

Abstract
Light and heavy chain deposition disease (LHCDD) is a rare complication of monoclonal gammopathy. In all documented cases, LHCDD is the association of deposits of a monoclonal light chain with a normal heavy chain, especially in the kidneys. We describe here a 78-year-old woman whose renal biopsy showed nodular glomerulosclerosis, initially diagnosed as diabetic nephropathy. Detailed kidney biopsy immunofluorescence study corrected the diagnosis to γ1-κ-LHCDD. Advanced immunoblot analysis showed deletion of CH1 in the both blood and kidney heavy chain. We report here, to our knowledge, the first case of γ1 LHCDD associated with a deletion of CH1.
Keywords: chronic kidney disease, MGRS, MIDD, monoclonal gammopathy, multiple myeloma
Background
Recently, the concept of monoclonal gammopathy of renal significance (MGRS) has emerged [1], including all renal diseases related to monoclonal gammopathy, and underlying the role of the B-cell clone in the pathogeny of this disease. Monoclonal immunoglobulin deposition disease (MIDD) is part of MGRS, and is a rare complication of monoclonal gammopathy, in which a monoclonal light chain (light chain deposition disease LCDD), heavy chain (heavy chain deposition disease HCDD) or both (LHCDD) deposit in basement membranes, particularly in the kidneys [2–5]. LHCDD can mimic diabetic nephropathy, showing nodular glomerulosclerosis on renal biopsy, thus emphasizing the necessity of careful immunofluorescence study of these kidney biopsies.
Deletion of the constant domain of heavy chain CH1 has been found in all described cases of γ1 HCDD [6, 7], and this deletion probably participates in the pathogenesis of HCDD, allowing the heavy chain to get out of the endoplasmic reticulum without associated light chain [8]. Today, no one can say if the toxic heavy chain and light chain in LHCDD are secreted by one or two clones, and deletion of CH1 has not been described yet in LHCDD.
We report here a case of γ1-κ-LHCDD with deletion of CH-1.
Case report
A 78-year-old woman, with an uneventful medical history was referred because of hypertension and leg oedema of 5-months duration. Physical examination revealed exertional dyspnoea, overhydration and high blood pressure (200/100 mmHg). Serum creatinine was 400 µmol/L. Urinanalysis revealed proteinuria (+++) and haematuria (++). Haemoglobin was 8 g/dL, glucose 1 g/L.
Serological tests were negative for anti-nuclear, anti-neutrophil cytoplasmic, anti-GBM, hepatitis B and C antibodies (Ab) and cryoglobulins. Complement factors, CH50, C3 and C4, were with in the normal range. Ultrasound analysis showed large kidneys (11 cm on both sides). Renal biopsy retrieved a cortex of 10 glomeruli, none of which was sclerosed. Light microscopy showed hyperlobulated glomeruli, with mostly argyrophilic, periodic acid-Schiff (PAS)-positive and Congo-red negative, nodular glomerulosclerotic lesions. Immunofluorescence was not performed. According to the histological findings, the diagnosis of diabetic glomerulosclerosis was initially retained. She was started on haemodialysis and antihypertensive treatment.
She was then referred to our nephrology department. Since she had no past history of known diabetes and glycated haemoglobin was in the normal range (5.2%, in the absence of genetic haemoglobinopathy), a second kidney biopsy was performed. It contained 15 glomeruli, 5 of which were sclerosed. The nodular glomerulosclerosis was still present; mesangial nodules were composed of PAS-positive membrane-like material, but methanamine silver (Jones) negative, a feature distinctive from diabetic nephropathy (Figure 1A). Congo-red analysis was still negative. Immunofluorescence analysis with anti-IgG, -IgM and -IgA Ab showed only IgG deposition in glomerular mesangium as well as a linear pattern along tubular BM and arterial walls (Figure 1B). Anti-κ Ab showed intense deposits with the same topography that IgG deposition (Figure 1C), whereas lambda staining was negative (not shown). Staining with subclass γ1 Ab showed strong tubular BM deposits (data not shown), and no labelling was observed with anti-CH1 Ab (Figure 1D), whereas stainings for CH2 and CH3 were positive (Figure 1E and F). Unfortunately, no glomeruli were present in the slides stained for CH1-CH3. Nevertheless the clear tubular BM staining was observed for CH2 and CH3, whereas no tubular staining was observed for CH1, undoubtedly indicating truncated γ1 chain deposits.
Fig. 1.
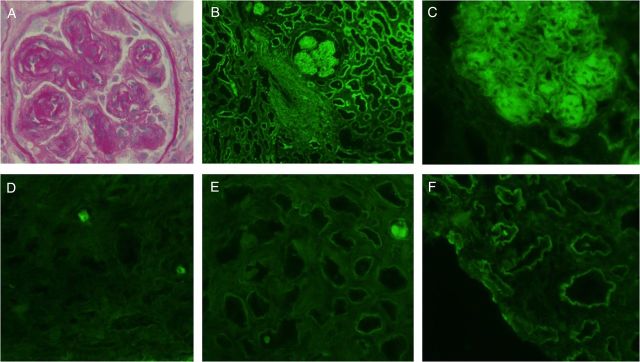
(A) Light microscopy of a representative nodular glomerulosclerosis: the glomerulus is lobulated, associated with mesangial nodular formation. Note double-contour aspects. The tubular basement membranes are thickened (PAS); (B) direct immunofluorescence staining for IgG heavy chain: strong staining on the tubular basement membranes, glomerular nodules and arterial walls; (C) immunofluorescence staining for κ light chains shows strong immunostaining on glomerular nodules, mesangial areas and tubular basement membrane; (D) immunofluorescence staining for the γ chain constant domain CH1; (E) immunofluorescence staining for the γ chain constant domain CH2; (F) immunofluorescence staining for the γ chain constant domain CH3.
High-resolution serum electrophoresis revealed hypo-albuminaemia (25 g/L), 2 monoclonal spikes (one at 1.4 g/L and one at the limit of detection), and a decreased level of gammaglobulins (2 g/L) (Figure 2A). Immunoblot analysis revealed the presence of two monoclonal IgG Kappa, the fast-migrating one being unreactive with anti-CH1 Ab (Figure 2B).
Fig. 2.
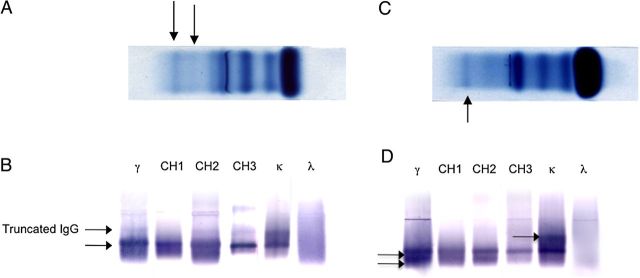
(A) Serum electrophoresis, showing the presence of 2 monoclonal spikes of Ig: the fast migrating Ig is the truncated Ig; (B) Serum immunoblots were revealed with monoclonal Ab against IgG1 subclass CH2 domain (clone NL16, γ1CH2), γ chain CH1 (clone ZB8), γ chain CH2 (clone HP6018), γ chain CH3 (clone HP6017), kappa (clone HP6053) and lambda (clone HP6054) chain constant regions. Results show the presence of 2 monoclonal IgG1 kappa, the upper one is truncated (no reactivity with CH1 antibody), the lower one reacts with all the monoclonal Ab specific to the 3 heavy chain constant domains; (C) urine electrophoresis, showing the presence of 1 monoclonal spike. The second spike is not detected with this technique; (D) urine immunoblots were revealed with monoclonal Ab against IgG1 subclass CH2 domain (clone NL16, γ1CH2), γ chain CH1 (clone ZB8), γ chain CH2 (clone HP6018), γ chain CH3 (clone HP6017), kappa (clone HP6053) and lambda (clone HP6054) chain constant regions.
In the urines, protein electrophoresis (2.94 g/L) showed albumin (60%), one monoclonal microspike in the gammaglobulin fraction and diffuse gammaglobulins (Figure 2C). Immunoblot study showed the presence of two complete monoclonal IgG1 kappa and monoclonal kappa light chains in tiny amounts (Figure 2D).
Serum concentration of free kappa and lambda chains was 490 mg/L and 59 mg/L, respectively (kappa/lambda =8.3).
Cytological analysis of the bone marrow aspiration showed 8% non-dystrophic plasma cells.
Taken together, these data suggested plasma cell secretion of a CH1-deleted γ1 heavy chain associated with a κ-monoclonal light chain, responsive for LHCDD.
The patient was treated by dexamethasone and melphalan (three cycles) while pursuing haemodialysis. There was no haematological nor renal response 5 months later.
The patient died of unknown cause after 12 months of evolution.
Discussion
Recently, expert consensus has described the concept of monoclonal gammopathy of renal significance (MGRS), underlying the pathogenic link between a monoclonal component and the kidney injuries [1]. LHCDD is a part of MGRS, and is very interesting to study because today, nobody can assess if this deposition disease is caused by one or two clones (one secreting the heavy chain and the other the light chain). It is interesting to note that in our patient, blood-free light chain assessment was abnormal, showing the presence of a monoclonal component.
It is noteworthy that LHCDD usually presents as a glomerular syndrome [3, 5], and that histological features can, as in our case, be confounded with diabetic nephropathy. Nevertheless, systematic use of immunofluorescence study with appropriate Ab should avoid diagnosis errors.
LHCDD is usually a variant of LCDD [3, 5]: the association of a monoclonal light chain with a normal heavy chain; whereas in our case, it was a truncated heavy chain with deletion of CH1 that was associated with a light chain. Then, even if there were two monoclonal spikes of IgGκ, whether HC and LC were secreted by two different plasma cell clones or the same clone has not been demonstrated.
Actually, deletion of the CH1 domain is found in the deposited or circulating HC in all patients with γ1-HCDD studied [6], and is, at least partially, responsible for the deposition in the kidneys [6]. It is known that normal heavy chains associate post-translationally with Ig-binding protein (BIP) in the endoplasmic reticulum [8]. The light chains later assemble with heavy chains, and the complex is transferred to the Golgi for further processing and secretion. As the binding site of BIP is located in the CH1 domain, when a mutant heavy chain lacks CH1 domain, it fails to associate with BIP and thus may be prematurely secreted in the circulation and deposit in the kidneys.
Therapeutic approaches for MIDD are not consensual as clinical trials are sparse. Few patients (under 65 years old) with LHCDD have been treated with high-dose chemotherapy and peripheral blood stem cell transplantation [9] with good tolerance and efficiency. Recently, recommendations by expert consensus have been made about MGRS treatment [10]. Recommended treatment must target the B-cell clone responsible for the deposition disease, and new anti-myeloma agent, like bortezomib, could become the reference treatment for LHCDD.
In summary, LHCDD is a rare disease, and its pathophysiology is not yet well understood. It is highly likely that deletion of CH1 participates in the pathophysiology of this disease, allowing the free heavy chain to circulate in the blood stream and deposit in the kidney. Nevertheless, it is not known whether the light chain responsible for LHCDD is secreted by the same clone or not. Further experimental studies are needed to better understand this phenomenon and it could be helpful, for example, to analyse systematically CH1 presence in LHCDD.
Conflict of interest statement
None of the authors reports conflict of interest. The results presented in this paper have not been published previously in whole or part.
References
- 1.Leung N, Bridoux F, Hutchison CA, et al. Monoclonal gammopathy of renal significance: when MGUS is no longer undetermined or insignificant. Blood 2012; 120: 4292–4295 [DOI] [PubMed] [Google Scholar]
- 2.Preud'homme JL, Aucouturier P, Touchard G, et al. Monoclonal immunoglobulin deposition disease (Randall type). Relationship with structural abnormalities of immunoglobulin chains. Kidney Int 1994; 46: 965–972 [DOI] [PubMed] [Google Scholar]
- 3.Lin J, Markowitz GS, Valeri AM, et al. Renal monoclonal immunoglobulin deposition disease: the disease spectrum. J Am Soc Nephrol 2001; 12: 1482–1492 [DOI] [PubMed] [Google Scholar]
- 4.Ronco PM, Alyanakian MA, Mougenot B, et al. Light chain deposition disease: a model of glomerulosclerosis defined at the molecular level. J Am Soc Nephrol 2001; 12: 1558–1565 [DOI] [PubMed] [Google Scholar]
- 5.Nasr SH, Valeri AM, Cornell LD, et al. Renal monoclonal immunoglobulin deposition disease: a report of 64 patients from a single institution. Clin J Am Soc Nephrol 2012; 7: 231–239 [DOI] [PubMed] [Google Scholar]
- 6.Moulin B, Deret S, Mariette X, et al. Nodular glomerulosclerosis with deposition of monoclonal immunoglobulin heavy chains lacking C(H)1. J Am Soc Nephrol 1999; 10: 519–528 [DOI] [PubMed] [Google Scholar]
- 7.Aucouturier P, Khamlichi AA, Touchard G, et al. Brief report: heavy-chain deposition disease. N Engl J Med 1993; 329: 1389–1393 [DOI] [PubMed] [Google Scholar]
- 8.Hendershot L, Bole D, Kohler G, et al. Assembly and secretion of heavy chains that do not associate posttranslationally with immunoglobulin heavy chain-binding protein. J Cell Biol 1987; 104: 761–767 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Royer B, Arnulf B, Martinez F, et al. High dose chemotherapy in light chain or light and heavy chain deposition disease. Kidney Int 2004; 65: 642–648 [DOI] [PubMed] [Google Scholar]
- 10.Fermand JP, Bridoux F, Kyle RA, et al. How I treat monoclonal gammapathy of renal significance (MGRS). Blood 2013; 122: 3583–3590 [DOI] [PubMed] [Google Scholar]