

Abstract
Ceramide, the basic structural unit of sphingolipids, controls the balance between cell growth and death by inducing apoptosis. We have previously shown that accumulation of ceramide, triggered by hydrogen peroxide (H2O2) or by short-chain ceramide analogs, induces apoptosis of lung epithelial cells. Here we elucidate the link between caspase-3 activation, at the execution phase, and ceramide accumulation, at the commitment phase of apoptosis in A549 human lung adenocarcinoma cells. The induction of ceramide accumulation by various triggers of ceramide generation, such as H2O2, C6-ceramide, or UDP-glucose-ceramide glucosyltransferase inhibitor DL-threo-1-phenyl-2-decanoylamino-3-morpholino-1-propanol, triggered the activation of caspase-3. This ceramide elevation also induced the cleavage of the death substrate poly(ADP-ribose) polymerase and was followed by apoptotic cell death. Ceramide-mediated apoptosis was blocked by a general caspase inhibitor, Boc-D-fluoromethylketone, and by overexpression of the antiapoptotic protein Bcl-2. Notably, overexpression of Bcl-2 reduced the basal cellular levels of ceramide and prevented the induction of ceramide generation by C6-ceramide, which implies ceramide generation as a possible target for the antiapoptotic effects of Bcl-2.
Keywords: Bcl-2 overexpression, hydrogen peroxide, sphingolipid signaling
SPHINGOLIPIDS ARE SIGNALING molecules capable of modulating the balance between cell growth and apoptotic cell death (23, 30). Ceramide, the basic structural unit of sphingolipids, plays an important role in activating cell death signals that are initiated by stress stimuli such as chemotherapeutic agents, cytokines, and ionizing radiation (13). Although the accumulation of ceramide may occur due to enhanced de novo synthesis (5), in most cases one or more of cellular sphingomyelinases are activated, which hydrolyze sphingomyelin to yield ceramide and phosphorylcholine (13). Our laboratory recently demonstrated that exposure of lung epithelial cells to the reactive oxidant hydrogen peroxide (H2O2) directly activates the endogenous membrane neutral sphingomyelinase (nSMase) to induce ceramide accumulation and apoptosis (6, 10). The observation that membrane-permeable synthetic ceramides (10, 16) or natural ceramide generated by transfected bacterial sphingomyelinase (36) induces apoptosis supports the role of ceramide as a second messenger that signals programmed cell death (11).
The target molecules and terminal effectors of the ceramide-mediated apoptotic pathway have not been completely defined and in lung cells are unknown. Recent studies indicate that specific protease activation by ceramide may play a crucial role in the apoptotic process. These proteases, termed caspases for cysteinyl aspartate-specific protease (3), are members of a cysteine protease family. During apoptosis, the inactive precursors of the caspases are cleaved at Asp-X sites, thereby generating the active protease. Activation of effector caspases, such as caspase-3 (cysteine protease protein-32/Yama/Apopain), an event initiated by the numerous stimuli that induce apoptosis, leads to the proteolytic cleavage of key proteins such as poly(ADP-ribose) polymerase (PARP). Studies with ceramide analogs (31) or chemotherapeutic agents (15, 28) suggest that ceramide accumulation induces the activation of caspase-3. In contrast, Metkar et al. (20) showed that ceramide-induced apoptosis in Fas-resistant cells is caspase-3 independent, whereas Takeda et al. (32) provided evidence that caspase-3 participates in nSMase activation to induce ceramide production. It is also unclear how prosurvival Bcl-2 family members prevent caspase-3 activation and apoptosis (1) and whether the modulation of ceramide is involved. For example, several studies demonstrated that overexpression of Bcl-2 obstructs ceramide-mediated cell death without altering ceramide generation (8, 35), whereas others suggested that the antiapoptotic effect of Bcl-2 might occur via the modulation of ceramide production as well as by preventing ceramide-mediated caspase activation (29, 34). In addition, recent studies demonstrated that exogenous ceramide can downregulate Bcl-2 expression and phosphorylation (27). In A549 lung adenocarcinoma cells it is not known whether ceramide accumulation precedes caspase-3 activation or vice versa, or whether caspase-3 activation is necessary for apoptosis induction due to ceramide accumulation. Moreover, how Bcl-2 influences the ceramide pathway and caspase activation in the A549 cell line has not been examined.
Targeting the ceramide metabolic pathway is an attractive strategy to induce lung cancer cells to become apoptotic. However, a better understanding of the mechanism(s) leading to ceramide-mediated apoptosis is required. The present study sought to establish whether the stimulation of endogenous ceramide generation per se suffices to trigger caspase-3 activation and apoptosis induction in A549 lung cancer cells. It also examined the influence of Bcl-2 overexpression on ceramide accumulation, caspase-3 activation, and apoptosis induction.
MATERIALS AND METHODS
Materials
Cell culture growth media, transfection reagents, phosphate-buffered saline (PBS), and fetal bovine serum (FBS) were obtained from Life Technologies (Grand Island, NY). Zeocin and the expression vector pTracer-CMV2 were from Invitrogen (Carlsbad, CA). C2-ceramide, C6-ceramide, and cardiolipin were from Matreya (Pleasant Gap, PA). DL-threo-1-phenyl-2-decanoylamino-3-morpholino-1-propanol (PDMP) was from Biomol (Plymouth Meeting, PA). Recombinant sn-1,2-diacylglycerol kinase (Escherichia coli) and the general caspase inhibitor Boc-D-fluoromethylketone (FMK) were purchased from Calbiochem (La Jolla, CA). Bradford protein assay and Western analysis reagents and equipment were from Bio-Rad (Hercules, CA). SuperSignal chemiluminescence substrate was from Pierce (Rockford, IL). Anti-PARP antibody, Anti-Bcl-2 antibody, and annexin V/7-amino-actinomycin (7-AAD) staining reagents were from BD Pharmingen (San Diego, CA). Anticleaved caspase-3 antibody was from Cell Signaling Technology (Beverly, MA). The ApopNexin and Tunnel apoptosis detection kits were from Intergen (Purchase, NY). [γ-32P]ATP (25 mCi/ml) was purchased from ICN Biomedical (Costa Mesa, CA). The caspase-3 activity kit was from Roche. Methanol, chloroform, and all other solvents were obtained from Fisher Scientific (Houston, TX). All other chemical reagents were from Sigma Chemical (St. Louis, MO).
Methods
Cell culture
Human lung adenocarcinoma A549 cells (ATCC) were grown in F12K media supplemented with 10% FBS and penicillin-streptomycin. Cells were seeded at a density of 1–5 × 106 cells/cm2 and incubated at 37°C in a humidified atmosphere containing 5% CO2. Treatments were performed in F12K media supplemented with 1% FBS (for C6-ceramide and PDMP) or in PBS containing 4.5 mM glucose (“pulse” treatment for H2O2). After treatments, the cells were removed from the culture plates by incubation with 0.05% trypsin-EDTA and washed twice with ice-cold PBS, and the cell pellets were used for the different assays.
Determination of cellular ceramide levels by diacylglycerol kinase assay
Ceramide was quantified by the diacylglycerol kinase assay as previously described (6, 10). In brief, cells were extracted with methanol-chloroform-1 N HCl (100:100:1, vol/vol/vol). Where indicated, 10 nmol of C2-ceramide were added to the lipid extract as an internal standard. The lipids in the organic phase were dried under vacuum and were resuspended in 100 µl of the reaction mixture containing [γ-P32]ATP and incubated at room temperature (RT) for 1 h. The reactions were terminated by extraction of lipids with 1 ml of methanol-chloroform-1 N HCl, 170 µl of buffered saline solution, and 30 µl of 0.1 M EDTA. The lower organic phase was dried under vacuum, and the lipids were resolved by thin-layer chromatography (TLC) on silica gel 60 plates (Whatman) using a solvent of chloroform-methanol-acetic acid (65:15:5, vol/vol/vol). 32P-labeled ceramide 1-phosphate was detected and quantified using Storm 860 PhosphorImager scanner and ImageQuant software, respectively (Molecular Dynamics). We calculated absolute levels of endogenous ceramide (pmol ceramide/µg protein) by comparing the corresponding band intensities of the samples with ceramide standards. All treatments for ceramide analysis were done in duplicate; each experiment was repeated two or more times.
Quantification of apoptosis
Apoptosis was mainly evaluated by the ApopNexin Apoptosis Detection Kit (Intergen). Apoptotic cells were detected by annexin V binding to phosphatidyl serine, which is translocated to the outer leaflet of the plasma membrane of apoptotic cells. Approximately 1.5 × 104 cells were resuspended in 1× ApopNexin binding buffer (Intergen) and incubated with phycoerythrin (PE)-conjugated annexin V (ApopNexin PE) and the fluorescent DNA-binding dye 7-AAD for 15 min at 4°C in the dark. The cells were analyzed by flow cytometry using the PE signal detector (FL1) and the 7-AAD signal detector (FL2) in a FACScan flow cytometer equipped with a doublet discriminating module (Becton Dickinson). Each experiment was repeated two or more times, and a minimum of 5,000 cells were selected for each measurement. Cells negative for both annexin V and 7-AAD staining are live cells; annexin V-positive and 7-AAD-negative stained cells are early apoptotic cells; 7-AAD- and annexin V-positive stained cells are necrotic and/or late apoptotic cells; and 7-AAD-positive and annexin V-negative stained cells are necrotic cells.
Apoptosis was also determined by terminal deoxynucleotidyl transferase (TdT)-mediated dUTP nick-end labeling (TUNEL), using the ApopTag Plus Fluorescein In-Situ Apoptosis detection kit (Intergen). Cells were fixed in freshly diluted 1% paraformaldehyde in PBS for 10 min at RT. Fixed cells were laid on Superfrost/plus microscope slides (Fisher) and incubated at RT to allow evaporation of all liquid. Cells were washed with PBS and incubated with TdT enzyme and digoxigenin-conjugated dUTP, at 37°C, for 1 h in a humidity chamber. The reaction was stopped, and the cells were incubated with fluorescein-conjugated antidigoxigenin antibodies for 30 min in RT, in the dark. Then, the cells were counterstained by propidium iodide and viewed under a fluorescent microscope with a filter for FITC (excitation 490 nm and emission 550 nm). Apoptotic cells appear bright green. Experiments were repeated twice, and ~500 cells were screened for each measurement.
Generation of stably transfected A549 cell lines
Human Bcl-2 cDNA was transferred from pSSV-Bcl-2 plasmid [a generous gift from Dr. R. Kumar (Department of Molecular and Cellular Oncology, The University of Texas, M. D. Anderson Cancer Center, Houston, TX)] into the mammalian expression vector pTracer-CMV2 (Invitrogen) using the restriction enzyme EcoRI (pBcl-2/zeo). A549 cells grown on 35-mm plates were transfected with the pBcl-2/zeo or with an empty pTracer-CMV2 vector using LipofectAmine Plus reagent (Life Technologies) according to the instructions provided by the manufacturer. Twenty-four hours after transfection, cells were trypsinized, transferred to 100-mm plates, and selected for Zeocin resistance (500 µg/ml Zeocine in growth media). Resistant colonies were trypsinized, pooled, and maintained in 100 µg/ml of Zeocin. A549 cells stably transfected with cDNA fragment containing full-length human glucosylceramide synthase (GCS) were a generous gift from Dr. B. Ogretmen (Department of Biochemistry and Molecular Biology, Medical University of South Carolina, Charleston, SC).
Caspase-3 activity assay
Caspase-3 activity was measured using a caspase-3 activity assay kit (Roche). This assay is based on a fluorometric immunosorbent enzyme assay performed in a multititer plate, according to the manufacturer’s instructions. Briefly, a 96-well multititer plate was coated with human caspase-3 monoclonal antibodies and blocked to prevent nonspecific binding. Then, cellular lysates (~2 × 106 cells) were incubated in the coated wells for 1.5 h at 37°C. After a washing step, the caspase-3-specific substrate Ac-Asp-Glu-Val-Asp-7-amino-4-(trifluoromethyl)coumarin (DEVD-AFC) was added and cleaved by the active enzymes to generate free fluorescent AFC. Fluorescence was measured at λmax = 505 nm after 3 h of incubation with the substrate and normalized by the protein content of each sample. Each treatment was done in duplicate; two or more independent experiments were conducted.
Western blotting
Western blot analysis was done as described previously (25). In brief, cells were lysed in Triton X buffer [1% Triton X, 5 mM EDTA, 5 mM EGTA, and protease inhibitor cocktail (Sigma) in PBS], and samples containing equal amounts of protein were resolved by SDS-PAGE and transferred to nitrocellulose membranes (Bio-Rad). The membranes were probed with the appropriate primary antibodies, followed by horseradish peroxidase-conjugated secondary antibodies (Jackson) and enhanced chemiluminescence reaction (Pierce).
RESULTS
Ceramide Elevation Induces Apoptosis in A549 Cells
We previously reported (6, 10, 19) that cellular ceramide levels are rapidly elevated during oxidative stress-induced apoptosis via the activation of the membrane nSMase. To establish a common role for elevated ceramide levels in the apoptosis of lung epithelial cells, we tested whether exposure of cells to various means of ceramide generation affects apoptosis induction in airway epithelial A549 cancer cells. First, C6-ceramide, a short-chain ceramide analog that induces ceramide accumulation (10, 16), was examined (Fig. 1, A and B). Then, the effect of PDMP on ceramide levels was tested (Fig. 1C). PDMP potently prevents the synthesis of glucosylceramide from uridine diphosphate-glucose and ceramide by inhibiting UDP-glucose-ceramide glucosyltransferase, thereby elevating endogenous ceramide levels (24).
Fig. 1.

Endogenous ceramide levels are elevated during C6-ceramide or DL-threo-1-phenyl-2-decanoylamino-3-morpholino-1-propanol (PDMP) treatment. Determination of ceramide levels in lipid extracts of A549 cells treated with 50 µM C6-ceramide (A and B) or 50 µM PDMP (C) for the indicated time points. Lipids were assayed for ceramide by the diacylglycerol (DAG) kinase assay, as described in materials and methods. The reaction products were separated by thin-layer chromatograhy (TLC) followed by PhosphorImager scanner analysis as demonstrated in A and B. The values in A and C are presented as percent (%) of control untreated cells and represent means + SE.
Figure 1, A and B, demonstrates TLC separation of 32P-labeled ceramide extracted from cells treated with C6-ceramide. First we calculated the mean levels of basal ceramide in intact cells from multiple experiments (see materials and methods). Those levels were determined as ~42.2 ± 6.2 pmol/µg protein and referenced for further experiments as 100% of control. As shown in Fig. 1A, exposure of A549 cell cultures to 50 µM C6-ceramide formed a time-dependent increase in endogenous ceramide, inducing a 2.5-fold increase within 3 h. Figure 1B demonstrates that the TLC analysis distinguishes between the exogenously added C6-ceramide and the endogenously generated cellular ceramide, which enabled the quantification of endogenous ceramide without interference from the ceramide analog. In addition, Fig. 1C shows that, 1 h following treatment with 50 µM PDMP, the levels of ceramide in A549 cells increased by nearly twofold, and they remained elevated for hours. This persistent elevation in ceramide levels is recognized as being closely related to the apoptotic responses (12).
We then tested whether the addition of a cell-permeable ceramide analog or PDMP can cause apoptosis in A549 cells. Changes in the apoptotic population of A549 cells were measured by flow cytometry by the annexin V-labeling assay (Fig. 2). Cells that were annexin V positive and 7-AAD negative were considered as early apoptotic, whereas the annexin V-positive and 7-AAD-positive population contained both late apoptotic and necrotic cells, which were not distinguishable from each other and were therefore excluded. Figure 2 presents flow cytometric analyses performed on control, C6-ceramide-treated, and PDMP-treated A549 cells and demonstrates that either C6-ceramide exposure (Fig. 2, A and B) or PDMP treatment (Fig. 2B) increases the population of apoptotic cells. Exposure to 50 µM C6-ceramide increased the early apoptotic population about sevenfold during a 48-h period (Fig. 2A), whereas exposure to 50 µM PDMP elevated the apoptotic population approximately sevenfold during the first 24 h of treatment (Fig. 2B). In contrast, C6-dihydroceramide, a compound that resembles ceramide but lacks the trans-double bond, did not induce apoptosis in A549 cells (Fig. 2C), indicating that the induction of apoptosis by C6-ceramide is specific. Together, these data suggest that accumulation of cellular ceramide by either C6-ceramide or PDMP treatments could trigger apoptosis in A549 cells.
Fig. 2.

Ceramide induces apoptosis. A: cells were treated with 50 µM C6-ceramide for 24 or 48 h. B: cells were treated with 50 µM PDMP or 50 µM C6-ceramide for 24 h. C: cells were treated with 50 µM C6 or 50 µM C6-dihydroceramide for 24 h. To assess apoptosis, cells were stained with annexin V-phycoerythrin (PE) and 7-amino-actinomycin (7-AAD) and then evaluated by FACS analysis, as described in materials and methods. Values in A represent 3 independent experiments and are presented as percent (%) of total cells counted (5,000). Values in B and C are presented as percent (%) of control untreated cells. Scale bars represent the relative fluorescence intensity of the indicated dyes.
Ceramide Elevation Precedes Caspase-3 Activation in A549 Cells
The role of caspases in the execution phase of apoptosis is well established (17). Our previous observation that exposure of primary human tracheobronchial cells or transformed A549 airway epithelial cells to H2O2 induces a rapid accumulation of cellular ceramide and increases the apoptotic populations of these cells (6, 10, 19) led us to examine the effects of ceramide accumulation on caspase activation, focusing on caspase-3, the last caspase to be activated in the execution phase of apoptosis. Accordingly, for the present studies, ceramide accumulation was induced by exposure to H2O2 (Fig. 3), C6-ceramide (Fig. 4), or PDMP (Fig. 5).
Fig. 3.

Pulse exposure of A549 cells to H2O2 induces caspase-3 activation and apoptosis. A: A549 cells were exposed for 1 h to increasing concentrations (50–250 µM) of H2O2 followed by incubation for 24 h in media supplemented with 10% FBS. Apoptosis was determined by flow cytometry, as described in materials and methods. Top: illustration of annexin V/7-AAD staining of A549 cells, untreated (control) or treated with 150 µM H2O2. Values are presented as percent (%) of total cells counted (5,000). Scale bars represent the relative fluorescence intensity of the indicated dyes. Bottom: the values are presented as percent (%) of control, untreated cells and represent means + SE. B: A549 cells were exposed to 150 µM H2O2 for 1 h followed by incubation for the indicated time points with media containing 10% FBS. Caspase-3 activity was quantified by fluorometric immunosorbent enzyme assay (FIENA) as described in materials and methods. The values are presented as percent (%) of control untreated cells. C: A549 cells were induced as in B for the indicated time points. Then, cells were lysed and proteins were analyzed by SDS-PAGE and immunoblotted using antipoly(ADP-ribose) polymerase (anti-PARP, top) or anticleaved caspase-3 (bottom) antibodies. Arrowheads indicate the migration distance of PARP and caspase-3 cleaved forms.
Fig. 4.

Induction of ceramide accumulation by C6-ceramide enhances caspase-3 activation. A: A549 cells were treated with 50 µM C6-ceramide for the indicated time points. Caspase-3 activity (○) and apoptosis (●) were determined as described in materials and methods. B: cells were treated with C6-ceramide as in A. At the indicated time points, cells were lysed and proteins from each time point were analyzed by SDS-PAGE and immunoblotted using anti-PARP (top) or anticleaved caspase-3 (bottom) antibodies. C: cells were treated with 50 µM C6-ceramide (C6-Cer) or 50 µM C6-dihydroceramide (C6-dihydro-Cer) for 12 h followed by assessment of caspase-3 activity by FIENA as described in materials and methods. The values are presented as percent (%) of control untreated cells and represent means + SE. D: A549 cells stably transfected with empty vector or with vector containing glucosylceramide synthase (GCS) cDNA were treated with 50 µM C6-ceramide for 3 or 24 h to detect endogenous ceramide or cleaved caspase-3, respectively. Ceramide levels (top) were analyzed by the DAG kinase assay as described in materials and methods. Caspase-3 cleavage (bottom) was detected by immunoblotting as described in B.
Fig. 5.

Induction of ceramide accumulation by PDMP enhances caspase-3 activation. A549 cells were treated with 50 µM PDMP in 1% FBS for 24 h. A: cells were lysed and proteins were analyzed by SDS-PAGE and immunoblotted using anti-PARP (top) or anticleaved caspase-3 (bottom) antibodies. B: caspase-3 activity was detected by FIENA as described in materials and methods. The values are presented as percent (%) of control untreated cells and represent means + SE.
A549 cells were pulsed for 1 h with various doses of H2O2, and then the pulse solution was replaced, and the cells were incubated in growth media for the indicated time points. As demonstrated in Fig. 3A, these H2O2 pulse treatments of A549 cells increased the population of early apoptotic cells in a dose-dependent manner, reaching a maximum at 150 µM H2O2 after 24 h of incubation. In parallel, we assayed caspase-3 (-like) activity as demonstrated by its ability to cleave a specific substrate (DEVD-AFC) and generate a fluorogenic product (Fig. 3B). As shown, caspase-3 (-like) activity increased fourfold and reached a maximum 12 h following the H2O2 pulse treatment (Fig. 3B). To further assess the time course for caspase-3 activation, we also assayed for its cleavage, as indicated by the appearance of the 17-kDa activated form of caspase-3 (Fig. 3C, bottom, arrowhead). To further address caspase-3 activity, we assayed the cleavage of the 116-kDa PARP polypeptide, a substrate of caspase-3, which produces a specific 85-kDa apoptotic fragment (Fig. 3C, top, arrowhead). As shown, 12 h after treatment with H2O2, both caspase-3 and PARP cleavages were maximal. These results suggest that ceramide accumulation, which mainly occurs within the first 30 min following H2O2 exposure (6, 10, 19), precedes the caspase-3 and PARP cleavage signals of apoptosis by at least several hours. Thus the loss of cell membrane integrity, as determined by annexin V binding (Fig. 3A), does not likely contribute to ceramide accumulation, since it occurs several hours after the increase in endogenous ceramide levels.
Figure 4 shows that exposure to 50 µM C6-ceramide mimicked H2O2 in the generation of typical apoptotic changes, as demonstrated by caspase-3 activation and PARP cleavage. Again, the rise in the general apoptotic cell numbers clearly lagged behind the increase in caspase-3 (-like) activity. As shown in Fig. 4A, apoptotic cells started to accumulate only after 12 h of exposure to C6-ceramide, at time points when caspase-3 (-like) activity had already reached a fourfold increase. This was confirmed by Western analyses of caspase-3 and PARP cleavages (Fig. 4B), which demonstrated that both the active form of caspase-3 and the cleaved form of PARP have accumulated after 8 h of treatment with C6-ceramide. In contrast, treatment with 50 µM dihydroceramide resulted in no caspase-3 activation (Fig. 4C).
To further demonstrate that the accumulation of endogenous ceramide (rather than C6-ceramide) was the causative signal for apoptosis, we measured the effect of GCS, which converts ceramide to glucosylceramide, on ceramide accumulation and on caspase-3 cleavage. Overexpression of GCS was recently shown to suppress endogenous ceramide levels in A549 cells and to prevent the inhibitory effects of C6-ceramide on telomerase activity (22). A549 cells stably transfected with empty vector or vector containing full-length GCS cDNA (22) were exposed to 50 µM C6-ceramide and analyzed for ceramide levels and caspase-3 cleavage. Figure 4D shows that both endogenous ceramide and caspase-3 cleavage were attenuated in GCS-overexpressing cells. These data demonstrate that endogenous ceramide levels correlate well with the apoptotic response and suggest that ceramide accumulation is the causative signal for apoptosis.
Similar to C6-ceramide treatment, when cells were treated with the inhibitor of ceramide catabolism PDMP, caspase-3 activation and PARP cleavage were observed (Fig. 5). Again, the accumulation of endogenous ceramide, caused by PDMP (Fig. 1), preceded the onset of caspase-3 activation (Fig. 5) and the increase in PDMP-induced apoptosis (Fig. 2).
Ceramide-Mediated Apoptosis in A549 Cells Is Dependent on the Activation of Caspases and Is Inhibited by Bcl-2
To examine the role of caspases in oxidative stress and ceramide-induced apoptosis, we utilized the general caspase inhibitor Boc-D-FMK, which irreversibly blocks the catalytic site of a wide variety of caspases. As anticipated, pretreatment of A549 cells with 50 µM Boc-D-FMK followed by treatment with 150 µM H2O2 inhibited the induction of caspase-3 activation by H2O2 (Fig. 6A). Boc-D-FMK also reduced the number of early apoptotic cells detected 24 h after H2O2 exposure as determined by the fluorescent TUNEL assay (Fig. 6B). Furthermore, exposure of A549 cells to 50 µM C6-ceramide in the presence of 50 µM Boc-D-FMK abrogated the induction of apoptosis by C6-ceramide (Fig. 6C), demonstrating that caspases play a major role in the apoptotic pathway mediated not only by H2O2 but also by the C6-ceramide analog.
Fig. 6.
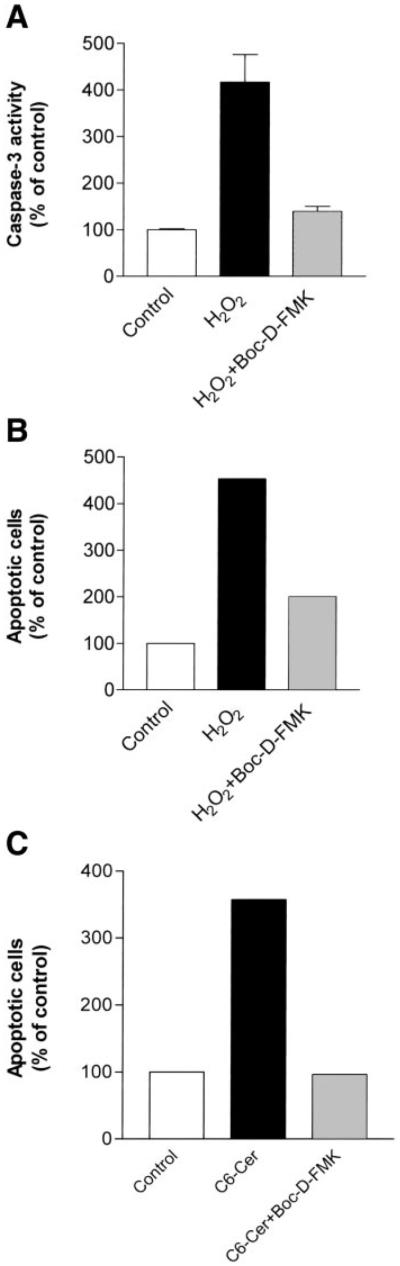
Ceramide-mediated apoptosis is dependent on caspase activation. A549 cells were preincubated in the absence (solid bars) or presence (gray bars) of 50 µM Boc-D-fluoromethylketone (FMK) for 2 h. Next, cells were treated with either 150 µM H2O2 (A, B) or 50 µM C6-ceramide (C). A: caspase-3 activity was determined by FIENA 12 h after the induction of treatments. The values are presented as percent (%) of control untreated cells and represent means + SE. B and C: apoptosis was detected and quantified by the fluorescent terminal deoxynucleotidyl transferase-mediated dUTP nick-end labeling assay 24 h after the induction of treatments, as described in materials and methods. The values resent the percent (%) of apoptotic cells, out of 300 cells counted.
As ceramide generation due to treatment with H2O2, C6-ceramide, or PDMP occurred in cells before caspase-3 activation and apoptosis induction, it became important to verify that the increases in ceramide levels were specific events preceding apoptosis. For that reason we used Bcl-2, an antiapoptotic molecule that has been shown to protect ALL-697 leukemia cells from ceramide-induced apoptosis (35). A549 cells overexpressing Bcl-2 or empty vector controls (Fig. 7A) were treated with H2O2 (Fig. 7, B–D) or with C6-ceramide (Fig. 7, B and C). Control cells showed an increase in caspase-3 (-like) activity and elevation in apoptosis in response to both treatments, but Bcl-2-overexpressing cells displayed resistance to either H2O2 or C6-ceramide-induced cell death.
Fig. 7.
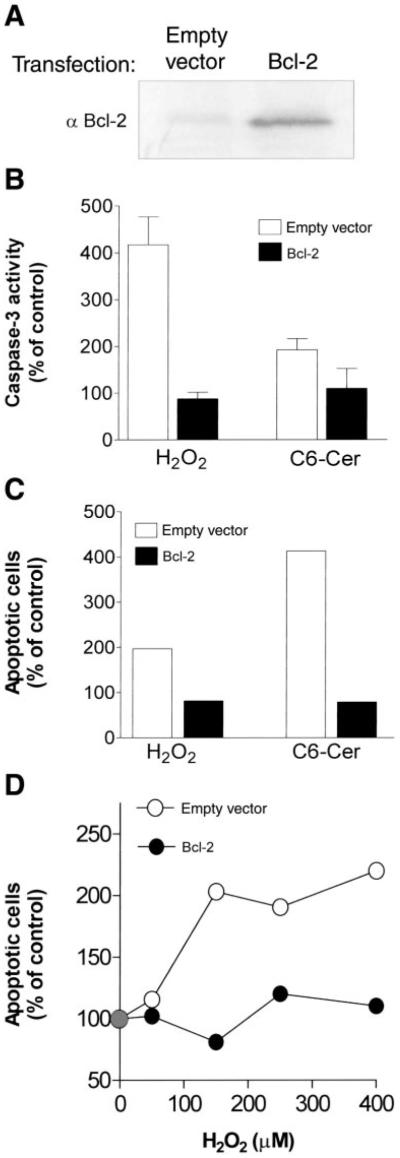
Overexpression of Bcl-2 inhibits both caspase-3 activation and apoptosis induction by H2O2 or C6-ceramide. A: A549 cells stably transfected either with an empty vector or with vector containing human Bcl-2 cDNA were lysed and analyzed by SDS-PAGE and Western immunoblotting using anti-Bcl-2 antibody. B and C: cells transfected with empty vector and Bcl-2 overexpressing A549 cells were treated with 150 µM H2O2 or 50 µM C6-ceramide. Caspase-3 activity (B) was determined by FIENA after 12 h, whereas apoptosis (C) was detected by flow cytometry after 24 h. D: cells transfected with empty vector (○) and Bcl-2 overexpressing (●) A549 cells were treated with 50–400 µM H2O2 and apoptosis was measured (by flow cytometry) after 24 h. Values are presented as the percent (%) of control untreated cells.
Because we have demonstrated that endogenous ceramide elevation drives the cell to death, we next examined whether Bcl-2 could interfere with ceramide generation. Endogenous ceramide levels were measured in response to C6-ceramide in control and in Bcl-2-overexpressing A549 cells. Ceramide levels increased significantly in control cells at 1 and 3 h (Fig. 8A). However, Bcl-2-overexpressing cells treated similarly with C6-ceramide showed only a slight increase in ceramide levels. Moreover, Bcl-2-overexpressing cells displayed lower levels of ceramide not only under apoptosis-inducing treatments, such as exposure to C6-ceramide, but also under normal growth conditions. As shown in Fig. 8B, basal ceramide levels in Bcl-2-overexpressing cells were ~2.5-fold lower than in cells transfected with empty vector, suggesting that the inhibition of ceramide production may be a possible mechanism underlying the antiapoptotic effects of Bcl-2 in A549 cells.
Fig. 8.

Overexpression of Bcl-2 inhibits endogenous ceramide generation induced by C6-ceramide. Quantitive determination of ceramide in lipid extracts of cells transfected with empty vector (open bars) and Bcl-2 overexpressing (solid bars) A549 cells. A: cells were treated with 50 µM C6-ceramide for the indicated time points. Lipids were analyzed for ceramide content by the DAG kinase assay, as described in materials and methods. The reaction products were separated by TLC followed by PhosphorImager scanner analysis. B: cells were lysed, and cellular lipids were extracted. Then, 10 nmol C2-ceramide (C2) were added to the lipid extraction mixture as an internal standard. Ceramide levels were quantified as in A. Top: representative PhosphorImager scanner illustration of ceramide levels in empty vector- and Bcl-2-transfected cells. Values in A and B are presented as percent (%) of control untreated cells and represent means ± SE.
DISCUSSION
The present study in the A549 cell line provides mechanistic insights into the role of ceramide accumulation in human lung adenocarcinoma and determines the time course and the order of the subsequent steps, which culminate in apoptosis induction (Fig. 9).
Fig. 9.

Model for the apoptotic events during ceramide-mediated apoptosis in transformed A549 lung epithelial cells. Caspase-3 is activated following the induction of ceramide accumulation by diverse inducing agents. This results in the cleavage of PARP and other possible target molecules, followed by apoptotic cell death. Bcl-2 functions to inhibit the activation of caspase-3 and may also prevent ceramide production.
The balance between pro- and antiapoptotic growth signaling is influenced by the ratio between ceramide and its derivatives, such as sphingosine 1-phosphate or glucosylceramide (23, 30). Thus targeting ceramide metabolic cell death may be an attractive clinical treatment strategy for lung cancer therapy (30). The role of ceramide in apoptosis, however, has generated considerable debate. Although there is growing evidence that ceramide is a cellular signal providing commitment to apoptosis (12), one report, among others, argues that “ceramide does not really fit anywhere on the map of signaling mechanisms that lead to apoptosis” (14). Moreover, whereas caspases are widely described as the executioners of the program for cell death, the literature also conflicts with respect to the placement of ceramide relative to caspases in the apoptotic cascade (8, 32). Nevertheless, as Radin recently noted (23), there is much evidence that exposure of cancer cells to ceramide, or to reagents that increase ceramide, induces apoptosis; this underscores the potential of the ceramide pathway as a specific target for cancer therapy.
Both exogenously supplied C6-ceramide or H2O2 and the inhibition of ceramide catabolism by PDMP treatment rapidly increased endogenous ceramide accumulation and induced apoptosis in A549 cells (Figs. 1–3). The enzymes that mediate ceramide accumulation can be divided into three groups: one that promotes ceramide accumulation via sphingomyelin degradation, a second that enhances de novo ceramide biosynthesis, and a third that prevents the catabolic removal of ceramide. We have previously shown that H2O2 stimulates ceramide generation via nSMase hydrolysis of sphingomyelin (6, 10). We now observe that C6-ceramide induces endogenous ceramide generation, likely occurring due to recycling of the C6-ceramide sphingosine backbone, which requires the catalytic activity of ceramide synthase, as was recently demonstrated by Ogretmen et al. (21). On the other hand, PDMP has been established as an inhibitor of ceramide glucosyltransferase and therefore inhibits ceramide metabolism (24). H2O2, C6-ceramide, PDMP, or GCS was utilized to target these three enzymatic groups, respectively, and each successfully modulated ceramide accumulation and apoptosis in our cell system. Therefore, different stimuli acting at diverse sites to stimulate ceramide accumulation were able to trigger apoptosis, which supports the hypothesis that an increase in ceramide levels, per se, is sufficient to initiate the apoptotic cascade in A549 cells.
How ceramide generation and accumulation affect or are affected by caspase(s) is unclear, and in lung cells, is entirely unknown. When caspase-3 was identified as the mammalian analog of the Caenorhabditis elegans CED-3 gene product, it was suggested that this protease could be a common effector for all apoptotic pathways (26). Recent evidence has demonstrated, however, that mice with a homozygous deletion of the caspase-3 gene still had normal development of all organs except for the brain (18), indicating that apoptotic pathways could be effective independently of caspase-3. Notably, it was also found that a potent inhibitor of caspase-3 activation was ineffective in preventing ceramide-induced apoptosis in U-937 cells (4). In addition, it has been shown that caspase-3-deficient MCF-7 cells were able to complete nuclear apoptosis in response to ceramide, and no PARP cleavage was observed (17). These studies support the notion that caspase-3 is not essential for apoptosis induction by the ceramide pathway and weaken the claim that PARP cleavage is a universal marker for apoptosis. Together, these reports suggest a new caspase-3-free apoptotic pathway triggered by ceramide.
However, in the present study, ceramide accumulation due to sphingomyelin hydrolysis, ceramide synthesis, or inhibition of UDP-glucose-ceramide glucosyltransferase induced caspase-3 activation and PARP cleavage (Figs. 3–5), and pretreatment of A549 cells with inhibitors of effector caspases prevented caspase-3 activation and apoptosis (Fig. 6). The observation that the cleaved forms of both caspase-3 and PARP were detectable after the observed ceramide increase (Fig. 4B) suggests that the ceramide elevation occurs earlier in the apoptotic cascade. These results agree with what has been shown with nonlung cell systems (7, 34) and indicate that ceramide accumulation was not merely a consequence of cell death, which suggests that ceramide accumulation per se may serve as an initial trigger for apoptosis, although not sufficient to induce apoptosis without activation of the downstream caspase(s) signal.
To further explore where endogenous ceramide generation is situated in the apoptotic-signaling cascade, we investigated the role of Bcl-2 overexpression on the ceramide-activated pathway. The antiapoptotic activity of Bcl-2 has been thought to be due to its ability to inhibit caspase-3 activity (26). The role of Bcl-2 overexpression in modulating the ceramide pathway, however, is controversial. For example, Tepper et al. (33) showed that Bcl-2 overexpression reduced CD95-induced ceramide accumulation in Jurkat cells stably transfected with the human Bcl-2 cDNA, whereas Jaffrezou et al. (16) demonstrated that Bcl-2 had no effect on ceramide generation induced by C6-ceramide in the myeloid cell line HL60/Bcl-2, genetically engineered to overexpress Bcl-2. Only a few reports demonstrate that Bcl-2 regulates ceramide formation (29, 34), whereas most studies indicate that Bcl-2 blocks apoptosis but does not affect ceramide generation (2, 8, 9, 35). Our results demonstrate that Bcl-2 protects against H2O2- and C6-ceramide-induced caspase-3 activation and cell death (Fig. 7). We further show that Bcl-2 inhibited ceramide generation in response to inducers of apoptosis and also reduced the basal cellular levels of ceramide production (Fig. 8), which suggests that the modulation of the ceramide pathway may be a target for the antiapoptotic effects of Bcl-2. Additional studies are required to precisely determine whether Bcl-2 prevents caspase activation directly or by inhibiting ceramide generation (see Fig. 9).
In summary, herein we provide evidence supporting the hypothesis that an increase in ceramide levels, per se, suffices to trigger apoptosis. Moreover, we define the time course for the major steps in ceramide-mediated apoptosis of transformed A549 lung epithelial cells and suggest an order for these apoptotic events (Fig. 9). We conclude that ceramide elevation is a major cause for apoptosis induction and not just an outcome of cell death and that it fits well upstream on the map of signaling mechanisms that lead to apoptosis in these airway epithelial cells.
Acknowledgments
We thank Elaine Chu for critical reading of the manuscript. This work was supported by Tobacco Related Disease Research
Program (TRDRP) Grant 10FT-0134 (T. Ravid), in part by TRDRP Grant 8RT-0098 (T. Goldkorn), and by National Heart, Lung, and Blood Institute Grant HL-66189 (T. Goldkorn).
Footnotes
The costs of publication of this article were defrayed in part by the payment of page charges. The article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
REFERENCES
- 1.Adams JM, Cory S. The Bcl-2 protein family: arbiters of cell survival. Science. 1998;281:1322–1326. doi: 10.1126/science.281.5381.1322. [DOI] [PubMed] [Google Scholar]
- 2.Allouche M, Bettaieb A, Vindis C, Rousse A, Grignon C, Laurent G. Influence of Bcl-2 overexpression on the ceramide pathway in daunorubicin-induced apoptosis of leukemic cells. Oncogene. 1997;14:1837–1845. doi: 10.1038/sj.onc.1201023. [DOI] [PubMed] [Google Scholar]
- 3.Alnemri ES, Livingston DJ, Nicholson DW, Salvesen G, Thornberry NA, Wong WW, Yuan J. Human ICE/CED-3 protease nomenclature. Cell. 1996;87:171. doi: 10.1016/s0092-8674(00)81334-3. [DOI] [PubMed] [Google Scholar]
- 4.Belaud-Rotureau MA, Lacombe F, Durrieu F, Vial JP, Lacoste L, Bernard P, Belloc F. Ceramide-induced apoptosis occurs independently of caspases and is decreased by leupeptin. Cell Death Differ. 1999;6:788–795. doi: 10.1038/sj.cdd.4400552. [DOI] [PubMed] [Google Scholar]
- 5.Bose R, Verheij M, Haimovitz-Friedman A, Scotto K, Fuks Z, Kolesnick R. Ceramide synthase mediates daunorubicin-induced apoptosis: an alternative mechanism for generating death signals. Cell. 1995;82:405–414. doi: 10.1016/0092-8674(95)90429-8. [DOI] [PubMed] [Google Scholar]
- 6.Chan C, Goldkorn T. Ceramide path in human lung cell death. Am J Respir Cell Mol Biol. 2000;22:460–468. doi: 10.1165/ajrcmb.22.4.3376. [DOI] [PubMed] [Google Scholar]
- 7.Chen L, Kim TJ, Pillai S. Inhibition of caspase activity prevents anti-IgM induced apoptosis but not ceramide generation in WEHI 231 B cells. Mol Immunol. 1998;35:195–205. doi: 10.1016/s0161-5890(98)00044-3. [DOI] [PubMed] [Google Scholar]
- 8.Dbaibo GS, Perry DK, Gamard CJ, Platt R, Poirier GG, Obeid LM, Hannun YA. Cytokine response modifier A (CrmA) inhibits ceramide formation in response to tumor necrosis factor (TNF)-alpha: CrmA and Bcl-2 target distinct components in the apoptotic pathway. J Exp Med. 1997;185:481–490. doi: 10.1084/jem.185.3.481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.El-Assaad W, El-Sabban M, Awaraji C, Abboushi N, Dbaibo GS. Distinct sites of action of Bcl-2 and Bcl-xL in the ceramide pathway of apoptosis. Biochem J. 1998;336:735–741. doi: 10.1042/bj3360735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Goldkorn T, Balaban N, Shannon M, Chea V, Matsukuma K, Gilchrist D, Wang H, Chan C. H2O2 acts on cellular membranes to generate ceramide signaling and initiate apoptosis in tracheobronchial epithelial cells. J Cell Sci. 1998;111:3209–3220. doi: 10.1242/jcs.111.21.3209. [DOI] [PubMed] [Google Scholar]
- 11.Goldkorn T, Dressler KA, Muindi J, Radin NS, Mendelsohn J, Menaldino D, Liotta D, Kolesnick RN. Ceramide stimulates epidermal growth factor receptor phosphorylation in A431 human epidermoid carcinoma cells. Evidence that ceramide may mediate sphingosine action. J Biol Chem. 1991;266:16092–16097. [PubMed] [Google Scholar]
- 12.Hannun YA. Functions of ceramide in coordinating cellular responses to stress. Science. 1996;274:1855–1859. doi: 10.1126/science.274.5294.1855. [DOI] [PubMed] [Google Scholar]
- 13.Hannun YA, Luberto C. Ceramide in the eukaryotic stress response. Trends Cell Biol. 2000;10:73–80. doi: 10.1016/s0962-8924(99)01694-3. [DOI] [PubMed] [Google Scholar]
- 14.Hofmann K, Dixit VM. Ceramide in apoptosis–does it really matter? Trends Biochem Sci. 1998;23:374–377. doi: 10.1016/s0968-0004(98)01289-4. [DOI] [PubMed] [Google Scholar]
- 15.Jaffrezou JP, Levade T, Bettaieb A, Andrieu N, Bezombes C, Maestre N, Vermeersch S, Rousse A, Laurent G. Daunorubicin-induced apoptosis: triggering of ceramide generation through sphingomyelin hydrolysis. EMBO J. 1996;15:2417–2424. [PMC free article] [PubMed] [Google Scholar]
- 16.Jaffrezou JP, Maestre N, de Mas-Mansat V, Bezombes C, Levade T, Laurent G. Positive feedback control of neutral sphingomyelinase activity by ceramide. FASEB J. 1998;12:999–1006. doi: 10.1096/fasebj.12.11.999. [DOI] [PubMed] [Google Scholar]
- 17.Janicke RU, Sprengart ML, Wati MR, Porter AG. Caspase-3 is required for DNA fragmentation and morphological changes associated with apoptosis. J Biol Chem. 1998;273:9357–9360. doi: 10.1074/jbc.273.16.9357. [DOI] [PubMed] [Google Scholar]
- 18.Kuida K, Zheng TS, Na S, Kuan C, Yang D, Karasuyama H, Rakic P, Flavell RA. Decreased apoptosis in the brain and premature lethality in CPP32-deficient mice. Nature. 1996;384:368–372. doi: 10.1038/384368a0. [DOI] [PubMed] [Google Scholar]
- 19.Lavrentiadou SN, Chan C, Kawcak TN, Ravid T, Tsaba A, van der Vliet A, Rasooly R, Goldkorn T. Ceramide-mediated apoptosis in lung epithelial cells is regulated by glutathione. Am J Respir Cell Mol Biol. 2001;25:676–684. doi: 10.1165/ajrcmb.25.6.4321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Metkar SS, Anand M, Manna PP, Naresh KN, Nadkarni JJ. Ceramide-induced apoptosis in fas-resistant Hodgkin’s disease cell lines is caspase independent. Exp Cell Res. 2000;255:18–29. doi: 10.1006/excr.1999.4773. [DOI] [PubMed] [Google Scholar]
- 21.Ogretmen B, Pettus BJ, Rossi MJ, Wood R, Usta J, Szulc Z, Bielawska A, Obeid LM, Hannun YA. Biochemical mechanisms of the generation of endogenous long chain ceramide in response to exogenous short chain ceramide in the A549 human lung adenocarcinoma cell line. Role for endogenous ceramide in mediating the action of exogenous ceramide. J Biol Chem. 2002;277:12960–12969. doi: 10.1074/jbc.M110699200. [DOI] [PubMed] [Google Scholar]
- 22.Ogretmen B, Schady D, Usta J, Wood R, Kraveka JM, Luberto C, Birbes H, Hannun YA, Obeid LM. Role of ceramide in mediating the inhibition of telomerase activity in A549 human lung adenocarcinoma cells. J Biol Chem. 2001;276:24901–24910. doi: 10.1074/jbc.M100314200. [DOI] [PubMed] [Google Scholar]
- 23.Radin NS. Killing cancer cells by poly-drug elevation of ceramide levels: a hypothesis whose time has come? Eur J Biochem. 2001;268:193–204. doi: 10.1046/j.1432-1033.2001.01845.x. [DOI] [PubMed] [Google Scholar]
- 24.Radin NS, Vunnam RR. Inhibitors of cerebroside metabolism. Methods Enzymol. 1981;72:673–684. doi: 10.1016/s0076-6879(81)72057-3. [DOI] [PubMed] [Google Scholar]
- 25.Ravid T, Avner R, Polak-Charcon S, Faust JR, Roitelman J. Impaired regulation of 3-hydroxy-3-methylglutaryl-coenzyme A reductase degradation in lovastatin-resistant cells. J Biol Chem. 1999;274:29341–29351. doi: 10.1074/jbc.274.41.29341. [DOI] [PubMed] [Google Scholar]
- 26.Reed JC. Bcl-2 and the regulation of programmed cell death. J Cell Biol. 1994;124:1–6. doi: 10.1083/jcb.124.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ruvolo PP, Deng X, Ito T, Carr BK, May WS. Ceramide induces Bcl2 dephosphorylation via a mechanism involving mitochondrial PP2A. J Biol Chem. 1999;274:20296–20300. doi: 10.1074/jbc.274.29.20296. [DOI] [PubMed] [Google Scholar]
- 28.Sawada M, Nakashima S, Banno Y, Yamakawa H, Hayashi K, Takenaka K, Nishimura Y, Sakai N, Nozawa Y. Ordering of ceramide formation, caspase activation, and Bax/ Bcl-2 expression during etoposide-induced apoptosis in C6 glioma cells. Cell Death Differ. 2000;7:761–772. doi: 10.1038/sj.cdd.4400711. [DOI] [PubMed] [Google Scholar]
- 29.Sawada M, Nakashima S, Banno Y, Yamakawa H, Takenaka K, Shinoda J, Nishimura Y, Sakai N, Nozawa Y. Influence of Bax or Bcl-2 overexpression on the ceramide-dependent apoptotic pathway in glioma cells. Oncogene. 2000;19:3508–3520. doi: 10.1038/sj.onc.1203699. [DOI] [PubMed] [Google Scholar]
- 30.Senchenkov A, Litvak DA, Cabot MC. Targeting ceramide metabolism–a strategy for overcoming drug resistance. J Natl Cancer Inst. 2001;93:347–357. doi: 10.1093/jnci/93.5.347. [DOI] [PubMed] [Google Scholar]
- 31.Smyth MJ, Perry DK, Zhang J, Poirier GG, Hannun YA, Obeid LM. prICE: a downstream target for ceramide-induced apoptosis and for the inhibitory action of Bcl-2. Biochem J. 1996;316:25–28. doi: 10.1042/bj3160025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Takeda Y, Tashima M, Takahashi A, Uchiyama T, Okazaki T. Ceramide generation in nitric oxide-induced apoptosis. Activation of magnesium-dependent neutral sphingomyelinase via caspase-3. J Biol Chem. 1999;274:10654–10660. doi: 10.1074/jbc.274.15.10654. [DOI] [PubMed] [Google Scholar]
- 33.Tepper AD, de Vries E, van Blitterswijk WJ, Borst J. Ordering of ceramide formation, caspase activation, and mitochondrial changes during CD95− and DNA damage-induced apoptosis. J Clin Invest. 1999;103:971–978. doi: 10.1172/JCI5457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yoshimura S, Banno Y, Nakashima S, Takenaka K, Sakai H, Nishimura Y, Sakai N, Shimizu S, Eguchi Y, Tsujimoto Y, Nozawa Y. Ceramide formation leads to caspase-3 activation during hypoxic PC12 cell death. Inhibitory effects of Bcl-2 on ceramide formation and caspase-3 activation. J Biol Chem. 1998;273:6921–6927. doi: 10.1074/jbc.273.12.6921. [DOI] [PubMed] [Google Scholar]
- 35.Zhang J, Alter N, Reed JC, Borner C, Obeid LM, Hannun YA. Bcl-2 interrupts the ceramide-mediated pathway of cell death. Proc Natl Acad Sci USA. 1996;93:5325–5328. doi: 10.1073/pnas.93.11.5325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zhang P, Liu B, Jenkins GM, Hannun YA, Obeid LM. Expression of neutral sphingomyelinase identifies a distinct pool of sphingomyelin involved in apoptosis. J Biol Chem. 1997;272:9609–9612. doi: 10.1074/jbc.272.15.9609. [DOI] [PubMed] [Google Scholar]