

Abstract
The EGF Receptor (EGFR) is a proto-oncogene commonly dysregulated in several cancers including non-small cell lung cancer (NSCLC) and, thus, is targeted for treatment using tyrosine kinase inhibitors (TKIs) such as Erlotinib. However, despite the efficacy observed in NSCLC patients harboring oncogenic variants of the EGFR, general ineffectiveness of TKIs in NSCLC patients who are current and former smokers necessitates identification of novel mechanisms to overcome this phenomenon. Previously, we showed that NSCLC cells harboring either wild-type (WT) EGFR or oncogenic mutant (MT) L858R EGFR become resistant to the effects of TKIs when exposed to cigarette smoke (CS), evidenced by their auto-phosphorylation and prolonged downstream signaling. Here, we present Src as a target mediating CS-induced resistance to TKIs in both WT EGFR and L858R MT EGFR expressing NSCLC cells. First, we show that CS exposure of A549 cells leads to time-dependent activation of Src which then abnormally binds to the WT EGFR causing TKI resistance, contrasting previous observations of constitutive binding between inactive Src and TKI-sensitive L858R MT EGFR. Next, we demonstrate that Src inhibition restores TKI sensitivity in CS-exposed NSCLC cells, preventing EGFR auto-phosphorylation in the presence of Erlotinib. Furthermore, we show that over-expression of a dominant-negative Src (Y527F/K295R) restores TKI sensitivity to A549 exposed to CS. Importantly, the TKI resistance that emerges even in CS-exposed L858R EGFR expressing NSCLC cells could be eliminated with Src inhibition. Together, these findings offer new rationale for using Src inhibitors for treating TKI-resistant NSCLC commonly observed in smokers.
Keywords: EGFR, Src, lung cancer, cigarette smoke, TKI resistance, oxidative stress, NSCLC
INTRODUCTION
Despite extensive preclinical and clinical studies, lung cancer accounts for more than one million deaths per year and its prognosis remains meager, with only a 5 to 15% 5-year survival rate (1). In response, advanced molecular studies identified the EGFR as a key target in non-small cell lung cancer (NSCLC) (2, 3) that becomes over-expressed in the bronchial epithelium of smokers (4-6). EGFR is a member of the ErbB family of receptor tyrosine kinases (RTKs), which includes ErbB2, ErbB3, and ErbB4, that autophosphorylate themselves upon homo- or heterodimerization after ligand binding, acting as mediators of proliferation and survival (7). EGFR overexpression is observed in tumors from more than 60% of patients with metastatic NSCLC and is correlated with poor prognosis (8). Though smoking has been established as the most important cause of lung cancer, it has been recently demonstrated that somatic mutations of the EGFR are responsible for approximately 10 to 25% of all non-smoking related lung cancer cases (9, 10). These initial findings triggered the development of tyrosine kinase inhibitors (TKIs) such as Gefitinib and Erlotinib that eventually became standard therapies for NSCLC (11).
Many oncologists currently treat NSCLC patients with promising small molecule TKIs, such as Gefitinib (Iressa®) or Erlotinib (Tarceva®), that reversibly inhibit kinase activity via competitive binding to the ATP binding site of the EGFR. A high response rate to TKIs was observed in women and in never-smokers with adenocarcinoma, specifically in the Japanese population (12), who were subsequently demonstrated to harbor specific somatic mutations (L858R substitution and Δ746-750 deletion) in the kinase domain of the EGFR, resulting in a constitutively activated EGFR that provides a selective growth advantage to the affected lung cells (13-17). However, it has been recently observed that the same EGFR mutations are not limited to adenocarcinoma from female never-smokers; a large number (40%) of these EGFR mutations are actually found in adenocarcinoma tumor specimens from both men and women who are current and former smokers (18). Yet, patients who are smokers harboring TKI-sensitive EGFR mutations were not rigorously screened and tested in any clinical trial in order to evaluate whether or not they would benefit from TKI treatment (19). It is also remarkable, though still an anecdotal observation, that patients receptive to TKI therapy acquire resistance to TKIs if they began smoking (20).
We have shown before that Gefitinib and Erlotinib treatments cannot inhibit the EGFR activation/auto-phosphorylation in NSCLC cells exposed to cigarette smoke. Therefore, smoking-related TKI resistance of NSCLC cells exists and is attributed to post-translational changes in the EGFR conformation and signaling (21). We presented evidence for a novel, active EGFR conformation caused by oxidative stress from cigarette smoke (CS) exposure. Unlike the canonical EGF-induced conformation, it does not dimerize, and it interacts strongly with Src, resulting in ligand-independent EGFR activation that is resistant to inhibition by TKIs. Importantly, we established that CS exposure renders TKIs ineffective in L858R EGFR-transformed cells, thus sustaining clonal growth of lung tumors even in the presence of TKI drugs (21). Notably, this effect was observed for several TKIs (Gefitinib, Erlotinib and AG1478) and with different cell lines (A549, HCC827 and NIH-3T3 over-expressing L858R EGFR).
Src, the first proto-oncogene described, is a key participant downstream of the EGFR family. Phosphorylation of several tyrosine residues within the EGFR has been shown to be increased following Src overexpression both in vitro and in vivo, indicating that Src is needed for full biological response following EGF stimulus (22, 23). Furthermore, using chimeric EGF/ErbB2 receptors (22, 24), it was demonstrated that Src specifically associates with ErbB2, but not with wild-type EGFR or other ErbB family members. However, mutant EGFRs isolated from lung adenocarcinomas have the capacity to associate with Src and these EGFR mutants require Src kinase activity for transformation (24, 25).
Here we demonstrate that CS/oxidative stress leads to robust Src phosphorylation/activation which becomes aberrantly associated with EGFR in NSCLC cells. We used Src mutants and inhibitors to analyze the role of Src in generating resistance to TKIs in EGFR-overexpressing NSCLC cells exposed to cigarette smoke. Our findings suggest that the association of the EGFR with activated Src is critical for the TKI resistant phenotype observed in both the WT- and L858R MT EGFR-expressing cells during CS exposure, supporting our theory that simultaneous targeting of Src during TKI treatment may overcome smoking/ oxidative stress-related resistance to TKIs in NSCLC.
MATERIALS AND METHODS
Cell culture, treatments, and reagents
A549 adenocarcinoma (ATCC), NCI-H3255 (generous gifts from Dr. Philip Mack, University of California at Davis), NIH-3T3 (generous gifts from Dr. Hamid Band, University of Nebraska Medical Center; (25)), and CHO cells have been employed in this study. All the cell lines used in this study were previously characterized by others, as reported (25-27); in addition, we verified the correct expression of WT and L858R EGFR mutants in all the cell lines (the mRNA of EGFR was sequenced after RT-PCR and extraction of the PCR product from agarose gel). A549 cells were cultured in F12K medium (GIBCO) supplemented with 10% Fetal Bovine Serum (FBS; GIBCO) and 1% Pen/Strep (GIBCO). NCI-H3255 cells were cultured in RPMI medium (GIBCO) supplemented with 10% FBS and 1% Pen/Strep. NIH 3T3 cells stably expressing wild type EGFR or L858R mutant EGFR, CHO cells were cultured in DMEM medium (GIBCO) supplemented with 10% FBS and 1% Pen/Strep.
Transfections of wild type EGFR, kinase dead K721M EGFR, oncogenic L858R EGFR, constitutively-active (CA) Y527F Src-GFP, and dominant-negative (DN) Y527F/K295R Src-GFP constructs were performed using Lipofectamine 2000 Transfection reagent (Invitrogen) according to the manufacturer's protocol. CA-Src-GFP and DNSrc-GFP constructs were kindly provided by Dr. Yoav Henis (Tel Aviv University). Cells were transfected at ~80% confluence in either 35 or 60 mm dishes and treated after ~24 hrs.
For treatments, DMEM medium supplemented with 20 mM HEPES, pH 7.4 was used. EGF was added directly into treatment medium at a final concentration of 100 ng/ml. AG1478 (Cell Signaling), Erlotinib (Tarceva®) and Dasatinib (Sprycel®) were dissolved in dimethyl sulfoxide (DMSO) and then added to the treatment medium at a final concentration of 1 μM. PP1 and PP2 were dissolved in DMSO and then added to the treatment medium at a final concentration of 5 μM. DMSO was added at a final concentration of <0.1% to control-untreated (-) cells.
Cells were collected by scraping directly in lysis buffer: 1% NP-40 (Igepal, from Sigma), 50 mM Tris, 10% Glycerol, 0.02% NaN3, 150 mM NaCl, pH 7.4, containing a cocktail of phosphatase and protease inhibitors (Sigma) as well as 1 mM NaF and Na3VO4. Lysates were passed 5 times through a 30 gauge needle prior to centrifugation and further processing of the samples (either immunoprecipitation or immuno-blotting). All the other reagents were from Sigma, unless differently specified.
Cigarette smoke exposure
Serum-starved cells were exposed to cigarette smoke (CS) gas phase as described before (28). Cells were placed in a vacuum oven with a chamber volume of 0.45 ft3 at a constant temperature of 37°C. A negative pressure was generated by vacuum (~10 Hg) and smoke from one cigarette (University of Kentucky, 2R4F) was drawn into the chamber by equilibrating the pressure inside the chamber with atmospheric pressure through a valve. For some experiments (requiring prolonged exposure to CS-induced oxidative stress) medium pre-conditioning with CS was used: 6 ml of medium were put in a 50 ml syringe, which was further used to aspirate ~60cc of CS puff through a valve-equipped tube. The CS-conditioning of the medium was allowed (inside the syringe) for 30’ at 37°C; then the CS-conditioned medium was diluted (or not) with fresh medium, prior to be used in the experiments.
Immunoprecipitation (IP)
200-400 μg of total protein extracts were incubated for 3 h with 2-4 μg of antibodies (Abs): anti (α) 528 (against EGFR) or αSrc (Santa Cruz Biotech). 50 μl of 50% protein A-agarose bead complexes (Repligen) were added to the samples and incubated for 90 min. Four washes (by sequential centrifugation and re-suspension) with the NP-40-lysis buffer were done prior to re-suspending the immuno-precipitates in the loading dye for SDS-PAGE, as described before (28, 29).
SDS-PAGE and Immunoblotting (IB)
6-12% acrylamide gels were used in a two cell system (BioRad) for 1-4 hrs at 100 V. 20-100 μg total protein extracts or the IP samples were loaded into each well of the SDS-PAGE in the presence of dithiothreitol (DTT) reducing loading dye. After SDS-PAGE separation, proteins were transferred to a nitrocellulose membrane and “blocked” with 5% skim milk in Tris buffered saline with 0.05% Tween-20 (TBS-T) for 120 min. or overnight, as described (29). Primary antibodies were incubated in 5% milk-TBS-T for 2 hrs at room temperature. Secondary antibodies, either goat αmouse or goat αrabbit horseradish peroxidase (HRP)-conjugated (Jackson ImmunoResearch), were incubated for 90 min. at room temperature at 1:10,000 dilution in 5% milk-TBS-T. Bands were imaged using enhanced chemiluminescence (Supersignal® West Pico Luminol Enhancer; Thermo Scientific). Extensive washes with TBS-T were done in between each step. When performing sequential IBs to assess the specific sites of EGFR phosphorylation, antibodies were stripped off the membranes using stripping buffer (Restore™ PLUS; Thermo Scientific) in between of each IB. Primary antibodies used in this study for IBs were: α2232 (αEGFR, Cell Signaling, 1:1000), αSrc (Santa Cruz Biotech, 1:1000), αphospho-Y416 Src (Cell Signaling, 1:1000), αp-Y20 (Santa Cruz Biotech, 1:3000), αp-Y1173 EGFR (Santa Cruz Biotech, 1:1000), and αp-Y1086 EGFR, αp-Y1068 EGFR, and αp-Y845 EGFR (Cell Signaling, 1:1000).
Cell viability assay
NSCLC Cell viability was measured by the standard/ common 3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) method. Briefly, the cells were cultured and treated in phenol red-free medium which was supplemented with 1mg/ ml MTT. The conversion of the yellow tetrazole (MTT) into purple formazan was carried out for 45’ in the cell incubator. The reaction was terminated and the formazan was solubilized by the addition of isopropanol containing 4mM HCl and 0.1% NP40 (in a volume equal to that of the reaction medium). Absorbance of the solution was measured by spectrophotometry at 540 nm. The negative control consisted of a sample with no cells/ no conversion of tetrazole.
Statistical analysis
Each experiment (IB, IP/IB) was repeated at least three times and the shown images are representative. The plotted data are reported as mean ± standard deviations (St-Devs). Statistical significance were determined by Student's t-test and p-value <0.05 considered statistically significant.
RESULTS
EGFR resistance to TKIs in NSCLC exposed to cigarette smoke: Src activation and binding to EGFR
To address why CS exposure renders the EGFR resistant to TKI treatment in NSCLC cells (21), we tested the role of Src in this phenomenon. Previously, we demonstrated that Src is consistently phosphorylated/activated under oxidative stress (29). Fig. 1 presents the kinetics of Src activation, shown by the phosphorylation of Tyr416 (pY416-Src), in NSCLC cells exposed to tobacco smoke from 1 cigarette compared to that of EGF stimulation (100 ng/ml) for 30 min. As shown in Fig. 1, Src is robustly activated by CS exposure but not by EGF. Furthermore, the CS-induced activation of Src was not limited to A549 but was also observed in several cell types utilized for our current study including NIH-3T3 stably expressing L858R EGFR (Sup. Fig. 1A, B) and CHO cells (Sup. Fig. 1C).
Figure 1. Src is aberrantly activated/phosphorylated by cigarette smoke (CS) exposure compared to its activation state during EGF stimulation of cells.
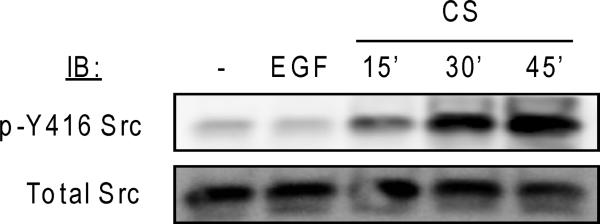
Serum starved A549 cells were incubated (or not) for 30’ with EGF (100 ng/ml) or for 15‘, 30’ and 45’ CS (smoke from 1 cigarette); p-Y416 (active) Src and total Src were measured by IB in 50 μg of total proteins.
We first tested whether these two activated tyrosine kinases, Src and EGFR, interact under CS. Serum-starved A549 cells were incubated with 1 μM Erlotinib for 30 minutes and subsequently exposed to either EGF (100 ng/ml; 15 minutes) or CS (1 cigarette; 30 minutes). Cells were lysed, immunoprecipitated (IPed) for the EGFR or Src, and immunoblotted (IBed) for pY416-Src, pY1173-EGFR, and total EGFR. As shown in Fig. 2, the CS-activated EGFR is not only unaffected by Erlotinib but also abnormally binds the activated Src under these conditions. Furthermore, Erlotinib did not prevent Src activation nor its binding to the EGFR during stimulation by CS (Fig. 2A). However, minimal-to-no binding of Src to EGFR could be observed under EGF stimulation thus indicating that Src may have some role in imparting the TKI-resistant phenotype to EGFR-overexpressing cells (Fig. 2B)
Figure 2. WT EGFR is bound by Src and is resistant to TKI upon CS exposure (both in the presence and absence of TKI).

Serum starved A549 cells were incubated (or not) with 1 μM Erlotinib for 30’ and exposed to 100 ng/ml EGF or smoke from 1 cigarette (CS) for additional 30’. EGFR was IPed from the total cell lysates and the immuno-precipitates were IBed for p-Y416 (active) Src (A), total EGFR and p-Y1173 EGFR (B).
CS-induced TKI resistance is eliminated by inhibition of Src activity
Following the observation that Src binds to the aberrantly-activated EGFR during CS exposure, we next tested whether Src activity plays a role in the WT EGFR's resistance to TKIs under CS stimuli. Serum-starved A549 cells were incubated (or not) with 10 μM of the Src inhibitors PP1 and PP2 (5 μM PP1 + 5 μM PP2) for 30 minutes and then treated (or not) with EGF or CS for an additional 30 minutes. Cells were lysed and immuno-blotted (IBed) for pY416-Src and total protein. As demonstrated in Sup. Fig. 2, Src inhibitors PP1 and PP2 could quench the observed activation of Src by CS in A549 (as a result of inhibiting its tyrosine kinase activity required for its self-activation).
We next assessed whether Src inhibition could restore TKI sensitivity to NSCLC cells exposed to CS. Serum-starved A549 cells were incubated (or not) for 30 min. with 1 μM Erlotinib and 10 μM PP1-PP2 (5 μM PP1 + 5 μM PP2) or a combination of them, and then treated with EGF or CS for additional 30 min., as indicated in Fig. 3. EGFR was IPed from the total cell lysates and IBed for total receptor (EGFR), total EGFR tyrosine-phosphorylation (p-YEGFR) and phosphorylation level of specific tyrosines (p-Y1173, p-Y1068, p-Y845). Fig. 3A shows that CS induces a strong activation/ auto-phosphorylation of EGFR, as presented by the receptor phosphorylation at Y1173 and Y1068. Src-dependent trans-phosphorylation site Y845 of EGFR was also phosphorylated, as expected (21, 28). With PP1 andPP2, the CS-dependent phosphorylation of Y845 of EGFR was markedly reduced, while the receptor auto-phosphorylation on Y1173 and Y1068 remained intact. Importantly, when the TKI treatment (Erlotinib) was combined with Src inhibition,auto-phosphorylation of the receptor was significantly inhibited, not only following EGF stimulation, but also upon the exposure of cells to CS (Fig. 3A). Quantitative values obtained from the analysis of three independent IP/IB experiments evaluating the p-Y1173 signal normalized to total EGFR are shown in Fig. 3B. Similar results were observed in NSCLC cells treated with Erlotinib and Dasatinib, a FDA-approved Src inhibitor (not shown). Our findings strongly suggest that the activation of Src controls the EGFR resistance to TKI, which emerges during CS exposure in lung epithelial cells (21).
Figure 3. Src inhibition restores TKI sensitivity to WT EGFR in NSCLC cells exposed to CS (even though Src is still bound to EGFR).

A. Serum starved A549 were incubated (or not) for 30’ with 1 μM Erlotinib and 10 μM PP1-PP2 (5-5 μM), combined (or not), and then exposed to either EGF or CS for additional 30’, as before. EGFR was IPed from the total cell lysates and IBed for total receptor, total tyrosine phosphorylation level (p-YEGFR) and specific Tyr site phosphorylation levels (p-Y1173, p-Y1068, p-Y845). B. The graphic shows the quantification of EGFR p-Y1173, normalized to total EGFR in three independent experiments. NS stands for “Not statistically significant”. * = p < 0.05 in respect to control (-).
TKI-resistant phenotype emerges when Src is bound to the EGFR in its active, “open” state
We next tested whether the CS-induced interaction between EGFR and Src (21) is dependent on Src activity. Serum-starved A549 cells were incubated with 10 μM PP1-PP2 for 30 min. and then treated with smoke, as before. Cells were lysed, Src was IPed and the immuno-precipitates were IBed for total Src and total EGFR. As shown in Sup. Fig. 3A, EGFR was pull down together with Src in the presence of Src inhibitors, demonstrating that Src kinase activity is not mandatory for physical binding between Src and EGFR that occurs under CS exposure, similar to findings from a previous study with H2O2-induced oxidative stress (29).
Subsequently, we investigated whether EGFR kinase activity is required for its binding to Src. To test this and to circumvent the fact that the kinase activity of the EGFR cannot be inhibited by TKIs upon CS exposure (as shown above in Fig. 2B), we transiently over-expressed either wild type (WT) or a kinase-dead (KD) mutant EGFR (K721A-EGFR) in CHO cells which do not endogenously express significant amounts of EGFR or other ErbB family members. 24h after transfection, cells were exposed to either EGF or CS, as before, and EGFR was IPed and IBed for pY416-Src, pY1173-EGFR, and total EGFR. Intriguingly, though the KD EGFR cannot be activated/ phosphorylated by either EGF or CS xposure, active Src still directly binds to the KD EGFR following CS exposure (Sup. Fig. 3B), thus demonstrating that the kinase activity of EGFR and its subsequent auto-phosphorylation is not essential for its interaction with Src.
To further confirm the requirement of Src in the emergence of EGFR resistance to TKIs, GFP-fused dominant-negative Src (DN-Src; K295R/Y527F) or a constitutively active form of Src (CA-Src; Y527F) were transiently expressed in A549 cells. Binding of these two Src constructs to EGFR was tested in the presence of EGF or CS exposure. Intriguingly, our co-IP studies demonstrated that both GFP-tagged Src constructs (CA- and DN-Src) bind to EGFR in the presence and absence of CS exposure (Fig. 4), reiterating our previous observations that Src remained bound to the aberrantly-activated WT EGFR under CS stimulation despite Src inhibition. Moreover, it reaffirmed that an “open” conformation of Src is sufficient for its interaction/binding to the WT EGFR.
Figure 4. CS-dependent TKI resistance of WT EGFR harboring NSCLC cells is caused by Src activation.

A549 cells were transiently-transfected with WT EGFR and either GFP-tagged Dominant-Negative (DN) Src or GFP-tagged Constitutively-Active (CA) Src; 24h post-transfection the cells were serum starved and incubated for 30’ with 1μM Erlotinib and then exposed to either EGF or CS, as before. A. EGFR was IPed from the total cell lysates and IBed for total Src (GFP) and total receptor. B. EGFR was IPed from the total cell lysates and IBed for total receptor, total tyrosine (Y) phosphorylation level (p-YEGFR) and specific Y site phosphorylation levels (p-Y1173, p-Y1068, p-Y845).
Next, we further tested whether Src activity is required for the EGFR's resistance to Erlotinib/TKIs under CS exposure by transiently over-expressing either DN-Src or CA-Src in A549 for 24h then subsequently incubating the cells with 1 μM Erlotinib for 30 minutes and exposing them to either EGF or CS, as before. Fig. 4 shows that the DN-Src could sensibly improve the efficacy of Erlotinib in inhibiting EGFR auto-phosphorylation during CS exposure, compared to the unobstructed CS-induced EGFR Tyr phosphorlyation in cells expressing the CA-Src treated with Erlotinib (Fig. 4).
Overall, these data demonstrate that CS-induced Src activation controls TKI sensitivity of EGFR. In WT EGFR-overexpressing NSCLC cells exposed to CS, resistance to TKIs is being developed post-translationally not only because WT EGFR is aberrantly activated, but primarily because Src is also abnormally activated and binds the WT EGFR under CS exposure. Indeed, this acquired TKI-resistant phenotype reliably disappears upon Src inhibition (for example by PP1-2 or Dasatinib treatment or overexpression of DN-Src) despite still being bound to the CS-activated EGFR.
Phosphorylation of Tyr845-EGFR does not control its CS-induced TKI Resistance
Having demonstrated that Src inhibition restored TKI sensitivity to WT EGFR-expressing NSCLC cells, we investigated whether phosphorylation of Y845-EGFR, a Tyr phosphorylation site canonically linked to Src activity (25, 30), plays a role in generating the TKI-resistant phenotype observed during CS exposure. To test this, NIH-3T3 cells stably over-expressing either WT or Y845F MT EGFR were incubated with Erlotinib and exposed to either EGF or CS, as before. Intriguingly, we observed no differences in TKI sensitivity/resistance between WT and Y845F MT EGFR, presented in Sup. Fig. 4, indicating that trans-phosphorylation on Y845 does not control the CS-generated TKI resistant phenotype of EGFR. Given the potential for trans-phosphorylation of EGFR by other ErbB family members or by endogenous EGFR in NIH-3T3 cells, we further confirmed this finding in CHO cells which do not express endogenous EGFR or any other ErbB member. Again, no differences between WT and Y845F MT EGFR were observed in terms of CS-induced EGFR TKI resistance, while CS-activated Src remained bound to EGFR regardless of the presence of intact Y845 (not shown).
CS-induced TKI resistance in NSCLC cells expressing TKI-sensitive L858R MT EGFR is reversed by Src inhibition
The discovery that somatic mutant variants of the EGFR (L858R substitution and Δ746-750 deletion) were uniquely sensitized to TKIs drives further TKI drug development and investigations into conformation states of the EGFR. Previous studies have shown that interactions between WT EGFR and Src was generally not stable (31) but the interactions between Src and mutant EGFRs, which harbor unique structural differences in the activation loop of the kinase domain (32), were more stable. Furthermore, we previously demonstrated that the TKI-sensitive L858R MT EGFR, which is constitutively-active, became resistant to TKIs upon CS exposure (21).
Subsequently, we tested whether the half maximal (50%) inhibitory concentration (IC50) of the TKI Erlotinib against L858R EGFR was affected by CS exposure. For this we used NIH-3T3 cells stably over-expressing the human L858R EGFR MT; the cells were incubated for 30’ with increasing concentrations of Erlotinib (from 10−12 to 10−4 M) and then exposed (or not) for additional 30’ to CS; after the treatments, the level of receptor auto-phosphorylation on Y1173 was measured (by IB). Fig. 5 shows that CS exposure not only increases the IC50 of Erlotinib against the L858R EGFR MT (~25 times), but further reduces Erlotinib maximal inhibition of the receptor (~5 times). As mentioned before (21), the stability/ integrity of the drug itself was not affected by CS exposure.
Figure 5. A. CS exposure increases the IC50 of erlotinib (TKI) against L858R EGFR MT, and also reduces erlotinib maximal inhibition of the receptor.

Serum-starved NIH-3T3 cells stably over-expressing L858R EGFR MT were incubated for 30’ with erlotinib at different concentrations (as indicated, in Molarity (M)) and then exposed (or not) to CS for additional 30’, as before. 30-50 μg of proteins from the cell lysates were analyzed by IB for pY1173 and EGFR signal. A. the graphic shows densitometry values (IB) of pY1173/ EGFR signal over increasing erlotinib concentrations (in logarithmic scale), reported as % of the signal obtained in cells not exposed to erlotinib (-) (either control or CS-exposed cells). The estimated IC50(s) of erlotinib and maximal inhibition values are indicated by the arrows (for either control or CS-exposed cells). B. A sample IB is shown.
We next tested whether Src controlled the TKI-resistant phenotype also observed for the TKI-sensitive L858R MT EGFR. Initially, NIH-3T3 + L858R MT EGFR cells were incubated with 1 μM AG1478 (TKI) for 30 min. then exposed to EGF or CS (or not), as before. Cell lysates were collected and IPed for either Src or EGFR and IB-ed for pY1173-EGFR, total EGFR, and total Src. Besides previous reports that Src binds to the L858R MT EGFR (25, 33), the CS-activated L858R MT EGFR showed greater Src binding than that under EGF or no stimulation, shown in Sup. Fig. 5A. As expected, the constitutively-active L858R MT EGFR remained sensitive to the TKI under control conditions and EGF stimulation, demonstrated by suppressed phosphorylation of several Tyr residues, but became resistant to the TKI under CS exposure (Sup. Fig. 5B).
Subsequently, we treated H3255 NSCLC cells (harboring L858R EGFR MT) with both Erlotinib and PP1/PP2 under CS exposure to further test our theory. Serum starved H3255 cells were incubated with Erlotinib, PP1-PP2, or a combination of them for 30 min. and subsequently treated with EGF or CS, as before. EGFR was IPed from the cell lysates and IBed for total EGFR and receptor auto-phosphorylation on Y1173 and Y1068. Fig. 6A shows that the L858R MT EGFR of H3255 cells is constitutively phosphorylated on Y1173 and Y1068, as expected (21), which is further increased by both EGF and CS treatments, and that Erlotinib treatment could abolish the receptor auto-phosphorylation on Y1173 and Y1068 in control and EGF stimulation conditions. However, when these cells were exposed to CS, Erlotinib had reduced efficacy, as expected. Importantly, the combination of TKI with Src inhibitiors could block the activity and auto-phosphorylation of the EGFR in the CS-exposed cells much better than either drug alone (Fig. 6A). This further indicates a critical role of Src activity in controlling TKI sensitivity of NSCLC cells in a setting of CS exposure and potentially other sources of oxidative stress for both WT-EGFR and constitutively active/TKI-sensitive L858R EGFR (see model in Fig. 6B).
Figure 6. A. Inhibition of SFKs improves TKI sensitivity of kinase activated EGFR mutant (L858R MT EGFR) harboring NSCLC cells during CS exposure.

Serum starved H3255 cells were incubated (or not) for 30’ with 1 μM Erlotinib and 10 μM PP1-PP2 (5-5 μM), combined (or not), and then exposed to either EGF or CS for additional 30’, as before. EGFR was IPed from the total cell lysates and IBed for total receptor and specific p-Y1173 and p-Y1068. B. Modeling structure/function alteration of EGFR which lead to TKI resistance upon binding of active c-Src to the receptor following exposure to CS-induced oxidative stress.
Notably, we have previously demonstrated by the soft/agar assay that CS exposure abolishes the TKI-dependent inhibition of anchorage independent growth of EGFR-transformed cells (21). In order to further assess the biological relevance of our biochemical findings, we tested the effects of TKI and CS exposure treatments on H3255 NSCLC cells proliferation/viability; such cells are dependent on their L858R EGFR mutation for proliferation and survival. Indeed, it has been shown before by others that H3255 cells rapidly undergo apoptosis when exposed to TKIs (34). As shown now, in Fig. 7, incubation of H3255 cells with TKI (1μM Erlotinib) caused a marked decrease in cell proliferation/viability (compared to control). Importantly, while the CS exposure also reduced H3255 cell viability, compared to control condition/growth, such CS exposure entirely canceled the inhibitory effect of TKI (Fig. 7). Moreover, Dasatinib treatment reduced cell viability of CS-treated cells, implicating Src activity as survival/ proliferative mechanism during CS/oxidative stress exposure. Notably, Dasatinib treatment was additive to TKI in reducing cell proliferation/viability in the presence and absence of CS exposure, providing rationale for combinatory therapy using both EGFR and Src inhibitors in treating NSCLC both in smokers and non-smokers.
Figure 7. Effects on H3255 NSCLC cell viability by erlotinib, CS exposure and dasatinib.

NSCLC cells (H3255) were continuously cultured with medium pre-conditioned (or not) by a 30’ CS exposure (prepared as described under the methods) in the presence or absence of 1 μM erlotinib (TKI) and 0.1 μM dasatinib (Das.). Cell viability was assessed by MTT assay. The data are plotted as % of the initial MTT control value (untreated cells at the time point zero).
DISCUSSION
Our recently published observations demonstrated that both WT and L858R MT EGFR become resistant to TKIs (a number of TKIs were tested: AG1478, Erlotinib, and Gefitinib) (Sup. Fig. 6) in NSCLC cells exposed to CS-induced oxidative stress (21). We provided evidence that (i) the CS-activated EGFR is resistant to TKIs by post-translational mechanism(s) and not by random somatic mutations, (ii) such CS-dependent TKI-resistant phenotype is also observed in terms of activation of Akt/protein kinase B and Erk (Extracellular regulated protein kinase), as well as TKI-unresponsive clonal growth of transformed cells in soft agar, (iii) CS did not chemically “damage” and reduce the effectiveness of the TKIs used (21), (iv) the CS-activated EGFR may present a novel, active conformation that differs from the “conventional”, ligand (EGF-induced) one and also from that of the oncogenic mutant EGFR, and (v) CS-activated Src strongly binds to the EGFR (21). Following these critical observations, we investigated the role of Src in EGFR's resistance to TKI in NSCLC cells exposed to CS-induced oxidative stress (21, 29).
Initially, we found that Src, a proto-oncogene and tyrosine kinase often over-expressed in cancer (35-39), was robustly activated and bound to both the WT and L858R MT EGFR during CS exposure (Fig. 2, Fig. 5) (21), even in the presence of TKIs (Fig. 2). In addition, we have discovered that (i) Src inhibition restores TKI sensitivity of WT EGFR-expressing NSCLC during CS exposure (suppressing the phosphorylation of Y1068 and Y1173 of EGFR) (Fig. 3), (ii) a dominant-negative form of Src sensitizes WT EGFR-expressing NSCLC to TKI during CS exposure (Fig. 4) and (iii) Src inhibition abolishes TKI resistance also in L858R EGFR-harboring NSCLC cells during CS exposure (Fig. 6A). Notably, however, a constitutively-active form of Src alone is not sufficient for causing EGFR resistance to TKI until exposed to CS, suggesting that both EGFR and Src should be aberrantly activated by CS in order to cause TKI resistance. Nevertheless, given the prevalence of smoking and both WT and “activating” MT EGFR expression in NSCLC, the fore-mentioned studies provide stronger scientific rationale for a novel combinatory therapy using both EGFR and Src inhibitors for treating NSCLC, especially in smokers.
Furthermore, we strongly believe in the high clinical value of these studies. Defining the impact of oxidative stress generated by tobacco smoking on TKI treatments, and the respective mechanism involved, will serve as a message to patients who continue smoking despite undergoing TKI therapy and also guide physicians for decision relating to drug regiments and dosage recommendations. This is of great importance whenever a patient continues smoking or is subjected to conditions of high lung oxidative stress. Notably, lung cancer incidence is significantly elevated in patients with COPD (chronic obstructive pulmonary diseases), who present chronic oxidative stress (40) and aberrantly activated EGFR (41, 42) in their lungs.
As recently described by Mitchell et al (19), we still do not know whether lung cancer patients carrying the “TKI-sensitive” EGFR mutations and being former or current smokers would benefit from a TKI therapy (in comparison to those that are never-smokers). However, in this manuscript we present evidence indicating that tobacco smoking/ oxidative stress could affect the efficacy of TKIs’ inhibition of both EGFR phosphorylation and cell viability of NSCLC. Moreover, this occurs even if the cells carry a TKI-sensitive EGFR mutation, such as L858R. Importantly, we have previously reported that CS exposure affects EGFR activation/ phosphorylation specifically via generation of H2O2-induced oxidative stress (28). Consistently, we confirmed that direct exposure of NSCLC cells to H2O2 affects EGFR active conformation rendering the receptor resistant to TKIs (29).
Src and other members of its family (Src Family Kinases; SFKs) have been thoroughly studied in several cancers, including NSCLC (43-45). Recently, studies by Band and colleagues established novel cooperative roles for Src in supporting oncogenic mutant EGFR-dependent transformation of airway epithelial cells via the endocytotic recycling pathway and constitutive phosphorylation of Tyr845 in MT EGFRs (25, 30, 46). Importantly, several Src/SFKs are significantly activated in the airways of NSCLC patients actively smoking compared to those who were never or former smokers (45). Masaki et al had similar observations that both Src activity and expression are elevated in malignant lung tissue samples, particularly in adenocarcinomas, compared with surrounding normal lung parenchyma from the same patients (47).
Previous molecular studies show that Src interacts with EGFR in two unique ways that somehow involve the activation loop of the EGFR kinase. Src is known to phosphorylate Tyr845 (Y845) of both WT and MT EGFRs, a tyrosine residue found on the activation loop of the EGFR kinase domain (25, 48). However, past mutagenic studies showed that the phosphorylation of Y845 was not required for activation of the EGFR kinase, though its phosphorylation by Src assists in maximal biological activity by EGF stimulation (48, 49). Recently, Shaw and colleagues presented convincing data demonstrating that phosphorylation of Y845 aids in stabilizing a critical salt-bridge interaction between Lys721 and Glu738 required for catalytic activity of the EGFR (50). Following these studies, we tested whether Y845 phosphorylation may be important for TKI resistance under CS. However, this appeared to not be the case (Sup. Fig. 4) as the Y845F MT EGFR continued to be activated by CS (pY1173), both in the presence and absence of TKIs. Given that Src activity is involved in generating EGFR TKI resistance (Fig. 3), it is possible that aberrant activation, and conformation of the EGFR under CS exposure (21, 29) may be caused by Src-dependent phosphorylation of other EGFR tyrosines, which is currently being investigated in our laboratory.
Canonically, only ErbB2/Her2, and not WT EGFR, directly interacts with Src despite ErbB2 and EGFR sharing over 95% homology in amino acid sequence of the kinase domain. Previous studies by Muller and colleagues clearly demonstrated that interactions between Src and ErbB2 are conformation-dependent, requiring the structure of the catalytic kinase domain of ErbB2 for binding (31). Similarly, Band and colleagues showed that Tyr845 of EGFR was required for binding and oncogenic cooperativity of Src to both L858R substitution mutant and Δ746-750 deletion mutant EGFRs (25). However, interactions between WT EGFR and Src under normal conditions (unstimulated or EGF-stimulated) appear to be transient and/or non-existent, which intrigued us when we observed that Src strongly binds to the WT EGFR only during exposure to CS-induced oxidative stress (Fig. 2) (21, 29). Our results show that this aberrant interaction between Src and EGFR continues to occur during CS exposure despite Src inhibition (Sup. Fig. 3A) or a kinase-dead EGFR (Sup. Fig. 3B), suggesting that neither Src or EGFR kinase activity is solely responsible for their interaction, but rather, their conformations which may be initiated and/or sustained by CS-induced oxidative stress (See “Model” in Fig. 6B).
Though structural studies on Src are ongoing, the active conformation(s) of Src triggered by oxidative stress are incompletely resolved. The phosphorylation of Y527-Src (pY527) is important for down-regulation of Src activity, because it renders Src inactive due to a “closed” conformation involving Src's SH2 phospho-Tyr binding domain (51, 52). Substituting Tyr527 for a phenylalanine (F) forfeits this negative-regulatory function thereby leaving the Src in an “open” conformation (24, 53). Muller and group showed that an “open” Src conformation induced by the Y527F mutation was sufficient for Src binding to ErbB2 (24). Intriguingly, our current study mirrored that finding, shown in Fig. 4A, that Y527F-Src (CA-Src) and Y527F/K295R (DN-Src), which are both in the “open” state, constitutively bind to the EGFR regardless of stimuli (EGF; CS). Ultimately, understanding the mechanism of how CS aberrantly evokes an active conformation in both Src and EGFR will provide clues for designing new inhibitors specifically for treatment of NSCLC in smokers.
Functionally, we have shown before by the soft/agar assay that CS exposure eradicates the TKI-dependent inhibition of anchorage independent growth of EGFR-transformed cells (21). Additionally, others have shown previously that TKIs induce apoptosis in H3255 cells (34). Our data demonstrated that Erlotinib (TKI) induces a drastic reduction in cell viability (as expected). This was shown further to be reduced by Dasatinib co-treatment. However, the TKI effect was significantly counteracted by CS exposure (Fig. 7). It is interesting that at the same time, Dasatinib treatment alone reduced the cell viability of CS-treated cells, implicating Src activity as a survival/ proliferative mechanism during CS/oxidative stress exposure. Dasatinib treatment was additive with TKI in reducing cell viability in the presence and absence of CS exposure, providing the rationale for a combinatory therapy using both EGFR and Src inhibitors to treat NSCLC both in smokers and non-smokers.
In summary, we provide insight into the initial structure/function alterations of EGFR and Src following exposure to CS. We present for the first time a post-translational mechanism accounting for drug (TKI) resistance development in NSCLC cells, offering not only a paradigm shift in the way we think of cancer therapy resistance, but also a specific target, Src, in the context of tobacco smoke/oxidative stress-related lung cancer. Specifically, the results of our investigation into Src as a molecular target for acquired drug resistance to TKIs in NSCLC provide strong support for continued evaluation of combinatory therapies for treatment of NSCLC, particularly in patients who are exposed to lung oxidative stress (such as in smokers) and thus harbor aberrantly activated EGFR and Src. However, it should be noted that expression of KRAS mutants or other oncogenes may de novo overcome the need of EGFR and Src signaling for cell survival and proliferation limits the patient population where such a combined therapy targeting both EGFR and Src should be considered. Further studies should elucidate the dynamic interaction/cooperation between EGFR and Src during CS-induced oxidative stress, which may lead to novel opportunities for therapeutic intervention.
Supplementary Material
ACKNOWLEDGEMENTS
We thank Dr. H. Band (University of Nebraska Medical Center) for providing the NIH-3T3 cell line stably over-expressing EGFR (WT and L858R MT EGFR). We thank Dr. Philip Mack (University of California, Davis) for providing the NCI-H3255 NSCLC cells. We thank Dr. Yoav Henis (Tel Aviv University) for providing the Src constructs (CA-Y527F and DN-Y527F/K295R).
GRANT SUPPORT
Financial Support: this work was supported by grants from the National Institutes of Health (HL-66189 to T.Goldkorn) and from the Tobacco-Related Disease Research Program (TRDRP) (17RT-0131 to T.Goldkorn and 20FT-0087 to S.Filosto).
Abbreviations used are
- *
p < 0.05
- ~
about
- α
anti-
- Δ
delta (deletion)
- Ab or Abs
antibody or antibodies
- CA
constitutive-active
- CHO
Chinese Hamster Ovarian
- CS
cigarette smoke
- DN
dominant-negative
- EGF
epidermal growth factor
- EGFR
epidermal growth factor receptor
- H2O2
hydrogen peroxide
- HAE
human airway epithelial
- KD
Kinase Dead
- IB, or IBed
immuno-blotting or immuno-blotted
- IP or IPed
immuno-precipitation or immuno-precipitated
- min.
minutes
- MT
mutant
- NS
Non statistically Significant
- NSCLC
non-small cell lung cancer
- p-
phosphorylated
- St-Devs
standard deviations
- TKIs
tyrosine kinase inhibitor(s)
- Y or Tyr-
tyrosine
- WT
wild type
Footnotes
The authors declare no conflict of interest.
REFERENCES
- 1.Jemal A, Murray T, Samuels A, Ghafoor A, Ward E, Thun MJ. Cancer statistics, 2003. CA Cancer J Clin. 2003;53:5–26. doi: 10.3322/canjclin.53.1.5. [DOI] [PubMed] [Google Scholar]
- 2.Rusch V, Klimstra D, Venkatraman E, Pisters PW, Langenfeld J, Dmitrovsky E. Overexpression of the epidermal growth factor receptor and its ligand transforming growth factor alpha is frequent in resectable non-small cell lung cancer but does not predict tumor progression. Clinical cancer research : an official journal of the American Association for Cancer Research. 1997;3:515–22. [PubMed] [Google Scholar]
- 3.Hirsch FR, Varella-Garcia M, Bunn PA, Jr., Di Maria MV, Veve R, Bremmes RM, et al. Epidermal growth factor receptor in non-small-cell lung carcinomas: correlation between gene copy number and protein expression and impact on prognosis. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 2003;21:3798–807. doi: 10.1200/JCO.2003.11.069. [DOI] [PubMed] [Google Scholar]
- 4.Polosa R, Prosperini G, Leir SH, Holgate ST, Lackie PM, Davies DE. Expression of c-erbB receptors and ligands in human bronchial mucosa. Am J Respir Cell Mol Biol. 1999;20:914–23. doi: 10.1165/ajrcmb.20.5.3308. [DOI] [PubMed] [Google Scholar]
- 5.Barsky SH, Roth MD, Kleerup EC, Simmons M, Tashkin DP. Histopathologic and molecular alterations in bronchial epithelium in habitual smokers of marijuana, cocaine, and/or tobacco. J Natl Cancer Inst. 1998;90:1198–205. doi: 10.1093/jnci/90.16.1198. [DOI] [PubMed] [Google Scholar]
- 6.O'Donnell RA, Richter A, Ward J, Angco G, Mehta A, Rousseau K, et al. Expression of ErbB receptors and mucins in the airways of long term current smokers. Thorax. 2004;59:1032–40. doi: 10.1136/thx.2004.028043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Linggi B, Carpenter G. ErbB receptors: new insights on mechanisms and biology. Trends in cell biology. 2006;16:649–56. doi: 10.1016/j.tcb.2006.10.008. [DOI] [PubMed] [Google Scholar]
- 8.Sharma SV, Bell DW, Settleman J, Haber DA. Epidermal growth factor receptor mutations in lung cancer. Nat Rev Cancer. 2007;7:169–81. doi: 10.1038/nrc2088. [DOI] [PubMed] [Google Scholar]
- 9.Parkin DM, Bray F, Ferlay J, Pisani P. Global cancer statistics, 2002. CA Cancer J Clin. 2005;55:74–108. doi: 10.3322/canjclin.55.2.74. [DOI] [PubMed] [Google Scholar]
- 10.Mountzios G, Fouret P, Soria JC. Mechanisms of Disease: signal transduction in lung carcinogenesis -- a comparison of smokers and never-smokers. Nat Clin Pract Oncol. 2008;5:610–8. doi: 10.1038/ncponc1181. [DOI] [PubMed] [Google Scholar]
- 11.Dancey JE. Predictive factors for epidermal growth factor receptor inhibitors--the bull's-eye hits the arrow. Cancer cell. 2004;5:411–5. doi: 10.1016/s1535-6108(04)00122-9. [DOI] [PubMed] [Google Scholar]
- 12.Yatabe Y. EGFR mutations and the terminal respiratory unit. Cancer Metastasis Rev. 2010;29:23–36. doi: 10.1007/s10555-010-9205-8. [DOI] [PubMed] [Google Scholar]
- 13.Lynch TJ, Bell DW, Sordella R, Gurubhagavatula S, Okimoto RA, Brannigan BW, et al. Activating mutations in the epidermal growth factor receptor underlying responsiveness of non-small-cell lung cancer to gefitinib. N Engl J Med. 2004;350:2129–39. doi: 10.1056/NEJMoa040938. [DOI] [PubMed] [Google Scholar]
- 14.Paez JG, Janne PA, Lee JC, Tracy S, Greulich H, Gabriel S, et al. EGFR mutations in lung cancer: correlation with clinical response to gefitinib therapy. Science. 2004;304:1497–500. doi: 10.1126/science.1099314. [DOI] [PubMed] [Google Scholar]
- 15.Minna JD, Gazdar AF, Sprang SR, Herz J. Cancer. A bull's eye for targeted lung cancer therapy. Science. 2004;304:1458–61. doi: 10.1126/science.1099578. [DOI] [PubMed] [Google Scholar]
- 16.Weinstein IB. Cancer. Addiction to oncogenes--the Achilles heal of cancer. Science. 2002;297:63–4. doi: 10.1126/science.1073096. [DOI] [PubMed] [Google Scholar]
- 17.Shigematsu H, Gazdar AF. Somatic mutations of epidermal growth factor receptor signaling pathway in lung cancers. Int J Cancer. 2006;118:257–62. doi: 10.1002/ijc.21496. [DOI] [PubMed] [Google Scholar]
- 18.D'Angelo SP, Pietanza MC, Johnson ML, Riely GJ, Miller VA, Sima CS, et al. Incidence of EGFR exon 19 deletions and L858R in tumor specimens from men and cigarette smokers with lung adenocarcinomas. J Clin Oncol. 2011;29:2066–70. doi: 10.1200/JCO.2010.32.6181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mitchell P, Mok T, Barraclough H, Strizek A, Lew R, van Kooten M. Smoking history as a predictive factor of treatment response in advanced non-small-cell lung cancer: a systematic review. Clinical lung cancer. 2012;13:239–51. doi: 10.1016/j.cllc.2011.08.003. [DOI] [PubMed] [Google Scholar]
- 20.Lara P. (UC Davis School of Medicine) Personal anecdotal communication [Google Scholar]
- 21.Filosto S, Becker CR, Goldkorn T. Cigarette smoke induces aberrant EGF receptor activation that mediates lung cancer development and resistance to tyrosine kinase inhibitors. Mol Cancer Ther. 2012;11:795–804. doi: 10.1158/1535-7163.MCT-11-0698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Maa MC, Leu TH, McCarley DJ, Schatzman RC, Parsons SJ. Potentiation of epidermal growth factor receptor-mediated oncogenesis by c-Src: implications for the etiology of multiple human cancers. Proceedings of the National Academy of Sciences of the United States of America. 1995;92:6981–5. doi: 10.1073/pnas.92.15.6981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tice DA, Biscardi JS, Nickles AL, Parsons SJ. Mechanism of biological synergy between cellular Src and epidermal growth factor receptor. Proc Natl Acad Sci U S A. 1999;96:1415–20. doi: 10.1073/pnas.96.4.1415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Marcotte R, Zhou L, Kim H, Roskelly CD, Muller WJ. c-Src associates with ErbB2 through an interaction between catalytic domains and confers enhanced transforming potential. Mol Cell Biol. 2009;29:5858–71. doi: 10.1128/MCB.01731-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chung BM, Dimri M, George M, Reddi AL, Chen G, Band V, et al. The role of cooperativity with Src in oncogenic transformation mediated by non-small cell lung cancer-associated EGF receptor mutants. Oncogene. 2009;28:1821–32. doi: 10.1038/onc.2009.31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Girard L, Zochbauer-Muller S, Virmani AK, Gazdar AF, Minna JD. Genome-wide allelotyping of lung cancer identifies new regions of allelic loss, differences between small cell lung cancer and non-small cell lung cancer, and loci clustering. Cancer research. 2000;60:4894–906. [PubMed] [Google Scholar]
- 27.Giard DJ, Aaronson SA, Todaro GJ, Arnstein P, Kersey JH, Dosik H, et al. In vitro cultivation of human tumors: establishment of cell lines derived from a series of solid tumors. J Natl Cancer Inst. 1973;51:1417–23. doi: 10.1093/jnci/51.5.1417. [DOI] [PubMed] [Google Scholar]
- 28.Khan EM, Lanir R, Danielson AR, Goldkorn T. EGF receptor exposed to cigarette smoke is aberrantly activated and undergoes perinuclear trafficking. FASEB J. 2008;22:910–7. doi: 10.1096/fj.06-7729com. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Filosto S, Khan E, Tognon E, Becker C, Ashfaq M, Ravid T, et al. EGF receptor exposed to oxidative stress acquires abnormal phosphorylation and aberrant activated conformation that impairs canonical dimerization. PLoS ONE. 2011;6:e23240. doi: 10.1371/journal.pone.0023240. Epub 2011 Aug 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Dimri M, Naramura M, Duan L, Chen J, Ortega-Cava C, Chen G, et al. Modeling breast cancer-associated c-Src and EGFR overexpression in human MECs: c-Src and EGFR cooperatively promote aberrant three-dimensional acinar structure and invasive behavior. Cancer research. 2007;67:4164–72. doi: 10.1158/0008-5472.CAN-06-2580. [DOI] [PubMed] [Google Scholar]
- 31.Kim H, Chan R, Dankort DL, Zuo D, Najoukas M, Park M, et al. The c-Src tyrosine kinase associates with the catalytic domain of ErbB-2: implications for ErbB-2 mediated signaling and transformation. Oncogene. 2005;24:7599–607. doi: 10.1038/sj.onc.1208898. [DOI] [PubMed] [Google Scholar]
- 32.Zhang X, Gureasko J, Shen K, Cole PA, Kuriyan J. An allosteric mechanism for activation of the kinase domain of epidermal growth factor receptor. Cell. 2006;125:1137–49. doi: 10.1016/j.cell.2006.05.013. [DOI] [PubMed] [Google Scholar]
- 33.Yang S, Park K, Turkson J, Arteaga CL. Ligand-independent phosphorylation of Y869 (Y845) links mutant EGFR signaling to stat-mediated gene expression. Exp Cell Res. 2008;314:413–9. doi: 10.1016/j.yexcr.2007.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tracy S, Mukohara T, Hansen M, Meyerson M, Johnson BE, Janne PA. Gefitinib induces apoptosis in the EGFRL858R non-small-cell lung cancer cell line H3255. Cancer Res. 2004;64:7241–4. doi: 10.1158/0008-5472.CAN-04-1905. [DOI] [PubMed] [Google Scholar]
- 35.Irby RB, Yeatman TJ. Role of Src expression and activation in human cancer. Oncogene. 2000;19:5636–42. doi: 10.1038/sj.onc.1203912. [DOI] [PubMed] [Google Scholar]
- 36.Yeatman TJ. A renaissance for SRC. Nature reviews Cancer. 2004;4:470–80. doi: 10.1038/nrc1366. [DOI] [PubMed] [Google Scholar]
- 37.Burgess DJ. Breast cancer: SRC hits the mark. Nature reviews Cancer. 2011;11:314–5. doi: 10.1038/nrc3062. [DOI] [PubMed] [Google Scholar]
- 38.Finn RS. Targeting Src in breast cancer. Annals of oncology : official journal of the European Society for Medical Oncology / ESMO. 2008;19:1379–86. doi: 10.1093/annonc/mdn291. [DOI] [PubMed] [Google Scholar]
- 39.Asim M, Siddiqui IA, Hafeez BB, Baniahmad A, Mukhtar H. Src kinase potentiates androgen receptor transactivation function and invasion of androgen-independent prostate cancer C4-2 cells. Oncogene. 2008;27:3596–604. doi: 10.1038/sj.onc.1211016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Rahman I. The role of oxidative stress in the pathogenesis of COPD: implications for therapy. Treat Respir Med. 2005;4:175–200. doi: 10.2165/00151829-200504030-00003. [DOI] [PubMed] [Google Scholar]
- 41.Ganesan S, Unger BL, Comstock AT, Angel KA, Mancuso P, Martinez FJ, et al. Aberrantly activated EGFR contributes to enhanced IL-8 expression in COPD airways epithelial cells via regulation of nuclear FoxO3A. Thorax. 2013;68:131–41. doi: 10.1136/thoraxjnl-2012-201719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Woodruff PG. Novel outcomes and end points: biomarkers in chronic obstructive pulmonary disease clinical trials. Proc Am Thorac Soc. 2011;8:350–5. doi: 10.1513/pats.201101-015RM. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sudol M. From Rous sarcoma virus to plasminogen activator, src oncogene and cancer management. Oncogene. 2011;30:3003–10. doi: 10.1038/onc.2011.38. [DOI] [PubMed] [Google Scholar]
- 44.Giaccone G, Zucali PA. Src as a potential therapeutic target in non-small-cell lung cancer. Annals of oncology : official journal of the European Society for Medical Oncology / ESMO. 2008;19:1219–23. doi: 10.1093/annonc/mdn048. [DOI] [PubMed] [Google Scholar]
- 45.Zhang J, Kalyankrishna S, Wislez M, Thilaganathan N, Saigal B, Wei W, et al. SRC-family kinases are activated in non-small cell lung cancer and promote the survival of epidermal growth factor receptor-dependent cell lines. Am J Pathol. 2007;170:366–76. doi: 10.2353/ajpath.2007.060706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Chung BM, Raja SM, Clubb RJ, Tu C, George M, Band V, et al. Aberrant trafficking of NSCLC-associated EGFR mutants through the endocytic recycling pathway promotes interaction with Src. BMC Cell Biol. 2009;10:84. doi: 10.1186/1471-2121-10-84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Masaki T, Igarashi K, Tokuda M, Yukimasa S, Han F, Jin YJ, et al. pp60c-src activation in lung adenocarcinoma. Eur J Cancer. 2003;39:1447–55. doi: 10.1016/s0959-8049(03)00276-4. [DOI] [PubMed] [Google Scholar]
- 48.Biscardi JS, Maa MC, Tice DA, Cox ME, Leu TH, Parsons SJ. c-Src-mediated phosphorylation of the epidermal growth factor receptor on Tyr845 and Tyr1101 is associated with modulation of receptor function. J Biol Chem. 1999;274:8335–43. doi: 10.1074/jbc.274.12.8335. [DOI] [PubMed] [Google Scholar]
- 49.Gotoh N, Tojo A, Hino M, Yazaki Y, Shibuya M. A highly conserved tyrosine residue at codon 845 within the kinase domain is not required for the transforming activity of human epidermal growth factor receptor. Biochemical and biophysical research communications. 1992;186:768–74. doi: 10.1016/0006-291x(92)90812-y. [DOI] [PubMed] [Google Scholar]
- 50.Shan Y, Eastwood MP, Zhang X, Kim ET, Arkhipov A, Dror RO, et al. Oncogenic mutations counteract intrinsic disorder in the EGFR kinase and promote receptor dimerization. Cell. 2012;149:860–70. doi: 10.1016/j.cell.2012.02.063. [DOI] [PubMed] [Google Scholar]
- 51.Boggon TJ, Eck MJ. Structure and regulation of Src family kinases. Oncogene. 2004;23:7918–27. doi: 10.1038/sj.onc.1208081. [DOI] [PubMed] [Google Scholar]
- 52.Parsons SJ, Parsons JT. Src family kinases, key regulators of signal transduction. Oncogene. 2004;23:7906–9. doi: 10.1038/sj.onc.1208160. [DOI] [PubMed] [Google Scholar]
- 53.Shvartsman DE, Donaldson JC, Diaz B, Gutman O, Martin GS, Henis YI. Src kinase activity and SH2 domain regulate the dynamics of Src association with lipid and protein targets. The Journal of cell biology. 2007;178:675–86. doi: 10.1083/jcb.200701133. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.