

Abstract
OBJECTIVE
Immune intervention trials in recent-onset type 1 diabetes would benefit from biomarkers associated with good therapeutic response. In the previously reported randomized placebo-controlled anti-CD3 study (otelixizumab; GlaxoSmithKline), we tested the hypothesis that specific diabetes autoantibodies might serve this purpose.
RESEARCH DESIGN AND METHODS
In the included patients (n = 40 otelixizumab, n = 40 placebo), β-cell function was assessed as area under the curve (AUC) C-peptide release during a hyperglycemic glucose clamp at baseline (median duration of insulin treatment: 6 days) and every 6 months until 18 months after randomization. (Auto)antibodies against insulin (I[A]A), GAD (GADA), IA-2 (IA-2A), and ZnT8 (ZnT8A) were determined on stored sera by liquid-phase radiobinding assay.
RESULTS
At baseline, only better preserved AUC C-peptide release and higher levels of IAA were associated with better preservation of β-cell function and lower insulin needs under anti-CD3 treatment. In multivariate analysis, IAA (P = 0.022) or the interaction of IAA and C-peptide (P = 0.013) independently predicted outcome together with treatment. During follow-up, good responders to anti-CD3 treatment (i.e., IAA+ participants with relatively preserved β-cell function [≥25% of healthy control subjects]) experienced a less pronounced insulin-induced rise in I(A)A and lower insulin needs. GADA, IA-2A, and ZnT8A levels were not influenced by anti-CD3 treatment, and their changes showed no relation to functional outcome.
CONCLUSIONS
There is important specificity of IAA among other diabetes autoantibodies to predict good therapeutic response of recent-onset type 1 diabetic patients to anti-CD3 treatment. If confirmed, future immune intervention trials in type 1 diabetes should consider both relatively preserved functional β-cell mass and presence of IAA as inclusion criteria.
Introduction
Type 1 diabetes is a chronic T cell–mediated autoimmune disease ultimately leading to a major loss of insulin-secreting β-cells, hyperglycemia due to insulinopenia, and—if not well controlled—life-threatening complications (1). Humanized nonmitogenic Fc-mutated monoclonal anti-CD3 antibodies—hOKT3γ1(Ala-Ala) (teplizumab; Macrogenics) (2,3) and ChAglyCD3 (otelixizumab) (4,5)—could slow disease progression by targeting activated T lymphocytes in recent-onset type 1 diabetic patients, but preservation of functional β-cell mass was transient and largely confined to individuals with relatively intact C-peptide secretion and young age (<27 years) at diagnosis (2–5). Likewise, the efficacy of several other immune interventions in recent-onset diabetes was highest in participants with younger age at inclusion, shorter disease duration, or higher residual insulin-producing capacity at the start of treatment (1,6).
Future trials, particularly if planned at the preclinical stage, would benefit from biomarkers that identify responders to a given intervention. This would avoid exposing nonresponders needlessly to immunomodulators with potentially harmful adverse effects (1,7,8). Diabetes autoantibodies are obvious candidates in this respect because (changes in) antibody status or levels have been associated with clinical outcome in islet or pancreas transplantation protocols and in the oral arm of the DPT-1 trial (9,10). Taking advantage of the data and sample base from the previously reported first randomized placebo-controlled anti-CD3 study originally designed to test the safety and β-cell preserving effects of otelixizumab in recent-onset type 1 diabetes (4), we wanted to test the hypothesis that specific autoantibody profiles at diagnosis might predict the efficacy of a short course (6 days) of anti-CD3 treatment. In the original study, only the presence of islet cell antibodies (ICA) and/or GADA positivity were examined as potential predictive autoantibody markers (4). We therefore measured autoantibodies against insulin (IAA), GAD (GADA), insulinoma-associated protein-2 (IA-2A), and zinc transporter 8 (ZnT8A) at clinical onset in participants in this study (4). We investigated whether autoantibody levels could help identify individuals who benefited most from otelixizumab treatment in terms of preservation of functional β-cell mass, determined as area under the curve (AUC) of second-phase glucose-stimulated C-peptide release during a hyperglycemic clamp in addition to already established factors (4,5), and might thus serve as independent predictors of clinical outcome. In addition, we investigated whether treatment with anti-CD3 influenced the natural history of diabetes antibody patterns after diagnosis (i.e., the declining trend of GADA, IA-2A, and ZnT8A and the insulin treatment–induced rise in insulin antibodies [IA]) (11–13).
Research Design and Methods
Patient Selection and Treatment
Eighty recent-onset type 1 diabetic patients were included in a randomized phase 2 placebo-controlled trial (4) (trial number NCT00627146) (Supplementary Fig 1). They were selected according to the following criteria: age 12–39 years, positivity for ICA and/or GADA, random plasma C-peptide level ≥0.2 nmol/L at a glycemia of 10.0–13.9 mmol/L, treatment with insulin for ≤4 weeks before enrollment, polyuria for <6 months, <10% weight loss during the previous 6 months, and positivity for Epstein-Barr virus IgG. Patients received an infusion of ChAglyCD3 (otelixizumab, n = 40) or placebo (n = 40), administered during 2–4 h on 6 consecutive days (64 mg cumulative dose in the first 4 patients; 48 mg cumulative dose in the following 36 patients). Treatment was randomized according to trial center (four in Belgium, one in Germany), age (<15 or ≥15 years) and presence or absence of ICA (4). The initial protocol was planned for an 18-month study. Efficacy and safety data were reported previously (4). Written informed consent was obtained from each patient.
The primary end point was second-phase AUC C-peptide release (min 60–140) during the hyperglycemic clamp test (4). Because the number of participants who underwent hyperglycemic clamp testing during a 48-month follow-up was insufficient to use clamp-derived C-peptide release as a primary end point (n = 36) (5), we limited the current study on autoantibody predictor ability to 18 months of follow-up. The anti-CD3–treated group and the placebo group did not differ statistically in clinical or laboratory characteristics (Table 1) or in the expression of susceptible or protective HLA haplotypes (data not shown) (4).
Table 1.
Characteristics of patients at screening and start of the treatment
| Variable | Placebo group (n = 40) | ChAglyCD3 (n = 40) | P |
|---|---|---|---|
| At screening | |||
| Men | 26 (65) | 25 (63) | 0.816 |
| Age (years)* | 26 (7) | 27 (7) | 0.596 |
| Time of insulin treatment (days) | 6 (2–12) | 7 (3–14) | 0.432 |
| IAA+ | 15 (38) | 10 (25) | 0.228 |
| GADA+ | 37 (93) | 34 (85) | 0.288 |
| IA-2A+ | 21 (53) | 23 (58) | 0.653 |
| ZnT8A+ | 19 (48) | 20 (50) | 0.823 |
| ≥1 autoAb+ | 39 (98) | 38 (95) | 0.556 |
| ≥2 autoAb+ | 28 (70) | 27 (68) | 0.809 |
| IAA levels in IAA+ patients (% tracer binding) | 1.3 (0.7–1.9) | 0.9 (0.7–1.3) | 0.276 |
| GADA levels in GADA+ patients (WHO units/mL) | 532 (267–3,551) | 279 (110–1,585) | 0.088 |
| IA-2A levels in IA-2A+ patients (WHO units/mL) | 271 (93–1,407) | 620 (95–1,852) | 0.411 |
| ZnT8A levels in ZnT8A+ patients (index) | 0.17 (0.08–0.38) | 0.32 (0.10–0.54) | 0.275 |
| At start of treatment | |||
| Time since diagnosis (days) | 25 (19–27) | 21 (18–24) | 0.113 |
| Time of insulin treatment (days) | 20 (13–25) | 20 (16–24) | 0.769 |
| Insulin dose (units ⋅ day−1 ⋅ kg−1)* | 0.38 (0.20) | 0.46 (0.27) | 0.201 |
| HbA1c (%)* | 8.1 (1.5) | 8.6 (1.5) | 0.059 |
| HbA1c (mmol/mol)* | 65 (16) | 71 (16) | |
| C-peptide release under glucose clamping (nmol ⋅ L−1 ⋅ min−1) | |||
| AUC min 60–140* | 0.61 (0.29) | 0.54 (0.30) | 0.160 |
Data are mean (SD)*, n (%), or median (IQR).
Patient Follow-up
Patients received intensive insulin therapy for the entire period, and at least once every 3 months, doses were adjusted according to metabolic needs. Outpatient study visits occurred every 3 months at the trial center or at the local hospital. The parameters recorded included type and dose of insulin, body weight, home blood glucose levels, concomitant medication, and adverse events (4). HbA1c levels were determined every 3 months, and if the patient agreed, a glucose clamp was performed at baseline and every 6 months thereafter (4). The C-peptide concentration was determined by time-resolved immunofluorescence assay before and at 60, 90, 120, and 140 min after the start of the hyperglycemic phase of the clamp. The induced C-peptide release was expressed as AUC per minute or in multivariate analysis as mean C-peptide (i.e., the average of the C-peptide concentrations at 60, 90, 120, and 140 min). In patients who were C-peptide negative at a previous visit, the clamp test was not done and a zero value was filled in for subsequent time points. C-peptide negativity was defined as AUC C-peptide ≤0.03 nmol ⋅ L−1 ⋅ min−1 during the hyperglycemic clamp (4).
Diabetes Autoantibodies
Autoantibodies were determined at screening (i.e., after a median [interquartile range {IQR}] duration of insulin treatment of 6 [3–14] days) and at 6, 12, and 18 months after the start of the anti-CD3 treatment by liquid-phase radiobinding assays. IAA and IA were determined by a modification (14) of the Palmer assay (15) using acid charcoal extraction of serum to deplete IAA and IA from bound endogenous or exogenous insulin. Briefly, sera were incubated with acid charcoal to disrupt circulating complexes of antibodies with endogenous or exogenous insulin and to absorb unbound insulin. After neutralization and centrifugation, the insulin-free supernatants were incubated in duplicate with radioactive-labeled human recombinant insulin in the presence and absence of excess cold insulin (final concentration 1.76 × 10−6 mol/L). Next, the formed immune complexes were precipitated using polyethylene glycol. Unbound cold or labeled insulin was removed by a washing step, and the radioactivity of the polyethylene glycol precipitate was measured, averaged, and expressed as percentage added tracer bound (24,000 cpm/tube) after correction for displacement. The latter was considered complete if the counts could be lowered to the level of IAA-negative control samples (typically 200–250 cpm), and this was the case for all analyzed samples.
Strongly IA-positive samples were diluted until their binding signal fell into the linear range of the assay, and the results were multiplied by the dilution factor. This explains why some sera had levels exceeding 100% tracer binding. This assay performed best in the Fourth Immunology of Diabetes Workshop on Insulin Autoantibodies (16). Like all assays for IAA or IA, this method is unable to distinguish between antibodies against endogenous or exogenous insulin (17). GADA, IA-2A, and ZnT8A levels were determined as described before (18) and expressed as World Health Organization (WHO) units/mL by comparison with the WHO standard serum (GADA, IA-2A) (19) or as a relative index by comparison with an in-house positive standard serum (ZnT8A) (20).
In all but three samples at screening, IAA could also be measured by a modified microassay (21). Briefly, serum samples were incubated with 125I-labeled human insulin in the presence or absence of an excess unlabeled insulin (Actrapid; Novo Nordisk; final concentration: 2.24 × 10−5 mol/L), and immune complexes were precipitated with a mixture of 20% glycine pretreated protein A-Sepharose and 8% ethanolamine pretreated protein G-Sepharose. The differences in counts were expressed as arbitrary units (aU) by comparison with an in-house positive standard serum.
cDNAs for the preparation of radioligands by in vitro transcription-translation encoded the intracellular portion of IA-2 (gift of M. Christie, King’s College School of Medicine and Dentistry, London, U.K.), full-length 65kDa GAD (gift of Ǻ. Lernmark when at the University of Washington, Seattle, WA), and the fused Arg325 and Trp325 carboxy-termini of ZnT8A (gift of J.C. Hutton, Barbara Davis Center for Childhood Diabetes, Aurora, CO). Cutoff values for antibody positivity were determined as the 99th percentile of autoantibody levels in >250 nondiabetic control subjects and amounted to ≥0.60% tracer binding for I(A)A, ≥23.0 WHO units/mL for GADA, ≥1.4 WHO units/mL for IA-2A, ≥0.039 index for ZnT8A, and ≥0.48 aU for the I(A)A microassay. The (auto)antibodies were determined by technicians blinded to the participants’ identification and treatment regimen.
Statistical Analysis
Data were stored at the Belgian Diabetes Registry. Statistical differences between groups were assessed by the Mann-Whitney U test (unpaired) and Wilcoxon test (paired) for continuous data and a χ2 test for categorical data. The change in mean clamp-derived C-peptide release versus baseline was selected as the end point (4). To evaluate the effect of potential prognostic factors on this end point, forward stepwise multiple regression analysis was used. In the original anti-CD3 protocol, the predefined prognostic factors were age, sex, body weight, BMI, duration of insulin therapy at screening, status of ICA and GADA (positive vs. negative), and HLA-DQ (protective vs. nonprotective) at screening, as well as HbA1c level and clamp-derived C-peptide release (above or below the median [P50] of all 80 included patients) at start of the treatment. In the present multivariate analysis, we included only the variables that were significant in previous analyses (i.e., initial clamp-derived C-peptide release, which was the only significant predictor of C-peptide preservation at month 18 in the original multivariate analysis) (4) and age (predictor of insulin needs at month 48) (5), together with the newly determined antigen-specific autoantibody levels at screening (IAA, GADA, IA-2A, and ZnT8A). Values below the limit of detection (0.3% tracer binding for IAA) were assigned a value of 0.2%. The parameters with a P value of ≤0.05 in univariate analysis were further tested in multiple linear regression analysis performed in a stepwise fashion. Statistical analysis was performed separately in the placebo-treated and the ChAglyCD3-treated group as well as in the entire study group. Statistical tests were performed two-sided at the 5% level of significance with SPSS 20.0 software (IBM SPSS Statistics, Armonk, NY) and the multivariate analyses with SAS 9.2 software (SAS Institute, Inc., Cary, NC), and the figures were generated with GraphPad Prism 5.00 software (GraphPad Software, Inc., San Diego, CA). No adjustments were made for multiplicity.
Results
Baseline Characteristics
Otelixizumab-treated and placebo-treated new-onset patients had similar baseline characteristics. The groups did not differ in age and sex distribution, duration of insulin treatment, prevalence (alone or in combination) and levels (in case of positivity) of autoantibodies at screening, or in HbA1c level, C-peptide release during hyperglycemic clamp, time since diagnosis, or in duration and dosage of insulin therapy at the start of the trial (Table 1). The median (IQR) time since start insulin treatment of the entire group was 6 (3–14) days; hence, most samples were taken within 2 weeks of insulin treatment. Moreover, IAA levels at screening were not significantly different from the levels at the start of treatment within 4 weeks of diagnosis (P = 0.214 by Wilcoxon test) and can thus safely be assumed to represent true IAA (14).
IAA Levels at Diagnosis Predict a Better Response to Anti-CD3 Treatment
We examined by multiple linear regression whether baseline IAA levels could serve as independent predictor(s) of the response to anti-CD3 therapy apart from residual β-cell function or age at diagnosis (4,5). Higher baseline IAA levels were thus found to independently predict better preservation of functional β-cell mass at month 18, expressed as a lesser change in clamp-derived C-peptide release from baseline and reflected by a negative standardized coefficient β (Table 2). In the placebo group, we confirmed (4) that clamp-derived baseline C-peptide levels above the median independently predicted better outcome. In addition, higher IAA levels at screening came out as the strongest independent predictor, whereas none of the tested variables appeared predictive in the ChAglyCD3-treated group (Table 2). Similar results, albeit with slightly higher P values, were obtained when IAA levels were determined with an I(A)A microassay (not shown) (21). When all participants were considered together in multivariate analysis, IAA levels at screening remained the only independent predictor of better outcome, apart from anti-CD3 administration (model 1, Table 2). A significant interaction between higher IAA levels and C-peptide release was observed (model 2, Table 2). Age was not retained as an independent predictor of clinical outcome at 18 months, which agrees with our previous analysis at this time point (4). Results were similar when I(A)A levels at the start of the trial were used instead of IAA levels at screening (not shown).
Table 2.
Forward multiple linear regression with the change in C-peptide concentration over 18 months as a dependent variable
| Independent variables |
Change in mean C-peptide concentration (start-month 18) |
||||
|---|---|---|---|---|---|
| Univariate |
Multivariate | Multivariate | |||
| model 1 |
model 2 |
||||
| P | P | β | P | β | |
| Placebo-treated patients | |||||
| Age* | 0.519 | NM | NM | ||
| C-peptide (P50)† | 0.033 | 0.050 | −0.354 | NM | |
| GADA* | 0.395 | NM | NM | ||
| ZnT8A* | 0.988 | NM | NM | ||
| IA-2A* | 0.586 | NM | NM | ||
| IAA* | 0.024 | 0.024 | −0.397 | NM | |
| IAA × C-peptide (P50)† | 0.016 | — | 0.016 | −0.422 | |
| ChAglyCD3-treated patients | |||||
| Age* | 0.383 | NM | NM | ||
| C-peptide (P50)† | 0.830 | NM | NM | ||
| GADA* | 0.807 | NM | NM | ||
| ZnT8A* | 0.333 | NM | NM | ||
| IA-2A* | 0.359 | NM | NM | ||
| IAA* | 0.894 | NM | NM | ||
| IAA × C-peptide (P50)† | 0.677 | NM | NM | ||
| All patients | |||||
| Age* | 0.519 | NM | NM | ||
| C-peptide (P50)† | 0.033 | NM | NM | ||
| GADA* | 0.899 | NM | NM | ||
| ZnT8A* | 0.710 | NM | NM | ||
| IA-2A* | 0.207 | NM | NM | ||
| IAA* | 0.009 | 0.022 | −0.264 | NM | |
| Treatment‡ | 0.005 | 0.020 | 0.268 | 0.023 | 0.260 |
| IAA × C-peptide (P50)† | 0.004 | — | 0.013 | −0.286 | |
β, standardized coefficient; NM, not selected for entry in the multivariate model. Boldface text indicates P < 0.05.
Variable at screening.
Variable at start of the treatment, AUC 60–140 min < P50 (0) or ≥P50 (1) of the entire group.
Placebo-treated (0), ChAglyCD3-treated (1).
Figure 1 illustrates that the results of the multivariate analysis should be interpreted in the sense that in the placebo-treated group, patients with higher IAA levels (above or equal to the P50 of the entire patient group, corresponding to ≥0.50% tracer binding) or with higher clamp-derived AUC C-peptide release at baseline (≥P50 of the whole group corresponding to ≥25% of healthy control subjects) (4) experienced a rapid loss of functional β-cell mass versus baseline, which was preferentially slowed by anti-CD3 treatment to the same level as observed in the other subgroups. Figure 1A shows the significantly less rapid decline in AUC C-peptide release, and Fig. 1B shows the associated less rapid increase in insulin needs induced by anti-CD3 treatment in patients with IAA ≥P50 but not in patients with IAA <P50 (Fig. 1C and D). Likewise, anti-CD3 treatment preserved clamp-derived C-peptide release and the associated lower insulin needs, compared with the placebo group, in individuals with AUC C-peptide release at inclusion ≥P50 of the entire group (Fig. 1E and F) but not in those with initial values <P50 (Fig. 1G and H), as already shown in the original study report (4). The C-peptide–preserving effect of anti-CD3 treatment was largely confined to individuals with both AUC C-peptide and IAA levels ≥P50 of the whole patient group, hereby confirming the interaction observed in multivariate analysis (Fig. 2). IAA+ and IAA− anti-CD3–treated patients did not differ in the development or in the levels of anti-otelixizumab antibodies as determined by two different methods (not shown) (22).
Figure 1.
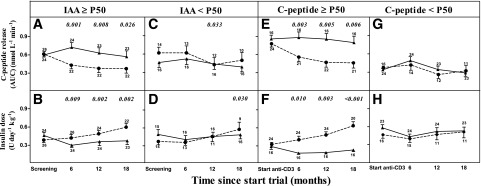
Efficacy tests for AUC C-peptide release (panels A, C, E, and G) and insulin dose (panels B, D, F, and H) during 18-month follow-up in placebo-treated (●) and ChAglyCD3-treated (▲) patients stratified according to IAA levels at screening above (panels A and B) or below (panels C and D) the median of all 80 patients or for clamp-derived AUC C-peptide release above (panels E and F) or below (panels G and H) the median value of the entire group. Values represent means ± SE. The small numbers next to the whiskers indicate the number of observations at the respective times. Significant P values for differences in change in AUC C-peptide from baseline between anti-CD3 and placebo groups are shown in italic above the relevant times.
Figure 2.

AUC C-peptide release during 18-month follow-up in placebo-treated (●) and ChAglyCD3-treated (▲) patients stratified according to clamp-derived C-peptide release and IAA levels at screening. Panel A: Patients with clamp-derived AUC C-peptide and IAA above the respective median value of the whole group. Panel B: Patients with clamp-derived C-peptide and/or IAA below the respective median values of the whole group. Values represent means ± SE. The small numbers next to the whiskers indicate the number of observations at the respective times. Significant P values for differences in change in AUC C-peptide from baseline between anti-CD3 and placebo groups are shown in italic above the relevant times.
Influence of Anti-CD3 Treatment on Diabetes (Auto)Antibody Levels During Follow-up
The natural history of GADA, IA-2A and ZnT8A autoantibody levels after diabetes onset was not meaningfully altered by otelixizumab treatment. The levels tended overall to progressively decrease with time after diagnosis, in line with previous observations (12,13), and this decline occurred at a similar rate in both treatment arms during the 18-month study period (Supplementary Fig. 2). Neither did both groups differ in autoantibody kinetics after screening when stratified according to baseline residual C-peptide release during hyperglycemic clamp or age (≥P50 or <P50 of entire group, not shown), parameters previously shown to predict clinical outcome (4,5). In contrast, the rise in insulin-induced IA was less pronounced under otelixizumab treatment during the 18-month study period, but only in initially IAA+ patients with relatively preserved functional β-cell mass at diagnosis (≥P50 AUC C-peptide release at baseline) (Supplementary Fig. 3). It was associated with less insulin use (not shown).
Conclusions
This study identified elevated IAA levels at screening of type 1 diabetes as an independent predictor of the efficacy of anti-CD3 (otelixizumab) treatment to preserve residual β-cell mass for 18 months. Its predictive value significantly interacts with a relatively preserved β-cell mass at baseline, previously recognized as a predictor of good functional outcome (4). IAA+ new-onset patients with relatively preserved β-cell function are therefore proposed as candidates of choice for future immune intervention trials in type 1 diabetes. If confirmed prospectively in sufficiently powered studies, specific cutoffs may be determined for IAA levels, similar to existing guidelines for C-peptide (maximally stimulated C-peptide ≥0.2 nmol/L during a mixed-meal tolerance test) (23), above which good responders to anti-CD3 treatment might by identified and thereby further paving the way to personalized immune interventions. Anti-CD3 treatment did not influence the natural history of other autoantibodies after diagnosis but was associated with a lesser rise in I(A)A levels selectively in patients who responded well to anti-CD3 therapy and required less insulin.
The strengths of the study include the randomized, placebo-controlled nature of the otelixizumab trial (4) in new-onset diabetes with a very short duration of disease (<4 weeks insulin treatment) compared with other studies (1–7), the measurement of functional β-cell mass by hyperglycemic clamps—the gold standard (24)—to derive objective outcome measures and stratification criteria, the assessment of functional outcome as change versus baseline, as well as the use of the slightly adapted (14) original Palmer I(A)A assay (15) with acid charcoal extraction to strip IAA or IA from bound insulin before incubation with insulin tracer and with correction of each individual IAA result for nonspecific binding in the presence of an excess cold insulin. Because most participants were sampled for antibodies within 2 weeks from start of insulin treatment (median 6 days), the reported IAA levels represent true autoantibodies. The otelixizumab trial was originally powered to detect differences in C-peptide release but not to test the present hypothesis. This retrospective study is limited by the number of patients included in the trial (n = 80) and the IAA prevalence, leading to a relatively low number of participants in subgroups, particularly when stratifying for two variables in each treatment arm. Nevertheless, stratification according to initial IAA status and β-cell function allowed disclosing differences between subgroups, which were confirmed by multivariate analysis in the entire patient group.
How might a better efficacy of anti-CD3 in IAA+ patients with relatively preserved AUC C-peptide tentatively be explained? First, individuals with relatively preserved β-cell mass at diagnosis, corresponding to ≥25% of healthy control subjects (4), have of course the potential to lose more β-cells than those in whom most cells have already been destroyed at inclusion. Next, the presence of IAA is associated with younger age at diagnosis of type 1 diabetes, higher prevalence of multiple autoantibodies and HLA class II susceptibility genotypes, and more prominent insulitis (1,12,25,26). Likewise, younger age at first autoantibody appearance (in general IAA) and higher levels of IAA, but not of GADA or IA-2A, are associated with more rapid progression to diabetes in risk groups (27). This suggests that in general, IAA+ individuals experience a more severe underlying autoimmune reactivity, with insulin autoimmunity playing a key role in the disease pathogenesis (25,28,29). In support, almost half of the clonable T cells from pancreatic draining lymph nodes of individuals with type 1 diabetes recognized an insulin epitope in a DR4-restricted manner (30).
Anti-CD3 treatment is believed to primarily target pathogenic T cells, considered to be the main effectors of β-cell destruction (31), in the context of a primed and ongoing immune response, while preserving regulatory T cells (32). Conceivably, IAA+ patients in a more aggressive disease phase may benefit most from otelixizumab treatment as a result of diminished autoreactive T-cell activity and/or a relative predominance of regulatory T cells. Anti-CD3 treatment also caused a transient partial depletion in B lymphocytes (4). As suggested in the rituximab trial (33), B lymphocytes may contribute to the disease process by augmenting local immune response through cytokines and/or antigen presentation to T lymphocytes.
Our results do not allow us to unambiguously identify the nature and extent of the mechanisms underlying our observations. They have, however, excluded that differences in the development of anti-otelixizumab antibodies could be responsible for the observed differences in outcome between IAA+ and IAA− patients, as was already previously shown not to be the case for differences between patients with initially high or low AUC C-peptide (22). Finally, we consider it unlikely that IAA levels represent a surrogate for younger age because they outperformed age for the prediction of functional outcome in multivariate analysis; moreover, in this study, IAA prevalence and levels did not differ according to age in either treatment arm (not shown). Our findings may thus help explain why age at diagnosis has been associated with better efficacy in several immune interventions in type 1 diabetes (1–7).
In support of the ability of IAA, but not of the other autoantibodies, to predict better responsiveness to immune modulation, oral administration of insulin significantly delayed onset of type 1 diabetes in first-degree relatives with high IAA levels for as long as the treatment continued (10). Likewise, preexisting IAA predicted efficacy of oral insulin in combination with anti-CD3 treatment to cure autoimmune diabetes in NOD mice (34). In contrast, a clinical trial with teplizumab, another humanized anti-CD3 monoclonal antibody similar to otelixizumab, failed to identify IAA at baseline (within 8 weeks after diagnosis) as a predictor of good clinical response (8). The reasons for the discrepant results may relate to the lower numbers of participants (22 responders vs. 27 nonresponders) than in the current study. Moreover, our observations may have benefitted from the better reproducibility and accuracy of hyperglycemic clamp–derived measures of functional β-cell mass compared with parameters derived from other stimulation tests, as reflected by their superior predictive value for clinical outcome in pretype 1 diabetes and their close correlation with implanted β-cell mass in islet transplantation or histopathological findings at clinical disease onset (26,35–37).
We have shown that anti-CD3 treatment did not alter the natural history of progressively declining GADA, IA-2A, and ZnT8A levels during an 18-month period after diagnosis, whereas it selectively suppressed the insulin-induced rise in I(A)A in the subgroup of initially IAA+ patients with relatively preserved C-peptide at diagnosis and better clinical outcome. Current I(A)A assays do not allow us to establish to what extent this lesser IA rise is due to a suppression of insulin (auto)immunity, possibly via the mechanisms discussed above, or just the consequence of a lower cumulative insulin dose (17). Nevertheless, our follow-up results are in line with observations in several other immune interventions that have been reported to selectively influence I(A)A levels in insulin-treated patients in association with better outcome. For example, this was the case for rituximab in recent-onset type 1 diabetes (28) and for rapamycin in long-standing disease (38).
In conclusion, our results have identified the presence of IAA shortly after diagnosis as a powerful predictor of good responsiveness to anti-CD3 treatment. This observation warrants the measurement of IAA in future trials before intervention in recent-onset type 1 diabetes. If confirmed, the added value of baseline IAA in combination with sufficient residual C-peptide at diagnosis (4) can be specified with a higher power. It may also lead to the definition of cutoffs for IAA levels above which the prediction of a successful metabolic outcome can be improved, thereby paving the way to more personalized immune interventions. A meta-analysis of previously completed effective immune interventions with enrollment shortly after diagnosis may already indicate the potential of IAA levels.
Supplementary Material
Article Information
Acknowledgments. The authors gratefully acknowledge the several contributions to this work of their late mentor John C. Hutton, PhD, and gratefully acknowledge the expert technical assistance of coworkers at the central unit and the clinical biology reference laboratory of the Belgian Diabetes Registry (V. Baeten, T. De Mesmaeker, H. Dewinter, N. Diependaele, S. Exterbille, T. Glorieux, C. Groven, T. Haulet, A. Ivens, D. Kesler, F. Lebleu, M. Van Molle, S. Vanderstraeten, and A. Walgrave from the Department of Clinical Chemistry and Radio-immunology, University Hospital Brussels-UZ Brussel, Brussels, Belgium, and E. Quartier, G. Schoonjans from the Diabetes Research Center, Vrije Universiteit Brussel). The authors are grateful to Dr. G. Hale (when at Therapeutic Antibody Centre, Oxford, U.K.) for performing the analyses on prevalence and titers of anti-otelixizumab antibodies and for providing the results (22). The authors sincerely thank all participating patients and all health care workers who contributed to their recruitment and follow-up in the otelixizumab trial (list provided in Keymeulen et al. [4]).
Funding. The otelixizumab study was supported by the JDRF (grants 4-2001-434 and 4-2005-1327). The autoantibody measurements for the present report were performed in the context of the EU-FP7 project 241883 and project G.0868.11 of the Research Foundation Flanders (FWO-Vlaanderen, Belgium). B.K. and I.W. are Senior Clinical Investigators for the Research Foundation Flanders. K.V. is supported by EU-FP7 project 241883. H.W.D. and J.M.W. received funding from National Institutes of Health (R01-DK-052068, R56-AI-094389) and JDRF (4-2007-1056). The sponsor of the study had no role in study design, data collection, data analysis, data interpretation, or writing of the report.
Duality of Interest. H.W.D. and J.M.W. are coinventors of U.S. patent No. 12/521,022 based on PCT/US2007/089125. No other potential conflicts of interest relevant to this article were reported.
Author Contributions. S.D., B.K., D.G.P., and F.K.G. designed the study; searched the literature; collected, analyzed, and interpreted data; and wrote the manuscript. L.K. and I.W. analyzed and interpreted data and provided statistical and writing assistance. A.V.D., E.V.B., U.V.d.V., P.G., K.V., H.W.D., and J.M.W. participated in data collection and provided writing assistance. All authors approved the final version. F.K.G. is the guarantor of this work, and, as such, had full access to all the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis.
Footnotes
This article contains Supplementary Data online at http://care.diabetesjournals.org/lookup/suppl/doi:10.2337/dc14-1575/-/DC1.
References
- 1.Gorus FK, Keymeulen B, Veld PA, Pipeleers DG. Predictors of progression to Type 1 diabetes: preparing for immune interventions in the preclinical disease phase. Expert Rev Clin Immunol 2013;9:1173–1183 [DOI] [PubMed] [Google Scholar]
- 2.Herold KC, Hagopian W, Auger JA, et al. Anti-CD3 monoclonal antibody in new-onset type 1 diabetes mellitus. N Engl J Med 2002;346:1692–1698 [DOI] [PubMed] [Google Scholar]
- 3.Sherry N, Hagopian W, Ludvigsson J, et al.; Protégé Trial Investigators . Teplizumab for treatment of type 1 diabetes (Protégé study): 1-year results from a randomised, placebo-controlled trial. Lancet 2011;378:487–497 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Keymeulen B, Vandemeulebroucke E, Ziegler AG, et al. Insulin needs after CD3-antibody therapy in new-onset type 1 diabetes. N Engl J Med 2005;352:2598–2608 [DOI] [PubMed] [Google Scholar]
- 5.Keymeulen B, Walter M, Mathieu C, et al. Four-year metabolic outcome of a randomised controlled CD3-antibody trial in recent-onset type 1 diabetic patients depends on their age and baseline residual beta cell mass. Diabetologia 2010;53:614–623 [DOI] [PubMed] [Google Scholar]
- 6.Skyler JS, Ricordi C. Stopping type 1 diabetes: attempts to prevent or cure type 1 diabetes in man. Diabetes 2011;60:1–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ludvigsson J; Linköping Diabetes Immune Intervention Study Group . The role of immunomodulation therapy in autoimmune diabetes. J Diabetes Sci Tech 2009;3:320–330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Herold KC, Gitelman SE, Ehlers MR, et al.; AbATE Study Team . Teplizumab (anti-CD3 mAb) treatment preserves C-peptide responses in patients with new-onset type 1 diabetes in a randomized controlled trial: metabolic and immunologic features at baseline identify a subgroup of responders. Diabetes 2013;62:3766–3774 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Piemonti L, Everly MJ, Maffi P, et al. Alloantibody and autoantibody monitoring predicts islet transplantation outcome in human type 1 diabetes. Diabetes 2013;62:1656–1664 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Vehik K, Cuthbertson D, Ruhlig H, Schatz DA, Peakman M, Krischer JP; DPT-1 and TrialNet Study Groups . Long-term outcome of individuals treated with oral insulin: diabetes prevention trial (DPT-1) oral insulin trial. Diabetes Care 2011;34:1585–1590 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ludvigsson J, Binder C, Mandrup-Poulsen T. Insulin autoantibodies are associated with islet cell antibodies; their relation to insulin antibodies and B-cell function in diabetic children. Diabetologia 1988;31:647–651 [DOI] [PubMed] [Google Scholar]
- 12.Gorus FK. Diabetes registries and early biological markers of insulin-dependent diabetes mellitus. Diabetes Metab Rev 1997;13:247–274 [DOI] [PubMed] [Google Scholar]
- 13.Wenzlau JM, Walter M, Gardner TJ, et al. Kinetics of the post-onset decline in zinc transporter 8 autoantibodies in type 1 diabetic human subjects. J Clin Endocrinol Metab 2010;95:4712–4719 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gorus FK, Sodoyez JC, Pipeleers DG, Keymeulen B, Foriers A, Van Schravendijk CF. Detection of autoantibodies against islet amyloid polypeptide in human serum. Lack of association with type 1 (insulin-dependent) diabetes mellitus, or with conditions favouring amyloid deposition in islets. The Belgian Diabetes Registry. Diabetologia 1992;35:1080–1086 [DOI] [PubMed] [Google Scholar]
- 15.Srikanta S, Ricker AT, McCulloch DK, Soeldner JS, Eisenbarth GS, Palmer JP. Autoimmunity to insulin, beta cell dysfunction, and development of insulin-dependent diabetes mellitus. Diabetes 1986;35:139–142 [DOI] [PubMed] [Google Scholar]
- 16.Greenbaum CJ, Palmer JP, Kuglin B, Kolb H. Insulin autoantibodies measured by radioimmunoassay methodology are more related to insulin-dependent diabetes mellitus than those measured by enzyme-linked immunosorbent assay: results of the Fourth International Workshop on the Standardization of Insulin Autoantibody Measurement. J Clin Endocrinol Metab 1992;74:1040–1044 [DOI] [PubMed] [Google Scholar]
- 17.Franke B, Galloway TS, Wilkin TJ. Developments in the prediction of type 1 diabetes mellitus, with special reference to insulin autoantibodies. Diabetes Metab Res Rev 2005;21:395–415 [DOI] [PubMed] [Google Scholar]
- 18.De Grijse J, Asanghanwa M, Nouthe B, et al.; Belgian Diabetes Registry . Predictive power of screening for antibodies against insulinoma-associated protein 2 beta (IA-2beta) and zinc transporter-8 to select first-degree relatives of type 1 diabetic patients with risk of rapid progression to clinical onset of the disease: implications for prevention trials. Diabetologia 2010;53:517–524 [DOI] [PubMed] [Google Scholar]
- 19.Lernmark A, Kolb H, Mire-Sluis T. Towards a World Health Organization (WHO) approved standard sample for islet cell antibodies, GAD65 and IA-2 autoantibodies. Diabetologia 1999;42:381–382 [DOI] [PubMed] [Google Scholar]
- 20.Wenzlau JM, Juhl K, Yu L, et al. The cation efflux transporter ZnT8 (Slc30A8) is a major autoantigen in human type 1 diabetes. Proc Natl Acad Sci U S A 2007;104:17040–17045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Williams AJ, Norcross AJ, Chandler KA, Bingley PJ. Non-specific binding to protein A Sepharose and protein G Sepharose in insulin autoantibody assays may be reduced by pre-treatment with glycine or ethanolamine. J Immunol Methods 2006;314:170–173 [DOI] [PubMed] [Google Scholar]
- 22.Hale G, Rebello P, Al Bakir I, et al. Pharmacokinetics and antibody responses to the CD3 antibody otelixizumab used in the treatment of type 1 diabetes. J Clin Pharmacol 2010;50:1238–1248 [DOI] [PubMed] [Google Scholar]
- 23.Palmer JP. C-peptide in the natural history of type 1 diabetes. Diabetes Metab Res Rev 2009;25:325–328 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.DeFronzo RA, Tobin JD, Andres R. Glucose clamp technique: a method for quantifying insulin secretion and resistance. Am J Physiol 1979;237:E214–E223 [DOI] [PubMed] [Google Scholar]
- 25.Jasinski JM, Eisenbarth GS. Insulin as a primary autoantigen for type 1A diabetes. Clin Dev Immunol 2005;12:181–186 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Pipeleers D, Ling Z. Pancreatic beta cells in insulin-dependent diabetes. Diabetes Metab Rev 1992;8:209–227 [DOI] [PubMed] [Google Scholar]
- 27.Steck AK, Johnson K, Barriga KJ, et al. Age of islet autoantibody appearance and mean levels of insulin, but not GAD or IA-2 autoantibodies, predict age of diagnosis of type 1 diabetes: diabetes autoimmunity study in the young. Diabetes Care 2011;34:1397–1399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yu L, Herold K, Krause-Steinrauf H, et al.; Type 1 Diabetes TrialNet Anti-CD20 Study Group . Rituximab selectively suppresses specific islet antibodies. Diabetes 2011;60:2560–2565 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Atkinson MA. The pathogenesis and natural history of type 1 diabetes. Cold Spring Harb Perspect Med 2012;2:a007641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kent SC, Chen Y, Bregoli L, et al. Expanded T cells from pancreatic lymph nodes of type 1 diabetic subjects recognize an insulin epitope. Nature 2005;435:224–228 [DOI] [PubMed] [Google Scholar]
- 31.Martin S, Wolf-Eichbaum D, Duinkerken G, et al. Development of type 1 diabetes despite severe hereditary B-lymphocyte deficiency. N Engl J Med 2001;345:1036–1040 [DOI] [PubMed] [Google Scholar]
- 32.Chatenoud L, Warncke K, Ziegler AG. Clinical immunologic interventions for the treatment of type 1 diabetes. Cold Spring Harb Perspect Med 2012;2:a007716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Pescovitz MD, Greenbaum CJ, Krause-Steinrauf H, et al.; Type 1 Diabetes TrialNet Anti-CD20 Study Group . Rituximab, B-lymphocyte depletion, and preservation of beta-cell function. N Engl J Med 2009;361:2143–2152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mamchak AA, Manenkova Y, Leconet W, et al. Preexisting autoantibodies predict efficacy of oral insulin to cure autoimmune diabetes in combination with anti-CD3. Diabetes 2012;61:1490–1499 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Balti EV, Vandemeulebroucke E, Weets I, et al. Hyperglycemic clamp and oral glucose tolerance test for 3-year prediction of clinical onset in persistently autoantibody-positive offspring and siblings of type 1 diabetic patients. J Clin Endocrinol Metab. 18 November 2014 [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- 36.Vandemeulebroucke E, Keymeulen B, Decochez K, et al.; Belgian Diabetes Registry . Hyperglycaemic clamp test for diabetes risk assessment in IA-2-antibody-positive relatives of type 1 diabetic patients. Diabetologia 2010;53:36–44 [DOI] [PubMed] [Google Scholar]
- 37.Keymeulen B, Gillard P, Mathieu C, et al. Correlation between beta cell mass and glycemic control in type 1 diabetic recipients of islet cell graft. Proc Natl Acad Sci U S A 2006;103:17444–17449 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Piemonti L, Maffi P, Monti L, et al. Beta cell function during rapamycin monotherapy in long-term type 1 diabetes. Diabetologia 2011;54:433–439 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.