a predicted NF-κB binding motif in fragment 44-b and generation of a mutant version. (B) EMSAs show binding of NF-κB to 44-b in vitro. The DNA–protein complex generated when 16HBE14o- nuclear extract was incubated with a 32P-labeled 44-b probe was competed by 50× excess of unlabeled 44-b probe and an NF-κB consensus probe but not by mutant versions (Table E3). Incubation of the complex with an antibody specific for NF-κB (p65) generated a supershift (GR antibody = isotype control). (C) LPS treatment of 16HBE14o- cells increases NF-κB occupancy at the binding site in the CFTR promoter (−0.5 kb) but not at DHS-44kb. ChIP was performed with an antibody specific for NF-κB (p65). Black bars: 200 ng/ml LPS in PBS. White bars: PBS. Data normalized to IgG; error bars represent SEM of at least two ChIP experiments.
NF-κB (p65) Binds to a NF-κB Motif in DHS-44(279) In Vitro but Not In Vivo
First, an in silico approach was taken to identify transcription factors interacting with the DHS-44(279) sequence. Inspection of ENCODE ChIP-seq data (35) revealed a NF-κB binding site within fragment 44-b at chr7:117075514–117075933 (hg 19) in the GM12878 lymphoblastoid cell line. This corresponds to a predicted NF-κB motif at chr7:117075702–117075716. EMSAs showed that the DNA–protein complex formed by 44-b and 16HBE14o- nuclear extracts was effectively competed by an unlabeled competitor containing the NF-κB binding consensus (Table E3) (36). This competition was destroyed by a single nucleotide mutation at the NF-κB motif in the competitor (Figure 2B). Moreover, mutation of the NF-κB site in 44-b repressed its ability to compete the EMSA complex (Figures 2A and 2B). A supershift experiment, with an antibody specific for NF-κB (p65), also effectively shifted the complex. However, we were unable to confirm NF-κB binding to this element in vivo in airway epithelial cells. Using the p65 antibody in a ChIP experiment with LPS-treated 16HBE14o- cells, no enrichment of this factor was seen within the DHS-44kb region (Figure 2C). In contrast, 3-fold enrichment of p65 was seen at the NF-κB element previously reported in the CFTR promoter (Figure 2C) (37). These data suggest that although the NF-κB motif within 44-b binds to NF-κB (p65) in vitro, it is not functional in 16HBE14o- airway epithelial cells. Hence, we next looked for additional transcription factors binding to DHS-44(279) in these cells.
Bach1 Binds to an ARE within DHS-44(279) In Vitro
In addition to the factors predicted to bind to 44-b by MatInspector (38) in silico analysis, inspection of ENCODE ChIP-seq data (35, 39) showed a large number of transcription factors binding to 44-a and 44-c in cell types that were not relevant to this study. To identify interacting proteins, the critical nucleotides necessary for DNA–protein complex formation were revealed by scanning mutagenesis (Figures 3A and 3B; Table E3). Serial mutations of up to four nucleotides were generated at the in vivo footprint sites in probes 44-a and 44-c (Table E3). Next, each mutant was used as an unlabeled competitor oligonucleotide in EMSA reactions with 32P-labeled 44-a or 44-c and 16HBE14o- nuclear extract (Figures E1B and E1C). Critical nucleotides were identified by loss of competition in the EMSA reactions. To examine the impact of these critical nucleotides on the DHS-44(279) enhancer activity, the same bases were mutated in the pGL3 245 plasmids containing DHS-44(279) at the enhancer site (Table E2). Mutant and wild-type constructs were transfected into 16HBE14o- cells, and luciferase activities were measured (Figure 3D). Mutation of the critical nucleotides in 44-a and 44-b individually decreased enhancer activity by 50% compared with the wild-type sequence. However, the mutation that abolished competition in 44-c (Figure 3A) did not alter DHS-44(279) enhancer activity in (Figure 3D). These data suggest that the cis-acting elements at 44-a and 44-b are more important than 44-c in the regulatory mechanism. When all three mutations were generated together in DHS-44(279), its enhancer activity dropped to 15% of wild-type levels (Figure 3D; 3xmut).
Figure 3.
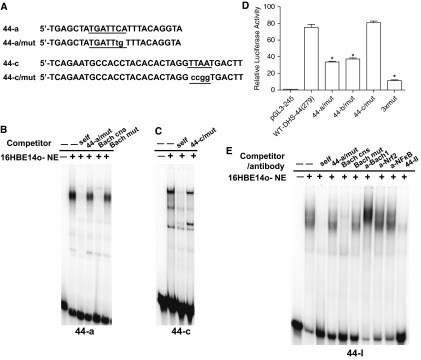
BTB and CNC homology 1, basic leucine zipper transcription factor (Bach1) binds to an antioxidant response element within DHS-44(279) in vitro. (A) Scanning mutagenesis identified the critical nucleotides for transcription factor binding at 44-a and 44-c. (B, C, and E) EMSA experiments with 32P-labeled 44-a (B), 44-c (C), or 44-I (E) probes and 16HBE14o- nuclear extract. The DNA–protein complex generated with each probe was competed by 50× excess of the same unlabeled oligonucleotide but not by mutant versions (44-a/mut and 44-c/mut) (A, Table E3). The complex formed with the 44-a probe was also competed by an unlabeled Bach1 consensus oligonucleotide (39, 40) (Table E3) but not by a mutant version (B). (D) Mutagenesis of DHS-44(279) decreases its enhancer activity, confirming critical transcription factor binding sites. pGL3 constructs and transient reporter gene assays in 16HBE14o- cells as described in Figure 1. Mutant plasmids (44-a/mut and 44-c/mut are shown in A; 44-b/mut is shown in Fig. 2A) were compared with wild-type DHS-44(279). Data show luciferase activities relative to the CFTR basal promoter vector (= 1). Error bars represent SEM (n = 6). *P < 0.05 using unpaired t tests. (E) Binding of Bach1 to 44-I in vitro. The DNA–protein complexes formed with 32P-labeled 44-I probe were competed by 50× excess of unlabeled 44-I and Bach1 consensus probe but not by mutant versions (Table E3). A supershift is seen on incubation of the DNA–protein complex with an antibody specific for Bach1 (NF-κB or nuclear factor, erythroid 2-like 2, isotype controls).
A number of transcription factor binding sites are predicted to be destroyed by mutation of the critical nucleotides “TTAA” within 44-c. EMSA showed that competitor oligonucleotides containing consensus binding sites for HNF1, Six3, and ISL1 effectively competed the formation of the 44-c DNA–protein complex (data not shown). However, because mutation of the 44-c TTAA sequence in DHS-44(279) had no effect on luciferase expression (Figure 3B), we focused on the 44-a element.
The scanning mutagenesis approach identified binding sites for several transcription factors within the 44-a sequence, including paired box 6 (PAX6), down-regulator of transcription 1, TBP-binding (negative cofactor 2) (DR1), HMG box transcription factor 1 (HBP1), activator protein 1 (API), pre–B-cell leukemia homeobox 1 (PBX1/MEIS), and Bach1. Of these, only a consensus oligonucleotide for Bach1 (40, 41) effectively competed the DNA–protein complex formed by 44-a with 16HBE14o- nuclear proteins (Figures 3A and 3B). Moreover, the same consensus sequence with mutations in the Bach1 core did not compete. The DNA–protein complex formed by 44-a did not supershift when incubated with the antibody specific to Bach1 (data not shown), but, when using the longer probe 44-I that encompasses the 44-a element, a clear supershift was evident (Figure 3E). Moreover, when Bach1 was overexpressed in 16HBE14o- cells by transient transfection of a mouse Bach1 cDNA clone, a strong supershift was seen with the Bach1 antibody and probe 44-I (Figure E1D).
SFN Regulates CFTR Expression in Airway Epithelial Cells
The binding site for the Bach1 transcriptional repressor is an ARE, which is the critical motif in pathways that respond to oxidative stress. Under normal conditions, Bach1 binds to an ARE, but in response to oxidative stress it is displaced by Nrf2 (nuclear factor, erythroid 2–like 2), which recognizes the same sequence motif. SFN, a naturally occurring antioxidant, was used to trigger the cellular antioxidant response in 16HBE14o- cells. Two hours of 10 μM SFN treatment was sufficient to enable transfer of Nrf2 from the cytoplasm to the nucleus to activate gene expression (Figure 4A). Serum-starved 16HBE14o- cells and primary HBE cells were treated with 10 μM SFN for 0, 2, 4, and 6 hours, and CFTR mRNA levels were assayed at each time point. After 2 hours of SFN treatment, CFTR mRNA levels significantly increased to a maximum of 1.5-fold (P < 0.05), returned to pretreatment levels by 4 hours of SFN treatment, and then continued to fall to 70% of initial expression levels after 6 hours of SFN exposure (Figure 4B). CFTR expression levels in HBE cells showed a similar, though slower, response profile to SFN treatment (Figure 4C); the reduction after the 2-hour post-SFN peak only reached pretreatment levels by 6 hours. These data suggest that SFN regulates the CFTR expression in human airways.
Figure 4.

Sulforaphane (SFN) regulates CFTR expression in airway epithelial cells. (A) Western blot showing the relocation of nuclear factor, erythroid 2-like 2 (Nrf2) into the nucleus after 2 hours of SFN treatment in 16HBE14o- cells. (B and C) Two hours of SFN treatment increases CFTR mRNA levels in 16HBE14o- cells (B) and human bronchial epithelial cells (C). CFTR mRNA levels assayed by Taqman quantitative RT-PCR normalized to 18S RNA levels. (D) Recruitment of Bach1, Nrf2, and with v-Maf avian musculoaponeurotic fibrosarcoma oncogene homolog K (MafK) to DHS-44(279) during 6 hours of SFN treatment in 16HBE14o- cells. ChIP with antibodies specific for each factor is shown after 0, 2, 4, and 6 hours of SFN treatment. Data were normalized to IgG and combined from at least two ChIP experiments. In all panels, error bars represent SEM. *P < 0.05 using an unpaired t test.
To determine whether the SFN-triggered antioxidant response regulates CFTR expression directly through the DHS-44(279) cis-element, ChIP was used to measure the enrichment levels of the constitutive ARE binding factor Bach1, the SFN-induced ARE binding factor Nrf2, and their common partner MafK at this site (Figure 4D). Within 2 hours of SFN treatment, Bach1 levels at DHS-44(279) dropped 3-fold compared with basal conditions and then gradually rose over the subsequent after 4 hours. In contrast, the enrichment of Nrf2 increased 3.3-fold after 2 hours of SFN treatment and subsequently fell to basal levels within 6 hours of treatment. The MafK enrichment at DHS-44(279) was not significantly changed during 4 hours of SFN treatment, although its levels were approximately 1.3-fold lower by 6 hours after SFN exposure. We also assayed the enrichment of Bach1, Nrf2, and MafK at other airway selective DHS and at sites within the promoter region (Figure E2). Nrf2 was enriched after SFN treatment at the −3.4 kb DHS (20, 22) and −2 kb extended promoter region and at the −44 kb DHS. In contrast, constitutive Bach1 was enriched only at the DHS-44 site. The dynamic response of factors binding to DHS-44(279) after SFN treatment confirmed the direct role of the Nrf2-regulated antioxidant response at this site.
Overexpression of Bach1 and Nrf2 Alters CFTR Expression Levels and the Activity of the DHS-44kb Element
To further understand the mechanism of action of the ARE at DHS-44kb, we next evaluated the impact of Bach1 and Nrf2 on CFTR expression in airway epithelial cells.
Expression plasmids for human Nrf2 (hNrf2) or mouse Bach1 (mBach1) were transiently transfected into 16HBE14o- cells and shown by Western blot to produce high levels of the respective proteins (Figure 5A). Without SFN treatment, hNrf2 overexpression had no impact on CFTR mRNA levels in comparison to empty vector–transfected controls. However, 2 hours after SFN treatment, CFTR mRNA levels increased to 1.8-fold and remained elevated (1.3-fold) 4 hours after treatment (Figure 5B). These data suggest that increasing Nrf2 up-regulates CFTR expression. Bach1 overexpression initially decreased CFTR mRNA levels by approximately 30% in comparison to the vector-transfected controls (Figure 5B). However, 2 hours after SFN exposure in the same cells, CFTR mRNA levels increased by approximately 1.5-fold when compared with vector controls, suggesting that Nrf2 translocating to the nucleus rescued the repressive effect of mBach1 (Figure 5B).
Figure 5.
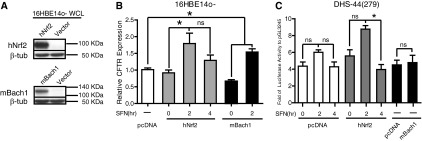
Overexpression of Bach1 and Nrf2 alters CFTR expression levels and the activity of the DHS-44kb element, where β-tubulin is the normalizer. (A) Western blots show overexpression of Nrf2 and Bach1 in whole cell lysate from 16HBE14o- cells. (B) After SFN treatment, overexpression of Nrf2 and Bach1 influences CFTR expression in 16HBE14o- cells. Nrf2 or Bach1 expression plasmids or vector alone (pcDNA3.1) were transfected into 16HBE14o- cells. After 48 hours, the cells were treated with 10 μM SFN for 0, 2, and 4 hours after cell harvest. CFTR mRNA levels assayed by Taqman quantitative RT-PCR (normalized to 18 s rRNA) are shown: pcDNA3.1 control (white bar), Nrf2 overexpression (gray bars), and Bach1 overexpression (black bars). Error bars represent SEM (n ≥ 6). *P < 0.05 using an unpaired t test. (C) Overexpression of Nrf2 influences the enhancer activities of DHS-44(279) in 16HBE14o- cells. pGL3 245 or pGL3 245 DHS-44(279) were cotransfected with Nrf2 or Bach1 expression plasmids or pcDNA3.1. After 48 hours, the cells were treated with 10 μM SFN for 0, 2, and 4 hours before cell harvest. Data show the relative enhancer activity of DHS-44(279) compared with the CFTR promoter (pGL3 245) when cotransfected with pcDNA3.1 (white bars) or Nrf2 (gray bars) followed by SFN treatment or Bach1 without SFN treatment (black bars). Error bars represent SEM (n ≥ 6). *P < 0.05 using an unpaired t test. ns, not significant.
The direct impact of Bach1 and Nrf2 on the DHS-44kb ARE was also evaluated. The pGL3 245 CFTR promoter–containing luciferase reporter vector and the same vector containing the DHS-44(279) fragment at the enhancer site were transiently transfected into 16HBE14o- cells together with either the hNrf2 or mBach1 cDNAs. Overexpression of hNrf2 increased DHS-44(279)–derived luciferase expression 1.2-fold without SFN treatment, in comparison to vector-transfected control cells. Two hours of SFN treatment further increased luciferase values 2.6-fold in the Nrf2-overexpressing cells but only roughly 1.6-fold of that in vector-transfected cells. These results suggest that the Nrf2-regulated antioxidant response affects CFTR expression, at least in part, through the DHS-44kb ARE. However, mBach1 overexpression did not significantly change luciferase activity driven by the DHS-44(279) enhancer element, suggesting that the mechanism of Bach1 repression of CFTR expression may be more complex.
Distal cis-Regulatory Elements Interact To Regulate CFTR Expression in the Airway
Two airway-selective cis-regulatory elements for CFTR map within 10 kb of each other at DHS-44kb and DHS-35kb. The two sites are always evident concurrently in human airway epithelial cells (20, 22), suggesting that they may be functionally linked. We showed that both DHS contain airway-selective enhancers, so we first investigated whether the two enhancers have cooperative activity. The 0.6-kb DHS-44kb region and the 1.4-kb DHS-35kb region were combined and inserted into the enhancer site of pGL3 245. The luciferase activity of the combined DHS fragments was compared with the individual elements after transient transfection into 16HBE14o- cells. The enhancement of luciferase values relative to the pGL3 245 construct was 6.4- and 10.1-fold respectively, for DHS-44kb and DHS-35kb. In combination, the two elements enhanced luciferase expression 27.7-fold (Figure 6A), implicating a moderate cooperative effect, as seen with other CFTR enhancer elements (16, 20).
Figure 6.

Distal cis-regulatory elements interact to regulate CFTR expression in the airway. (A) When combined in tandem in the same vector DHS −35 kb and −44 kb have a modest cooperative effect on enhancer activity in vitro. 16HBE14o- cells were transfected with pGL3B luciferase-reporter constructs containing the CFTR basal promoter and fragments at DHS −44 kb or −35 kb individually or combined in the enhancer site in the forward orientation. Enhancer activities are shown relative to the CFTR basal promoter–alone vector (= 1); error bars represent SEM (n = 6). *P ≤ 0.05 when the single fragments are compared with the combined fragments using unpaired t tests. (B) siRNA-mediated knockdown of nuclear factor Y (NF-Y) enhances CFTR expression in 16HBE14o- cells (from Ref. 22). Western blot shows the efficiency of nuclear transcription factor Y, subunit A (NF-YA) depletion by siRNA in comparison to nontargeting control (NC) in 16HBE14o- cells. Cells were lysed after 72 hours to evaluate CFTR mRNA expression by Taqman quantitative RT-PCR from total RNA as described in Figure 4 legend. Error bars represent SEM (n ≥ 9). *P < 0.001 using an unpaired t test. (C) NF-YA depletion alters Nrf2 enrichment at DHS-44kb in 16HBE14o- cells. Cells were reverse transfected for 72 hours with siRNA targeting human NF-YA (KD [knockdown], gray bars) or negative control (NC, white bars). ChIP with antibodies specific for Nrf2 followed by quantitative PCR analysis. Primers (Table E3) are located at multiple DHS 5′ to the CFTR promoter. Data normalized to IgG are from a single representative ChIP experiment (the experiment was performed twice). Error bars represent SEM. *P < 0.05 using an unpaired t test.
We previously showed that recruitment of NF-Y is critical to the mechanism of action of the DHS-35kb element (22) and that siRNA-mediated depletion of NF-YA and its subsequent loss from DHS-35 is accompanied by a significant increase in enrichment of p300, a histone acetyltransferase (42), at DHS-44kb (22). Because p300 is a marker of active enhancer elements, this result suggested that the regulatory element at DHS-44 might respond to loss of NF-YA at DHS-35 and increase its activity. To further investigate this potential control mechanism, Nrf2 occupancy at airway-selective regulatory elements was evaluated by ChIP in 16HBE14o- cells after siRNA-mediated depletion of NF-YA or delivery of a nontargeting siRNA (Figure 6C). Nrf2 enrichment increased approximately 2-fold after NF-YA depletion not only at DHS −44 kb but also at another airway-selective DHS-3.4kb (20) and within the CFTR promoter region (−2 and −0.5 kb) (Figure 6C). No change in occupancy of Nrf2 was seen at DHS-35kb. Inspection of the −3.4 kb DHS and CFTR promoter sequence identified several putative AREs. The increase in Nrf2 occupancy at these sites after NF-YA depletion was accompanied by a significant increase (∼ 1.7-fold) in CFTR expression (Figure 6B). These data suggest that multiple AREs within the CFTR locus respond to environmental stress to cause rapid activation of CFTR expression.
Discussion
The CFTR protein, a cAMP-regulated epithelial chloride channel, not only mediates chloride and bicarbonate transport but is also a regulator of other ion transport pathways. It has a major role in the maintenance of cellular homeostasis under normal conditions or subsequent to environmental stress. Oxidative stress plays an important part in the pathogenesis of many lung diseases, including asthma, chronic obstructive pulmonary disease, hypoxia-induced acute lung injury, pulmonary fibrosis, and lung cancer (41). It is thought that accumulation of reactive oxygen species and recurrent inflammation resulting from oxidative stress are major causes of lung damage in these disorders. Dysfunction of CFTR caused by oxidative stress is seen at mRNA, protein, and functional levels in studies of cigarette smokers (10, 44–47). The Keap1-Nrf2 antioxidant pathway plays a primary role in cellular defense against oxidative stress (48–50). Here we identify an ARE within a DHS at −44 kb upstream of the CFTR translation start site and show that CFTR expression is regulated by this element in human airway epithelial cells. The antioxidant transcriptional regulator Nrf2 has an important role at this ARE within 4 hours of an SFN-triggered antioxidant response. Under normal conditions, the ARE is occupied by a repressive Bach1/MafK heterodimer, but this complex is displaced by an activating Nrf2/MafK heterodimer under conditions of cellular oxidative stress. This dynamic regulation of factors binding at DHS-44kb alters CFTR expression levels and can be induced by SFN treatment in 16HBE14o- human airway epithelial cells. Also within the DHS-44kb region is a NF-κB site that binds this factor in vitro in gel shift assays but not in LPS-treated 16HBE14o- cells in vivo, as shown by ChIP. However, NF-κB is enriched at DHS-44kb in ChIP-seq data from several lymphoblastoid cell lines generated by the ENCODE consortium (51), suggesting that this binding site may be tissue- or cell-type specific. NF-κB acts as specific mediator in response to cytokines and to bacterial or viral antigens, which is in contrast to the different mechanism of activation of Nrf2 by oxidative stress. The DHS-44kb region may contain several cis-elements involved in the regulation of CFTR expression in response to various cellular stimuli, which may be mutually exclusive.
Subsequent to our identification of the functional ARE at DHS-44kb, we inspected the core sequences of other airway-selective DHS at −35 and −3.4 kb and within the promoter region where Nrf2 enrichment was observed after initiation of the antioxidant response (Figure E2). Significant Bach1 enrichment was only detected at DHS-44kb and not at the other sites before its displacement by Nrf2, suggesting that a conventional ARE is located at DHS-44kb (Figure 4B, Figure E2). Previous work (11) documented a YY1/ARE-like cis-acting element within the CFTR promoter that recruits a repressive YY1 and Nrf2 complex. However, there is no direct evidence that the ARE at the promoter has a role in controlling expression of CFTR subsequent to oxidative stress.
Bach1 acts as a constitutive repressor at the ARE within DHS-44kb under normal conditions, although our ChIP data suggest that it does not interact with the neighboring airway enhancer element at DHS-35kb. However, when NF-YA, one of the important factors driving the DHS-35 kb enhancer, was depleted by targeting with a specific siRNA, enhancer activity of the DHS-44 element increased, as measured by p300 and Nrf2 enrichment (Figure 6C). These data suggest a coordinated mechanism for monitoring airway CFTR expression levels under different conditions of environmental stress. This could ensure maintenance of normal transepithelial transport by long-range interaction of different cis-regulatory elements and potentially the recruitment of additional transcription factors complexes.
Acknowledgments
Acknowledgments
The authors thank Dr. K. Igarashi for the gift of mouse Bach1 cDNA plasmid (pCMV-FLAG Bach1 v.2) and anti-Bach1 antibody, Dr. S. Randell for HBE cells, and Dr. J. Browne for helpful discussions.
Footnotes
This work was supported by the National Institutes of Health grants R01 HL094585 and HD068901 (PI:AH) and the Cystic Fibrosis Foundation (PI:AH).
Author Contributions: Concept and design: Z.Z. and A.H. Experimentation: Z.Z. and S.-H.L. Analysis and interpretation: Z.Z. and A.H. Preparation of the manuscript: Z.Z., S.-H.L., and A.H.
Originally Published in Press as DOI: 10.1165/rcmb.2014-0263OC on September 26, 2014
This article has an online supplement, which is accessible from this issue's table of contents at www.atsjournals.org
Author disclosures are available with the text of this article at www.atsjournals.org.
References
- 1.Dhakshinamoorthy S, Jain AK, Bloom DA, Jaiswal AK. Bach1 competes with Nrf2 leading to negative regulation of the antioxidant response element (ARE)-mediated NAD(P)H:quinone oxidoreductase 1 gene expression and induction in response to antioxidants. J Biol Chem. 2005;280:16891–16900. doi: 10.1074/jbc.M500166200. [DOI] [PubMed] [Google Scholar]
- 2.Srisook K, Kim C, Cha YN. Molecular mechanisms involved in enhancing HO-1 expression: de-repression by heme and activation by Nrf2, the “one-two” punch. Antioxid Redox Signal. 2005;7:1674–1687. doi: 10.1089/ars.2005.7.1674. [DOI] [PubMed] [Google Scholar]
- 3.Sun J, Brand M, Zenke Y, Tashiro S, Groudine M, Igarashi K. Heme regulates the dynamic exchange of Bach1 and NF-E2-related factors in the Maf transcription factor network. Proc Natl Acad Sci USA. 2004;101:1461–1466. doi: 10.1073/pnas.0308083100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Prestera T, Talalay P. Electrophile and antioxidant regulation of enzymes that detoxify carcinogens. Proc Natl Acad Sci USA. 1995;92:8965–8969. doi: 10.1073/pnas.92.19.8965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Fahey JW, Talalay P. Antioxidant functions of sulforaphane: a potent inducer of Phase II detoxication enzymes. Food Chem Toxicol. 1999;37:973–979. doi: 10.1016/s0278-6915(99)00082-4. [DOI] [PubMed] [Google Scholar]
- 6.Kraft A, Lohnson DA, Johnson JA. Nuclear factor E2-related factor 2-dependent antioxidant response element activation by tert-butylhydroquinone and sulforaphane occurring preferentially in astrocytes conditions neurons against oxidative insult. J Neurosci. 2004;24:1101–1112. doi: 10.1523/JNEUROSCI.3817-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lee YJ, Lee SH. Sulforaphane induces antioxidative and antiproliferative responses by generating reactive oxygen species in human bronchial epithelial BEAS-2B cells. J Korean Med Sci. 2011;26:1474–1482. doi: 10.3346/jkms.2011.26.11.1474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Li W, Kong A-N. Molecular mechanisms of Nrf2-mediated antioxidant response. Mol Carcinog. 2009;48:91–104. doi: 10.1002/mc.20465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chen Y-T, Shi D, Yang D, Yan B. Antioxidant sulforaphane and sensitizer trinitrobenzene sulfonate induce carboxylesterase-1 through a novel element transactivated by nuclear factor-E2 related factor-2. Biochem Pharmacol. 2012;84:864–871. doi: 10.1016/j.bcp.2012.06.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cantin AM, Bilodeau G, Ouellet C, Liao J, Hanrahan JW. Oxidant stress suppresses CFTR expression. Am J Physiol Cell Physiol. 2006;290:C262–C270. doi: 10.1152/ajpcell.00070.2005. [DOI] [PubMed] [Google Scholar]
- 11.René C, Lopez E, Claustres M, Taulan M, Romey-Chatelain M-C. NF-E2-related factor 2, a key inducer of antioxidant defenses, negatively regulates the CFTR transcription. Cell Mol Life Sci. 2010;67:2297–2309. doi: 10.1007/s00018-010-0336-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Koh J, Sferra TJ, Collins FS. Characterization of the cystic fibrosis transmembrane conductance regulator promoter region: chromatin context and tissue-specificity. J Biol Chem. 1993;268:15912–15921. [PubMed] [Google Scholar]
- 13.Chou JL, Rozmahel R, Tsui LC. Characterization of the promoter region of the cystic fibrosis transmembrane conductance regulator gene. J Biol Chem. 1991;266:24471–24476. [PubMed] [Google Scholar]
- 14.Yoshimura K, Nakamura H, Trapnell BC, Dalemans W, Pavirani A, Lecocq JP, Crystal RG. The cystic fibrosis gene has a “housekeeping”-type promoter and is expressed at low levels in cells of epithelial origin. J Biol Chem. 1991;266:9140–9144. [PubMed] [Google Scholar]
- 15.Gillen AE, Harris A. Transcriptional regulation of CFTR gene expression. Front Biosci (Elite Ed) 2012;4:587–592. doi: 10.2741/401. [DOI] [PubMed] [Google Scholar]
- 16.Ott CJ, Blackledge NP, Kerschner JL, Leir S-H, Crawford GE, Cotton CU, Harris A. Intronic enhancers coordinate epithelial-specific looping of the active CFTR locus. Proc Natl Acad Sci USA. 2009;106:19934–19939. doi: 10.1073/pnas.0900946106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kerschner JL, Gosalia N, Leir S-H, Harris A. Chromatin remodeling mediated by the FOXA1/A2 transcription factors activates CFTR expression in intestinal epithelial cells. Epigenetics. 2014;9:557–565. doi: 10.4161/epi.27696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kerschner JL, Harris A. Transcriptional networks driving enhancer function in the CFTR gene. Biochem J. 2012;446:203–212. doi: 10.1042/BJ20120693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gosalia N, Neems D, Kerschner JL, Kosak ST, Harris A. Architectural proteins CTCF and cohesin have distinct roles in modulating the higher order structure and expression of the CFTR locus. Nucleic Acids Res. 2014;42:9612–9622. doi: 10.1093/nar/gku648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhang Z, Ott CJ, Lewandowska MA, Leir S-H, Harris A. Molecular mechanisms controlling CFTR gene expression in the airway. J Cell Mol Med. 2012;16:1321–1330. doi: 10.1111/j.1582-4934.2011.01439.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ott CJ, Blackledge NP, Leir S-H, Harris A. Novel regulatory mechanisms for the CFTR gene. Biochem Soc Trans. 2009;37:843–848. doi: 10.1042/BST0370843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhang Z, Leir S-H, Harris A. Immune mediators regulate CFTR expression through a bifunctional airway-selective enhancer. Mol Cell Biol. 2013;33:2843–2853. doi: 10.1128/MCB.00003-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cozens AL, Yezzi MJ, Kunzelmann K, Ohrui T, Chin L, Eng K, Finkbeiner WE, Widdicombe JH, Gruenert DC. CFTR expression and chloride secretion in polarized immortal human bronchial epithelial cells. Am J Respir Cell Mol Biol. 1994;10:38–47. doi: 10.1165/ajrcmb.10.1.7507342. [DOI] [PubMed] [Google Scholar]
- 24.Fogh J, Wright WC, Loveless JD. Absence of HeLa cell contamination in 169 cell lines derived from human tumors. J Natl Cancer Inst. 1977;58:209–214. doi: 10.1093/jnci/58.2.209. [DOI] [PubMed] [Google Scholar]
- 25.Smith AN, Barth ML, McDowell TL, Moulin DS, Nuthall HN, Hollingsworth MA, Harris A. A regulatory element in intron 1 of the cystic fibrosis transmembrane conductance regulator gene. J Biol Chem. 1996;271:9947–9954. doi: 10.1074/jbc.271.17.9947. [DOI] [PubMed] [Google Scholar]
- 26.Phylactides M, Rowntree R, Nuthall H, Ussery D, Wheeler A, Harris A. Evaluation of potential regulatory elements identified as DNase I hypersensitive sites in the CFTR gene. Eur J Biochem. 2002;269:553–559. doi: 10.1046/j.0014-2956.2001.02679.x. [DOI] [PubMed] [Google Scholar]
- 27.Mouchel N, Henstra SA, McCarthy VA, Williams SH, Phylactides M, Harris A. HNF1alpha is involved in tissue-specific regulation of CFTR gene expression. Biochem J. 2004;378:909–918. doi: 10.1042/BJ20031157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Suzuki H, Tashiro S, Sun J, Doi H, Satomi S, Igarashi K. Cadmium induces nuclear export of Bach1, a transcriptional repressor of heme oxygenase-1 gene. J Biol Chem. 2003;278:49246–49253. doi: 10.1074/jbc.M306764200. [DOI] [PubMed] [Google Scholar]
- 29.Mouchel N, Broackes-Carter F, Harris A. Alternative 5′ exons of the CFTR gene show developmental regulation. Hum Mol Genet. 2003;12:759–769. doi: 10.1093/hmg/ddg079. [DOI] [PubMed] [Google Scholar]
- 30.Bischof JM, Ott CJ, Leir S-H, Gosalia N, Song L, London D, Furey TS, Cotton CU, Crawford GE, Harris A. A genome-wide analysis of open chromatin in human tracheal epithelial cells reveals novel candidate regulatory elements for lung function. Thorax. 2012;67:385–391. doi: 10.1136/thoraxjnl-2011-200880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Pollard KS, Hubisz MJ, Rosenbloom KR, Siepel A. Detection of nonneutral substitution rates on mammalian phylogenies. Genome Res. 2010;20:110–121. doi: 10.1101/gr.097857.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lunter G, Ponting CP, Hein J. Genome-wide identification of human functional DNA using a neutral indel model. PLoS Comput Biol. 2006;2:e5. doi: 10.1371/journal.pcbi.0020005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lunter G, Ponting CP, Hein J. Genome-wide identification of human functional DNA using a neutral indel model. PLoS Comput Biol. 2006;2:e5. doi: 10.1371/journal.pcbi.0020005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Boyle AP, Song L, Lee B-K, London D, Keefe D, Birney E, Iyer VR, Crawford GE, Furey TS. High-resolution genome-wide in vivo footprinting of diverse transcription factors in human cells. Genome Res. 2011;21:456–464. doi: 10.1101/gr.112656.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wang J, Zhuang J, Iyer S, Lin X-Y, Greven MC, Kim B-H, Moore J, Pierce BG, Dong X, Virgil D, et al. Factorbook.org: a Wiki-based database for transcription factor-binding data generated by the ENCODE consortium. Nucleic Acids Res. 2013;41:D171–D176. doi: 10.1093/nar/gks1221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Han Z, Boyle DL, Aupperle KR, Bennett B, Manning AM, Firestein GS. Jun N-terminal kinase in rheumatoid arthritis. J Pharmacol Exp Ther. 1999;291:124–130. [PubMed] [Google Scholar]
- 37.Brouillard F, Bouthier M, Leclerc T, Clement A, Baudouin-Legros M, Edelman A. NF-κ B mediates up-regulation of CFTR gene expression in Calu-3 cells by interleukin-1β. J Biol Chem. 2001;276:9486–9491. doi: 10.1074/jbc.M006636200. [DOI] [PubMed] [Google Scholar]
- 38.Quandt K, Frech K, Karas H, Wingender E, Werner T. Matlnd and Matlnspector: new fast and versatile tools for detection of consensus matches in nucleotide sequence data. Nucleic Acids Research. 1995;23:4878–4884. doi: 10.1093/nar/23.23.4878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wang J, Zhuang J, Iyer S, Lin X, Whitfield TW, Greven MC, Pierce BG, Dong X, Kundaje A, Cheng Y, et al. Sequence features and chromatin structure around the genomic regions bound by 119 human transcription factors. Genome Res. 2012;22:1798–1812. doi: 10.1101/gr.139105.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sasaki S, Ito E, Toki T, Maekawa T, Kanezaki R, Umenai T, Muto A, Nagai H, Kinoshita T, Yamamoto M, et al. Cloning and expression of human B cell-specific transcription factor BACH2 mapped to chromosome 6q15. Oncogene. 2000;19:3739–3749. doi: 10.1038/sj.onc.1203716. [DOI] [PubMed] [Google Scholar]
- 41.Muto A, Hoshino H, Madisen L, Yanai N, Obinata M, Karasuyama H, Hayashi N, Nakauchi H, Yamamoto M, Groudine M, et al. Identification of Bach2 as a B-cell-specific partner for small maf proteins that negatively regulate the immunoglobulin heavy chain gene 3′ enhancer. EMBO J. 1998;17:5734–5743. doi: 10.1093/emboj/17.19.5734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Vo N, Goodman RH. CREB-binding protein and p300 in transcriptional regulation. J Biol Chem. 2001;276:13505–13508. doi: 10.1074/jbc.R000025200. [DOI] [PubMed] [Google Scholar]
- 43.Park HS, Kim SR, Lee YC. Impact of oxidative stress on lung diseases. Respirology. 2009;14:27–38. doi: 10.1111/j.1440-1843.2008.01447.x. [DOI] [PubMed] [Google Scholar]
- 44.Kreindler JL, Jackson AD, Kemp PA, Bridges RJ, Danahay H. Inhibition of chloride secretion in human bronchial epithelial cells by cigarette smoke extract. Am J Physiol Lung Cell Mol Physiol. 2005;288:L894–L902. doi: 10.1152/ajplung.00376.2004. [DOI] [PubMed] [Google Scholar]
- 45.Raju SV, Jackson PL, Courville CA, McNicholas CM, Sloane PA, Sabbatini G, Tidwell S, Tang LP, Liu B, Fortenberry JA, et al. Cigarette smoke induces systemic defects in cystic fibrosis transmembrane conductance regulator function. Am J Respir Crit Care Med. 2013;188:1321–1330. doi: 10.1164/rccm.201304-0733OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Braun AP.Cigarette smoke and calcium conspire to impair CFTR function in airway epithelia Channels (Austin)In press) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Moran AR, Norimatsu Y, Dawson DC, MacDonald KD. Aqueous cigarette smoke extract induces a voltage-dependent inhibition of CFTR expressed in Xenopus oocytes. Am J Physiol Lung Cell Mol Physiol. 2014;306:L284–L291. doi: 10.1152/ajplung.00163.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Linker RA, Lee DH, Ryan S, van Dam AM, Conrad R, Bista P, Zeng W, Hronowsky X, Buko A, Chollate S, et al. Fumaric acid esters exert neuroprotective effects in neuroinflammation via activation of the Nrf2 antioxidant pathway. Brain. 2011;134:678–692. doi: 10.1093/brain/awq386. [DOI] [PubMed] [Google Scholar]
- 49.Scannevin RH, Chollate S, Jung MY, Shackett M, Patel H, Bista P, Zeng W, Ryan S, Yamamoto M, Lukashev M, et al. Fumarates promote cytoprotection of central nervous system cells against oxidative stress via the nuclear factor (erythroid-derived 2)-like 2 pathway. J Pharmacol Exp Ther. 2012;341:274–284. doi: 10.1124/jpet.111.190132. [DOI] [PubMed] [Google Scholar]
- 50.Gold R, Kappos L, Arnold DL, Bar-Or A, Giovannoni G, Selmaj K, Tornatore C, Sweetser MT, Yang M, Sheikh SI, et al. DEFINE Study Investigators. Placebo-controlled phase 3 study of oral BG-12 for relapsing multiple sclerosis. N Engl J Med. 2012;367:1098–1107. doi: 10.1056/NEJMoa1114287. [DOI] [PubMed] [Google Scholar]
- 51.Wang J, Zhuang J, Iyer S, Lin X-Y, Greven MC, Kim B-H, Moore J, Pierce BG, Dong X, Virgil D, et al. Factorbook.org: a Wiki-based database for transcription factor-binding data generated by the ENCODE consortium. Nucleic Acids Research. 2013;41:D171–D176. doi: 10.1093/nar/gks1221. [DOI] [PMC free article] [PubMed] [Google Scholar]