

Abstract
For several decades, acute kidney injury (AKI) was generally considered a reversible process leading to complete kidney recovery if the individual survived the acute illness. Recent evidence from epidemiologic studies and animal models, however, have highlighted that AKI can lead to the development of fibrosis and facilitate the progression of chronic renal failure. When kidney injury is mild and baseline function is normal, the repair process can be adaptive with few long-term consequences. When the injury is more severe, repeated, or to a kidney with underlying disease, the repair can be maladaptive and epithelial cell cycle arrest may play an important role in the development of fibrosis. Indeed, during the maladaptive repair after a renal insult, many tubular cells that are undergoing cell division spend a prolonged period in the G2/M phase of the cell cycle. These tubular cells recruit intracellular pathways leading to the synthesis and the secretion of profibrotic factors, which then act in a paracrine fashion on interstitial pericytes/fibroblasts to accelerate proliferation of these cells and production of interstitial matrix. Thus, the tubule cells assume a senescent secretory phenotype. Characteristic features of these cells may represent new biomarkers of fibrosis progression and the G2/M-arrested cells may represent a new therapeutic target to prevent, delay or arrest progression of chronic kidney disease. Here, we summarize recent advances in our understanding of the biology of the cell cycle and how cell cycle arrest links AKI to chronic kidney disease.
INTRODUCTION
Acute kidney injury (AKI) has long been thought to be a reversible process whereby the kidney had the ability to completely recover after an ischemic or a toxic insult that results in lethal cellular damage. It has become clear, however, during the last decade that evolving evidence from animal models and human epidemiologic studies have linked AKI to chronic kidney disease (CKD) [1–4]. Furthermore, AKI can precipitate end-stage renal disease when the baseline glomerular filtration rate (GFR) is already decreased [5, 6]. This relationship between AKI and CKD is bidirectional as CKD predisposes to AKI [4].
The pathophysiological processes brought into play after AKI to restore a functional nephron are partially known. After injury, tubular cells, and especially proximal tubular cells, lose their polarity and brush border [7]; membrane proteins such as β-integrins are mislocated [8, 9] and some tubule cells die particularly if the injury is sustained [10]. During the normal process of repair after AKI, surviving tubular cells undergo dedifferentiation, then migrate along the basement membrane, proliferate and finally differentiate to restore a functional nephron [11–13]. It is now accepted that in many cases, however, this extraordinary ability to completely recover after injury does not occur and AKI leads to abnormal repair with persistent parenchymal inflammation, fibroblast proliferation and excessive deposition of extracellular matrix [10] (Figure 1). Several risk factors for the development of CKD after AKI have been described including the kind of insult, the duration of exposure and the GFR before injury [1, 3, 4, 14]. It is also likely that aging represents an important risk factor [15].
FIGURE 1:

Normal and abnormal repair after AKI. After injury, tubular cells, and especially proximal tubular cells, lose their polarity and brush border; membrane proteins and tubule cells die if the injury is sustained. During the normal process of repair after AKI, surviving tubular cells undergo dedifferentiation, then migrate along the basement membrane, proliferate and finally differentiate to restore a functional nephron. However, in some conditions, the recovery process after injury becomes maladaptive and AKI leads to abnormal repair with persistent parenchyma inflammation, fibroblast proliferation and excessive deposition of extracellular matrix. CTGF, connective tissue growth factor; TGF-β1, transforming growth factor beta-1.
The mechanisms involved in the development of fibrosis have not been completely deciphered. While there has been recognition of tubule cell involvement in fibrosis, much of the attention on the tubular epithelial cell in this process has been focused on epithelial to mesenchymal transformation (EMT) whereby epithelial cells are proposed to transdifferentiate to myofibroblasts [16]. This concept has been brought into question more recently, however, by a number of studies [12, 17], including those using lineage tracing, that fail to find evidence of transdifferentiation [17, 18]. As the focus has moved away from EMT, there has been a renewed interest in paracrine actions of the tubules which contribute to inflammation and activation of interstitial fibroblasts and perivascular pericytes [19]. We propose that cellular senescence plays a major role in the pathophysiology of CKD. Acute tubular injury, and its associated effects on the epithelial cell, can lead to a maladaptive repair and a chronic inflammatory state. DNA damage can lead to senescence. Kidney injury secondary to ischemia/reperfusion or toxins can lead to DNA damage. In addition, however, there are a number of other factors that can lead to cell cycle arrest and tubular cell senescence in the absence of DNA damage. Repeated proliferation and recurrent exposure to reactive oxygen species, as might be characteristic of repeated insults underlying CKD and/or the aging process, can lead to telomere shortening and senescence [20]. Senescent cells are very metabolically active and are relatively resistant to apoptosis. Our laboratory has reported that severe AKI leads to tubular cell cycle arrest in the G2/M phase of the cell cycle with activation of the ‘senescence-associated secretory phenotype’ (SASP). This results in secretion of pro-proliferative and profibrotic factors, which can then lead to chronic inflammation, proliferation of pericytes/perivascular fibroblasts which then become myofibroblasts and deposition of collagen with fibrosis, subsequent vascular rarefaction and CKD [2]. Here we will review the evidence for cell cycle involvement during AKI, the links between injury, potential DNA damage and G2/M arrest, and suggest new therapeutic approaches and targets that derive from recent studies.
CHARACTERISTICS OF THE CELL CYCLE
Cell division is the result of a tightly regulated sequence of events leading to the birth of two daughter cells. It consists of four distinct phases with specific characteristics (G0–G1, S, G2 and M) (Figure 2). G0 is a resting stage where the cell has left the cell cycle and is not in the process of cell division. While it has generally been considered that under normal conditions tubular epithelial cells are in G0, there are some data in rats suggesting that tubular epithelial cells are poised in G1 [21, 22]. When cells enter into the process of cell division from G0, the G1 phase is characterized by cell growth in size and synthesis of mRNA and proteins required for DNA duplication. This phase is followed by the S phase where the DNA is replicated. Then, the cell enters into the G2 phase, which is a phase of rapid cell growth and protein synthesis before the mitosis phase (M phase). During the M phase, cell growth stops and there is an ordered progression to cell division.
FIGURE 2:
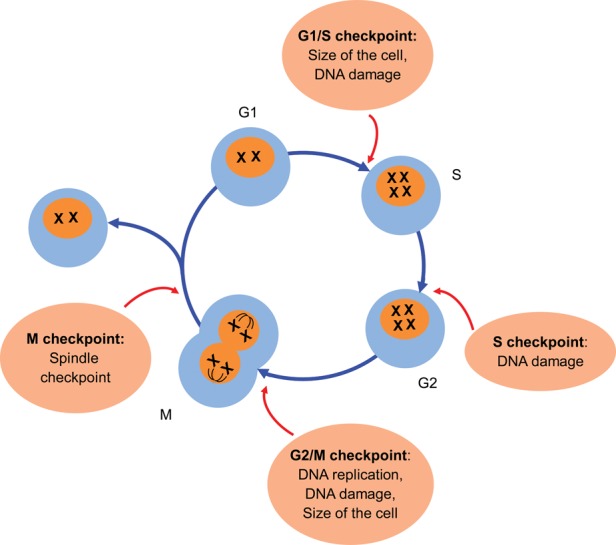
Cell cycle and checkpoints. Cell division is the result of a tightly regulated sequence of events leading to the birth of two daughter cells. It consists of four distinct phases with specific changes (G0–G1, S, G2 and M). The progression through the cell cycle is controlled by cyclic proteins, called cyclins, cyclin-dependent kinases and their inhibitors.
The progression through the cell cycle is finely controlled by cyclic proteins, called cyclins, cyclin-dependent kinases (CDKs) and their inhibitors (CDK inhibitors, CKIs, such as p15, p16INK4a, p21 and p27) [23]. The cyclins were identified in the early 1980s by their cyclic oscillations during the sea urchin cell cycle [24]. At baseline, when CDKs are uncoupled from cyclins, they have very low levels of kinase activity [23]. Binding of a CDK to the cyclin leads to the formation of a complex that leads to CDK activation and serine/threonine phosphorylation of their downstream targets to facilitate progression through the cell cycle [23]. It has become increasingly recognized that cyclins, CDKs and CKIs do more than regulate the cell cycle. They play important roles in transcription, epigenetic regulation and metabolism, and some of these effects are unrelated to kinase activity or cyclin/CDK complex formation [25]. While these additional effects to do not directly affect the cell cycle machinery, they do relate to proliferation and repair.
Importantly, to ensure the fidelity of the cell cycle process, many quality control steps called checkpoints are present during cell division [26] (Figure 2). These checkpoints respond to problems that have to be fixed before allowing safe progression through the cell cycle. For example, the G1 checkpoint ensures that the size of the cell is large enough to progress into the S phase and checks for the presence of DNA damage [26]. The intra-S phase checkpoint is activated by the occurrence of DNA damage during the S phase or by unrepaired DNA damage that escapes the G1/S checkpoint [26]. The G2 checkpoint verifies three important things: (i) that DNA is replicated; (ii) that all replication errors have been repaired and (iii) that the size of the cell is sufficient enough to divide [26]. The last checkpoint is the spindle checkpoint which ensures that chromosomes are well aligned on the spindle, ready for mitosis [26]. All these checkpoints have some degree of redundancy, but each of them has some relative specificity. How each checkpoint is controlled provides insights into the complexity of this important system that regulates movement through the cell cycle (Figure 3).
FIGURE 3:

Pathways involved to block cell cycle progression. Brief summary of the different pathways involved to block cell cycle progression during the G2/M phase, including either DNA damage (black arrows) or cytokine pathway (red arrows). TGF-β, transforming growth factor beta; ATM, ataxia telangiectasia mutated; ATR, ataxia telangiectasia and Rad3-related; Chk, checkpoint kinase; Cdc, cell division cycle; MAP kinases, mitogen-activated protein kinases; CDK, cyclin-dependent kinases; Rb, retinoblastoma protein.
The regulatory pathways involved in the G1/S checkpoint are multiple, complex and interdependent. Among them, one pathway involves the Ataxia telangiectasia mutated (ATM)/ataxia telangiectasia and Rad3-related protein (ATR) and a second involves the p16INK4a protein, a CKI [27]. When DNA damage is detected, ATM and/or ATR, which belong to the phosphatidylinositol 3-kinase family, are activated and phosphorylate several downstream targets, including p53 and checkpoint kinase 2 (Chk2) [28, 29]. Chk2, in turn, phosphorylates and inactivates Cdc25a phosphatase [28]. This phosphorylation leads to the nuclear exclusion and proteolytic degradation of Cdc25a. Phosphorylated p53 results in its stabilization and accumulation. With phosphorylation p53 leads to the transcription of p21, a protein belonging to the family of CDIs, which in turn inhibits the Cdk2/cyclin E complex and favors cyclin E degradation, preserving the association of the tumor suppressor retinoblastoma protein (Rb) and the transcription factor E2F [30]. A second pathway involved in the G1/S checkpoint is activated by several mechanisms including replicative senescence and the transforming growth factor-β (TGF-β ) [31]. This pathway involves p16INK4a31. During the cell cycle, Cdk4/6 interacts with cyclin D1, leading to the phosphorylation of Rb. This relieves the inhibition of the transcription factor E2F, which in turn favors the expression of cyclin E that binds Cdk2 and allows the G1–S phase transition [32]. p16 binds to Cdk4/6, blocking Cdk4/6 interaction with cyclin D1 and hence blocking cell cycle progression [32].
The S checkpoint involves two different pathways that slow down and can interrupt ongoing DNA synthesis, both of which are controlled by the ATM/ATR signaling machinery. The first one involves the activation of Chk2, which in turn inhibits Cdc25a, as previously described, and Cdk2 activity, leading to the blocking of Cdc45 onto chromatin, which is required for the recruitment of DNA polymerase α [33]. The second one is more complex and is thought to be important in the arrest of DNA replication in response to ionizing radiation [33]. This pathway involves ATM/NBS1 (Nibrin)/BRCA1 (Breast Cancer 1)/FANCD2 (Fanconi Anemia Group 2)/SMC1 (Structural Maintenance of Chromosomes 1). Hence, after DNA damage, NBS1 and BRCA1 migrate to the sites of DNA breaks where ATM is recruited and activated leading to the phosphorylation of SMC1 [33]. This activation favors cell cycle arrest and the recovery process [33].
The G2/M checkpoint, often activated by DNA damage, is an important checkpoint at a decision point of the cell to progress through mitosis. Depending on the type of DNA damage, the ATM/Chk2/Cdc25 signal transduction pathway [34] and/or the ATR/Chk1/Cdc25 pathway [35, 36] is/are activated to arrest the cell cycle. DNA damage is not the only way to induce G2 arrest. Indeed, many types of stress can induce the activation of p38 MAPK/MK2 and subsequent inactivation of Cdc25 [37]. Similarly, TGF-β1 has been recently shown to induce G2/M arrest in kidney proximal tubule cells through p21 induction in a model of CKD [38].
Several markers can be used to identify the different phases of the cell cycle in cells and tissues. Proliferative cell nuclear antigen (PCNA), a cofactor of the DNA polymerase δ, is mainly expressed during the G1, S and G2 phases of the cell cycle [2]. Its expression is very weak during the M phase. PCNA is upregulated in the late G1 phase, is maximal during the S phase and then declines during the G2/M phase. Ki-67 is expressed during all phases of the cell cycle [39]. Quiescent cells in the G0 phase do not express Ki-67 [40]. 5-Bromo-2′-deoxyuridine (BrdU) is a synthetic nucleoside that is an analog of thymidine that is incorporated into the newly synthesized DNA during the S phase of the cell cycle. BrdU is characterized by its long-term retention in dividing cells with passage to their daughter cells.
The G2/M phase can be detected by the phosphorylation of histone H3 on the residue Ser10 (p-H3) [2]. Furthermore, G2 versus M phase can be differentiated according to the staining patterns of p-H3 where G2 is characterized by patchy staining and M phase by a more uniform staining pattern [2]. Finally, the number of cells in the G1 phase can be quantitated by subtracting the number of cells in the S phase (BrdU positive) and G2/M phase (p-H3 positive) from the total number of proliferating cells (Ki-67 positive) [2].
THE CELL CYCLE IN KIDNEY PHYSIOLOGY AND PATHOPHYSIOLOGY
During normal physiological conditions, tubular epithelial cells divide at a very low rate as revealed by PCNA and Ki-67 immunostaining [41, 42]. This tightly regulated proliferation rate allows the kidney to replace the very few cells that die daily under normal conditions. After AKI, however, the rate of dividing cells dramatically increases to replace necrotic/apoptotic cells. This is particularly prevalent in the S3 segment of the proximal tubules [10, 42, 43]. In the S3 segment, cells are particularly sensitive to injury due to: (i) high metabolic demand, (ii) relative hypoxia in the outer medulla and (iii) exposure to high concentrations of intra-tubular toxins due to water absorption of glomerular filtrate in the upstream S1 and S2 segments in the cortex. After mild injury, surviving tubular cells proliferate to cover the exposed basal membrane and restore the cell number [12, 42, 43]. In combination with proliferation, these cells also dedifferentiate transiently, expressing embryologic markers such as vimentin [42, 43], and will then redifferentiate into specialized tubular cells leading to the restoration and repair of the nephron [13].
Few cell cycle regulators have been studied during the recovery phase after AKI. Among them, p21 (CIP1/WAF1) was one of the first proteins explored. Early after AKI, p21, which is downstream of p53, is upregulated both at the mRNA and protein levels in the kidney [44]. Interestingly, the effect of p21 seems to be different during AKI or CKD progression. Indeed, p21 seems to be protective during AKI, as assessed by the more pronounced kidney dysfunction, more severe kidney damage and the higher rate of mortality rate observed in p21−/− mice compared with wild-type mice [45, 46]. In contrast, p21−/− mice developed less pronounced histologic lesions after sub-total nephrectomy with enhanced tubular proliferation compared with wild-type mice [47]. p53, which regulates the transcription of p21, was also found to be upregulated in the kidney after AKI and its inhibition or gene deletion reduces kidney lesions [2, 48–51]. In contrast to the moderate amount of information related to p53 and p21, very few data are available regarding other proteins that regulate the cell cycle. After ischemic injury, mRNA and protein levels of cyclins D1, D3 and B, mRNA level of cyclin A, protein levels and the activities of CDK4 and CDK2 increase with a temporal relationship consistent with tubular cell proliferation [52]. The precise role of each cyclin and its regulators during AKI deserves a good deal more investigation.
DNA DAMAGE AND G2/M ARREST DURING AKI
We have identified a role for the cell cycle in maladaptive repair after AKI. We proposed that epithelial cell cycle arrest can lead to fibrosis progression, thus providing a pathophysiological link between acute injury and CKD [2]. As stated above, after mild tubular injury, survivor epithelial cells enter into the cell cycle and proliferate to regenerate a structurally and functionally repaired nephron [12, 13]. This repair process can be maladaptive, however, especially if the injury is more severe, involves DNA damage or occurs on the backdrop of chronic injury and increased cell senescence as may occur with chronic injury or in aging. Maladaptive repair will lead to incomplete structural and functional restoration of kidney tissue with persistent low grade inflammation, activation of perivascular fibroblasts (pericytes) [17], accumulation of myofibroblasts, vascular rarefaction [53], intermittent chronic ischemia, increased production of interstitial matrix and development of fibrosis. Indeed, under particularly stressful conditions, some tubular cells stay arrested in the G2/M phase leading to the production of fibrotic factors such as connective tissue growth factor and TGF-β [2, 38, 54, 55]. In this setting, the percentage of cells that have entered the cell cycle that undergo G2/M arrest correlates with the development of fibrosis [2, 38, 54, 55]. Moreover, pharmacologic intervention which leads to an increased number of tubular cells arrested in the G2/M phase after AKI worsens kidney fibrosis, whereas interventions which enhance the movement through G2/M are associated with less fibrosis [2, 38, 54–57]. Cells trapped in G2/M may represent a new histologic biomarker for CKD progression.
Thus, although the epithelial cell does not transdifferentiate into a myofibroblast, the damaged proximal epithelial cell can contribute in important ways to the fibrosis process via paracrine mechanisms that are potentiated by a state of accelerated senescence characterized by G2/M arrest and production of profibrotic factors. We demonstrated this by using multiple models of AKI, including mild and moderate bilateral ischemia/reperfusion, unilateral ischemia/reperfusion, aristolochic acid administration and unilateral ureteral obstruction [2]. We developed histochemical approaches to map the cell cycle progression in vivo as described previously in this review. There were large differences in the distribution of cell cycle stage over time (42 days) between insults that were mild and those that were severe leading to fibrosis [2] (Figure 4). G2/M-arrested proximal tubular epithelial cells activated the c-jun NH2-terminal kinase (JNK) signaling cascade that acts to upregulate profibrotic cytokine production. The fibrotic fate of the injured kidney can partially be abrogated with a JNK inhibitor, a p53 inhibitor or the removal of the contralateral kidney. The latter approach was employed because of the potential facilitation of repair in the remaining kidney due to contralateral nephrectomy.
FIGURE 4:
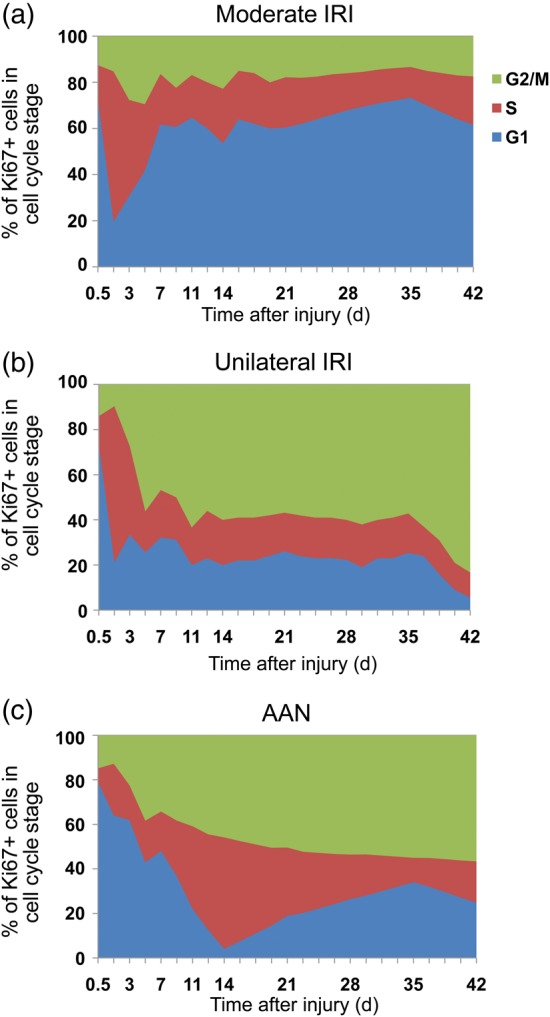
Cell cycle distribution of tubular cells in different models of AKI. Cell cycle distribution (G1, S and G2/M) of tubular cells in moderate ischemia–reperfusion injury (IRI) (a), unilateral ischemia reperfusion injury (UIRI) (b) and acute aristolochic acid toxic nephropathy (AAN) (c) models as a function of time after the insult. Extract from [2].
The G2/M phase arrest can, at least in part, be related to DNA damage associated with severe injury secondary to severe ischemia, aristolochic acid exposure or unilateral ureteral obstruction. The presence of DNA damage after these insults was consistent with the measured activation of the ATM/ATR pathway with implications for their downstream targets, Chk1 and Chk2 [2]. It is important to note that activation of ATM/ATR pathways with downstream G2/M arrest and secretion of fibrotic factors has been observed in rodent models, but may also occur in human kidney diseases. Indeed, patients with FAN1 (which encodes the Fanconi anemia-associated nuclease 1 protein) mutation develop progressive karyomegalic interstitial nephritis characterized by tubular atrophy, tubular microcysts and fibrosis [58]. FAN1 is a protein involved in DNA repair after damage. Cells exhibiting the FAN1 mutation [58] or FAN1 depletion [59] are more susceptible to DNA damage and genome instability, and are arrested in late G2 phase of the cell cycle [58]. An important role played by DNA damage in kidney diseases and fibrosis has also been recently reinforced by the discovery of a mutation in human genes encoding for proteins involved in DNA repair process such as the ATM pathway [60]. This leads to a nephronophthisis-related ciliopathy phenotype with DNA damage and an aging-degenerative profile [60]. Finally, in G2/M arrested cells, DNA damage and repair processes, assessed by the expression of γ-H2AX, correlate with fibrosis progression in kidney transplant recipients [58].
Interestingly, DNA damage is also observed under physiological conditions in the kidney without fibrosis. Indeed, DNA damage is found in the renal inner medulla where cells are physiologically exposed to a high interstitial concentration of NaCl [61]. When the interstitial concentration of NaCl in the medulla increases, DNA damage occurs but repair is rapid [61]. This rapid repair contrasts with maladaptive repair after injury described above. In fact, it has been shown that high concentration of NaCl induces double-strand DNA breaks in specific regions of the genome [62]. Importantly, these regions, called ‘gene deserts’, are void of genes, a fact which may explain the reduced consequences of such DNA breaks [62]. It is tempting to speculate that during AKI, DNA breaks occur in important genes for cell survival, and certainly their localization in the genome deserves more investigation.
It is also of note that aging is associated with a decreased capacity to repair and regenerate injured tissues including kidneys [51, 63]. Indeed, cellular senescence is a state of irreversible growth arrest that can also occur when oncogenic DNA damage occurs. These cells are metabolically active, secreting pro-inflammatory cytokines, chemokines and proteases. In the context of cancer, this state of cell senescence that is associated with secretion of inflammatory and profibrotic factors has been labeled the ‘SASP’. This cell state can cause local and systemic inflammation, disrupt tissue architecture and reinforce G2/M arrest through cytokine secretion [20, 64]. Finally, as we have previously mentioned, DNA damage is not the only way to block cells in the G2/M phase, and many factors such as IL-8 [65], secreted by the senescent cell itself, or a neighboring one [66], bind to CXCR2 [65] and activate several pathways including NF-κB [65] and the p38 MAPK/MK2 pathway [37, 65]. Therefore, ischemic or toxic injury to the kidney can also result in G2/M arrest via mechanisms independent of DNA damage.
G2/M-ARRESTED CELLS AS A NEW THERAPEUTIC TARGET
This newly recognized contributor to the pathophysiology of fibrosis progression opens new therapeutic opportunities and, similar to anticancer strategies, this G2/M checkpoint emerges as an attractive therapeutic target. One approach to therapy targeting cell cycle arrest involves preventing cells from activating the downstream Chks that trigger G2/M arrest. Approaches could include blockade of the ATM pathway, which we have shown results in decreased G2/M arrest and decreased profibrotic growth factor release from cells treated with aristolochic acid. It is also very interesting that it has been shown that genetic deletion of one allele of the p65 subunit of NF-κB or pharmacologic inhibition of the NF-κB-activating kinase, IKK, prevented oxidative DNA damage, delayed cellular senescence and delayed age-related manifestations in a progeroid mouse where accelerated aging is the result of defective DNA repair [67].
A second therapeutic approach is to help cells overcome the G2/M checkpoint [2, 54]. This strategy has already been successfully used to reduce kidney fibrosis in rodent models. Indeed, either pharmacologic interaction, by using p53 inhibition [2, 50], histone deacetylase inhibitors [54], or a ‘mechanical’ approach using unilateral nephrectomy of the healthy kidney after unilateral ischemia reperfusion injury [2, 68] resulted in reduced fibrosis. In the latter case, it has been long recognized in the rat [68] that the repair process after unilateral ischemic injury is characterized by loss of functional nephrons and kidney atrophy. If the contralateral kidney is then removed (even 2 weeks after the ischemic episode), the post-ischemic kidney increases in size and its creatinine clearance markedly increases due to increase in single-nephron GFR as well as recruitment of additional functional nephrons. With these various maneuvers, the exact mechanisms that push the cell to enter into the cell cycle are not always known, and the outcome of these cells during and after mitosis is unclear.
A third approach may be to facilitate apoptosis of senescent cells. A key question is why cells arrested in G2/M do not undergo apoptosis? There are likely other systems brought into play which block apoptosis. These pathways are likely maladaptive and profibrotic and, when identified, may themselves represent additional therapeutic targets to reduce the progression of CKD.
A fourth way to counteract the effects of G2/M cell cycle arrest is to block pathways that are involved in profibrotic cytokine secretion. This will leave these cells in a state that we would call ‘anergic’, similar to auto-reactive B cells after selection in the thymus. Indeed, the JNK pathway is activated in G2/M-arrested cells, and blocking this pathway, either in vitro or in vivo, reduces the amount of profibrotic cytokine secretion [2]. It is important to note that this approach did not modify the prevalence of G2/M cells [2], but rather affected c-Jun as a downstream mediator of the profibrotic effect.
A fifth approach to the treatment of cellular senescence an its consequences would be to selectively deplete the senescent cells. Of course, if the targeting is not specific this could potentially result in loss of cells, which normally do not divide or divide at very low rates, such as neurons. Nevertheless, a novel method has been used to selectively remove senescent cells using a genetic approach [64]. Cell cycle-arrested cells express elevated levels of p21 and p16INK4a. Baker et al. created a transgenic mouse in which cells expressing p16INK4a were selectively depleted by administration of a drug. In a BubR1 progeroid mouse background, life-long removal of p16INK4a-positive cells delayed onset of adipose tissue, skeletal muscle and eye phenotypes and, when depleted in the adult, the treatment prevented the progression of already established age-related disorders [64]. Using non-genetic approaches potentially appropriate for use in humans could rely on using antibodies or small molecules to selectively target molecules that are specifically expressed on senescent cells.
In conclusion, recent progress in our understanding of the pathophysiology of AKI has emphasized the important role of tubular cell cycle arrest in the process of maladaptive repair. These findings open new avenues to better understand, prevent and slow down or arrest chronic fibrosis progression and progressive CKD.
CONFLICT OF INTEREST STATEMENT
None declared.
ACKNOWLEDGEMENTS
G.C. is supported by research grants from Novartis, Baxter, Fresenius Medical Care, Astellas, Alexion Pharmaceuticals, Philippe Foundation, Inc., Bettencourt Schueller Foundation, Société Française de Néphrologie and Assistance Publique-Hôpitaux de Paris. Studies described in the laboratory of J.V.B. were supported by US NIH-NIDDK (grants DK39773 and DK72381) and also by the Harvard Stem Cell Institute.
REFERENCES
- 1.Basile DP, Donohoe D, Roethe K, et al. Renal ischemic injury results in permanent damage to peritubular capillaries and influences long-term function. Am J Physiol Renal Physiol 2001; 281: F887–F899 [DOI] [PubMed] [Google Scholar]
- 2.Yang L, Besschetnova TY, Brooks CR, et al. Epithelial cell cycle arrest in G2/M mediates kidney fibrosis after injury. Nat Med 2010; 16: 535–543 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hsu CY. Yes, AKI truly leads to CKD. J Am Soc Nephrol 2012; 23: 967–969 [DOI] [PubMed] [Google Scholar]
- 4.Coca SG, Singanamala S, Parikh CR. Chronic kidney disease after acute kidney injury: a systematic review and meta-analysis. Kidney Int 2012; 81: 442–448 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Khosla N, Soroko SB, Chertow GM, et al. Preexisting chronic kidney disease: a potential for improved outcomes from acute kidney injury. Clin J Am Soc Nephrol 2009; 4: 1914–1919 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Liangos O, Wald R, O'Bell JW, et al. Epidemiology and outcomes of acute renal failure in hospitalized patients: a national survey. Clin J Am Soc Nephrol 2006; 1: 43–51 [DOI] [PubMed] [Google Scholar]
- 7.Sutton TA, Molitoris BA. Mechanisms of cellular injury in ischemic acute renal failure. Semin Nephrol 1998; 18: 490–497 [PubMed] [Google Scholar]
- 8.Gailit J, Colflesh D, Rabiner I, et al. Redistribution and dysfunction of integrins in cultured renal epithelial cells exposed to oxidative stress. Am J Physiol 1993; 264: F149–F157 [DOI] [PubMed] [Google Scholar]
- 9.Zuk A, Bonventre JV, Brown D, et al. Polarity, integrin, and extracellular matrix dynamics in the postischemic rat kidney. Am J Physiol 1998; 275: C711–C731 [DOI] [PubMed] [Google Scholar]
- 10.Bonventre JV, Yang L. Cellular pathophysiology of ischemic acute kidney injury. J Clin Invest 2011; 121: 4210–4221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bonventre JV. Dedifferentiation and proliferation of surviving epithelial cells in acute renal failure. J Am Soc Nephrol 2003; 14(Suppl 1): S55–S61 [DOI] [PubMed] [Google Scholar]
- 12.Humphreys BD, Valerius MT, Kobayashi A, et al. Intrinsic epithelial cells repair the kidney after injury. Cell Stem Cell 2008; 2: 284–291 [DOI] [PubMed] [Google Scholar]
- 13.Humphreys BD, Czerniak S, DiRocco DP, et al. Repair of injured proximal tubule does not involve specialized progenitors. Proc Natl Acad Sci USA 2011; 108: 9226–9231 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chawla LS, Kimmel PL. Acute kidney injury and chronic kidney disease: an integrated clinical syndrome. Kidney Int 2012; 82: 516–524 [DOI] [PubMed] [Google Scholar]
- 15.Rosner MH. The pathogenesis of susceptibility to acute kidney injury in the elderly. Curr Aging Sci 2009; 2: 158–164 [DOI] [PubMed] [Google Scholar]
- 16.Iwano M, Plieth D, Danoff TM, et al. Evidence that fibroblasts derive from epithelium during tissue fibrosis. J Clin Invest 2002; 110: 341–350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Humphreys BD, Lin SL, Kobayashi A, et al. Fate tracing reveals the pericyte and not epithelial origin of myofibroblasts in kidney fibrosis. Am J Pathol 2010; 176: 85–97 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Asada N, Takase M, Nakamura J, et al. Dysfunction of fibroblasts of extrarenal origin underlies renal fibrosis and renal anemia in mice. J Clin Invest 2011; 121: 3981–3990 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kaissling B, Lehir M, Kriz W. Renal epithelial injury and fibrosis. Biochim Biophys Acta 2013; 1832: 931–939 [DOI] [PubMed] [Google Scholar]
- 20.Tchkonia T, Zhu Y, van Deursen J, et al. Cellular senescence and the senescent secretory phenotype: therapeutic opportunities. J Clin Invest 2013; 123: 966–972 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Vogetseder A, Picard N, Gaspert A, et al. Proliferation capacity of the renal proximal tubule involves the bulk of differentiated epithelial cells. Am J Physiol Cell Physiol 2008; 294: C22–C28 [DOI] [PubMed] [Google Scholar]
- 22.Witzgall R. Are renal proximal tubular epithelial cells constantly prepared for an emergency? Focus on ‘the proliferation capacity of the renal proximal tubule involves the bulk of differentiated epithelial cells’. Am J Physiol Cell Physiol 2008; 294: C1–C3 [DOI] [PubMed] [Google Scholar]
- 23.Morgan DO. Cyclin-dependent kinases: engines, clocks, and microprocessors. Annu Rev Cell Dev Biol 1997; 13: 261–291 [DOI] [PubMed] [Google Scholar]
- 24.Evans T, Rosenthal ET, Youngblom J, et al. Cyclin: a protein specified by maternal mRNA in sea urchin eggs that is destroyed at each cleavage division. Cell 1983; 33: 389–396 [DOI] [PubMed] [Google Scholar]
- 25.Lim S, Kaldis P. Cdks, cyclins and CKIs: roles beyond cell cycle regulation. Development 2013; 140: 3079–3093 [DOI] [PubMed] [Google Scholar]
- 26.Johnson DG, Walker CL. Cyclins and cell cycle checkpoints. Annu Rev Pharmacol Toxicol 1999; 39: 295–312 [DOI] [PubMed] [Google Scholar]
- 27.Kastan MB, Bartek J. Cell-cycle checkpoints and cancer. Nature 2004; 432: 316–323 [DOI] [PubMed] [Google Scholar]
- 28.Matsuoka S, Huang M, Elledge SJ. Linkage of ATM to cell cycle regulation by the Chk2 protein kinase. Science 1998; 282: 1893–1897 [DOI] [PubMed] [Google Scholar]
- 29.Hirao A, Kong YY, Matsuoka S, et al. DNA damage-induced activation of p53 by the checkpoint kinase Chk2. Science 2000; 287: 1824–1827 [DOI] [PubMed] [Google Scholar]
- 30.Dulic V, Kaufmann WK, Wilson SJ, et al. p53-dependent inhibition of cyclin-dependent kinase activities in human fibroblasts during radiation-induced G1 arrest. Cell 1994; 76: 1013–1023 [DOI] [PubMed] [Google Scholar]
- 31.Datto MB, Li Y, Panus JF, et al. Transforming growth factor beta induces the cyclin-dependent kinase inhibitor p21 through a p53-independent mechanism. Proc Natl Acad Sci USA 1995; 92: 5545–5549 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Russo AA, Tong L, Lee JO, et al. Structural basis for inhibition of the cyclin-dependent kinase Cdk6 by the tumour suppressor p16INK4a. Nature 1998; 395: 237–243 [DOI] [PubMed] [Google Scholar]
- 33.Falck J, Mailand N, Syljuasen RG, et al. The ATM-Chk2-Cdc25A checkpoint pathway guards against radioresistant DNA synthesis. Nature 2001; 410: 842–847 [DOI] [PubMed] [Google Scholar]
- 34.Xu B, Kim ST, Lim DS, et al. Two molecularly distinct G(2)/M checkpoints are induced by ionizing irradiation. Mol Cell Biol 2002; 22: 1049–1059 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zhao H, Piwnica-Worms H. ATR-mediated checkpoint pathways regulate phosphorylation and activation of human Chk1. Mol Cell Biol 2001; 21: 4129–4139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Brown EJ, Baltimore D. Essential and dispensable roles of ATR in cell cycle arrest and genome maintenance. Genes Dev 2003; 17: 615–628 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Astuti P, Pike T, Widberg C, et al. MAPK pathway activation delays G2/M progression by destabilizing Cdc25B. J Biol Chem 2009; 284: 33781–33788 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wu CF, Chiang WC, Lai CF, et al. Transforming growth factor beta-1 stimulates profibrotic epithelial signaling to activate pericyte-myofibroblast transition in obstructive kidney fibrosis. Am J Pathol 2013; 182: 118–131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Scholzen T, Gerdes J. The Ki-67 protein: from the known and the unknown. J Cell Physiol 2000; 182: 311–322 [DOI] [PubMed] [Google Scholar]
- 40.Gerdes J, Lemke H, Baisch H, et al. Cell cycle analysis of a cell proliferation-associated human nuclear antigen defined by the monoclonal antibody Ki-67. J Immunol 1984; 133: 1710–1715 [PubMed] [Google Scholar]
- 41.Nadasdy T, Laszik Z, Blick KE, et al. Proliferative activity of intrinsic cell populations in the normal human kidney. J Am Soc Nephrol 1994; 4: 2032–2039 [DOI] [PubMed] [Google Scholar]
- 42.Witzgall R, Brown D, Schwarz C, et al. Localization of proliferating cell nuclear antigen, vimentin, c-Fos, and clusterin in the postischemic kidney. Evidence for a heterogenous genetic response among nephron segments, and a large pool of mitotically active and dedifferentiated cells. J Clin Invest 1994; 93: 2175–2188 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lin F, Moran A, Igarashi P. Intrarenal cells, not bone marrow-derived cells, are the major source for regeneration in postischemic kidney. J Clin Invest 2005; 115: 1756–1764 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Megyesi J, Udvarhelyi N, Safirstein RL, et al. The p53-independent activation of transcription of p21 WAF1/CIP1/SDI1 after acute renal failure. Am J Physiol 1996; 271: F1211–6 [DOI] [PubMed] [Google Scholar]
- 45.Megyesi J, Safirstein RL, Price PM. Induction of p21WAF1/CIP1/SDI1 in kidney tubule cells affects the course of cisplatin-induced acute renal failure. J Clin Invest 1998; 101: 777–782 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Nowak G, Price PM, Schnellmann RG. Lack of a functional p21WAF1/CIP1 gene accelerates caspase-independent apoptosis induced by cisplatin in renal cells. Am J Physiol Renal Physiol 2003; 285: F440–F450 [DOI] [PubMed] [Google Scholar]
- 47.Megyesi J, Price PM, Tamayo E, et al. The lack of a functional p21(WAF1/CIP1) gene ameliorates progression to chronic renal failure. Proc Natl Acad Sci USA 1999; 96: 10830–10835 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wei Q, Dong G, Yang T, et al. Activation and involvement of p53 in cisplatin-induced nephrotoxicity. Am J Physiol Renal Physiol 2007; 293: F1282–F1291 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Molitoris BA, Dagher PC, Sandoval RM, et al. siRNA targeted to p53 attenuates ischemic and cisplatin-induced acute kidney injury. J Am Soc Nephrol 2009; 20: 1754–1764 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zhou L, Fu P, Huang XR, et al. Activation of p53 promotes renal injury in acute aristolochic acid nephropathy. J Am Soc Nephrol 2010; 21: 31–41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Clements ME, Chaber CJ, Ledbetter SR, et al. Increased cellular senescence and vascular rarefaction exacerbate the progression of kidney fibrosis in aged mice following transient ischemic injury. PLoS ONE 2013; 8: e70464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Park SK, Kang SK, Lee DY, et al. Temporal expressions of cyclins and cyclin dependent kinases during renal development and compensatory growth. Kidney Int 1997; 51: 762–769 [DOI] [PubMed] [Google Scholar]
- 53.Grgic I, Campanholle G, Bijol V, et al. Targeted proximal tubule injury triggers interstitial fibrosis and glomerulosclerosis. Kidney Int 2012; 82: 172–183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Cianciolo Cosentino C, Skrypnyk NI, Brilli LL, et al. Histone deacetylase inhibitor enhances recovery after AKI. J Am Soc Nephrol 2013; 24: 943–953 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Tang J, Liu N, Tolbert E, et al. Sustained activation of EGFR triggers renal fibrogenesis after acute kidney injury. Am J Pathol 2013; 183: 160–172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Li J, Dai CX, Sun H, et al. Protective effects and mechanisms of curcumin on podophyllotoxin toxicity in vitro and in vivo. Toxicol Appl Pharmacol 2012; 265: 190–199 [DOI] [PubMed] [Google Scholar]
- 57.Gasparitsch M, Arndt AK, Pawlitschek F, et al. RAGE-mediated interstitial fibrosis in neonatal obstructive nephropathy is independent of NF-kappaB activation. Kidney Int 2013; 84: 911–919 [DOI] [PubMed] [Google Scholar]
- 58.Zhou W, Otto EA, Cluckey A, et al. FAN1 mutations cause karyomegalic interstitial nephritis, linking chronic kidney failure to defective DNA damage repair. Nat Genet 2012; 44: 910–915 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.MacKay C, Declais AC, Lundin C, et al. Identification of KIAA1018/FAN1, a DNA repair nuclease recruited to DNA damage by monoubiquitinated FANCD2. Cell 2010; 142: 65–76 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Chaki M, Airik R, Ghosh AK, et al. Exome capture reveals ZNF423 and CEP164 mutations, linking renal ciliopathies to DNA damage response signaling. Cell 2012; 150: 533–548 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Sheen MR, Kim SW, Jung JY, et al. Mre11-Rad50-Nbs1 complex is activated by hypertonicity. Am J Physiol Renal Physiol 2006; 291: F1014–F1020 [DOI] [PubMed] [Google Scholar]
- 62.Dmitrieva NI, Cui K, Kitchaev DA, et al. DNA double-strand breaks induced by high NaCl occur predominantly in gene deserts. Proc Natl Acad Sci USA 2011; 108: 20796–20801 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Westhoff JH, Schildhorn C, Jacobi C, et al. Telomere shortening reduces regenerative capacity after acute kidney injury. J Am Soc Nephrol 2010; 21: 327–336 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Baker DJ, Wijshake T, Tchkonia T, et al. Clearance of p16Ink4a-positive senescent cells delays ageing-associated disorders. Nature 2011; 479: 232–236 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Acosta JC, O'Loghlen A, Banito A, et al. Chemokine signaling via the CXCR2 receptor reinforces senescence. Cell 2008; 133: 1006–1018 [DOI] [PubMed] [Google Scholar]
- 66.Acosta JC, Banito A, Wuestefeld T, et al. A complex secretory program orchestrated by the inflammasome controls paracrine senescence. Nat Cell Biol 2013; 15: 978–990 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Tilstra JS, Robinson AR, Wang J, et al. NF-kappaB inhibition delays DNA damage-induced senescence and aging in mice. J Clin Invest 2012; 122: 2601–2612 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Finn WF. Enhanced recovery from postischemic acute renal failure. Micropuncture studies in the rat. Circ Res 1980; 46: 440–448 [DOI] [PubMed] [Google Scholar]