

Abstract
Reactive oxygen species (ROS) are mediators of lung injury, and glutathione (GSH) is the major nonprotein antioxidant that protects the cell from oxidative stress. We have recently shown that H2O2 induces ceramide-mediated apoptosis in human lung epithelial cells. We hypothesized that ROS-mediated depletion of GSH plays a regulatory role in ceramide generation, and thus in the induction of apoptosis. Our present studies demonstrate that GSH at physiologic concentrations (1 to 10 mM) inhibits ceramide production in a time- and dose-dependent manner in A549 human alveolar epithelial cells. On the other hand, buthionine-sulfoximine–mediated depletion of intracellular GSH induces elevation of ceramide levels and apoptosis. In addition, GSH blocks H2O2-mediated induction of intracellular ceramide generation and apoptosis. These effects were not mimicked by oxidized GSH (GSSG) or other thiol antioxidants, such as dithiothreitol and 2-mercaptoethanol. Moreover, increase of intracellular H2O2, mediated by inhibition of catalase by aminotriazole, also induces ceramide generation and apoptosis. These effects were blocked by N-acetylcysteine. Our results suggest that GSH depletion may be the link between oxidative stress and ceramide-mediated apoptosis in the lung.
Reactive oxygen species (ROS) are generated as a byproduct of normal aerobic cell metabolism. Because excessive accumulation of ROS (oxidative stress) is toxic, aerobic cells have evolved sensitive and effective antioxidant defenses that tightly regulate intracellular levels of ROS; nonprotein thiols, such as glutathione (GSH), are the major defense. The lung epithelium is constantly exposed to high concentrations of oxygen and oxidants, and is thus a primary target for ROS. Therefore, lung epithelial cells are fortified with high intracellular and extracellular levels of antioxidants (1, 2). However, massive amounts of ROS are generated in conditions such as inflammation or exposure to cigarette smoke, air pollutants, and drugs. Consequently, the oxidation–reduction (redox) state of the cell is altered, an oxidant-antioxidant imbalance results, and overall tissue integrity is threatened (1–4). Although a link between reactive oxidants, such as hydrogen peroxide (H2O2), and epithelial injury has been established, the mechanisms leading to epithelial cell dysfunction have just started becoming clear. To cope with oxidative stress the cell launches several responses, including apoptosis.
Apoptosis is an evolutionary conserved process of preprogrammed and tightly regulated cell death. When damaged cells undergo apoptosis, phagocytes are recruited at the site to remove apoptotic cells, thus reducing inflammatory response and tissue damage. The mechanism(s) by which oxidative stress induces apoptosis is still mainly unknown and the subject of intense research. Ceramide is a membrane sphingolipid that has recently emerged as a second messenger involved in the induction of apoptosis (5–9). It can be generated by the de novo biosynthetic pathway, which is catalyzed by ceramide synthase. Alternatively, ceramide can be generated as a result of sphingomyelin (SM) hydrolysis by sphingomyelinases (SMases) in the hydrolytic pathway, which is the major source for ceramide in cellular responses to extracellular signaling (6, 7, 9–11).
Several types of SMases that are distinguished by their optimum pH, cellular localization, and ion dependence have been identified; these include lysosomal and secreted acidic SMases and membrane-bound, Mg2+-dependent and cytosolic, Mg2+-independent neutral SMases (N-SMases) (9, 12). We have shown recently that H2O2 acts on the plasma membrane of tracheobronchial and airway epithelial cells to activate a Mg2+-dependent N-SMase, generate ceramide, and induce apoptosis (5, 6). In addition, cell permeant short-chain ceramide analogs, such as C6-ceramide, induce apoptosis in several cell systems, including lung epithelial cells (5, 6, 10, 13). These data substantiate the role of ceramide in the apoptotic signaling pathways.
In vivo and in vitro studies have shown that GSH, the most abundant nonprotein thiol in mammalian cells, plays a key role in defense against oxidant-induced apoptosis and injury (2, 4, 14–16). Extracellular supplementation of GSH or N-acetylcysteine (NAC), a known precursor of GSH, prevents oxidant-induced apoptosis (14, 16–19), whereas depletion of GSH has been suggested to be an early event that precedes the onset of apoptosis induced by various agents (14, 19). How depletion of GSH transmits apoptotic signals is unknown. One potential mechanism involves the release of the GSH inhibitory effect on N-SMase, followed by the increase in cellular ceramide levels and apoptosis (17, 19, 20).
The lung is continuously exposed to oxidants and is therefore armed with antioxidants. Both lung epithelial cells and the epithelial lining fluid (ELF) have relatively high concentrations of GSH. We have previously shown that oxidants such as H2O2 induce apoptosis in lung epithelial cells by modulating the ceramide pathway (5, 6). In the present study, we focus on the role of GSH, the main anti-oxidant in lung epithelium, in modulating ceramide-mediated apoptosis in lung epithelial cells. This study demonstrates the involvement of GSH in the modulation of ceramide generation and apoptosis in alveolar epithelial cells. We show that low GSH levels were required for ceramide production, whereas high GSH levels inhibit the generation of ceramide. The decreased levels of GSH and increased levels of ceramide correlate with the induction of apoptosis in these lung epithelial cells. Moreover, GSH and NAC, but not other thiol-containing antioxidants or oxidized GSH (GSSG), inhibit H2O2-mediated induction of ceramide and apoptosis. Taken together, these results suggest a novel role for ROS and GSH in regulating ceramide-mediated apoptosis.
Materials and Methods
Materials
Cell culture growth media, buffers, and fetal bovine serum (FBS) were obtained from Life Technologies, Inc. (Grand Island, NY). C6-ceramide and cardiolipin were from Matreya Inc. (Pleasant Gap, PA). Recombinant sn-1,2-diacylglycerol kinase (Escherichia coli) and monobrobimane (mBBr) were purchased from Calbiochem (La Jolla, CA). The ApopNexin Apoptosis Detection and Tunnel apoptosis kits were from Intergen Co. (Purchase, NY). [γ32P]adenosine triphosphate (ATP) (25 mCi/ml) was purchased from ICN Biomedical (Costa Mesa, CA). Bis-benzimide (Hoechst 33258), L-D-buthionine sulfoximine (BSO), aminotriazole (ATZ), GSH, GSSG, NAC, dithiothreitol (DTT), 2-mercaptoethanol, and all chemical reagents were from Sigma Chemical Co. (St. Louis, MO). Microscope slides, methanol, chloroform, and all other solvents were obtained from Fisher Scientific (Houston, TX).
Cell Culture
Human alveolar epithelial (A549) cells (American Type Culture Collection, Rockville, MD) were grown in F12K medium supplemented with 10% FBS and penicillin-streptomycin. Primary airway epithelial cells were grown as previously described (6). Briefly, tracheas were isolated from primate lungs provided by the Primate Center at the University of California, Davis (Davis, CA). Tissues were immersed in Eagle’s minimum essential medium with 0.1% protease for 24 h at 4°C. Dissociated cells were grown in F12 medium supplemented with transferrin (5 μg/ml), hydrocortisone (0.1 μM), choleratoxin (0.2 μg/ml), insulin (5 μg/ml), epidermal growth factor (10 μg/ml), and bovine hypothalamus extract. Primary cells were further passaged once or twice. Cells were seeded at a density of 1 to 5 × 106 cells/cm2 (A549) or 1 to 5 × 103 cells/cm2 (primaries) and incubated at 37°C in a humidified atmosphere of 5% CO2 air. Treatments were performed in F12 medium supplemented with 1% FBS or growth factor-deprived F12 medium. After treatments, the cells were removed from the culture plates by incubation with 0.05% trypsin–ethylenediaminetetraacetic acid (EDTA), washed twice with ice-cold phosphate-buffered saline (PBS) and the cell pellets were used for the different assays.
Determination of Cellular Ceramide Levels by Diacylglycerol Kinase Assay
Ceramide was quantified by the diacylglycerol (DAG) kinase assay as previously described (5, 6). Briefly, cells were extracted with methanol:chloroform:1 N HCl (100:100:1, vol/vol/vol). The lipids in the organic phase were dried under vacuum and were resuspended in 100 μl of the reaction mixture containing [γ-P32]ATP and incubated at room temperature for 1 h. The reactions were terminated by extraction of lipids with 1 ml methanol:chloroform:1 N HCl, 170 μl of buffered saline solution, and 30 μl of 0.1 M EDTA. The lower organic phase was dried under vacuum, and the lipids were resolved by thin layer chromatography (TLC) on silica gel 60 plates (Whatman) using a solvent of chloroform: methanol:acetic acid (65:15:5, vol/vol/vol). Ceramide 1-phosphate was detected by autoradiography, and incorporated 32P was quantified by densitometry scanning using a Molecular Dynamics Gel Scanner (Sunnyvale, CA).
Determination of Cellular GSH Levels
Low molecular mass thiols were determined by high performance liquid chromatography as described (21). Cell lysates were incubated with an equal volume of 2 mM mBBr, 20 mM N-ethylmorpholine (pH 8.0) and incubated for 5 min at room temperature, in the dark. The proteins were then precipitated by the addition of trichloroacetic acid to a final concentration of 5% and centrifuged at 3,000 × g for 3 min. The supernatants were injected on a 5-μm Spherisorb RP-18 column and eluted with 8% acetonitrile in 0.25% acetic acid, at a flow rate of 1 ml/min. GSH was detected using fluorescence detection (excitation, 394 nm; emission, 480 nm), and quantified using external standards.
Detection of Apoptosis by TUNEL Analysis
Apoptosis was also determined by terminal deoxynucleotidyl transferase (TdT)-mediated deoxyuridine triphosphate (dUTP)–biotin nick end-labeling (TUNEL), using the ApopTag Peroxidase kit (Intergen). The cells were fixed in 10% formalin for 30 min. The fixed cells were laid on superfrost/plus microscope slides (Fisher) and incubated at room temperature to allow evaporation of all liquid. The cells were then washed, treated with 3% H2O2 to quench any endogenous peroxidase activity, and equilibrated before incubation with the TdT enzyme in the presence of digoxigenin-conjugated dUTP at 37°C for 1 h. The antidigoxigenin peroxidase conjugate was applied for 30 min and the peroxidase substrate was applied and allowed to stain for 15 min before the cells were washed and counterstained with 0.5% (wt/vol) methyl green. The slides were washed in 100% n-butanol, and the samples were dehydrated in xylene before mounting.
Detection of Apoptosis by DNA Staining with Hoechst Dye
Apoptosis was also determined by in situ DNA fluorescence using the DNA fluorochrome bis-benzimide (Hoechst 33258) to bind to A-T regions of DNA. Cells were fixed twice in Carnoy’s fixative (3 parts methanol to 1 part glacial acetic acid), 5 min each time, and then allowed to air dry. The cells were stained with bis-benzimide (0.5 μg/ml) for 30 min, washed twice with distilled water, and mounted on a microscope slide. The slides were evaluated for apoptotic cells under a fluorescent microscope.
Quantitation of Apoptosis by Annexin V Flow Cytometry
Apoptosis was evaluated by the ApopNexin Apoptosis Detection Kit (Intergen). Apoptotic cells were detected by virtue of early changes in the plasma membrane (PM) phospholipid asymmetry. Annexin V binds to phosphatidyl serine, which in apoptotic cells is translocated to the outer leaflet of the PM. Approximately 15 × 104 cells were resuspended in 1× ApopNexin Binding Buffer (Intergen) and incubated with fluorescein isothiocyanate (FITC)–conjugated Annexin V (ApopNexin FITC) and the fluorescent DNA-binding dye propidium iodide (PI) for 15 min at 4°C in the dark. The cells were analyzed by flow cytometry using the FITC signal detector (FL1) and the PI signal detector (FL2) in a FAC-Scan flow cytometer equipped with a doublet discriminating module (Becton Dickinson & Co., Franklin Lakes, NJ). Cells negative for both Annexin V and PI staining are live cells; Annexin V–positive and PI-negative staining cells are undergoing early stages of apoptosis; PI- and Annexin V–positive staining cells are necrotic and/or late apoptotic cells; and PI-positive and Annexin V–negative staining cells are necrotic cells.
Results
Extracellular GSH Decreases Ceramide Levels in A549 Cells
To determine the role of GSH in the ceramide pathway in lung epithelial cells, we investigated the effect of GSH on cellular ceramide levels. A549 alveolar epithelial cells were incubated with different concentrations of GSH for different times, in the presence of 1% FBS. As shown in Figure 1, GSH inhibited ceramide generation in a dose- (Figure 1A) and time-dependent (Figure 1B) manner. Specifically, incubation of A549 cells with GSH at concentrations as low as 1 mM for 3 h dramatically decreased intracellular ceramide. Similarly, 5 mM GSH was sufficient to diminish ceramide levels within the first 2 h of GSH treatment. This GSH-mediated decrease in cellular ceramide levels indicates that GSH may play a regulatory role in lung epithelial cell ceramide homeostasis and suggests that GSH may mediate its antiapoptotic effects via inhibition of ceramide production.
Figure 1.

Extracellularly supplemented GSH inhibits ceramide generation in A549 cells. Autoradiography of ceramide in lipid extracts of A549 cells treated in the presence of regular medium supplemented with 1% serum with (A) increasing concentrations of GSH (0 to 5 mM) for 3 h or (B) 5 mM GSH for different incubation times. Incubations were terminated by washes with ice-cold PBS, and the cells were harvested with 0.05% trypsin-EDTA. Cellular lipids were extracted and assayed for ceramide by the DAG kinase assay, as described previously (5, 6). The reaction products were analyzed by TLC and autoradiography.
Exposure of A549 Cells to H2O2 Decreases Intracellular GSH Levels, Which Is Accompanied by Elevated Ceramide Levels and Induction of Apoptosis
We have previously shown that exposure of human tracheobronchial and airway epithelial cells to H2O2 induces an increase in cellular ceramide levels and apoptosis (5, 6). Because H2O2 and GSH appear to have opposite effects on ceramide production, we hypothesized that H2O2 may mediate its effects on ceramide generation, and therefore apoptosis, by depleting cellular GSH. To test this hypothesis, A549 cells were exposed to 250 μM H2O2. As demonstrated in Figure 2A, cellular GSH levels dropped to 70% within the first 15 min of exposure to H2O2 and reached maximal decrease (up to 50% of baseline levels) during the first hour. Then, after 4 h of incubation, GSH levels were replenished.
Figure 2.

H2O2 mediates induction of intracellular ceramide levels and apoptosis via depletion of intracellular GSH. A549 cells were incubated in medium supplemented with 1% serum with 250 μM H2O2 for the indicated times. (A). Cell lysates were analyzed for GSH as described in Materials and Methods. (B). Cellular lipids were extracted and assayed for ceramide by the DAG kinase assay (5, 6). The reaction products were analyzed by TLC and quantified using a phosphorimager. (C). To determine apoptosis, cells were stained with Annexin V-FITC and PI, and were evaluated by FACS analysis, as described in Materials and Methods. The values are represented as percent (%) of control, not treated, cells and represent mean ± SEM.
The decrease in cellular GSH was accompanied by elevation in ceramide levels. As shown in Figure 2B, exposure of cells to 250 μM H2O2 increased cellular ceramide in a time-dependent manner. This ceramide elevation apparently conditioned the cells to commit to apoptosis because after exposure of cells to H2O2 for only 1 h, followed by incubation with regular growth medium, the cells proceeded into apoptosis in a dose- and time-dependent manner (Figure 2C).
Extracellular GSH Inhibits H2O2-Induced GSH Depletion, Ceramide Generation, and Apoptosis in A549 Cells
Because GSH inhibits ceramide generation, it became important to determine the effect of GSH on H2O2 signaling in A549 alveolar epithelial cells. Preincubation of these cells with 10 mM GSH for 30 min, before the 30-min exposure to 250 μM H2O2, prevented an H2O2-mediated decrease in intracellular GSH levels (Figure 3A). Moreover, GSH prevented induction of ceramide generation by H2O2 (Figure 3B). Of note is that exposure of cells to GSH alone mediated a decrease in intracellular GSH rather than an increase (Figure 3A). This may be the result of an active GSH efflux mechanism (22), inhibition of GSH synthesis, or increased sequestration of GSH to specific subcellular compartments (23). It appears therefore that although both GSH and H2O2 have similar effects on cellular GSH levels, they have opposite effects on ceramide production. This suggests that GSH may elicit its inhibitory effects extracellularly. Indeed, extracellularly supplemented GSH was capable of inhibiting the apoptotic signaling triggered by H2O2 (Figure 3C). Therefore, we conclude that in A549 cells, H2O2 mediates induction of ceramide generation via GSH depletion, an effect that leads to apoptosis. These effects are efficiently prevented by extracellular supplementation of GSH.
Figure 3.

Extracellular supplementation of GSH prevents H2O2-induced GSH depletion, ceramide generation, and apoptosis in human lung epithelial cells. A549 cells were incubated in medium supplemented with 1% serum with 10 mM GSH for 30 min, followed by incubations with 250 μM H2O2 for an additional 30 min (A and B) or 24 h (C). (A). Cell lysates were analyzed for GSH as described in Materials and Methods. (B). Cellular lipids were extracted and assayed for ceramide by the DAG kinase assay (5, 6). The reaction products were analyzed by TLC and quantified using a phosphorimager. (C). Apoptotic cells were stained with Annexin V-PI and detected by FACS. The values are represented as percent (%) of control, not treated, cells.
NAC Prevents Elevation of Ceramide and Apoptosis Induced by ATZ
To test the effect of endogenously generated H2O2 on ceramide levels and apoptosis, we treated primary tracheobronchial cells with ATZ, which increases intracellular H2O2 levels by inhibiting endogenous catalase (18). The cells responded to ATZ with an immediate elevation of ceramide within the first 10 min of treatment, which reached a maximum of threefold in approximately 20 min (Figure 4A). Pretreatment of cells with 10 mM NAC, a well-known antioxidant and precursor of GSH (17, 18, 24), for 1 h inhibited ATZ-induced ceramide elevation. In addition, ceramide elevation was followed by an induction of apoptosis after treatment with 30 mM ATZ for 18 h (Figure 4B). Pretreatment of cells with 10 mM NAC for 1 h inhibited the apoptotic effects of ATZ.
Figure 4.

ATZ induces ceramide and apoptosis, effects efficiently inhibited by NAC. (A). Primate tracheal epithelial cells were incubated with 30 mM ATZ for the indicated times with (solid triangles) or without (solid circles) preincubation with 10 mM NAC for 1 h. Cellular lipids were analyzed for ceramide by the DAG kinase assay, as described in Materials and Methods. (B). Cells were incubated with 30 mM ATZ for 18 h with or without preincubation with 10 mM NAC for 1 h. After treatments, cells were evaluated for apoptosis by TUNEL. The values represent apoptotic cells (percent of total cells counted) where at least 300 cells were counted.
Inhibition of Ceramide Production Is an Intrinsic Property Specific for GSH
To determine whether inhibition of ceramide production is an intrinsic property of all antioxidants or is a specific property of GSH, we tested the effect of other thiol antioxidants as well as GSSG on the induction of ceramide levels by H2O2. A549 cells were preincubated for 1 h with 10 mM of DTT, 2-mercaptoethanol, or GSSG, followed by a 30-min incubation in the presence of 250 μM H2O2. GSH inhibited the H2O2-mediated GSH depletion (Figure 5A) and ceramide increase (Figure 5B). However, none of the nonphysiologic antioxidants, DTT, 2-mercaptoethanol, or GSSG, were successful in inhibiting an H2O2-mediated decrease in intracellular GSH or increase in ceramide. These data indicate that the effect of GSH is mediated by its antioxidant reduced form and further suggest that the ability to inhibit H2O2-induced ceramide generation is an intrinsic property of GSH and not all thiol antioxidants.
Figure 5.

GSH, but not GSSG or other antioxidants, inhibits ceramide generation induced by H2O2 in lung epithelial cells. A549 cells were preincubated for 1 h in medium supplemented with 1% serum in the presence or absence of 10 mM of GSH, GSSG, DTT, or 2-mercaptoethanol, followed by incubations with 250 μM H2O2 for an additional 30 min. The cells were rinsed with ice-cold PBS to terminate the treatments and harvested by trypsinization. GSH (A) and ceramide (B) levels were determined as described in Materials and Methods. Values represent mean ± SEM. *Mean of the group that was significantly different from the mean of the H2O2-treated group (P < 0.05). All other comparisons were statistically not significant (P > 0.05). Statistical analysis was performed using the Mann-Whitney, nonparametric, two-tailed test.
Depletion of GSH Increases Ceramide Levels and Apoptosis
To further investigate whether depletion of intracellular GSH could modulate ceramide levels, we used BSO, a widely used inhibitor of GSH synthesis. We investigated the effect of cellular GSH depletion by BSO on ceramide levels in A549 and primary tracheobronchial epithelial cells. BSO-mediated GSH depletion increased ceramide production in a dose- and time-dependent manner (Figures 6 and 7). Treatment of lung epithelial cells with 100 to 500 μM BSO for 24 h depleted intracellular GSH (Figure 6A), and dose-dependently increased ceramide levels (Figure 6B) as well as apoptosis in both A549 (Figure 6C) and primary cells (Figure 6D). In addition, exposure of cells to 250 μM BSO for 12 h or more markedly depleted cellular GSH pools (Figure 7A). In return, GSH depletion increased ceramide concentrations and the number of apoptotic cells, which reached a maximum after 48 h (Figure 7B). These results further support the pivotal role of GSH in the modulation of cellular ceramide levels and apoptosis in lung epithelial cells.
Figure 6.

BSO depletes cellular GSH and induces ceramide production and apoptosis in a dose-dependent manner. After treatments with the indicated concentrations of BSO for 24 h, A549 cells were washed with ice-cold PBS and collected by trypsinization. (A). The cells were lysed and cellular GSH content was determined as previously described (21). (B). Treated cells were extracted with methanol:chloroform:1 N HCl, and cellular ceramide levels were determined by the DAG kinase assay (6). (C). To determine apoptosis, cells were stained with Annexin V-FITC and PI, and were evaluated by FACS analysis, as described in Materials and Methods. Values represent mean ± SEM. (D). Primary cells, after treatments with 250 or 500 μM BSO for 24 h, were evaluated for apoptosis by staining with Hoechst, as described in Materials and Methods.
Figure 7.
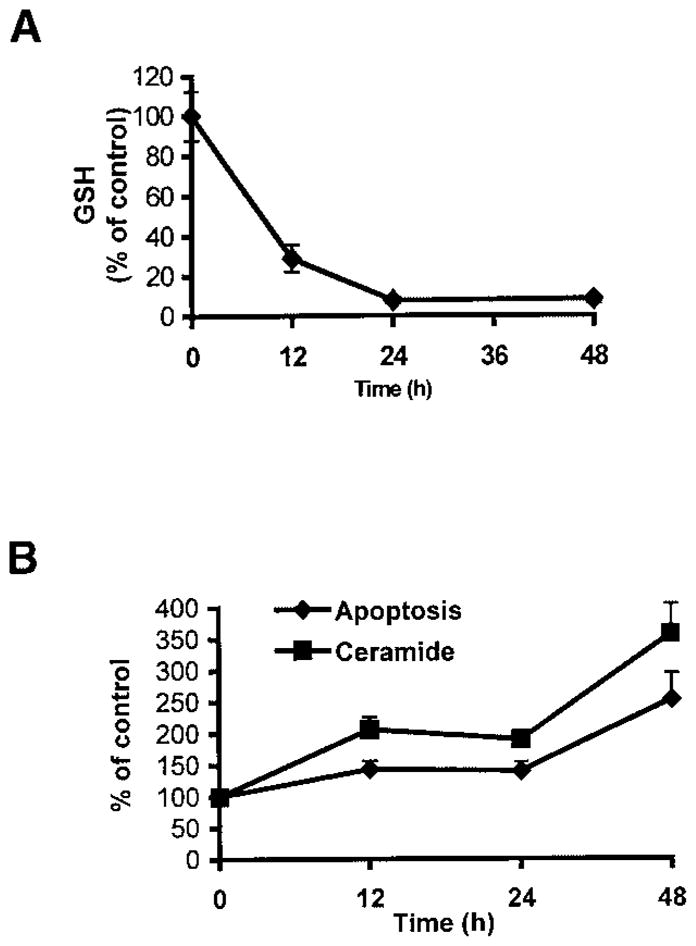
BSO depletes cellular GSH and induces ceramide production and apoptosis in a time-dependent manner. After treatments with 250 μM BSO for the indicated times, A549 cells were washed with ice-cold PBS and collected by trypsinization. (A). Cells were lysed and cellular GSH content was determined as previously described (21). (B). Treated cells were extracted with methanol:chloroform:1 N HCl, and cellular ceramide levels were determined by the DAG kinase assay. Treated cells were also stained with Annexin V-FITC and PI, and were evaluated for apoptosis, as described in Materials and Methods. Values represent mean ± SEM.
C6-Ceramide, Similar to H2O2, Increases Cellular Ceramide Levels and Induces Apoptosis
Membrane-permeant synthetic ceramide analogs have proven to be useful tools in studies for the role of ceramide in signal transduction and apoptosis; they mimic the effects of most ceramide pathway agonists (5, 6, 10, 25). We used the C6-ceramide analog to test whether it mimics the effects of ceramide elevation by H2O2. Treatment of A549 cells with 25 μM C6-ceramide elevated cellular ceramide levels up to 1.7-fold in a time-dependent manner (Figure 8A). However, unlike H2O2, the ceramide elevation induced by C6-ceramide was not accompanied by an immediate decrease in cellular GSH levels (Figure 8B, right panel) and therefore, a 1-h preincubation with GSH did not prevent the increase in ceramide levels induced by C6-ceramide (Figure 8B, left panel). These results indicate that once ceramide is elevated intracellularly by exogenous addition of ceramide analogs, extracellular supplementation of GSH does not affect modulation of cellular ceramide levels.
Figure 8.

C6-ceramide mediates ceramide generation without depletion of GSH. (A). A549 cells were treated with 25 μM C6-cer-amide for the indicated times. After treatment, cellular lipids were extracted and assayed for ceramide by the DAG kinase assay (5, 6). The reaction products were analyzed by TLC and quantified using a phosphorimager. (B). A549 cells were preincubated with (hatched bars) or without (solid bars) 10 mM GSH for 1 h before exposure to 25 μM C6-ceramide for 15 min. After treatment, cell lysates were analyzed for ceramide and GSH, as described in Materials and Methods. All values are represented as percent (%) of control.
To determine the apoptotic effects of C6-ceramide, A549 cells were exposed to 25 μM C6-ceramide for 12 or 24 h. As shown in Figure 9A, C6-ceramide induced apoptosis only after 24 h of exposure. Of note is that exposure of A549 cells to C6-ceramide for 12 or 24 h results in significantly decreased intracellular GSH levels (26). Therefore, as expected, complementation with 10 mM GSH inhibited the C6-ceramide induction of apoptosis (Figure 9B). These results suggest an additional role of GSH, downstream to ceramide generation in the ceramide-mediated apoptotic pathway, as discussed in subsequent text.
Figure 9.

C6-ceramide induces apoptosis, an effect efficiently inhibited by GSH. (A). A549 cells were treated with 25 μM C6-ceramide for the indicated times. (B). Cells were preincubated with (hatched bars) or without (solid bars) 10 mM GSH 1 h before exposure to 25 μM C6-ceramide for 24 h. After treatments, apoptosis was determined by Annexin V staining of apoptotic cells, as described in Materials and Methods. All values are represented as percent (%) of control.
Discussion
Ceramide generation has been identified as a key regulatory step in signaling cascades that lead to apoptosis in several systems (5–8, 10, 11, 18). In the last few years, the role of oxidants and antioxidants has become apparent in modulating ceramide-mediated apoptosis (5, 6, 16, 18–20). The lung is a primary organ targeted by oxidants and has therefore developed strong intracellular and extracellular antioxidant defense systems. Lung cells have elevated GSH levels that are modulated in response to oxidants (1–4); GSH concentrations can reach as high as 10 mM. Moreover, the alveolar ELF contains GSH at high concentrations—about 400 to 500 μM compared with the 0.5 to 5 μM present in blood plasma (27, 28). Pulmonary diseases such as cystic fibrosis and adult respiratory distress syndrome have been associated with changes in GSH concentration of ELF and apoptosis (2, 4, 29, 30). It is therefore of great importance to elucidate the mechanisms by which GSH modulates ceramide-mediated apoptosis in lung epithelial cells.
In the present study, we demonstrated that extracellular supplementation of GSH to A549 lung epithelial cells inhibited ceramide production in these cells, whereas depletion of intracellular GSH by H2O2 or BSO was paralleled with increased ceramide levels and apoptosis induction. When GSH was supplemented extracellularly, the H2O2-induced drop in cellular GSH was diminished and subsequently both ceramide elevation and apoptosis were prevented. These were all specific properties of GSH and not of other thiol-containing molecules. Importantly, ATZ mimicked the effects of H2O2 that were provided extracellularly, and NAC inhibited the effects of intracellularly generated H2O2. In addition, C6-ceramide mediated an elevation in cellular ceramide levels that was followed by an induction of apoptosis. These results suggest that in lung epithelial cells, H2O2 triggers the apoptotic pathway by inducing ceramide generation via depletion of GSH and that elevation of ceramide is sufficient and necessary for the induction of apoptosis.
In several systems, ROS generation has been shown to play an important and early role in ceramide-mediated apoptosis induced by serum starvation (31), anthracyclins such as daunorubicin (32), and cytokines such as tumor necrosis factor (TNF)-α (18). In these systems, generation of ROS precedes ceramide elevation, and interestingly, GSH depletion is frequently associated with these effects (18, 31), whereas supplementation of antioxidants such as GSH and NAC inhibits the induction of both ceramide levels and apoptosis (16–20). Therefore, it appears that generation of ROS and increase in ceramide may be the common effect of several diverse apoptotic stimuli. However, how GSH regulates ceramide levels is not yet established.
It is well documented that SMases are activated in response to several stimuli, thus initiating the SM/ceramide pathway. Recently it has been shown that one of these enzymes, the neutral Mg2+-dependent N-SMase, is regulated by GSH. GSH elicits a direct inhibitory effect on N-SMase from blood cells (17, 19, 20) and on purified N-SMase from brain cells (33). Moreover, drops in cellular GSH levels induced by TNF-α signaling precede activation of N-SMase (19). Our recent studies have suggested a role of a membrane-bound N-SMase in mediating the effects of H2O2 on ceramide production and apoptosis in bronchial and airway epithelial cells (5, 6). This information in combination with our present data showing that only GSH, but not GSSG or other antioxidants, inhibits ceramide production (Figure 5) suggest that GSH may be an inhibitor of N-SMase in human lung epithelial cells.
Interestingly, administration of GSH to A549 cells, in addition to decreased cellular ceramide, was also associated with a decrease in intracellular GSH (Figure 3A). This decrease in intracellular GSH could be the result of inhibition of GSH synthesis, sequestration of GSH in the nucleus (23), or induced efflux of GSH from the cells (22, 27). Under these experimental conditions, supplementation of GSH inhibited ceramide production efficiently (Figure 3B). On the other hand, depletion of intracellular GSH by H2O2 or BSO induced an increase in ceramide generation, followed by ceramide-mediated apoptosis, and these effects were prevented by the replenishment of GSH (Figures 2, 3, 6, and 7). These findings suggest that both extracellular and intracellular GSH may modulate ceramide production in the lung. The exact vectorial mode (extra- or intracellular) of GSH effects remains to be elucidated. This will be facilitated once the N-SMase, which is modulated by GSH, is molecularly characterized.
We propose that in lung epithelial cells, the PM-bound N-SMase may exist as an inactive form inhibited by high levels of both intra- and extracellular GSH present in ELF, thus maintaining low levels of ceramide (Figure 10). The inhibition of N-SMase may render lung cells less sensitive and less susceptible to oxidants, to which they are ordinarily exposed. This would increase the threshold for ceramide elevation required for the induction of apoptosis. However, once oxidant levels increase, they decrease GSH levels, thereby overcoming its inhibitory effect on N-SMase. Therefore, ceramide is elevated and the apoptotic pathway is initiated. This is further supported by our findings that the inhibitory effect on H2O2-induced ceramide production is specific for GSH and not for other thiol-containing molecules and most importantly not for GSSG (Figure 5). Therefore, oxidation of GSH by oxidants renders it incapable of inhibiting ceramide generation. It is interesting that even a short exposure of cells to H2O2 for 1 h, followed by growth in regular medium, is sufficient to induce apoptosis. This demonstrates that the events that control the fate of the cells occur within this hour, during which GSH is depleted and ceramide is generated (Figure 2).
Figure 10.
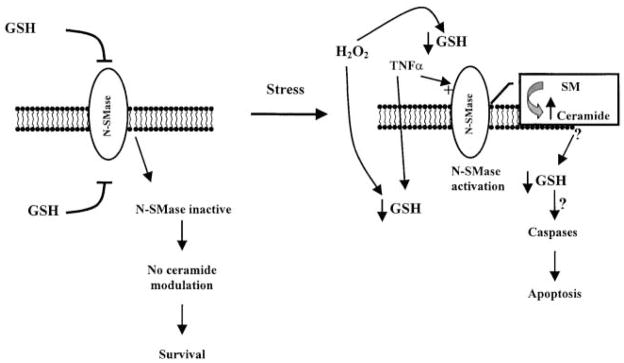
Schematic representation of the role of oxidants (H2O2) and antioxidants (GSH) in ceramide generation and apoptosis. GSH inhibits N-SMase activity, thus maintaining low ceramide levels (19, 20). Stress factors (i.e., extracellularly administered H2O2) result in ROS-mediated depletion of GSH. This may be a critical event in the induction of apoptosis by activation of N-SMase and generation of ceramide. However, supplementation of antioxidants, such as GSH, counteract this effect by inhibiting N-SMase activity. Therefore, the redox state of the cell determines the activity of N-SMase and the levels of ceramide, thus modulating the apoptotic pathway in lung epithelial cells.
Supplementation of GSH shortly before exposure to H2O2 was sufficient to inhibit the apoptotic effects of H2O2. It appears that providing GSH to replenish the decreased levels of GSH is sufficient to maintain ceramide below the threshold levels, thus preventing apoptosis. Once ceramide is increased, i.e., by administration of C6-ceramide, GSH can no longer prevent ceramide elevation (Figure 8B). However, it is still capable of protecting the cell from apoptotic cell death (Figure 9B). These results suggest that GSH may play a dual role in ceramide-mediated apoptosis: one role is at the initiation of the apoptotic pathway, where a decrease in GSH levels modulates ceramide generation, possibly via activation of N-SMase. GSH may also have an additional role downstream to ceramide generation, where depletion of GSH by ceramide elevation may modulate downstream targets, such as caspases. Several studies suggest that ceramide elevation may induce ROS generation in the apoptotic pathways (25, 34). Ceramide analogs have also been shown to induce ROS and apoptosis, effects efficiently inhibited by GSH (25, 32). In our system, even though short exposures to C6-ceramide induce elevation in intracellular ceramide levels, only long exposures (to C6-ceramide) decrease intracellular GSH levels (26). How C6-ceramide, a nonphysiologic ceramide analog, increases intracellular ceramide in lung epithelial cells is still unclear. However, the effects of this nonphysiologic analog can be compared with those of H2O2, a physiologic oxidant. We propose that the initial drop in GSH, which occurs within the first hour of exposure to H2O2, mediates ceramide elevation via activation of N-SMase, whereas the secondary GSH depletion may regulate caspases, such as caspase 3 (26), as well as ceramide generation via a positive feedback mechanism (10). Therefore, our ongoing studies focus on the molecular sites of ROS and ceramide generation in lung epithelial cells. We are in the process of elucidating the molecular mechanisms that link the redox state of the cell to N-SMase activation, ceramide production, and the execution phase of apoptosis in lung epithelial cells (submitted).
Acknowledgments
The authors would like to thank Dr. Adiel Barak and Edward A. Medina for useful discussions and for editing the manuscript, and Drs. Sharanya Reddy and Patrick Wong for assistance with the HPLC analysis. This work was supported by grant 8RT-0098 from the Tobacco Related Disease Research Program (T.G. and A.V.) and by grants HL47628 (A.V. and T.G.) and HL60812 (A.V. and T.G.) from the National Institutes of Health.
Abbreviations
- ATZ
aminotriazole
- BSO
L-D-buthionine sulfoximine
- DAG
diacylglycerol
- DTT
dithiothreitol
- EDTA
ethylenediaminetetraacetic acid
- ELF
epithelial lining fluid
- FACS
fluorescence-activated cell sorter
- FBS
fetal bovine serum
- FITC
fluorescein isothiocyanate
- GSH
reduced glutathione
- GSSG
oxidized glutathione
- NAC
N-acetylcysteine
- N-SMase
neutral sphingomyelinase
- PBS
phosphate-buffered saline
- PI
propidium iodide
- PM
plasma membrane
- ROS
reactive oxygen species
- SEM
standard error of the mean
- SMase
sphingomyelinase
- TLC
thin layer chromatography
References
- 1.Cross CE, van der Vliet A, O’Neill CA, Louie S, Halliwell B. Oxidants, antioxidants, and respiratory tract lining fluids. Environ Health Perspect. 1994;102(Suppl 10):185–191. doi: 10.1289/ehp.94102s10185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Rahman I, Li XY, Donaldson K, Harrison DJ, MacNee W. Glutathione homeostasis in alveolar epithelial cells in vitro and lung in vivo under oxidative stress. Am J Physiol. 1995;269(3 Pt 1):L285–L292. doi: 10.1152/ajplung.1995.269.3.L285. [DOI] [PubMed] [Google Scholar]
- 3.Mulier B, Rahman I, Watchorn T, Donaldson K, MacNee W, Jeffery PK. Hydrogen peroxide-induced epithelial injury: the protective role of intracellular nonprotein thiols (NPSH) Eur Respir J. 1998;11:384–391. doi: 10.1183/09031936.98.11020384. [DOI] [PubMed] [Google Scholar]
- 4.Rahman I, MacNee W. Oxidative stress and regulation of glutathione in lung inflammation. Eur Respir J. 2000;16:534–554. doi: 10.1034/j.1399-3003.2000.016003534.x. [DOI] [PubMed] [Google Scholar]
- 5.Goldkorn T, Balaban N, Shannon M, Chea V, Matsukuma K, Gilchrist D, Wang H, Chan C. H2O2 acts on cellular membranes to generate ceramide signaling and initiate apoptosis in tracheobronchial epithelial cells. J Cell Sci. 1998;111(Pt. 21):3209–3220. doi: 10.1242/jcs.111.21.3209. [DOI] [PubMed] [Google Scholar]
- 6.Chan C, Goldkorn T. Ceramide path in human lung cell death. Am J Respir Cell Mol Biol. 2000;22:460–468. doi: 10.1165/ajrcmb.22.4.3376. [DOI] [PubMed] [Google Scholar]
- 7.Hannun YA. The sphingomyelin cycle and the second messenger function of ceramide. J Biol Chem. 1994;269:3125–3128. [PubMed] [Google Scholar]
- 8.Kolesnick R, Hannun YA. Ceramide and apoptosis. Trends Biochem Sci. 1999;24:224–225. doi: 10.1016/s0968-0004(99)01408-5. [discussion 227] [DOI] [PubMed] [Google Scholar]
- 9.Perry DK. Ceramide and apoptosis. Biochem Soc Trans. 1999;27:399–404. doi: 10.1042/bst0270399. [DOI] [PubMed] [Google Scholar]
- 10.Jaffrezou JP, Maestre N, de Mas-Mansat V, Bezombes C, Levade T, Laurent G. Positive feedback control of neutral sphingomyelinase activity by ceramide. FASEB J. 1998;12:999–1006. doi: 10.1096/fasebj.12.11.999. [DOI] [PubMed] [Google Scholar]
- 11.Segui B, Andrieu-Abadie N, Adam-Klages S, Meilhac O, Kreder D, Garcia V, Bruno AP, Jaffrezou JP, Salvayre R, Kronke M, Levade T. CD40 signals apoptosis through FAN-regulated activation of the sphingomyelin-ceramide pathway. J Biol Chem. 1999;274:37251–37258. doi: 10.1074/jbc.274.52.37251. [DOI] [PubMed] [Google Scholar]
- 12.Levade T, Jaffrezou JP. Signalling sphingomyelinases: which, where, how and why? Biochim Biophys Acta. 1999;1438:1–17. doi: 10.1016/s1388-1981(99)00038-4. [DOI] [PubMed] [Google Scholar]
- 13.Barak A, Morse LS, Goldkorn T. Ceramide: a potential mediator of apoptosis in human retinal pigment epithelial cells. Invest Ophthalmol Vis Sci. 2001;42:247–254. [PubMed] [Google Scholar]
- 14.Beaver JP, Waring P. A decrease in intracellular glutathione concentration precedes the onset of apoptosis in murine thymocytes. Eur J Cell Biol. 1995;68:47–54. [PubMed] [Google Scholar]
- 15.Denisova NA, Fisher D, Provost M, Joseph JA. The role of glutathione, membrane sphingomyelin, and its metabolites in oxidative stress-induced calcium “dysregulation” in PC12 cells. Free Radic Biol Med. 1999;27:1292–1301. doi: 10.1016/s0891-5849(99)00163-x. [DOI] [PubMed] [Google Scholar]
- 16.Teramoto S, Tomita T, Matsui H, Ohga E, Matsuse T, Ouchi Y. Hydrogen peroxide-induced apoptosis and necrosis in human lung fibroblasts: protective roles of glutathione. Jpn J Pharmacol. 1999;79:37n–44n. doi: 10.1254/jjp.79.33. [DOI] [PubMed] [Google Scholar]
- 17.Yoshimura S, Banno Y, Nakashima S, Hayashi K, Yamakawa H, Sawada M, Sakai N, Nozawa Y. Inhibition of neutral sphingomyelinase activation and ceramide formation by glutathione in hypoxic PC12 cell death. J Neurochem. 1999;73:675–683. doi: 10.1046/j.1471-4159.1999.0730675.x. [DOI] [PubMed] [Google Scholar]
- 18.Singh I, Pahan K, Khan M, Singh AK. Cytokine-mediated induction of ceramide production is redox-sensitive: implications to proinflammatory cytokine-mediated apoptosis in demyelinating diseases. J Biol Chem. 1998;273:20354–20362. doi: 10.1074/jbc.273.32.20354. [DOI] [PubMed] [Google Scholar]
- 19.Liu B, Andrieu-Abadie N, Levade T, Zhang P, Obeid LM, Hannun YA. Glutathione regulation of neutral sphingomyelinase in tumor necrosis factor-alpha-induced cell death. J Biol Chem. 1998;273:11313–11320. doi: 10.1074/jbc.273.18.11313. [DOI] [PubMed] [Google Scholar]
- 20.Liu B, Hannun YA. Inhibition of the neutral magnesium-dependent sphingomyelinase by glutathione. J Biol Chem. 1997;272:16281–16287. doi: 10.1074/jbc.272.26.16281. [DOI] [PubMed] [Google Scholar]
- 21.Cotgreave IA, Moldeus P. Methodologies for the application of monobromobimane to the simultaneous analysis of soluble and protein thiol components of biological systems. J Biochem Biophys Methods. 1986;13:231–249. doi: 10.1016/0165-022x(86)90102-8. [DOI] [PubMed] [Google Scholar]
- 22.Ghibelli L, Coppola S, Rotilio G, Lafavia E, Maresca V, Ciriolo MR. Non-oxidative loss of glutathione in apoptosis via GSH extrusion. Biochem Biophys Res Commun. 1995;216:313–320. doi: 10.1006/bbrc.1995.2626. [DOI] [PubMed] [Google Scholar]
- 23.Voehringer DW. BCL-2 and glutathione: alterations in cellular redox state that regulate apoptosis sensitivity. Free Radic Biol Med. 1999;27:945–950. doi: 10.1016/s0891-5849(99)00174-4. [DOI] [PubMed] [Google Scholar]
- 24.Phelps DT, Deneke SM, Daley DL, Fanburg BL. Elevation of glutathione levels in bovine pulmonary artery endothelial cells by N-acetylcysteine. Am J Respir Cell Mol Biol. 1992;7:293–299. doi: 10.1165/ajrcmb/7.3.293. [DOI] [PubMed] [Google Scholar]
- 25.Garcia-Ruiz C, Colell A, Mari M, Morales A, Fernandez-Checa JC. Direct effect of ceramide on the mitochondrial electron transport chain leads to generation of reactive oxygen species: role of mitochondrial glutathione. J Biol Chem. 1997;272:11369–11377. doi: 10.1074/jbc.272.17.11369. [DOI] [PubMed] [Google Scholar]
- 26.Tsaba A, Ravid T, Lavrentiadou SN, GT Caspases and ceramide in the apoptosis of airway epithelial cells. ATS 97th International Conference.2001. [Google Scholar]
- 27.van Klaveren RJ, Demedts M, Nemery B. Cellular glutathione turnover in vitro, with emphasis on type II pneumocytes. Eur Respir J. 1997;10:1392–1400. doi: 10.1183/09031936.97.10061392. [DOI] [PubMed] [Google Scholar]
- 28.van der Vliet A, O’Neill CA, Cross CE, Koostra JM, Volz WG, Halliwell B, Louie S. Determination of low-molecular-mass antioxidant concentrations in human respiratory tract lining fluids. Am J Physiol. 1999;276(2 Pt 1):L289–L296. doi: 10.1152/ajplung.1999.276.2.L289. [DOI] [PubMed] [Google Scholar]
- 29.Kam PC, Ferch NI. Apoptosis: mechanisms and clinical implications. Anaesthesia. 2000;55:1081–1093. doi: 10.1046/j.1365-2044.2000.01554.x. [DOI] [PubMed] [Google Scholar]
- 30.Fine A, Janssen-Heininger Y, Soultanakis RP, Swisher SG, Uhal BD. Apoptosis in lung pathophysiology. Am J Physiol Lung Cell Mol Physiol. 2000;279:L423–L427. doi: 10.1152/ajplung.2000.279.3.L423. [DOI] [PubMed] [Google Scholar]
- 31.Esteve JM, Mompo J, Garcia de la Asuncion J, Sastre J, Asensi M, Boix J, Vina JR, Vina J, Pallardo FV. Oxidative damage to mitochondrial DNA and glutathione oxidation in apoptosis: studies in vivo and in vitro. FASEB J. 1999;13:1055–1064. doi: 10.1096/fasebj.13.9.1055. [DOI] [PubMed] [Google Scholar]
- 32.Mansat-de Mas V, Bezombes C, Quillet-Mary A, Bettaieb A, D’Orgeix AD, Laurent G, Jaffrezou JP. Implication of radical oxygen species in ceramide generation, c-Jun N-terminal kinase activation and apoptosis induced by daunorubicin. Mol Pharmacol. 1999;56:867–874. doi: 10.1124/mol.56.5.867. [DOI] [PubMed] [Google Scholar]
- 33.Chatterjee S, Han H, Rollins S, Cleveland T. Molecular cloning, characterization, and expression of a novel human neutral sphingomyelinase. J Biol Chem. 1999;274:37407–37412. doi: 10.1074/jbc.274.52.37407. [DOI] [PubMed] [Google Scholar]
- 34.Fernandez-Checa JC, Kaplowitz N, Garcia-Ruiz C, Colell A, Miranda M, Mari M, Ardite E, Morales A. GSH transport in mitochondria: defense against TNF-induced oxidative stress and alcohol-induced defect. Am J Physiol. 1997;273(1 Pt 1):G7–G17. doi: 10.1152/ajpgi.1997.273.1.G7. [DOI] [PubMed] [Google Scholar]