

Abstract
Renin, as part of the renin-angiotensin system, plays a critical role in the regulation of blood pressure, electrolyte homeostasis, mammalian renal development and progression of fibrotic/hypertrophic diseases. Renin gene transcription is subject to complex developmental and tissue-specific regulation. Initial studies using the mouse As4.1 cell line, which has many characteristics of the renin-expressing juxtaglomerular cells of the kidney, have identified a proximal promoter region (−197 to −50 bp) and an enhancer (−2866 to −2625 bp) upstream of the Ren-1c gene, which are critical for renin gene expression. The proximal promoter region contains several transcription factor-binding sites including a binding site for the products of the developmental control genes Hox. The enhancer consists of at least 11 transcription factor-binding sites and is responsive to various signal transduction pathways including cAMP, retinoic acid, endothelin-1, and cytokines, all of which are known to alter renin mRNA levels. Furthermore, in vivo models have validated several of these key components found within the proximal promoter region and the enhancer as well as other key sites necessary for renin gene transcription.
The renin-angiotensin system (RAS) plays a major physiological role in the regulation of systemic blood pressure and fluid/electrolyte balance. The RAS has also been shown to be required for normal mammalian renal development and has been implicated in the progression of fibrotic/hypertrophic diseases [40,16,7,70,36,52]. The aspartyl protease renin is responsible for initiating the enzymatic cascade which results in the production of angiotensin (ANG) II, the effector molecule of the RAS. Transcription of renin genes is subject to developmental and tissue-specific regulation [58]. The primary source of active renin in the circulation is the kidney. In the mouse kidney renin expression is first detected at embryonic day 14.5 in the earliest developing arteries [8]. As the renal arterial tree develops renin expression is found in the newly forming arterial branches where it is then progressively restricted to smaller arteries and arterioles until, in the adult, it is primarily expressed by a specialized group of myoepithelioid granulated cells located in the afferent arteriole called juxtaglomerular cells (JG). Other sights of renin expression in the mouse include submandibular gland, adrenal gland, testes, ovary, anterior prostate, brain, and fetal subcutaneous tissue.
Initial transgenic studies where ~4 kb of the mouse renin 5′ flanking sequence was fused to either SV40 T antigen or GFP have shown that this limited upstream sequence is sufficient to specify correct renin expression patterns in mouse embryonic, extraembryonic, and adult tissues, suggesting that most of the key regulatory regions reside within this region [22,21]. The isolation of a renin-producing cell line (As4.1) derived by transgene-mediated tumorigenesis using the SV40 t antigen driven off the 5′ renin flanking sequence has provided an important tool for identifying cis-acting elements and trans-acting factors important for renin gene expression. In addition, primary cultures of chorionic cells [25] as well as Calu-6 cells [50], which have both been found to express endogenous renin, are widely used to study the transcriptional regulation of the human renin gene.
More recently, a large body of research has been carried out using in vivo models to try and validate previous in vitro findings in regards to renin gene transcription. Included within these in vivo models is the use of transgenic mice where critical regions within the renin regulatory region have been abrogated. Interestingly, some discrepancies between results found in initial cell culture studies and current mouse models have been identified [15,64,74,75]. This review will focus on the identification of key components responsible for renin gene transcription using both in vitro and in vivo models, including discussion on any conflicts in data between the two valuable resources.
The Proximal Promoter Region
Using the As4.1 cell line, a proximal promoter region residing at approximately −200 to +6 upstream of the renin gene has been identified[3]. This region has been shown to be absolutely critical for maximal cell-specific expression of renin in mouse and human [3,48]. The proximal promoter region shares a high level of sequence homology between mouse, rat, and human including a completely conserved TATA box [7]. The mouse Ren-1c gene contains an ~500 bp insert located at −80 which is not present in rat or human [5].
The HOX-PBX-binding site
In vitro experiments using the As4.1 cell line have identified a short sequence within the Ren-1c promoter region located at −72 to −50, which is both required for renin expression and is highly conserved among mouse, rat, and human [47]. This short sequence is also present in the human proximal promoter and is important for basal and cAMP-induced promoter activity of the human renin gene in human chorionic cells [14]. Further analysis using the As4.1 cell line has identified that HOX9/10 paralog members can pair with PBX1b and bind the Ren-1c sequence from −72 to −50 with high affinity. Prep1 has also been shown to form a ternary complex with HOX and PBX on the Ren-1c promoter in vitro. Moreover, a mutation of a single base within either the HOX or PBX half site dramatically reduces the transcriptional activity of Ren-1c in the As4.1 cells, suggesting that both of the HOX and PBX sites are necessary for renin expression. To test the importance of the HOX-PBX binding site in vivo the two critical bases found in the HOX-PBX binding sequence were mutated within a BAC transgenic where the renin regulatory region is responsible for driving GFP expression (RenGFP). When mutated, no GFP expression was visualized within embryonic adrenal gland or gonadal artery as well as within the kidney at any developmental age including e16.5, juvenile, and adult [15]. Interestingly, GFP expression was visualized in the SMG of the adult transgenic animal. This result concludes that the HOX-PBX site, while absolutely essential for renin expression within the kidney, is not apparently necessary for expression within the SMG. Furthermore, Tanimoto et. al. generated a transgenic mouse model where a wildtype or a mutated form of the Ren-1c promoter could be excised within the same transgenic construct using a Cre/loxP-mediated system [64]. In this elegant study a single base mutation within the HOX half site was able to completely attenuate basal promoter activity of the Ren-1c gene within the kidney, but did not affect extra-renal Ren-1c expression. When crossed into a hypotensive renin-null mutant mouse model the transgenic line carrying the mutated HOX half site was unable to rescue the phenotype where as a transgenic construct containing a mutated CNRE, LxRα binding site, was able to regain normal renin expression levels and blood pressure homeostasis within the renin-null mouse(discussed further in LxRα section). These results further validate the critical role of the HOX-PBX binding site in regulating renin expression in the kidney as well as facilitating blood pressure regulation.
The results reviewed here strongly suggest that renin gene expression within the kidney is an immediate downstream target of class I Hox gene regulation. Furthermore, the generation of Angiotensin II, a hormone known to have growth factor activities, as well as pressor activity, is directly regulated by an important family of developmental genes responsible for regulating embryonic patterning. This intriguing correlation underlies the emerging realization of the roles played by RAS during renal development as well as pathophysiology of the kidney.
The HOX-PBX binding site within the Ren-1c promoter is targeted by the retinoblastoma (RB) tumor suppressor gene leading to an increase in renin promoter activity in human embryonic kidney cells, which do not express endogenous renin [62]. Further studies to identify whether RB partners with the HOX-PBX complex to regulate renin expression need to be pursued.
The PPAR-γ binding site
Recently the human renin gene was shown to be regulated by the transcription factor peroxisome proliferating-activated receptor-γ (PPAR-γ) in human Calu-6 through a nontypical Pal3 (palindrome with a 3-bp spacer) binding site located from −148 to −134 [66]. The human renin Pal3 site binds PPARγ and the retinoid X receptor-α from Calu-6 nuclear extracts and is necessary for the PPARγ agonist rosiglitazone-mediated activation of human renin transcription [67]. Furthermore, the human renin Pal3 site is critical for PPAR-γ-dependent regulation of renin gene expression by mediating maximal transcription activation at low cellular levels of PPARγ. Similar to the other binding sites within the human promoter, the renin Pal3 side resides in an evolutionarily conserved region of the promoter, however the mouse and rat renin Pal3 elements have been shown to be transcriptionally silent in response to the PPARγ activation.
Further Binding Sites Identified in the Proximal Promoter Region
Within the proximal promoter region of the Ren-1c gene several other important transcriptional factor-binding sites have been identified upstream of the HOX-PBX binding domain [45]. Deletion of the region from −197 to −70 of the proximal promoter within a construct containing 4.1kb of the Ren-1c 5′ flanking sequence almost completely abrogates renin transcriptional activity in As4.1 cells [45]. Within this region, six cis-acting elements have been identified including two NFI-binding sites, an Sp1/Sp3-binding site, and three other sites Pb, Pc, and Pd, which all show affinity for nuclear proteins and contribute significantly to high-level renin expression in As4.1 cells [45].
Interesting, all of these binding sites, excluding Pb, are located within the M3 insertion region (−564 to −80), which is not present in the rat or human promoter. Therefore, the binding of these transcription factors may increase the transcriptional activity of the mouse renin promoter compared to that of rat and human, which is in agreement with higher circulating levels of renin found in mice when compared to levels in rats and humans[44].
Early studies using human chorionic cells have also identified a Ets-binding site, a cAMP-responsive element (CRE), a binding site for ARP-1 (COUP-TFII), as well as two unidentified sites within the proximal promoter region of the human renin gene [3]. The CRE located within the human promoter region has been shown to bind several transcription factors including CREB and ATF1 in renin-expressing Calu-6 cells [72]. The CRE site has also been shown to be important for basal and cAMP-induced promoter activity of the human renin gene in human chorionic cells [14].
The LxRa-Binding Site
The nuclear receptor LXRα has been postulated to mediate the cAMP response through a CNRE (cAMP and negative regulatory elements) motif residing at position −128 to −115 in the human renin gene and at −611 to 599 in the mouse Ren-1D gene [18,19,60,61]. In vivo analysis has demonstrated that LxRα and LxRβ can regulate renin expression through direct interaction with the renin promoter [37]. Furthermore, using LXRα and LXRβ null mice, a lower basal renin level as well as a blunted adrenergic response was observed identifying the cAMP/LXRα signaling pathway as being critical for adrenergic control of renin expression [37]. More recently, LXRα activation in mesenchymal stem cells (MSC) stimulates renin expression and induces MSCs to differentiate into renin-secreting, JG-like cells [35]. However, expression profiling of the natural renin-expressing cell of the adult kidney isolated from our RenGFP transgenic animals show very low levels of LXRα expression using Affymetrix microarray analysis, qPCR, and massively parallel signature sequencing (2004, Gross unpublished). Similarly, comparison of gene expression profiles of the renin expressing cell to other cells of the kidney did not identify LxRα as distinctive to the renin cell, conveying low to non-existent levels throughout kidney cell types with no significant enrichment in the renin-expressing cell population [4] (and correspondence with Dr. RA Gomez). Finally, in vivo deletion of the LXRα binding motif within the mouse promoter had no effect on either basal expression or the regulation of the renin gene, further contradicting the physiological significance of the CNRE [64]. Given the conflicting results, the relevance of the LxRα motif in regards to renin gene regulation remains somewhat equivocal.
The Enhancers
The Renal Enhancer
A 242-bp enhancer element (−2866 to −2625) has been identified in the Ren-1c 5′ flanking sequence in As4.1 cells [48]. The Ren-1c enhancer is capable of stimulating transcriptional activity by >50-fold in an orientation-independent fashion. The renal enhancer is also found in both the rat and human promoter. The human enhancer is located ~11kb upstream of the transcriptional start site and shows 71% identity with the mouse enhancer [55,71], whereas the rat enhancer is located at −5868 to −5615 bp with 85% sequence homology to the mouse enhancer. Through interaction with the renal enhancer transcription of the Ren-1c gene has been shown to be down regulated by endothelin [45,54], mechanical stretch [53], cyctolic calcium [23], as well as inflammatory cytokines [49,2,65]. Specifically, the cytokines oncostatin M, IL-6, and IL-1β inhibit renin gene expression through interaction with this enhancer [46].
The downstream portion of the enhancer contains a CRE and an adjacent E-box, which are the most critical sites for providing basal expression of the Ren-1c gene in As4.1 cells. Mutation in either of these sites results in almost complete loss of enhancer activity in As4.1 cells [41]. The CRE and E-box have previously been shown to bind the CREB/CREM and USF1/USF2 transcription factors, respectively. Furthermore CRE mediates the stimulatory effect of the cAMP/PKA cascade on renin expression. The transcriptional effects of cAMP are mediated through the binding of CREB to the CRE in the renin promoter. This is facilitated by the coactivators CBP and p300, can bind the phosphorylated form of CREB [1,32]. More recently ATF2 has been shown to bind to the CRE site repressing renin expression by drifting the transcriptional control of the renin gene away from CREB [8]. Furthermore, PPARγ can potentiate the stimulatory effect of cAMP on renin gene expression via trans-activation of adenylate cyclase-6 (AC6), which is targeted by PPARγ through a functional Pal3 sequence [11].
A region of the Ren-1cenhancer, which includes the CRE, E-box, and upstream TGACC element, has been identified as the target sequence for inhibitory effects of cytokines. However, mutation of each of the three sites does not lead to the abolishment of the inhibitory effect, suggesting that all three of the sites are important. Through interaction with the renal enhancer the inhibitory effect of IL-1β on renin gene expression has been shown to act through a mechanism involving the Erk-STAT3 pathway [31]. Down regulation of the Ren-1c gene is also mediated through tumor necrosis factor-α(TNF-α) [68]. Todorov et al have suggested that this inhibition is mediated by the CRE in the enhancer. The transcription factor NFκB, which is activated by TNF-α treatment, can form a complex with proteins binding the CRE. Furthermore, TNF-α inhibits renin gene expression by decreasing the transactivating capacity of NFκB-p65 and partially by attenuating CREB1 binding to the CRE of the Ren-1c enhancer [68]. Itani et al have demonstrated that renin gene expression can be negatively regulated by cellular reactive oxygen species through an NFκB-independent mechanism involving the renin enhancer and inhibition of transcriptional regulation by the cAMP response element [20]. This suggests that TNFα can inhibit renin gene expression though both a NFκB-dependent and an NFκB-independent mechanisms which involves the production of reactive oxygen species [20].
Li et al demonstrated that both renin mRNA and protein levels in the kidney are dramatically increased in Vitamin D receptor-null mice [29]. Treatment of As4.1 cells with vitamin D leads to a decrease in promoter activity of a transfected reporter construct containing 4.1kb of the Ren-1c 5′ flanking sequence[57]. The suppression of renin gene expression by 1,25(OH)2D3 has been shown to at least be partially due to blocking the formation of the CRE-CREB-CBP complex [73]. Furthermore, the transcriptional regulatory complex made up of CREB1, NCOR1, and VDR, which bind to the CRE-like domain in the renin enhancer, have been identified as being important to vitamin D receptor suppression of renin [39].
Downstream of the E-box within the Ren-1c enhancer are two TGACCT motifs, which are separated by 10bp and are homologous to the steroid receptor-binding site [44]. The transcription factors, retinoic acid receptors/retinoic X receptors have been shown to bind to these 2 sites [57]. Binding of these transcription factors in As4.1 cells not only stimulates basal enhancer activity but also the retinoic acid induction of Ren-1c expression. The orphan receptor EAR2 can bind to the TGACCT motifs, however it negatively regulates Ren-1c expression in As4.1 cells [30]. Furthermore, Rasd1 has been shown to interact with EAR2 inhibiting EAR2 transcriptional repression of renin [63]. Nr2f2 (Coup-TFII, Arp-1) can also negatively regulate renin promoter activity in response to physiological ques such as retinoic acid through interaction with this enhancer region [69]. The transcription factor PPAR-γ has also been reported to bind to this enhancer element [67]. In vivo experiments where PPARγ expression was selectively diminished in renin-expressing JG cells of the mouse kidney resulted in an increase in both renin expression and renin plasma levels, further validating the relevance of PPARγ in the control of renin gene regulation [10]. Calcium has also been shown to inhibit renin gene expression through interaction with the CRE and TGACCT motifs within the enhancer as well as through destabilization of renin mRNA [23].
An NF-Y-binding site has been identified close to the 3′ end of the renin enhancer, which overlaps with the downstream TGACCT motif [56]. Mutation of the NF-Y site increases enhancer activity, demonstrating that NF-Y negatively regulates renin gene transcription. Therefore binding of NF-Y to the enhancer may prevent binding of transcription factors to the TGACCT motif resulting in inhibition of enhancer activity [56].
At least seven additional transcription factor-binding sites have been identified residing in the more distal portion (−2866 to −2699) of the enhancer. One of these sites has been shown to bind the Wilm’s tumor suppressor WT1, which inhibits renin transcription [59]. The other six binding sites include 4 NFI-binding, Sp1/Sp3-binding, and an unknown transcription factor-binding site [42]. Mutational analysis has demonstrated that each of these six sites contributes to overall enhancer activity, whereas mutation of all six sites together results in a 90% decrease in Ren-1c expression. NFIX, the product of 1 of 4 homologous NFI genes, is the most highly expressed NFI mRNA in As4.1 cells, strongly suggesting a critical role of NFIX in regulating renin gene expression. Moreover, a direct interaction between NFIX and Sp1 has been reported [51]. It seems as though the cooperation between these 2 proteins is important for renin gene transcription because both the enhancer and proximal promoter of the Ren-1c gene contain adjacent NFI-binding and SP1/Sp3-binding sites [45,42].
Several different mouse models have been implemented to study the role of the renal enhancer on renin gene transcription in vivo. Using transgenic mice that express human renin from a 160kb P1 artificial chromosome (PAC) Zhou et al have demonstrated that the renal enhancer of the human renin gene is not critical for the stimulation of renin gene expression by angiotensin converting enzyme (ACE) inhibition [74]. Moreover, the enhCRE and CNRE have been shown to be dispensable for the cell-specific expression of the human renin gene in the afferent arteriole when AngII signaling is impaired [9]. This is in contrast to data generated using two different mouse models, one where a knockout of the endogenous renin locus (REKO mouse) was characterized [34] and another which implemented a Ren-1c containing Bacterial Artificial Chromosome (BAC) where the renin regulatory region was responsible for driving GFP and the enhancer was subsequently deleted in the BAC [15]. Using the REKO mouse model the renal enhancer was shown to be necessary for the full activation of renin transcription by the combination of a low-salt diet and an ACE inhibitor [34]. Similarly, a decreased response in GFP transgene expression was identified in the Ren-1c BAC transgenic animal containing the deleted enhancer when treated with the ACE inhibitor captopril [15]. The discrepancies between different mouse models may be a result of variation between mouse and human enhancer sequence or the ability of mouse transcription factors to carry out similar actions on the human renin promoter and enhancer [34]. Variation in the region deleted within the enhancer sequence of the different studies may also reflect the discrepancies observed between the various mouse models. Furthermore the human and mouse renal enhancers have been shown to be critical for basal transcription of renin within the kidney validating previous in vitro studies [74,34,15]. However, the exact mechanism in which the enhancer modulates levels of renin gene expression has not yet been fully elucidated. Using an in vitro model Morris has proposed that the enhancer increases the probability of achieving an active transcriptional state rather than regulating the level of promoter activity per cell [38]. Recent studies suggest that in vivo renin production is a function of the number of cells expressing renin rather than regulation of transcription within individual cells [33]. Although this evidence suggests an on/off mechanism of action further studies are necessary to fully understand how the enhancer regulates renin gene expression in normal and compromised physiological states. The extensive analysis of the renal enhancer has clearly identified its importance in maintaining baseline expression of renin as well as facilitating responses to various signal transduction pathways.
Chorionic Enhancer
The human renin 5′ flanking region contains a second enhancer located between −5777 and −5552 [13]. This enhancer is responsible for inducing human renin promoter activity ~60-fold in chorionic cells and is therefore known as the “chorionic” enhancer [13]. Using DNase I footprinting assays, three transcription factor binding sites have been identified within the chorionic enhancer, however the identity of these sites are as of yet unknown. The chorionic enhancer is not evolutionarily conserved and is trancriptionally silent in transgenic mice [75].
Interaction Between the Proximal Promoter Region and the Renal Enhancer
Using in vitro models a 242- base pair enhancer [48,41] consisting of a complex constellation of protein-DNA interaction sites, and a critical proximal promoter region[45] have been identified upstream of the renin gene. Although the exact physical location of the enhancer varies between species these key sites are highly conserved between mouse, rat, and human [44]. The human and mouse renal enhancers are critical for basal transcription of renin within the kidney [74,34,15], where the proximal promoter region is necessary for tissue specificity of renin gene expression [15,64]. Gomez et al have identified the histone acetyltransferases (HAT) CBP and p300 as being critical to maintenance of renin cell identity and structural integrity of the kidney [17]. The CBP and p300 HATs remodel chromatin into an active state within the enhancer region of the renin gene allowing transcription factors the ability to access key binding-sites necessary for activation of renin gene transcription. The exact mechanism of action responsible for bringing the enhancer and proximal promoter together to regulate renin gene expression is not fully understood. However, a highly conserved RBP-J recognition site, which utilizes the Notch signaling pathway and resides between the renal enhancer and proximal promoter element, has been proposed to be involved in mediating the crosstalk between the renal enhancer and proximal promoter region leading to renin gene transcription.
The Notch Signaling Pathway
Comparison of the human, rat, and mouse renin proximal promoter sequences led to the identification of a highly conserved sequence homologous to the recognition sequence for RBP-J/Su(H)/lAG1, a nuclear effector of the Notch signaling pathway. Notch, a transmembrane receptor, mediates cell-cell communication to determine cell fates and facilitates in the regulation of pattern formation [24]. When activated, through binding of its ligand, the intracellular domain of Notch (NIC) is released by proteolytic cleavage, translocates to the nucleus, and subsequently binds transcription factor RBP-J which then activates gene expression. Binding of NIC turns RBP-J from a repressor to an activator by replacing the RBP-J-bound corepressor complex with a co-activator complex.
As4.1 cell nuclear proteins that bind the putative rat renin RBP-J-binding site (−175 to −168) have been shown to contain RBP-J [43]. Furthermore, using Cos-7 cells, NIC activates transcription from a promoter containing multiple copies of the rat renin RBP-J-binding site [43]. An Ets-binding site has also been identified in the rat renin promoter, where the transcription factor Ets-1 can activate renin promoter activity at this site [43]. The intracellular domain of Notch, Ets1, and HOXD10-PBX1b-PREP1 has been shown to activate the rat renin promoter cooperatively in COS-7 cells [43]. These in vitro results strongly suggest that the renin gene is a downstream target of the Notch signaling pathway.
In vivo analysis of our RenGFP BAC transgenic line has identified Notch 3 co-localization with the GFP reporter in renal vasculature (figure 1). Co-localization of Notch 3 and Renin in the JG cell of the adult mouse kidney has also been identified by immunostaining as well as gene profiling of the renin expressing cell [4]. Furthermore, conditional deletion of the transcription factor RBP-J, the common downstream effector of all Notch receptors, in renin-expressing cells has been shown to diminish basal renin expression in the kidney, decrease circulating renin, lower blood pressure, as well as blunt the ability of smooth muscle cells along the kidney vasculature to regain the renin phenotype when homeostasis is threatened [6]. These results further emphasize the importance of this pathway on renin gene regulation in vivo.
Figure 1.
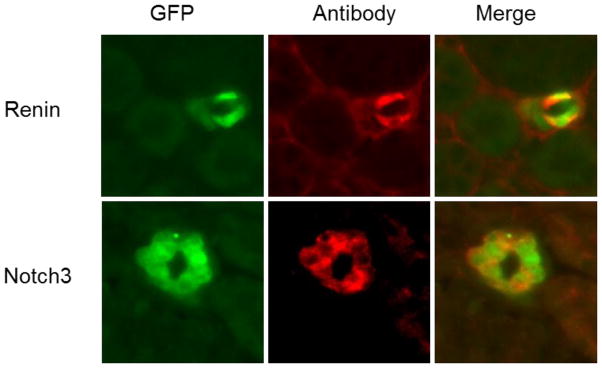
Colocalization of GFP with Renin and Notch3. GFP and the indicated antibody were separately imaged from immuno-stained frozen kidney sections of a 12 day old transgenic mouse as shown in the first and second columns respectively. The third column shows the merged images of the first two columns.
Pan et al have proposed that the RBP-J binding site is a transcriptional switch where activation of notch signaling sequesters the notch intra-cellular (NIC) domain into direct interaction with RBP-J turning the molecule from a repressor to an activator allowing for tissue-specific regulation of renin gene transcription though facilitating physical interaction between the enhancer and proximal promoter region [43]. The in vivo deletion of the RBP-J binding site within the renin regulatory region leads to a diminished level of renin expression suggesting that RBP-J functions to maintain the number of renin-expressing cells in the kidney [6]. Since RBP-J is known to have repressor and activator ability it stands to reason that deleting the site would cause a decrease in renin expression due to the loss of ability of N1C to form an activator complex in the renin expressing cell as proposed by Pan et al. Although much is known about these highly conserved sites, the exact mechanism at which the enhancer, proximal promoter region, and RBP-J site interact to activate renin gene transcription in a temporal and tissue – specific manner is not yet fully understood.
Perspectives
Many cis-acting elements responsible for the regulation of the renin genes have been identified using renin-expressing cell lines. The validation of many of these key sites have been characterized using various mouse model strategies. Expression profiling of the natural renin expressing cell of the mouse kidney has also led to validation and discovery of important factors responsible for the regulation of the renin genes in vivo. Although a detailed composite of the transcription factor binding sites has been developed and validated the map is far from being complete. Further studies using chromatin immunoprecipitation-sequencing, or ChIP-seq, could help to specifically validate the interaction of these key binding sites with previously identified and novel proteins.
Future studies are also necessary to further understand the tissue specific regulation of renin beyond the kidney. Specifically, renin transcripts within the submandibular gland (SMG) have been shown to initiate further upstream than those in the kidney and other tissues [12]. The presence of multiple transcriptional start sites within the SMG could allow for renin to be differentially regulated by different intracellular signals or transcription factors [58]. Furthermore, the HOX-PBX binding site, which is critical for renal renin expression, is not required for expression of renin within the SMG further supporting the model that alternate transcriptional start sites exist within the renin gene [15]. Further exploration of the alternate transcriptional start sites and their surrounding regulatory regions is necessary to fully understand the regulatory mechanisms acting on the control of renin expression within the SMG.
Moreover, the existence of local renin-angiotensin systems (RAS) in diverse tissues and organs has become increasingly recognized [27]. Included in these systems is the identification of a local RAS in the adult pancreas, which is involved in the regulation of normal exocrine and endocrine functions [26,28]. Recent data has identified a local RAS as being involved in regulating the functional maturation of the pancreatic progenitors toward the endocrine lineage [27]. Using a renin driven cre recombinase transgenic responsible for deleting p53 and Rb in renin-expressing cells of the developing mouse the Gross lab has created a highly metastatic islet cell carcinoma (Glenn and Gross in preparation for submission 2012) This highly penetrant tumor model arises from the endocrine lineage, specifically the glucagon expressing α cell, of the developing pancreas. Abrogation of the p53 and Rb alleles responsible for the initiation of disease are limited to renin-expressing cells, therefore expression of renin is present within the endocrine cells of the pancreas, which is of endodermal origin. Classically, renin-expressing cells have been found in tissues derived from the mesoderm. The specific time-point at which renin is expressed in the developing pancreas as well as the functional role of the gene during pancreatic development have yet to be elucidated. Further studies exploring the transcriptional regulation of renin as well as the role of local renin angiotensin systems within tissue and organ systems derived from the various germ layers are needed to fully comprehend the diverse functions of both renin and the RAS.
Acknowledgments
The authors’ research discussed in this review was supported by the NHLBI grant HL048459 (KWG) and the NCI grant CA121212 (KWG) and used core facilities supported in part by the NCI grant #P30CA016056.
References
- 1.Arany Z, Newsome D, Oldread E, Livingston DM, Eckner R. A family of transcriptional adaptor proteins targeted by the E1A oncoprotein. Nature. 1995;374 (6517):81–84. doi: 10.1038/374081a0. [DOI] [PubMed] [Google Scholar]
- 2.Baumann H, Wang Y, Richards CD, Jones CA, Black TA, Gross KW. Endotoxin-induced renal inflammatory response. Oncostatin M as a major mediator of suppressed renin expression. J Biol Chem. 2000;275 (29):22014–22019. doi: 10.1074/jbc.M002830200. [DOI] [PubMed] [Google Scholar]
- 3.Borensztein P, Germain S, Fuchs S, Philippe J, Corvol P, Pinet F. cis-regulatory elements and trans-acting factors directing basal and cAMP-stimulated human renin gene expression in chorionic cells. Circ Res. 1994;74 (5):764–773. doi: 10.1161/01.res.74.5.764. [DOI] [PubMed] [Google Scholar]
- 4.Brunskill EW, Sequeira-Lopez ML, Pentz ES, Lin E, Yu J, Aronow BJ, Potter SS, Gomez RA. Genes that confer the identity of the renin cell. J Am Soc Nephrol. 2011;22 (12):2213–2225. doi: 10.1681/ASN.2011040401. ASN.2011040401 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Burt DW, Nakamura N, Kelley P, Dzau VJ. Identification of negative and positive regulatory elements in the human renin gene. J Biol Chem. 1989;264 (13):7357–7362. [PubMed] [Google Scholar]
- 6.Castellanos Rivera RM, Monteagudo MC, Pentz ES, Glenn ST, Gross KW, Carretero O, Sequeira-Lopez ML, Gomez RA. Transcriptional regulator RBP-J regulates the number and plasticity of renin cells. Physiol Genomics. 2011;43 (17):1021–1028. doi: 10.1152/physiolgenomics.00061.2011. physiolgenomics.00061.2011 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Castrop H, Hocherl K, Kurtz A, Schweda F, Todorov V, Wagner C. Physiology of kidney renin. Physiol Rev. 2010;90 (2):607–673. doi: 10.1152/physrev.00011.2009. 90/2/607 [pii] [DOI] [PubMed] [Google Scholar]
- 8.Desch M, Hackmayer G, Todorov VT. Identification of ATF2 as a transcriptional regulator of renin gene. Biol Chem. 2011 doi: 10.1515/BC-2011-157. [DOI] [PubMed] [Google Scholar]
- 9.Desch M, Harlander S, Neubauer B, Gerl M, Germain S, Castrop H, Todorov VT. cAMP target sequences enhCRE and CNRE sense low-salt intake to increase human renin gene expression in vivo. Pflugers Arch. 2011;461 (5):567–577. doi: 10.1007/s00424-011-0956-z. [DOI] [PubMed] [Google Scholar]
- 10.Desch M, Schreiber A, Schweda F, Madsen K, Friis UG, Weatherford ET, Sigmund CD, Sequeira Lopez ML, Gomez RA, Todorov VT. Increased renin production in mice with deletion of peroxisome proliferator-activated receptor-gamma in juxtaglomerular cells. Hypertension. 2010;55 (3):660–666. doi: 10.1161/HYPERTENSIONAHA.109.138800. HYPERTENSIONAHA.109.138800 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Desch M, Schubert T, Schreiber A, Mayer S, Friedrich B, Artunc F, Todorov VT. PPARgamma-dependent regulation of adenylate cyclase 6 amplifies the stimulatory effect of cAMP on renin gene expression. Mol Endocrinol. 2010;24 (11):2139–2151. doi: 10.1210/me.2010-0134. me.2010-0134 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Field LJ, Philbrick WM, Howles PN, Dickinson DP, McGowan RA, Gross KW. Expression of tissue-specific Ren-1 and Ren-2 genes of mice: comparative analysis of 5′-proximal flanking regions. Mol Cell Biol. 1984;4 (11):2321–2331. doi: 10.1128/mcb.4.11.2321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Germain S, Bonnet F, Philippe J, Fuchs S, Corvol P, Pinet F. A novel distal enhancer confers chorionic expression on the human renin gene. J Biol Chem. 1998;273 (39):25292–25300. doi: 10.1074/jbc.273.39.25292. [DOI] [PubMed] [Google Scholar]
- 14.Germain S, Konoshita T, Philippe J, Corvol P, Pinet F. Transcriptional induction of the human renin gene by cyclic AMP requires cyclic AMP response element-binding protein (CREB) and a factor binding a pituitary-specific transacting factor (Pit-1) motif. Biochem J. 1996;316 ( Pt 1):107–113. doi: 10.1042/bj3160107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Glenn ST, Jones CA, Pan L, Gross KW. In vivo analysis of key elements within the renin regulatory region. Physiol Genomics. 2008;35 (3):243–253. doi: 10.1152/physiolgenomics.00017.2008. 00017.2008 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gomez RA. Role of angiotensin in renal vascular development. Kidney Int Suppl. 1998;67:S12–16. doi: 10.1046/j.1523-1755.1998.06703.x. [DOI] [PubMed] [Google Scholar]
- 17.Gomez RA, Pentz ES, Jin X, Cordaillat M, Sequeira Lopez ML. CBP and p300 are essential for renin cell identity and morphological integrity of the kidney. Am J Physiol Heart Circ Physiol. 2009;296 (5):H1255–1262. doi: 10.1152/ajpheart.01266.2008. 01266.2008 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Horiuchi M, Nakamura N, Tang SS, Barrett G, Dzau VJ. Molecular mechanism of tissue-specific regulation of mouse renin gene expression by cAMP. Identification of an inhibitory protein that binds nuclear transcriptional factor. J Biol Chem. 1991;266 (24):16247–16254. [PubMed] [Google Scholar]
- 19.Horiuchi M, Pratt RE, Nakamura N, Dzau VJ. Distinct nuclear proteins competing for an overlapping sequence of cyclic adenosine monophosphate and negative regulatory elements regulate tissue-specific mouse renin gene expression. J Clin Invest. 1993;92 (4):1805–1811. doi: 10.1172/JCI116770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Itani H, Liu X, Sarsour EH, Goswami PC, Born E, Keen HL, Sigmund CD. Regulation of renin gene expression by oxidative stress. Hypertension. 2009;53 (6):1070–1076. doi: 10.1161/HYPERTENSIONAHA.109.130633. HYPERTENSIONAHA.109.130633 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jones CA, Hurley MI, Black TA, Kane CM, Pan L, Pruitt SC, Gross KW. Expression of a renin/GFP transgene in mouse embryonic, extra-embryonic, and adult tissues. Physiol Genomics. 2000;4 (1):75–81. doi: 10.1152/physiolgenomics.2000.4.1.75. [DOI] [PubMed] [Google Scholar]
- 22.Jones CA, Sigmund CD, McGowan RA, Kane-Haas CM, Gross KW. Expression of murine renin genes during fetal development. Mol Endocrinol. 1990;4 (3):375–383. doi: 10.1210/mend-4-3-375. [DOI] [PubMed] [Google Scholar]
- 23.Klar J, Sigl M, Obermayer B, Schweda F, Kramer BK, Kurtz A. Calcium inhibits renin gene expression by transcriptional and posttranscriptional mechanisms. Hypertension. 2005;46 (6):1340–1346. doi: 10.1161/01.HYP.0000192025.86189.46. 01.HYP.0000192025.86189.46 [pii] [DOI] [PubMed] [Google Scholar]
- 24.Lai EC. Notch signaling: control of cell communication and cell fate. Development. 2004;131 (5):965–973. doi: 10.1242/dev.01074. [DOI] [PubMed] [Google Scholar]
- 25.Lang JA, Yang G, Kern JA, Sigmund CD. Endogenous human renin expression and promoter activity in CALU-6, a pulmonary carcinoma cell line. Hypertension. 1995;25 (4 Pt 2):704–710. doi: 10.1161/01.hyp.25.4.704. [DOI] [PubMed] [Google Scholar]
- 26.Lau T, Carlsson PO, Leung PS. Evidence for a local angiotensin-generating system and dose-dependent inhibition of glucose-stimulated insulin release by angiotensin II in isolated pancreatic islets. Diabetologia. 2004;47 (2):240–248. doi: 10.1007/s00125-003-1295-1. [DOI] [PubMed] [Google Scholar]
- 27.Leung KK, Liang J, Ma MT, Leung PS. Angiotensin II Type 2 Receptor Is Critical for the Development of Human Fetal Pancreatic Progenitor Cells into Islet-like Cell Clusters and Their Potential for Transplantation. Stem Cells. 2012;30 (3):525–536. doi: 10.1002/stem.1008. [DOI] [PubMed] [Google Scholar]
- 28.Leung PS. The physiology of a local renin-angiotensin system in the pancreas. J Physiol. 2007;580 (Pt 1):31–37. doi: 10.1113/jphysiol.2006.126193. jphysiol.2006.126193 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Li YC, Kong J, Wei M, Chen ZF, Liu SQ, Cao LP. 1,25-Dihydroxyvitamin D(3) is a negative endocrine regulator of the renin-angiotensin system. J Clin Invest. 2002;110 (2):229–238. doi: 10.1172/JCI15219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Liu X, Huang X, Sigmund CD. Identification of a nuclear orphan receptor (Ear2) as a negative regulator of renin gene transcription. Circ Res. 2003;92 (9):1033–1040. doi: 10.1161/01.RES.0000071355.82009.43. [DOI] [PubMed] [Google Scholar]
- 31.Liu X, Shi Q, Sigmund CD. Interleukin-1beta attenuates renin gene expression via a mitogen-activated protein kinase kinase-extracellular signal-regulated kinase and signal transducer and activator of transcription 3-dependent mechanism in As4.1 cells. Endocrinology. 2006;147 (12):6011–6018. doi: 10.1210/en.2006-0129. en.2006-0129 [pii] [DOI] [PubMed] [Google Scholar]
- 32.Lundblad JR, Kwok RP, Laurance ME, Harter ML, Goodman RH. Adenoviral E1A-associated protein p300 as a functional homologue of the transcriptional co-activator CBP. Nature. 1995;374 (6517):85–88. doi: 10.1038/374085a0. [DOI] [PubMed] [Google Scholar]
- 33.Machura K, Neubauer B, Steppan D, Kettl R, Gross A, Kurtz A. Role of blood pressure to mediate the influence of salt intake on renin expression in the kidney. Am J Physiol Renal Physiol. 2012 doi: 10.1152/ajprenal.00688.2011. ajprenal.00688.2011 [pii] [DOI] [PubMed] [Google Scholar]
- 34.Markus MA, Goy C, Adams DJ, Lovicu FJ, Morris BJ. Renin enhancer is crucial for full response in Renin expression to an in vivo stimulus. Hypertension. 2007;50 (5):933–938. doi: 10.1161/HYPERTENSIONAHA.107.096891. HYPERTENSIONAHA.107.096891 [pii] [DOI] [PubMed] [Google Scholar]
- 35.Matsushita K, Morello F, Wu Y, Zhang L, Iwanaga S, Pratt RE, Dzau VJ. Mesenchymal stem cells differentiate into renin-producing juxtaglomerular (JG)-like cells under the control of liver X receptor-alpha. J Biol Chem. 2010;285 (16):11974–11982. doi: 10.1074/jbc.M109.099671. M109.099671 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mehta PK, Griendling KK. Angiotensin II cell signaling: physiological and pathological effects in the cardiovascular system. Am J Physiol Cell Physiol. 2007;292 (1):C82–97. doi: 10.1152/ajpcell.00287.2006. 00287.2006 [pii] [DOI] [PubMed] [Google Scholar]
- 37.Morello F, de Boer RA, Steffensen KR, Gnecchi M, Chisholm JW, Boomsma F, Anderson LM, Lawn RM, Gustafsson JA, Lopez-Ilasaca M, Pratt RE, Dzau VJ. Liver X receptors alpha and beta regulate renin expression in vivo. J Clin Invest. 2005;115 (7):1913–1922. doi: 10.1172/JCI24594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Morris BJ. Fluorescence activated cell sorting of transiently transfected As4.1 cells shows renin enhancer directs on/off switching of renin promoter in vitro. Clin Exp Pharmacol Physiol. 2008;35 (4):367–371. doi: 10.1111/j.1440-1681.2008.04879.x. CEP4879 [pii] [DOI] [PubMed] [Google Scholar]
- 39.Nakane M, Ma J, Ruan X, Kroeger PE, Wu-Wong R. Mechanistic analysis of VDR-mediated renin suppression. Nephron Physiol. 2007;107 (2):p35–44. doi: 10.1159/000106792. 000106792 [pii] [DOI] [PubMed] [Google Scholar]
- 40.Nishimura H, Ichikawa I. What have we learned from gene targeting studies for the renin angiotensin system of the kidney? Intern Med. 1999;38 (4):315–323. doi: 10.2169/internalmedicine.38.315. [DOI] [PubMed] [Google Scholar]
- 41.Pan L, Black TA, Shi Q, Jones CA, Petrovic N, Loudon J, Kane C, Sigmund CD, Gross KW. Critical roles of a cyclic AMP responsive element and an E-box in regulation of mouse renin gene expression. J Biol Chem. 2001;276 (49):45530–45538. doi: 10.1074/jbc.M103010200. [DOI] [PubMed] [Google Scholar]
- 42.Pan L, Glenn ST, Jones CA, Gronostajski RM, Gross KW. Regulation of renin enhancer activity by nuclear factor I and Sp1/Sp3. Biochim Biophys Acta. 2003;1625 (3):280–290. doi: 10.1016/s0167-4781(03)00016-2. [DOI] [PubMed] [Google Scholar]
- 43.Pan L, Glenn ST, Jones CA, Gross KW. Activation of the rat renin promoter by HOXD10.PBX1b.PREP1, Ets-1, and the intracellular domain of notch. J Biol Chem. 2005;280 (21):20860–20866. doi: 10.1074/jbc.M414618200. M414618200 [pii] [DOI] [PubMed] [Google Scholar]
- 44.Pan L, Gross KW. Transcriptional regulation of renin: an update. Hypertension. 2005;45 (1):3–8. doi: 10.1161/01.HYP.0000149717.55920.45. 01.HYP.0000149717.55920.45 [pii] [DOI] [PubMed] [Google Scholar]
- 45.Pan L, Jones CA, Glenn ST, Gross KW. Identification of a novel region in the proximal promoter of the mouse renin gene critical for expression. Am J Physiol Renal Physiol. 2004;286 (6):F1107–1115. doi: 10.1152/ajprenal.00319.2003. [DOI] [PubMed] [Google Scholar]
- 46.Pan L, Wang Y, Jones CA, Glenn ST, Baumann H, Gross KW. Enhancer-dependent inhibition of mouse renin transcription by inflammatory cytokines. Am J Physiol Renal Physiol. 2005;288 (1):F117–124. doi: 10.1152/ajprenal.00333.2003. [DOI] [PubMed] [Google Scholar]
- 47.Pan L, Xie Y, Black TA, Jones CA, Pruitt SC, Gross KW. An Abd-B class HOX.PBX recognition sequence is required for expression from the mouse Ren-1c gene. J Biol Chem. 2001;276 (35):32489–32494. doi: 10.1074/jbc.M011541200. [DOI] [PubMed] [Google Scholar]
- 48.Petrovic N, Black TA, Fabian JR, Kane C, Jones CA, Loudon JA, Abonia JP, Sigmund CD, Gross KW. Role of proximal promoter elements in regulation of renin gene transcription. J Biol Chem. 1996;271 (37):22499–22505. doi: 10.1074/jbc.271.37.22499. [DOI] [PubMed] [Google Scholar]
- 49.Petrovic N, Kane CM, Sigmund CD, Gross KW. Downregulation of renin gene expression by interleukin-1. Hypertension. 1997;30 (2 Pt 1):230–235. doi: 10.1161/01.hyp.30.2.230. [DOI] [PubMed] [Google Scholar]
- 50.Pinet F, Corvol MT, Bourguignon J, Corvol P. Isolation and characterization of renin-producing human chorionic cells in culture. J Clin Endocrinol Metab. 1988;67 (6):1211–1220. doi: 10.1210/jcem-67-6-1211. [DOI] [PubMed] [Google Scholar]
- 51.Rafty LA, Santiago FS, Khachigian LM. NF1/X represses PDGF A-chain transcription by interacting with Sp1 and antagonizing Sp1 occupancy of the promoter. EMBO J. 2002;21 (3):334–343. doi: 10.1093/emboj/21.3.334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ruster C, Wolf G. Angiotensin II as a morphogenic cytokine stimulating renal fibrogenesis. J Am Soc Nephrol. 2011;22 (7):1189–1199. doi: 10.1681/ASN.2010040384. ASN.2010040384 [pii] [DOI] [PubMed] [Google Scholar]
- 53.Ryan MJ, Black TA, Gross KW, Hajduczok G. Cyclic mechanical distension regulates renin gene transcription in As4.1 cells. Am J Physiol Endocrinol Metab. 2000;279 (4):E830–837. doi: 10.1152/ajpendo.2000.279.4.E830. [DOI] [PubMed] [Google Scholar]
- 54.Ryan MJ, Black TA, Millard SL, Gross KW, Hajduczok G. Endothelin-1 increases calcium and attenuates renin gene expression in As4.1 cells. Am J Physiol Heart Circ Physiol. 2002;283 (6):H2458–2465. doi: 10.1152/ajpheart.00295.2002. [DOI] [PubMed] [Google Scholar]
- 55.Shi Q, Black TA, Gross KW, Sigmund CD. Species-specific differences in positive and negative regulatory elements in the renin gene enhancer. Circ Res. 1999;85 (6):479–488. doi: 10.1161/01.res.85.6.479. [DOI] [PubMed] [Google Scholar]
- 56.Shi Q, Gross KW, Sigmund CD. NF-Y antagonizes renin enhancer function by blocking stimulatory transcription factors. Hypertension. 2001;38 (3):332–336. doi: 10.1161/01.hyp.38.3.332. [DOI] [PubMed] [Google Scholar]
- 57.Shi Q, Gross KW, Sigmund CD. Retinoic acid-mediated activation of the mouse renin enhancer. J Biol Chem. 2001;276 (5):3597–3603. doi: 10.1074/jbc.M008361200. [DOI] [PubMed] [Google Scholar]
- 58.Sigmund CD, Gross KW. Structure, expression, and regulation of the murine renin genes. Hypertension. 1991;18 (4):446–457. doi: 10.1161/01.hyp.18.4.446. [DOI] [PubMed] [Google Scholar]
- 59.Steege A, Fahling M, Paliege A, Bondke A, Kirschner KM, Martinka P, Kaps C, Patzak A, Persson PB, Thiele BJ, Scholz H, Mrowka R. Wilms’ tumor protein (-KTS) modulates renin gene transcription. Kidney Int. 2008;74 (4):458–466. doi: 10.1038/ki.2008.194. ki2008194 [pii] [DOI] [PubMed] [Google Scholar]
- 60.Tamura K, Chen YE, Horiuchi M, Chen Q, Daviet L, Yang Z, Lopez-Ilasaca M, Mu H, Pratt RE, Dzau VJ. LXRalpha functions as a cAMP-responsive transcriptional regulator of gene expression. Proc Natl Acad Sci U S A. 2000;97 (15):8513–8518. doi: 10.1073/pnas.100519097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Tamura K, Chen YE, Tanaka Y, Sakai M, Tsurumi Y, Koide Y, Kihara M, Pratt RE, Horiuchi M, Umemura S, Dzau VJ. Nuclear receptor LXRalpha is involved in cAMP-mediated human renin gene expression. Mol Cell Endocrinol. 2004;224(1–2):11–20. doi: 10.1016/j.mce.2004.07.005. S0303-7207(04)00299-0 [pii] [DOI] [PubMed] [Google Scholar]
- 62.Tamura K, Umemura S, Nyui N, Yamaguchi S, Ishigami T, Hibi K, Yabana M, Kihara M, Fukamizu A, Murakami K, Ishii M. A novel proximal element mediates the regulation of mouse Ren-1C promoter by retinoblastoma protein in cultured cells. J Biol Chem. 1997;272 (27):16845–16851. doi: 10.1074/jbc.272.27.16845. [DOI] [PubMed] [Google Scholar]
- 63.Tan JJ, Ong SA, Chen KS. Rasd1 interacts with Ear2 (Nr2f6) to regulate renin transcription. BMC Mol Biol. 2011;12:4. doi: 10.1186/1471-2199-12-4. 1471-2199-12-4 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Tanimoto K, Sugiura A, Kanafusa S, Saito T, Masui N, Yanai K, Fukamizu A. A single nucleotide mutation in the mouse renin promoter disrupts blood pressure regulation. J Clin Invest. 2008;118 (3):1006–1016. doi: 10.1172/JCI33824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Todorov V, Muller M, Schweda F, Kurtz A. Tumor necrosis factor-alpha inhibits renin gene expression. Am J Physiol Regul Integr Comp Physiol. 2002;283 (5):R1046–1051. doi: 10.1152/ajpregu.00142.2002. [DOI] [PubMed] [Google Scholar]
- 66.Todorov VT, Desch M, Schmitt-Nilson N, Todorova A, Kurtz A. Peroxisome proliferator-activated receptor-gamma is involved in the control of renin gene expression. Hypertension. 2007;50 (5):939–944. doi: 10.1161/HYPERTENSIONAHA.107.092817. HYPERTENSIONAHA.107.092817 [pii] [DOI] [PubMed] [Google Scholar]
- 67.Todorov VT, Desch M, Schubert T, Kurtz A. The Pal3 promoter sequence is critical for the regulation of human renin gene transcription by peroxisome proliferator-activated receptor-gamma. Endocrinology. 2008;149 (9):4647–4657. doi: 10.1210/en.2008-0127. en.2008-0127 [pii] [DOI] [PubMed] [Google Scholar]
- 68.Todorov VT, Volkl S, Friedrich J, Kunz-Schughart LA, Hehlgans T, Vermeulen L, Haegeman G, Schmitz ML, Kurtz A. Role of CREB1 and NF{kappa}B-p65 in the down-regulation of renin gene expression by tumor necrosis factor {alpha} J Biol Chem. 2005;280 (26):24356–24362. doi: 10.1074/jbc.M502968200. M502968200 [pii] [DOI] [PubMed] [Google Scholar]
- 69.Weatherford ET, Liu X, Sigmund CD. Regulation of Renin Expression by the Orphan Nuclear Receptors Nr2f2 and Nr2f6. Am J Physiol Renal Physiol. 2012 doi: 10.1152/ajprenal.00362.2011. ajprenal.00362.2011 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Wolf G. Novel aspects of the renin-angiotensin-aldosterone-system. Front Biosci. 2008;13:4993–5005. 3058. doi: 10.2741/3058. [pii] [DOI] [PubMed] [Google Scholar]
- 71.Yan Y, Jones CA, Sigmund CD, Gross KW, Catanzaro DF. Conserved enhancer elements in human and mouse renin genes have different transcriptional effects in As4.1 cells. Circ Res. 1997;81 (4):558–566. doi: 10.1161/01.res.81.4.558. [DOI] [PubMed] [Google Scholar]
- 72.Ying L, Morris BJ, Sigmund CD. Transactivation of the human renin promoter by the cyclic AMP/protein kinase A pathway is mediated by both cAMP-responsive element binding protein-1 (CREB)-dependent and CREB-independent mechanisms in Calu-6 cells. J Biol Chem. 1997;272 (4):2412–2420. doi: 10.1074/jbc.272.4.2412. [DOI] [PubMed] [Google Scholar]
- 73.Yuan W, Pan W, Kong J, Zheng W, Szeto FL, Wong KE, Cohen R, Klopot A, Zhang Z, Li YC. 1,25-dihydroxyvitamin D3 suppresses renin gene transcription by blocking the activity of the cyclic AMP response element in the renin gene promoter. J Biol Chem. 2007;282 (41):29821–29830. doi: 10.1074/jbc.M705495200. M705495200 [pii] [DOI] [PubMed] [Google Scholar]
- 74.Zhou X, Davis DR, Sigmund CD. The human renin kidney enhancer is required to maintain base-line renin expression but is dispensable for tissue-specific, cell-specific, and regulated expression. J Biol Chem. 2006;281 (46):35296–35304. doi: 10.1074/jbc.M608055200. M608055200 [pii] [DOI] [PubMed] [Google Scholar]
- 75.Zhou X, Sigmund CD. Chorionic enhancer is dispensable for regulated expression of the human renin gene. Am J Physiol Regul Integr Comp Physiol. 2008;294 (2):R279–287. doi: 10.1152/ajpregu.00780.2007. 00780.2007 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]