

Abstract
Exposure to hydrogen peroxide (H2O2), one of the reactive oxidants in the gas phase of cigarette smoke (CS), induces aberrant phosphorylation of the epidermal growth factor receptor (EGFR), resulting in the lack of ubiquitination by c-Cbl, and impaired degradation. EGFR activation without the feedback regulation of normal degradation leads to uncontrolled cell growth and tumor promotion. Using immunoprecipitation, immunoblotting, and confocal microscopy, we now demonstrate that the pattern of EGFR activation by CS is similar to H2O2. We found that exposure of human airway epithelial cells to CS, as with exposure to H2O2, not only results in an increase in EGFR activation over time, but the EGFR activated by H2O2 or CS is neither ubiquitinated nor subsequently degraded due to its inability to bind the E3 ubiquitin ligase, c-Cbl, either directly or indirectly via the Grb2 adapter protein. Moreover, the stabilized H2O2- and CS-activated EGFR remains plasma membrane-bound, while a population of the receptor is trafficked to a perinuclear region. Concomitantly, CS exposure results in the activation of downstream Akt and ERK1/2 survival and proliferation pathways. Therefore, exposure to CS, like exposure to H2O2, results in prolonged signaling by the EGFR and may contribute to uncontrolled lung cell growth.
The epidermal growth factor (EGF) receptor (EGFR) is implicated in a number of cancers, and its oncogenic potential is linked to its inability to undergo clathrin-mediated endocytosis and lysosomal degradation (1, 2). The process of EGFR down-regulation is highly dependent on the ability of the E3 ubiquitin ligase, c-Cbl, to bind the receptor, thereby facilitating its entry into the clathrin-coated pits and lysosomal sorting (3–6). Recent evidence has also demonstrated the requirement of Grb2 in recruiting the RING domain of c-Cbl to the EGFR and for subsequent receptor entry into the clathrin-mediated endocytic pathway (7).
Our previous studies have demonstrated that oxidative stress induced by H2O2 causes the aberrant phosphorylation of the EGFR, where Tyr-1045, the c-Cbl binding site, is not phosphorylated and c-Cbl binding is abrogated (8). Therefore, under H2O2-induced oxidative stress, the EGFR is not only activated, but it is also stabilized due to its inability to enter the clathrin-mediated endocytic and lysosomal degradation pathways (8, 9). To further investigate the physiological relevance of oxidant-induced activation and stabilization of the EGFR, we focused our studies on cigarette smoke.
Among the plethora of deleterious chemicals found in cigarette smoke, H2O2 has been reported to be a significant constituent of the gas phase (10). We thus hypothesized that if mainstream cigarette smoke does indeed contain high amounts of H2O2, the effects of exposure on EGFR activation and stability should parallel those of H2O2 alone. To test this hypothesis, we utilized human airway epithelial cells in culture as a highly simplified model system in which to expose mainstream cigarette smoke. We examined EGFR phosphorylation, in association with c-Cbl, ubiquitination, and trafficking as determinants of receptor activation and stability. We also assessed the ability of cigarette smoke to activate downstream survival and proliferative signaling molecules such as Akt and extracellular signal-regulated kinase 1/2 (ERK1/2). The results herein demonstrate that the effects of cigarette smoke do indeed parallel those of H2O2, where the EGFR is aberrantly phosphorylated and stabilized due to the loss of c-Cbl binding and receptor ubiquitination. Moreover, the Akt and ERK1/2 pathways are also activated by cigarette smoke, thereby contributing to cellular events that may ultimately lead to hyperplasia and tumorigenesis.
MATERIALS AND METHODS
Cell culture
A549 human lung carcinoma cells from American Type Culture Collection (Manassas, VA, USA) were maintained in F-12K (Kaighn’s modification) nutrient mixture (Invitrogen, Carlsbad, CA, USA), supplemented with 10% fetal bovine serum (Invitrogen) and 1% penicillin/streptomycin. HBE1 human bronchial epithelial cells (generated by Dr. James Yankaskas, University of North Carolina, Chapel Hill, NC, USA and provided to us by Dr. Reen Wu, University of California, Davis, CA, USA) were maintained in DMEM and Ham’s F12 medium (1:1) supplemented with 5 mg/ml insulin, 5 mg/ml transferrin, 10 ng/ml EGF, 0.1 mM dexamethasone, 20 ng/ml cholera toxin, and 15 mg/ml bovine hypothalamus extract. Prior to treatments, cells at ~80% confluence were serum-starved overnight in F-12K medium containing 0.5% dialyzed fetal bovine serum or HBE1 medium without EGF.
Treatments
To generate H2O2, glucose oxidase (GO; type II from Aspergillus niger, 15,500 U/g; Sigma, St. Louis, MO, USA) was added to serum-free Dulbecco’s modified Eagle’s medium containing 25 mM glucose and 0.5% bovine serum albumin (Sigma). This medium was then preconditioned for 15 min at 37°C and added to cells for 15 min at 37°C. For incubation periods greater than 15 min, GO-containing medium was replaced every 15 min with fresh preconditioned medium. For EGF treatments, cells were incubated in the same medium supplemented with 100 ng/ml EGF (Upstate Biotechnology, Waltham, MA, USA).
Cigarette smoke exposure
The lids were removed from tissue culture dishes and serum-starved human airway epithelial (HAE; A549 or HBE1) cells were placed in a vacuum oven with a chamber volume of 0.45 cu ft, and the temperature was set at 37°C. A vacuum was generated (~25 in Hg) and smoke from one cigarette (University of Kentucky 2R4F) was drawn into the chamber by bleeding the chamber (a decrease of ~5 in Hg over ~30 s) through a short piece of tubing holding the cigarette. The chamber atmosphere was then lowered to 2.5 in Hg for the remainder of the exposure time.
H2O2 quantification
Measurement of H2O2 generated by 1 U/ml GO or smoke from 1 cigarette was performed by the method of Thurman et al. (11). Briefly, dishes of cells in Dulbecco’s modified Eagle’s medium (Invitrogen) containing 25 mM glucose and no phenol red were incubated with 1 U/ml GO or smoke from 1 cigarette. At the indicated times, 1-ml aliquots of medium were collected and 1 ml of 50% wt/vol trichloroacetic acid was added to each aliquot. The samples were centrifuged at 500 g for 10 min, and 0.2 ml of 10 mM ferrous ammonium sulfate and 0.1 ml of 2.5 M sodium thiocyanate (both from ICN Pharmaceuticals, Costa Mesa, CA) were added to the supernatant. Absorbance of the ferrithiocyanate complex was measured using a GENios Multi-Detection Reader (Tecan, Männedorf, Switzerland) at 480 nm and compared with standard curves obtained from dilutions of a standard H2O2 solution.
Apoptosis assessment
Cells were not treated (NT) or exposed to 1 U/ml GO or smoke from 1 cigarette for 45 min. At the end of the exposure, cells were washed with PBS and incubated in fresh cell culture medium for 24 or 48 h. Apoptosis was assessed by annexin V and propidium iodide (PI) staining as described previously (12).
Lysate preparation, immunoprecipitation, and immunoblotting
Lysate preparation and protein immunoprecipitation were performed as described by Bao et al. (13). After treatments, cells were extracted in solubilization buffer containing 50 mM Tris, pH 7.5, 150 mM NaCl, 10% glycerol, 1% Nonidet P-40, 1 mM EGTA, protease inhibitor cocktail (Sigma), and phosphatase inhibitor cocktail (Sigma). Lysates were cleared by centrifugation and 400 μg of protein in the supernatant were immunoprecipitated by overnight incubation with 4 μg anti-EGFR clone 225 monoclonal antibody (a generous gift from ImClone Systems Inc., New York, NY, USA) at 4°C, followed by protein A (Repligen Corp., Needham, MA, USA) precipitation for 1–2 h at 4°C. Immunoprecipitates were washed three times with HNTG buffer containing 20 mM HEPES, pH 7.5, 150 mM NaCl, 0.1% Triton X-100, and 10% glycerol, resolved by SDS-PAGE, and transferred to nitrocellulose membranes. Membranes were blocked for 1 h in Tris-buffered saline, pH 7.5, containing 0.5% Tween 20 and 5% nonfat milk, incubated overnight at 4°C with primary antibodies, followed by a 1 h incubation at room temperature with a 1:10,000 dilution of horseradish peroxidase-conjugated secondary antibodies (Jackson ImmunoResearch Laboratories, West Grove, PA, USA). Immunoreactive protein bands were detected with the SuperSignal West Pico Substrate (Pierce, Rockford, IL, USA). Blotting antibodies used were: anti-EGFR RK2 (generously provided by Dr. Joseph Schlessinger, New York University Medical Center, New York, NY, USA); anti-phosphotyrosine PY-20, anti-phosphotyrosine-1173, anti-Akt (Santa Cruz Biotechnology, Santa Cruz, CA, USA); anti-phosphotyrosines-845, 1045, and 1068, anti-phospho-Akt, anti-phospho-ERK1/2, anti-ERK1/2 (Cell Signaling Technology, Inc., Beverly, MA, USA); anti-phosphotyrosine-1086 (Calbiochem, La Jolla, CA, USA); anti-c-Cbl (Upstate, Charlottesville, VA, USA); anti-ubiquitin P4G7 (Covance, Princeton, NJ, USA).
Immunofluorescence
Immunofluorescence was performed as described previously (14). Briefly, A549 cells grown on coverslips were treated as indicated and fixed in 4% paraformaldehyde in phosphate-buffered saline (PBS). Cells were permeabilized for 15 min at room temperature with PBS containing 1% BSA, 0.2% Triton X-100, and 0.02% sodium azide; then the coverslips were blocked in PBS containing 1% BSA, 0.2% Nonidet P-40, 5% goat serum, and 0.02% sodium azide at room temperature for 1 h. Coverslips were then incubated for 1 h with anti-EGFR clone 528 (generously provided by John Mendelsohn, Memorial Sloan Kettering Cancer Center, New York, NY), followed by 1 h with Alexa Fluor 594 goat anti-mouse IgG (Molecular Probes, Eugene, OR, USA). Nuclei were stained with 1 μg/ml DAPI (Sigma) for 3 min. Coverslips were mounted onto glass slides using the ProLong Antifade Kit (Molecular Probes). Confocal microscopy at ×60 magnification was carried out using an Olympus FV1000 Fluoview confocal laser scanning microscope. Images are representative of at least 100 cells viewed in each of three separate experiments.
RESULTS
Cigarette smoke activates the EGFR in a dose- and time-dependent manner
Our previous studies demonstrated that the EGFR is activated when exposed to oxidative stress in the form of H2O2. Since cigarette smoke contains more than 1014 free radicals/oxidants per puff, we are now interested in determining the effects of cigarette smoke on EGFR activation and stability. Using human airway epithelial (HAE) cells (A549 lung adenocarcinoma and HBE1 papilloma virus-immortalized human bronchial epithelial cells), we show that exposure to cigarette smoke over the indicated time points or doses results in an increase in total EGFR phosphorylation in a time-and dose-dependent manner (Fig. 1). As with EGF andH2O2, cigarette smoke is able to induce EGFR phosphorylation significantly compared to nontreated cells.
Figure 1.

Cigarette smoke activates the EGFR in a dose- and time-dependent manner. Serum-starved human airway epithelial (HAE) cells were untreated (NT) or exposed to 100 ng/ml EGF for 15 min, 1 U/ml glucose oxidase (GO) for 45 min, and smoke from 1 cigarette at 37°C for the indicated time points (A) or smoke from the indicated amounts of cigarettes for 45 min (B). Cell lysates were separated by SDS-PAGE, transferred to nitrocellulose membranes, and immunoblotted with anti-phospho-tyrosine (PY20) and anti-EGFR (RK2) Abs. p-EGFR, phospho-EGFR.
Cigarette smoke and glucose oxidase generate comparable levels of H2O2
Since EGFR phosphorylation is induced by both cigarette smoke and H2O2 generated by GO, we wanted to determine the amount of H2O2 generated by cigarette smoke. Cell culture medium was exposed to smoke from one cigarette or incubated with 1 U/ml GO at 37°C for the indicated time points and the amount of H2O2 was determined as described in “Methods.” At 45 min, both GO and cigarette smoke produce ~600–700 μM H2O2 (Fig. 2A), as determined by the absorption of the red ferrithiocyanate complex formed in the presence of peroxides (11). At these concentrations of H2O2, cell toxicity becomes an issue. Therefore, we exposed cells to 1 U/ml GO or smoke from 1 cigarette for 45 min. Aliquots of medium were taken at the end of each exposure for H2O2 determination, and levels were in the 1.1–1.2 mM range for both GO and CS exposures (data not shown). Although the levels of H2O2 vary between experiments, the measurements have been performed numerous times with each experiment performed in triplicate, and the overriding outcome is that H2O2 measured from both GO and CS at the 30–60 min time points is consistently comparable. The cells were then washed with PBS and incubated with fresh medium for 24 or 48 h followed by annexin V and propidium iodide staining and flow cytometry for apoptosis assessment (Fig. 2B). Almost 90% of the cells were viable 48 h after GO exposure while ~30% were viable 48 h after cigarette smoke exposure. The apparent greater toxicity after smoke exposure may be attributed to the inherent complexity of cigarette smoke, which contains numerous chemicals, mutagens, and carcinogens compared to H2O2 alone, though this was not tested. Our hypothesis suggests that the surviving population of cells 48 h postexposure, with prolonged EGFR activation, may then have the potential to become hyperplastic.
Figure 2.
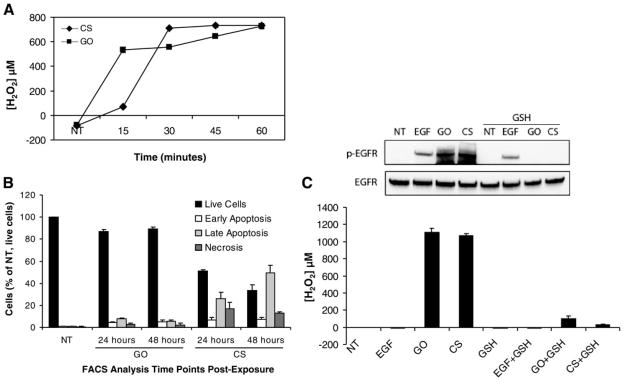
Cigarette smoke and glucose oxidase generate comparable levels of H2O2, which activates the EGFR and is inhibited by glutathione (GSH). A) H2O2 levels were measured in DMEM without phenol red, as described in Materials and Methods, after no treatment (NT) or exposure to 1 U/ml GO or smoke from 1 cigarette at 37°C for the indicated time points. The data points are presented as an average of triplicate measurements of each sample ± SDEV. B) Cells were not treated or exposed to 1 U/ml GO or smoke from 1 cigarette for 45 min, followed by washing with PBS and the addition of fresh growth medium. The cells were then incubated at 37°C, 5% CO2 for 24 or 48 h before staining with annexin V and propidium iodide for FACS analysis. C) Serum-starved HAE cells were not treated (NT) or pretreated with 10 mM GSH for 30 min followed by treatment with 100 ng/ml EGF for 15 min (±GSH), 1 U/ml GO for 30 min (±GSH), or smoke from 1 cigarette (CS) for 45 min (±GSH). One-milliliter aliquots of medium from each dish were taken in triplicate for H2O2 determination. Cell lysates were separated by SDS-PAGE, transferred to a nitrocellulose membrane, and immunoblotted with anti-phospho-tyrosine (PY20) and anti-EGFR (RK2) Abs. p-EGFR, phospho-EGFR.
To determine whether H2O2 from cigarette smoke is involved in EGFR activation, cells were pretreated with 10 mM glutathione (GSH) for 30 min followed by treatment with 100 ng/ml EGF for 15 min, 1 U/ml GO for 30 min, or smoke from one cigarette for 45 min. Aliquots of cell culture medium were taken from the dishes of exposed cells for measurement of H2O2, and the cells were then lysed for immunoblot analysis. H2O2 generated by either GO or cigarette smoke is almost completely ablated in the presence of GSH (Fig. 2C). Furthermore, there is a concomitant inhibition of GO- and cigarette smoke-induced EGFR phosphorylation in the presence of GSH, whereas activation by EGF is not affected (Fig. 2C).
Activation of the EGFR by H2O2 and cigarette smoke is ligand-independent
Previous studies have shown that cigarette smoke activation of the EGFR is due to the activation of metalloproteinases, which cleave and activate EGFR proligands (15–17). We examined this possibility here by pretreating HAE cells with a monoclonal antibody that specifically blocks the EGFR ligand binding site (mAb 225) followed by exposure to 100 ng/ml EGF for 15 min, 1 U/ml GO for 30 min, or smoke from one cigarette for 45 min. Although this antibody blocks the EGFR binding site, it also tends to have a limited ability to activate the EGFR by itself, as seen in the lane for 225 mAb pretreatment followed by no treatment (Fig. 3). However, when comparing EGF, GO and cigarette smoke exposures in the absence and presence of the 225 mAb, only EGF activation of EGFR is attenuated in the presence of the antibody (Fig. 3). Since GO and smoke activation of the EGFR appear unchanged in the presence of the 225 mAb, our results indicate that EGFR activation by cigarette smoke and H2O2 is ligand-independent.
Figure 3.
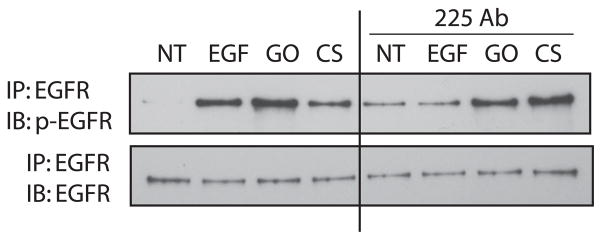
Cigarette smoke activation of the EGFR is ligand-independent. Serum-starved HAE cells were incubated in DMEM with and without 40 nM 225 mAb on ice for 1 h followed by the indicated treatments in the absence or presence of 225 mAb at 37°C. NT, no treatment; EGF, 100 ng/ml for 15 min; GO, 1 U/ml for 30 min; CS, smoke from 1 cigarette for 45 min. EGFR was immunoprecipitated (IP) with anti-EGFR mAb 225; precipitated proteins were separated by SDS-PAGE, transferred to a nitrocellulose membrane, and immunoblotted (IB) with anti-phospho-Tyr (PY20) and anti-EGFR (RK2) Abs. p-EGFR, phospho-EGFR.
Cigarette smoke exposure results in aberrant phosphorylation of the EGFR
We have previously shown that H2O2 exposure resulted in the aberrant phosphorylation of the EGFR (8). Because cigarette smoke generates an amount of H2O2 that is comparable to the level of H2O2 generated by GO in our previous studies (8, 9), we wanted to see whether the pattern of EGFR phosphorylation would be similar between the two treatments. Immunoblot analysis of the EGFR from HAE cells exposed to 100 ng/ml EGF for 15 min, 1 U/ml GO for 30 min, or smoke from 1 cigarette for 45 min shows that GO and smoke exposures result in similar phosphorylation patterns that are different from EGF exposure. Under GO and cigarette smoke, Tyr-845 is hyperphosphorylated, and Tyr-1045 is not phosphorylated (Fig. 4A). With EGF exposure, Tyr-1045 is strongly phosphorylated, whereas Tyr-845 is phosphorylated to a much lesser extent (Fig. 4A). Since Tyr-845 of EGFR is a known Src target, we next determined whether hyperphosphorylation of this site under cigarette smoke was due to Src kinase activation. Figure 4B shows that Src is activated by cigarette smoke in the absence of the Src kinase inhibitors PP1 and PP2 and is inhibited in the presence of these compounds. Concurrently, EGFR Tyr-845 is phosphorylated in the absence of PP1 or PP2, whereas the phosphorylation is inhibited in the presence of these inhibitors, suggesting that Src kinase activation is responsible for the hyperphosphorylation of this site during cigarette smoke exposure (Fig. 4B).
Figure 4.

Cigarette smoke exposure results in aberrant phosphorylation of the EGFR. A) Serum-starved HAE cells were not treated (NT) or exposed to 100 ng/ml EGF for 15 min, 1 U/ml GO for 45 min, or smoke from 1 cigarette (CS) for 45 min. B) Serum-starved HAE cells were not treated (NT) or pretreated with 25 μM PP1 or PP2 for 45 min followed by treatment with smoke from 1 cigarette for 30 min in the absence or presence of the inhibitors as indicated. Cell lysates were separated by SDS-PAGE, transferred to nitrocellulose membranes and immunoblotted with the indicated EGFR phospho-tyrosine-specific Abs, anti-EGFR (RK2) Ab, or Abs against phospho-Src and total Src. p-Tyr, phosphotyrosine; p-Src, phospho-Src.
EGFR exposed to cigarette smoke cannot bind c-Cbl and is not ubiquitinated
c-Cbl is an E3 ubiquitin ligase that plays a crucial role in down-regulating the EGFR. On EGFR activation, c-Cbl associates with phosphorylated Tyr-1045 and ubiquitinates the receptor, marking it for clathrin-mediated endocytosis and recognition by the lysosomal machinery, which results in receptor degradation and signal termination (3–6). EGFR exposed to 100 ng/ml EGF, its cognate ligand, is associated with c-Cbl and ubiquitinated. However, EGFR exposed to smoke from one cigarette for 45 min or to 1 U/ml GO for 30 min is not phosphorylated on Tyr-1045, which renders it unable to associate with c-Cbl and precludes it from being ubiquitinated (Fig. 5A). In addition, Grb2 has been shown to facilitate some binding of c-Cbl to the EGFR, but under H2O2 or smoke exposure, Grb2 and EGFR are no longer associated (Fig. 5B). Therefore, under oxidative stress/smoke exposure, c-Cbl also loses its ability to bind indirectly to the EGFR via Grb2.
Figure 5.

EGFR exposed to cigarette smoke cannot bind c-Cbl and is not ubiquitinated. Serum-starved HAE cells were untreated (NT) or incubated with 100 ng/ml EGF for 15 min, 1 U/ml GO for 30 min, or smoke from 1 cigarette (CS) for 45 min. Cells were lysed and A, the EGFR was immunoprecipitated (IP) from cell lysates using anti-EGFR mAb 225. B) EGFR was immunoprecipitated with anti-EGFR mAb 225 and Grb2 was immunoprecipitated with anti-Grb2 Ab. Immunoprecipitated proteins were separated by SDS-PAGE, transferred to a nitrocellulose membrane, and immunoblotted (IB) with the indicated Abs. Ub, ubiquitin; p-EGFR, phospho-EGFR.
EGFR exposed to cigarette smoke is not internalized as it is under EGF exposure
The process of EGFR down-regulation following ligand binding involves not only receptor phosphorylation and ubiquitination, but also entry of the receptor into clathrin-coated pits and trafficking through early, then late endosomes and into lysosomes. We previously demonstrated that the EGFR exposed to H2O2 was not internalized via clathrin-coated pits and was not degraded (8, 18). Here, we show that cigarette smoke exposure not only results in aberrant phosphorylation of the receptor, similar to H2O2 exposure (Fig. 3), but we also show that the EGFR is not internalized in the same manner as it is under EGF exposure. HAE cells exposed to 100 ng/ml EGF for 15 min show punctate staining, which is representative of internalized EGFR in vesicular structures, which were previously identified to colocalize with EEA1, an early endosomal marker (9), as viewed by laser scanning confocal microscopy (Fig. 6A). Exposure to 1 U/ml GO for 30 min or smoke from one cigarette for 60 min results in membrane staining of the receptor, similar to the untreated cells (Fig. 6A). With GO and cigarette smoke exposure, there is also some perinuclear accumulation of the EGFR. In our previous study, GO-generated H2O2 induced the clathrin-independent caveolae-mediated endocytosis of the EGFR, which was dependent on Src kinase phosphorylation of caveolin-1 (18). To support the possibility of caveolae-mediated EGFR endocytosis here, we demonstrate that caveolin-1 is phosphorylated under exposure to smoke from one cigarette for 30 min, and this is inhibited by a 45-min pretreatment with the Src inhibitors PP1 and PP2 (Fig. 6B).
Figure 6.
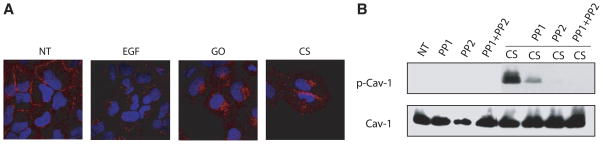
EGFR exposed to cigarette smoke is not internalized in the same manner as EGFR exposed to EGF. A) HAE cells grown to ~70% confluence on coverslips were serum-starved and left untreated (NT) or were exposed to 100 ng/ml EGF for 15 min, 1 U/ml GO for 30 min, or smoke from 1.5 cigarettes (CS) for 60 min. After fixation, the cells were incubated with anti-EGFR mAb 528 for 1 h followed by incubation with Alexa Fluor 594-conjugated Ab (red) for 1 h. Nuclei (blue) were stained with 1 μg/ml DAPI for 3 min. Cells were visualized by laser scanning confocal microscopy at ×60 magnification. Images are merged and are representative of at least 100 cells viewed in each of three separate experiments. B) Serum-starved HAE cells were not treated (NT) or pretreated with 25 μM PP1 or PP2 for 45 min followed by treatment with smoke from 1 cigarette for 30 min in the absence or presence of the inhibitors as indicated. Cell lysates were separated by SDS-PAGE, transferred to a nitrocellulose membrane and immunoblotted with anti-phospho-Cav-1 (Tyr-14), and total Cav-1 Abs. p-Cav-1, phosphocaveolin-1.
Cigarette smoke exposure leads to downstream survival and proliferation signaling
Two well-established mediators of cell proliferation and survival, ERK1/2 and Akt (also known as protein kinase B), are known to be involved in cell transformation when persistently activated (19–21). Reactive oxidant-induced activation of both ERK1/2 and Akt appear to be mediated mainly by growth factor receptors and activation of ERK1/2 by H2O2 in a variety of cells requires EGFR phosphorylation (22). Furthermore, the lack of EGFR turnover has been shown to mediate tumor promotion in nonneoplastic rat liver epithelial cells (23). Here, we show that exposure to 100 ng/ml EGF for 15 min, 1 U/ml GO for 30 min, or smoke from one cigarette for 45 min activates downstream ERK1/2 and Akt signaling (Fig. 7A) but only activation by GO and cigarette smoke is inhibited by GSH (data not shown), further suggesting that H2O2 is the compound in cigarette smoke that activates both the EGFR and these downstream signals. Furthermore, both EGFR and ERK1/2 phosphorylation persist for up to two hours after the removal of the treatment medium exposed to GO or cigarette smoke, whereas removal of EGF returns phosphorylation to near baseline levels at these time points (Fig. 7B). This further demonstrates that these signals may be prolonged due to the inability of the EGFR to be degraded under oxidative stress.
Figure 7.

Exposure to H2O2 and cigarette smoke results in prolonged EGFR activation and downstream proliferation signaling. Serum-starved HAE cells were untreated (NT) or incubated with 100 ng/ml EGF for 15 min, 1 U/ml GO for 30 min or smoke from 1 cigarette (CS) for 45 min. A) Cell lysates were separated by SDS-PAGE, transferred to a nitrocellulose membrane, and immunoblotted with anti-phospho-Tyr (PY20), anti-EGFR (RK2), anti-phospho-Akt, anti-Akt, anti-phospho-ERK1/2, and anti-ERK1/2 Abs. B) Cells were either lysed immediately or were washed with PBS, given fresh DMEM containing 0.5% BSA, and further incubated for 1 or 2 h at 37°C before lysis. Cell lysates were then separated by SDS-PAGE, transferred to a nitrocellulose membrane, and immunoblotted with anti-phospho-Tyr (PY20), anti-EGFR (RK2), anti-phospho-ERK1/2, and anti-ERK1/2 Abs. p-EGFR, phospho-EGFR; p-Akt, phospho-Akt; p-ERK1/2, phospho-ERK1/2.
DISCUSSION
The EGFR has been shown to be activated and aberrantly phosphorylated by H2O2, which results in the loss of c-Cbl-mediated ubiquitination of the receptor. This, in turn, prevents the clathrin-mediated internalization and subsequent degradation of the EGFR (8, 9, 18). Since H2O2 is known to be a major component of the gas phase of cigarette smoke (10), this study was undertaken to determine whether cigarette smoke has a similar effect as H2O2 on EGFR activation and trafficking.
Here, we demonstrate that cigarette smoke generates an amount of H2O2 that is comparable to the amount generated by GO, which was used in our previous studies (8, 9, 18). As such, we not only show that cigarette smoke can activate the EGFR in a dose- and time-dependent manner, but it also aberrantly phosphorylates the receptor. Moreover, the EGFR is not activated and very little H2O2 can be detected in cell culture medium exposed to GO or cigarette smoke in the presence of GSH, an antioxidant that specifically targets H2O2 and organic hydroperoxides. Although catalase targets H2O2 more specifically, we found that it was inhibited by cigarette smoke (data not shown). This phenomenon was also observed by Méndez-Álvarez et al. (24). Therefore, this report makes no claim that H2O2 is the sole component in cigarette smoke to cause the aberrant activation of the EGFR. Rather, we are using our previous knowledge of the effects of H2O2 on the EGFR to draw parallels to the effects of cigarette smoke on this receptor and suggest that CS-generated H2O2 has a major role in the aberrant activation of the EGFR. Whether other hydroperoxide species have a role in activating the EGFR remains to be determined.
We have also addressed the possibility that H2O2, either alone or as a component of cigarette smoke, is eliciting the activation of the EGFR by activating matrix metalloproteinases to cleave EGFR proligands, such as protransforming growth factor-α and amphiregulin, thereby inducing ligand-dependent EGFR activation (15–17). Preincubation of HAE cells with the EGFR neutralizing 225 mAb followed by exposure to cigarette smoke or H2O2 showed no inhibition of EGFR activation, whereas EGF activation of the receptor was inhibited, indicating that both cigarette smoke and H2O2 activation of the EGFR are ligand-independent (Fig. 3).
When HAE cells are exposed to either GO or cigarette smoke, the EGFR is phosphorylated at its known autophosphorylation sites (tyrosines 1068, 1086, and 1173), but is not phosphorylated at Tyr-1045, the c-Cbl binding site (Fig. 4). This has important ramifications on the stability and trafficking of the EGFR under oxidative stress. We and others have shown that both c-Cbl and Grb2 interactions with the EGFR are necessary for the ubiquitination and clathrin-mediated internalization of the EGFR (7, 18). Cigarette smoke not only abrogates the direct interaction of c-Cbl with the EGFR, via the loss of Tyr-1045 phosphorylation, but it also prevents the indirect interaction of c-Cbl with the EGFR via Grb2 (Fig. 5). Thus, we believe that the EGFR is precluded from entering clathrin-coated pits under exposure to cigarette smoke in the same way it is precluded from doing so under H2O2 exposure (18). Indeed we demonstrate by confocal microscopy the internalization of the EGFR under EGF treatment, but under GO and cigarette smoke, the EGFR remains at the plasma membrane (Fig. 6).
Interestingly, our confocal data show a population of the EGFR under cigarette smoke and H2O2 exposure accumulating in a perinuclear region (Fig. 6). We have observed this phenomenon in the past and demonstrated that under H2O2-induced oxidative stress, Src family kinases are activated, which then phosphorylate caveolin-1 and leads to the caveolae-mediated trafficking of the EGFR to a perinuclear region where it remains active and may thereby contribute to the proliferative signaling that is linked to tumorigenesis (18). Similar to H2O2, we not only demonstrate that cigarette smoke can induce the proliferative signaling molecules Akt and ERK1/2, but this signaling continues for at least two hours after the exposure (Fig. 7). Thus far, we have demonstrated that mainstream cigarette smoke, which produces significant amounts of H2O2 in cell culture medium during exposure, is behaving analogously to H2O2 generated by glucose oxidase. We have demonstrated that both cigarette smoke and GO-generated H2O2 induce the aberrant phosphorylation of the EGFR, which occurs in a ligand-independent manner, and precludes the receptor from being internalized in the same way as it is under EGF exposure (see schematic in Fig. 8). The parallel effects of H2O2 and cigarette smoke lead us to hypothesize that cigarette smoke is also inducing the caveolae-mediated trafficking of the EGFR to a perinuclear region where it will remain active and contribute to prolonged downstream proliferative signaling, which is currently under investigation.
Figure 8.

Schematic model of EGFR stabilization by cigarette smoke. Under EGF exposure, c-Cbl can bind directly and indirectly to the EGFR via phospho-Tyr-1045 and Grb2, respectively, allowing receptor ubiquitination, clathrin-mediated endocytosis, and lysosomal degradation. Under cigarette smoke exposure, EGFR Tyr-1045 is not phosphorylated and Grb2 can no longer interact with EGFR; therefore, the receptor does not follow the same degradation pathway that is induced by EGF. Instead, the EGFR is stabilized at the plasma membrane and also traffics to a perinuclear compartment where it may remain active and contribute to prolonged signaling. Ub, ubiquitin; circled P, phospho-Tyr.
Acknowledgments
This work was supported by grants from the Tobacco-Related Disease Research Program (13FT-0126 to E.M.K.) and the U.S. National Institutes of Health (T32 HL07013 to E.M.K.; HL-71871 and HL-66189 to T.G.).
References
- 1.Franklin WA, Veve R, Hirsch FR, Helfrich BA, Bunn PA., Jr Epidermal growth factor receptor family in lung cancer and premalignancy. Semin Oncol. 2002;29:3–14. doi: 10.1053/sonc.2002.31520. [DOI] [PubMed] [Google Scholar]
- 2.Eccles SA, Modjtahedi H, Box G, Court W, Sandle J, Dean CJ. Significance of the c-erbB family of receptor tyrosine kinases in metastatic cancer and their potential as targets for immunotherapy. Invasion Metastasis. 1994;14:337–348. [PubMed] [Google Scholar]
- 3.Huang F, Kirkpatrick D, Jiang X, Gygi S, Sorkin A. Differential regulation of EGF receptor internalization and degradation by multiubiquitination within the kinase domain. Mol Cell. 2006;21:737–748. doi: 10.1016/j.molcel.2006.02.018. [DOI] [PubMed] [Google Scholar]
- 4.Jiang X, Sorkin A. Epidermal growth factor receptor internalization through clathrin-coated pits requires Cbl RING finger and proline-rich domains but not receptor polyubiquitylation. Traffic. 2003;4:529–543. doi: 10.1034/j.1600-0854.2003.t01-1-00109.x. [DOI] [PubMed] [Google Scholar]
- 5.Mosesson Y, Shtiegman K, Katz M, Zwang Y, Vereb G, Szollosi J, Yarden Y. Endocytosis of receptor tyrosine kinases is driven by monoubiquitylation, not poly-ubiquitylation. J Biol Chem. 2003;278:21323–21326. doi: 10.1074/jbc.C300096200. [DOI] [PubMed] [Google Scholar]
- 6.Levkowitz G, Waterman H, Ettenberg SA, Katz M, Tsygankov AY, Alroy I, Lavi S, Iwai K, Reiss Y, Ciechanover A, Lipkowitz S, Yarden Y. Ubiquitin ligase activity and tyrosine phosphorylation underlie suppression of growth factor signaling by c-Cbl/Sli-1. Mol Cells. 1999;4:1029–1040. doi: 10.1016/s1097-2765(00)80231-2. [DOI] [PubMed] [Google Scholar]
- 7.Huang F, Sorkin A. Growth factor receptor binding protein 2-mediated recruitment of the RING domain of Cbl to the epidermal growth factor receptor is essential and sufficient to support receptor endocytosis. Mol Biol Cell. 2005;16:1268–1281. doi: 10.1091/mbc.E04-09-0832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ravid T, Sweeney C, Gee P, Carraway KL, III, Goldkorn T. Epidermal growth factor receptor activation under oxidative stress fails to promote c-Cbl mediated down-regulation. J Biol Chem. 2002;277:31214–31219. doi: 10.1074/jbc.M204677200. [DOI] [PubMed] [Google Scholar]
- 9.Ravid T, Heidinger JM, Gee P, Khan EM, Goldkorn T. c-Cbl-mediated ubiquitinylation is required for epidermal growth factor receptor exit from the early endosomes. J Biol Chem. 2004;279:37153–37162. doi: 10.1074/jbc.M403210200. [DOI] [PubMed] [Google Scholar]
- 10.Nakayama T, Kodama M, Nagata C. Generation of hydrogen peroxide and superoxide anion radical from cigarette smoke. Gann. 1984;75:95–98. [PubMed] [Google Scholar]
- 11.Thurman RG, Ley HG, Scholz R. Hepatic microsomal ethanol oxidation. Hydrogen peroxide formation and the role of catalase. Eur J Biochem. 1972;25:420–430. doi: 10.1111/j.1432-1033.1972.tb01711.x. [DOI] [PubMed] [Google Scholar]
- 12.Lavrentiadou SN, Chan C, Kawcak TN, Ravid T, Tsaba A, van der Vliet A, Rasooly R, Goldkorn T. Ceramide-mediated apoptosis in lung epithelial cells is regulated by glutathione. Am J Respir Cell Mol Biol. 2001;25:676–684. doi: 10.1165/ajrcmb.25.6.4321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bao J, Alroy I, Waterman H, Schejter ED, Brodie C, Gruenberg J, Yarden Y. Threonine phosphorylation diverts internalized epidermal growth factor receptors from a degradative pathway to the recycling endosome. J Biol Chem. 2000;275:26178–26186. doi: 10.1074/jbc.M002367200. [DOI] [PubMed] [Google Scholar]
- 14.Diamonti AJ, Guy PM, Ivanof C, Sweeney C, Carraway KL., III An RBCC protein implicated in maintenance of steady-state neuregulin receptor levels. Proc Natl Acad Sci U S A. 2002;99:2866–2287. doi: 10.1073/pnas.052709799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhang Q, Adiseshaiah P, Reddy SP. Matrix metalloproteinase/epidermal growth factor receptor/mitogen-activated protein kinase signaling regulate fra-1 induction by cigarette smoke in lung epithelial cells. Am J Respir Cell Mol Biol. 2005;32:72–81. doi: 10.1165/rcmb.2004-0198OC. [DOI] [PubMed] [Google Scholar]
- 16.Shao MXG, Nakanaga T, Nadel JA. Cigarette smoke induces MUC5AC mucin overproduction via tumor necrosis factor-alpha-converting enzyme in human airway epithelial (NCI-H292) cells. Am J Physiol Lung Cell Mol Physiol. 2004;287:L420–L427. doi: 10.1152/ajplung.00019.2004. [DOI] [PubMed] [Google Scholar]
- 17.Lemjabbar H, Li D, Gallup M, Sidhu S, Drori E, Basbaum C. Tobacco smoke-induced lung cell proliferation mediated by tumor necrosis factor alpha-converting enzyme and amphiregulin. J Biol Chem. 2003;278:26202–26207. doi: 10.1074/jbc.M207018200. [DOI] [PubMed] [Google Scholar]
- 18.Khan EM, Heidinger JM, Levy M, Lisanti MP, Ravid T, Goldkorn T. Epidermal growth factor receptor exposed to oxidative stress undergoes Src- and caveolin-1-dependent perinuclear trafficking. J Biol Chem. 2006;281:14486–14493. doi: 10.1074/jbc.M509332200. [DOI] [PubMed] [Google Scholar]
- 19.Vivanco I, Sawyers CL. The phosphatidylinositol 3-kinase AKT pathway in human cancer. Nat Rev Cancer. 2002;2:489–501. doi: 10.1038/nrc839. [DOI] [PubMed] [Google Scholar]
- 20.Luo J, Manning BD, Cantley LC. Targeting the PI3K-Akt pathway in human cancer: rationale and promise. Cancer Cell. 2003;4:257–262. doi: 10.1016/s1535-6108(03)00248-4. [DOI] [PubMed] [Google Scholar]
- 21.Vicent S, López-Picazo JM, Toledo G, Lozano MD, Torre W, Garcia-Corchón C, Quero C, Soria JC, Martín-Algarra S, Manzano RG, Montuenga LM. ERK1/2 is activated in non-small-cell lung cancer and associated with advanced tumours. Br J Cancer. 2004;90:1047–1052. doi: 10.1038/sj.bjc.6601644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Henson ES, Gibson SB. Surviving cell death through epidermal growth factor (EGF) signal transduction pathways: Implications for cancer therapy. Cell Signal. 2006;18:2089–2097. doi: 10.1016/j.cellsig.2006.05.015. [DOI] [PubMed] [Google Scholar]
- 23.Huang RP, Peng A, Golard A, Hossain MZ, Huang R, Liu YG, Boynton AL. Hydrogen peroxide promotes transformation of rat liver non-neoplastic epithelial cells through activation of epidermal growth factor receptor. Mol Carcinog. 2001;30:209–217. doi: 10.1002/mc.1030. [DOI] [PubMed] [Google Scholar]
- 24.Méndez-Álvarez E, Soto-Otero R, Sánchez-Sellero I, López-Rivadulla L. In vitro inhibition of catalase activity by cigarette smoke: relevance for oxidative stress. J Appl Tox. 1998;18:443–448. doi: 10.1002/(sici)1099-1263(199811/12)18:6<443::aid-jat530>3.0.co;2-p. [DOI] [PubMed] [Google Scholar]