

Abstract
Atypical haemolytic uraemic syndrome (aHUS) is a rare disease characterized by haemolytic microangiopathic anaemia, thrombocytopaenia and acute onset of renal failure, in the absence of Escherichia coli infection. Renal damage usually progresses to end-stage renal disease (ESRD), sometimes being accompanied by signs of extrarenal thrombotic microangiopathy (TMA). We report a case of full neurological and haematological recovery after eculizumab treatment in a patient with ESRD secondary to chronic aHUS refractory to plasmatherapy while she was under dialysis. It highlights the use of eculizumab for controlling extrarenal manifestations of aHUS in this population.
Keywords: atypical haemolytic uraemic syndrome, eculizumab, neurologic involvement, plasma exchange, thrombotic microangiopathy
Background
Atypical haemolytic uraemic syndrome (aHUS) is an orphan disease, with an incidence of 2–5 cases per million [1, 2]. aHUS can occur at any age and is probably underdiagnosed [3]. aHUS is characterized by haemolytic microangiopathic anaemia, thrombocytopaenia and acute renal failure (ARF), with signs or symptoms of thrombotic microangiopathy (TMA) in other organs [2]. It is due to uncontrolled activation of the alternative pathway of the complement system, caused by genetic mutation in complement genes (>60% of patients), anti-FH antibodies (5–6% of patients) and other mutations not related to the complement system recently described, as DKGE gene mutation or methylmalonic aciduria. Approximately one-third of patients remain without identified mutation [3–5]. Clinical manifestation of aHUS can be triggered by other complement-amplifying conditions (infection, pregnancy, organ transplant, malignant hypertension and systemic lupus erythematosus) that can increase the risk of misdiagnosis [6]. Onset of symptoms can be sudden and present as the full triad described above. However, clinical disease manifestation can also progress more slowly with mild anaemia, progressive renal insufficiency and oscillating thrombocytopaenia [7]. The kidney is the most frequent organ involved, although manifestations can occur in the brain (10–20% of cases), heart, lungs and gastrointestinal tract [6]. Diagnosis of aHUS relies on exclusion; no evidence of Shigatoxin-HUS, no criteria for thrombotic thrombocytopaenic purpura (normal ADAMTS13 activity) and no medical history indicating other causes of TMA (e.g. malignancy, drug use, etc.) [8]. Plasma exchange (PE) has been the standard first-line therapy, with variable efficacy. Published studies have shown that up to 40% of patients die or progress to end-stage renal disease (ESRD) during the first episode [2]. Eculizumab, a monoclonal antibody directed against C5 that blocks the cleavage of C5 into C5a and C5b, has demonstrated better efficacy and tolerability in the treatment of aHUS [9]. We report a case of aHUS with long evolution, subclinical haemolytic signs, progressive RF and neurological impairment resistant to PE, which showed a full reversal of neurological lesions after initiation of eculizumab therapy.
Case report
A 27-year-old female presented with hypertensive emergency and kidney failure. Family history: her sister had been diagnosed with membranoproliferative glomerulonephritis (MPGN) at age 14. Over the disease course, she had anaemia, increased lactate dehydrogenase (LDH) and decline in C3. Epilepsy was diagnosed and well controlled, but she developed neurological impairment with progressive mental retardation. Magnetic resonance imaging (MRI) revealed ischaemic lesions in small vessels and progressive renal failure developed over 11 years. She started peritoneal dialysis in 2010 and suffered further neurological impairment and died due to a respiratory infection in August, 2012. No definitive diagnosis of the underlying disease was established despite autopsy. Our 27-year-old patient was diagnosed with ARF, anaemia and thrombocytopaenia aged 1 year. She recovered spontaneously without sequalae, and no new episodes of anaemia, thrombocytopaenia, LDH and bilirubin increase nor renal impairment was observed during 14 years. At age 15, she presented with hyperuricaemia, non-nephrotic proteinuria (2 g/24 h), arterial hypertension [blood pressure (BP) 160/90 mmHg] and anaemia [haemoglobin (Hb) 11.5 g/dL], without thrombocytopaenia or schistocytes in peripheral blood. Serum creatinine (sCr) was 88.4 µmol/L, and creatinine clearance (ClCr) was 96 mL/min. Renal biopsy showed focal proliferative glomerulonephritis, affecting 28% of glomeruli, with mesangial matrix and cellularity proliferation, and focal hyaline material deposits, conditioning sclerosis in one of the glomeruli. On immunofluorescence, there were intense IgM and weak Cq deposits (Figure 1A). There was no evidence of TMA or endothelial damage. During the next 10 years, she maintained an anaemia that required erythropoietin-stimulating agents (ESA). Her sCr was stable, and no evidence of TMA was found (normal platelet count, no schistocytes, normal bilirubin) other than a slight and maintained increase in LDH possibly indicating ongoing haemolysis. She also required angiotensin-converting enzyme inhibitors for proteinuria treatment. When she was 26, arterial hypertension, proteinuria and anaemia worsened, and serum creatinine increased to 176.8 µmol/L, with poor response to antiproteinuric therapy. Five months later, she had a hypertensive emergency (BP 216/120 mmHg), with hypertonic hypertensive retinopathy on funduscopy, kidney failure (sCr 366.86 µmol/L), haemolytic microangiopathic anaemia (Hb 6.7 g/dL, LDH 350 UI/L, haptoglobin <5 mg/dL and schistocytes) and severe thrombocytopaenia (22 × 109/L). Coombs test and autoimmunity were negative, complement component C3 was normal (85 mg/dL) and ADAMTS13 activity was 83%. A misdiagnosis of TMA secondary to hypertensive emergency in the context of uncontrolled glomerulopathy was made. She required haemodialysis and PE was started. A kidney biopsy showed advanced TMA, with acute and subacute lesions over chronic vascular involvement (Figure 1B and Figure 2). After five PE sessions, haemolysis was controlled but renal function did not improve. The patient remained hypertensive on dialysis, requiring seven drugs to control BP (Table 1). Three months later, she presented with mild neurological symptoms (decreased intellectual performance, a self-limited episode of loss of consciousness) and a new episode of TMA with C3 consumption (68 mg/dL). An MRI was performed, showing several high-intensity subcortical white matter lesions in the frontal lobes, with the appearance of hypoxic-ischaemic or inflammatory lesions. Mild cerebral atrophy was also present (Figure 3A). Despite a neurological evaluation, no other aetiology than TMA was identified. Genetic investigation showed no mutation in FH, FI and MCP genes, but she is homozygotic for a risk haplotype (H3). Based on the patient's past history, the patient's sister bad evolution and the presence of a new episode of TMA while on dialysis, with better BP control, we reconsider our previous diagnosis of TMA secondary to malignant hypertension to aHUS being the primary cause and probably also in the sister. Diagnosis of aHUS was established and PE was restarted. After seven sessions without response and persistent haemolytic signs (marked thrombocytopaenia, anaemia, increased LDH and schistocytes in peripheral blood sample), eculizumab was initiated (900 mg/week, during 4 weeks followed by 1200 mg/14 days). The patient received Neisseria meningitidis vaccination and prophylactic penicillin for 15 days. After the first eculizumab dose, haemolysis resolved. After the third dose, a repeat MRI revealed improvement of the cerebral lesions (Figure 3B). After 18 months of eculizumab therapy, the patient is still requiring dialysis, but signs of haemolysis and neurological symptoms are completely absent. Anaemia is controlled (Hb 12.5 g/dL) with a lower ESA requirement (darbepoetin 30 μg/weekly). BP has improved after 2 months of eculizumab and normal BP is maintained with four antihypertensive drugs. She has completed her education and is waiting for a kidney transplant.
Fig. 1.
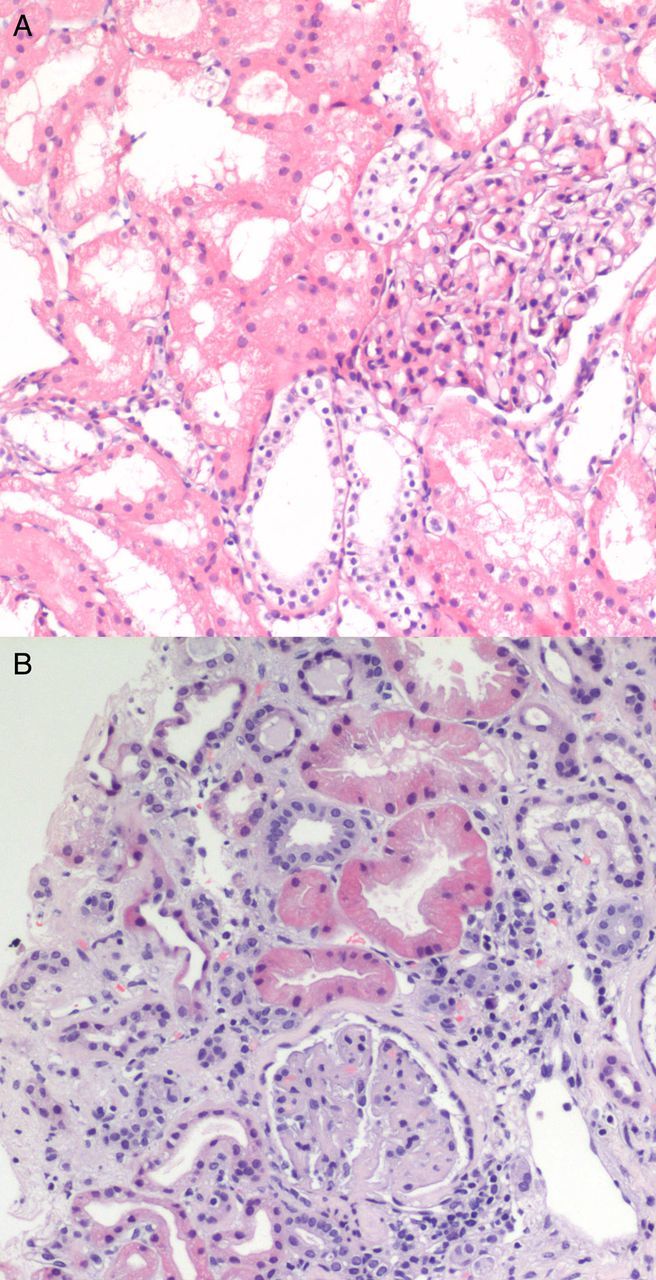
(A) First kidney biopsy. Focal proliferative glomerulonephritis, with mesangial matrix and cellular proliferation, and focal hyaline material deposits. No evidence of TMA or endothelial damage. (B) Second biopsy. Advanced TMA, with avascular glomeruli and mesangial sclerosis.
Fig. 2.

Second biopsy, arterioli detail. Signs of acute thrombotic microangiopathy with endoluminal collapse, endothelial and myointimal proliferation.
Table 1.
Changes in biochemical parameters and treatment
| Period | 2001–11 (clinical stability) | Dec 2011–Apr 2012 (clinical worsening) | May 2012 (hospital admission) | Jun 2012 (start dialysis) | Aug 2012 (dialysis, stability) | Oct 2012 (dialysis, neurological disorders) | Jun 2014 (dialysis, stability) |
|---|---|---|---|---|---|---|---|
| Scr (µmol/L) | 88.4 | 176.8 | 366.86 | 750 | |||
| Proteinuria (g/24 h) | 1.03 | 2.21 | 2.71 | n.d. | n.d. | n.d. | n.d. |
| BP (mmHg) | 140/85 | 160/90 | 216/120 | 190/100 | 145/80 | 170/110 | 140/80 |
| Hb (g/dL) | 12 ± 1 | 9 ± 0.5 | 6.7 | 8 ± 0.5 | 10.6 | 9.5 | 12.5 |
| Platelets (cells/109 µL) | 256 ± 40 | 231 ± 40 | 224 ± 30 | 22 ± 15 | 215 | 49 ± 20 | 157 ± 20 |
| LDH UI/L (normal range: 125–243) | 265 ± 10 | 300 ± 20 | 377 | 500 | 354 | 363 | 216 |
| Haptoglobin (mg/dL) | <5 | 125 | 79 | ||||
| Schistocytes (per HPF) | Absent | Absent | 2/HPF | 2/HPF | Absent | Absent | Absent |
| Treatment | |||||||
| Darbepoetin | 80 ± 20 µg/month | 80 µg/15d | 80 µg/15d | 150 µg/week | 10 µg/15d | 150 µg/week | 30 µg/week |
| ACEI | Ramipril 10 mg/BID | Ramipril 10 mg/BID | |||||
| ARBs | Irbesartan 300 mg/d | Irbesartan 300 mg/d | |||||
| Other antihypertensive | Amlodipine 5 mg/d Hydrochlorotiazide (HCTZ) 25 mg/d |
Amlodipine 5 mg/d HCTZ 25 mg/d Doxazosin 4 mg/BID Methyldopa 500 mg/BID |
Amlodipine 5 mg/d Torasemide 5 mg/d Doxazosin 4 mg/BID Methyldopa 500 mg/BID Nebivolol 5 mg/d |
Barnidipine 10 mg/BID Furosemide 80 mg/d HCTZ 25 mg/d Doxazosin 4 mg/BID Labetalol 100 mg/BID Hydralazine 50 mg/TID |
Barnidipine 10 mg/BID Labetalol 100 mg/BID Hydralazine 50 mg/TID Aliskiren 150 mg/d |
Barnidipine 10 mg/BID Furosemide 40 mg/d Doxazosin 4 mg/BID Atenolol 25 mg/BID |
Barnidipine 10 mg/BID Furosemide 40 mg/d Doxazosin 4 mg/BID Atenolol 25 mg/BID |
| PE | 5 sessions | 7 sessions | |||||
| Eculizumab | 900 mg/week | 1200 mg/14d | |||||
n.d., not documented; d, day; BID, twice a day; TID, three times a day.
Fig. 3.

Evolution of MRI before (A) and after (B). (A) MRI before eculizumab therapy. High-intensity subcortical white matter lesions (arrow). Signs of mild cerebral atrophy. (B) MRI 3 weeks after eculizumab therapy. Disappearance of white matter lesions (arrow).
Discussion
Apart from the efficacy of eculizumab for controlling neurological manifestations of aHUS, the present case highlights the difficulties in diagnosing aHUS when signs of the disease are mild (anaemia with subclinical haemolytic signs, gradual impairment of renal function and the absence of thrombocytopaenia) and progress slowly. The case also highlights the efficacy of eculizumab in a dialysis patient.
Usually, aHUS has an abrupt onset, with a complete clinical picture that helps the differential diagnosis [7]. Our case describes a chronic progressive evolution, with distinct clinical flares. The lack of thrombocytopaenia, schistocytes and worsening anaemia until the last episode of kidney failure made diagnosis difficult. Slow disease development has been described in 20% of patients, with proteinuria, elevated BP and progressive renal failure. In relapsing/remitting disease, many years can pass between flares. Up to 9 years has been reported in the literature [7]. In our case, the interval lasted over 20 years. Our patient was initially diagnosed with focal glomerulosclerosis with IgM deposits and non-nephrotic proteinuria (but she had a previous episode of ARF, probably due to TMA episode). Her sister was diagnosed with MPGN. The association between glomerulonephritis and aHUS has been described previously [10].
MPGN and aHUS may sometimes look similar on a biopsy and partially recovered TMA could possibly mimic chronic glomerulonephritis. The association between them has been made as both are diseases related to aberrant complement activation [11]. Proteinuria could be explained by endothelial dysfunction caused by TMA [12], but nephrotic syndrome itself can trigger aHUS by inducing endothelial injury in patients with genetic mutations [13, 14]. Moreover, alternative pathway dysregulation can manifest only as low-grade proteinuria and chronic kidney disease [13, 15]. Vasculitis can trigger aHUS by intensive complement activation or by endothelial damage in a patient with a genetic predisposition [10]. The association of IgM nephropathy and aHUS is novel. Nevertheless, IgM could activate the complement cascade [16], triggering aHUS. In our case, glomerulonephritis was not the initial event; she had a prior episode of ARF. It is possible that partially recovered TMA episodes mimic chronic glomerulonephritis on biopsy, as in the present case. The non-classical presentation in both sisters made diagnosis of aHUS as a primary disease complex. The biopsy findings were in addition not entirely conclusive for a diagnosis of aHUS, as the biopsy showed focal proliferative glomerulonephritis.
Genetic investigations identified no complement mutations in the patient. She is a carrier of a homozygotic risk polymorphism (H3 haplotype). The importance of risk polymorphisms in the development of aHUS is increasingly being recognized, and a lack of identified complement mutations is observed in 25–30% of patients [3, 17].
The similar evolution in the two sisters, and the good response to eculizumab in our patient, suggests a complement system disturbance, despite the absence of identified mutations. The evolution of the two patients supports the evidence that prognosis for aHUS with no identified mutation is as poor as in patients with identified complement factor mutations, in the absence of adequate therapy [17].
The prognosis for patients with aHUS before eculizumab was poor, with up to 40% of patients dying or progressing to ESRD during the first episode [2]. Outcomes have dramatically improved with the introduction of eculizumab [18, 19].
Renal function recovery has been described with eculizumab even after 6 months of dialysis [20]. In our case, kidney function did not improve despite treatment, probably due to the long-term nature of the organ damage causing irreversible damage and the delay in starting eculizumab therapy. In our patient, neurological damage progressed despite cessation of clinical haemolysis and ESRD. Therefore, the presence of extrarenal symptoms is critical when deciding to maintain anti-C5 therapy, regardless of renal replacement therapy requirement. Damage to extrarenal organs can progress in patients without renal function [21, 22]. This indicates subclinical activity, showing that an increase in platelet count is not always a reliable marker of recovery, highlighting the need for other biomarkers of disease activity [23]. In the present case, neurological damage and MRI lesions improved dramatically after starting eculizumab therapy. Rapid neurological recovery after eculizumab has been described previously, and it is maintained over time [20, 22, 24, 25].
In conclusion, nephrologists should be aware of the possibility of aHUS with a slow, low-grade chronic evolution that can be difficult to diagnose. Our case suggests that neurological involvement can be reversed with eculizumab that is becoming the standard of care in aHUS therapy, even in patients with aHUS on dialysis.
Conflict of interest statement
None declared.
Acknowledgements
Medical editing support provided by Bioscript Stirling Ltd, UK, and funded by Alexion Pharma International.
References
- 1.Noris M, Remuzzi G. Atypical hemolytic-uremic syndrome. N Engl J Med 2009; 361: 1676–1687 [DOI] [PubMed] [Google Scholar]
- 2.Loirat C, Frémeaux-Bacchi V. Atypical haemolytic uremic syndrome. Orphanet J Rare Dis 2011; 6: 60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Fremeaux-Bacchi V, Fakhouri F, Garnier A, et al. Genetics and outcome of atypical hemolytic uremic syndrome: a nationwide French series comparing children and adults. Clin J Am Soc Nephrol 2013; 8: 554–562 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lemaire M, Frémeaux-Bacchi V, Schaeffer K, et al. Recessive mutations in DKGE cause atypical haemolytic uremic syndrome. Nat Genet 2013; 45: 531–536 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Menni F, Testa S, Guez S. Neonatal atypical hemolytic uremic syndrome due to methylmalonic aciduria and homocystinuria. Pediatr Nephrol 2012; 27: 1401–1405 [DOI] [PubMed] [Google Scholar]
- 6.Loirat C, Noris M, Fremeaux-Bacchi V. Complement and the atypical hemolytic uremic syndrome in children. Pediatr Nephrol 2008; 23: 1957–1972 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sellier-Leclerc AL, Fremeaux-Bacchi V, Dragon-Durey MA, et al. Differential impact of complement mutations on clinical characteristics in atypicalhemolytic uremic syndrome. J Am Soc Nephrol 2007; 18: 2392–2400 [DOI] [PubMed] [Google Scholar]
- 8.Noris M, Mescia F, Remuzzi G. STEC-HUS, atypical HUS and TTP are all diseases of complement activation. Nat Rev Nephrol 2012; 8: 622–633 [DOI] [PubMed] [Google Scholar]
- 9.Legendre CM, Licht C, Muus P, et al. Terminal complement inhibitor eculizumab in atypical hemolytic-uremic syndrome. N Engl J Med 2013; 368: 2169–2181 [DOI] [PubMed] [Google Scholar]
- 10.Manenti L, Gnappi E, Vaglio A, et al. Atypical haemolyticuraemic syndrome with underlying glomerulopathies. A case series and a review of the literature. Nephrol Dial Transplant 2013; 28: 2246–2259 [DOI] [PubMed] [Google Scholar]
- 11.Wada T, Nangaku M. Novel roles of complement in renal diseases and their therapeutic consequences. Kidney Int 2013; 84: 441–450 [DOI] [PubMed] [Google Scholar]
- 12.Satchell S. The role of the glomerular endothelium in albumin handling. Nat Rev Nephrol 2013; 9: 717–725 [DOI] [PubMed] [Google Scholar]
- 13.Tkaczyk M, Czupryniak A, Owczarek D, et al. Markers of endothelial dysfunction in children with idiopathic nephrotic syndrome. Am J Nephrol 2008; 28: 197–202 [DOI] [PubMed] [Google Scholar]
- 14.Eremina V, Jefferson JA, Kowalewska J, et al. VEGF inhibition and renal thrombotic microangiopathy. N Engl J Med 2008; 358: 1129–1136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sethi S, Fervenza F, Zhang Y, et al. Secondary focal and segmental glomerulosclerosis associated with single-nucleotide polymorphisms in the genes encoding complement Factor H and C3. Am J Kidney Dis 2012; 60: 316–321 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Strassheim D, Renner B, Panzer S, et al. IgM contributes to glomerular injury in FSGS. J Am Soc Nephrol 2013; 24: 393–406 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Caprioli J, Noris M, Brioschi S, et al. The International Registry of Recurrent and Familial HUS/TTP. Genetics of HUS: the impact of MCP, CFH, and IF mutations on clinical presentation, response to treatment, and outcome. Blood 2006; 108: 1267–1279 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Greenbaum LA, Fila M, Tsimaratos M, et al. Eculizumab inhibits thrombotic microangiopathy and improves renal function in pediatric atypical hemolytic uremic syndrome patients [abstract]. In: XXIII ASN Kidney Week. Abstract SA-PO849, 2013 Nov 5-10, p. 821A American Society of Nephrology kidney Week, Atlanta, GA, 2013 [Google Scholar]
- 19.Fakhouri F, Hourmant M, Campistol JM, et al. Eculizumab inhibits thrombotic microangiopathy and improves renal function in adult patients with atypical hemolytic uremic syndrome [abstract]. In: America Society of Nephrology Kidney Week. Abstract FR-OR05, 2013 Nov 5-10, p. 49A-50-A ASN Kidney Week, Atlanta, GA, 2013 [Google Scholar]
- 20.Povey H, Vundru R, Junglee N, et al. Renal recovery with eculizumab in atypicalhemolytic uremic syndrome following prolonged dialysis. Clin Nephrol 2013 [DOI] [PubMed] [Google Scholar]
- 21.Békássy ZD, Kristoffersson AC, Cronqvist M, et al. Eculizumab in an anephric patient with atypical haemolytic uraemic syndrome and advanced vascular lesions. Nephrol Dial Transplant 2013; 28: 2899–2907 [DOI] [PubMed] [Google Scholar]
- 22.Salem G, Flynn JM, Cataland SR. Profound neurological injury in a patient with atypical haemolytic uremic syndrome. Ann Hematol 2013; 92: 557–558 [DOI] [PubMed] [Google Scholar]
- 23.Dorresteijn EM, van de Kar NC, Cransberg K. Eculizumab as rescue therapy for atypicalhemolytic uremic syndrome with normal platelet count. Pediatr Nephrol 2012; 27: 1193–1195 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hu H, Nagra A, Haq MR, et al. Eculizumab in atypical haemolytic uraemic syndrome with severe cardiac and neurological involvement. Pediatr Nephrol 2014; 29: 1103–1106 [DOI] [PubMed] [Google Scholar]
- 25.Ohanian M, Cable C, Halka K. Eculizumab safely reverses neurologic impairment and eliminates need for dialysis in severe atypical hemolytic uremic syndrome. Clin Pharmacol 2011; 3: 5–12 [DOI] [PMC free article] [PubMed] [Google Scholar]