

Abstract
During export in Escherichia coli, SecB, a homotetramer, structurally organized as a dimer of dimers, forms a complex with two protomers of SecA, which is the ATPase that provides energy to transfer a precursor polypeptide through the membrane via the SecYEG translocon. There are two areas of contact on SecB that stabilize the SecA:SecB complex: one, the flat sides of the SecB tetramer and the second, the C-terminal thirteen residues of SecB. These contacts within the complex are distributed asymmetrically. Breaking contact between SecA and the sides of SecB results in release of only one protomer of SecA yielding a complex of stoichiometry SecA1:SecB4. This complex mediates export; however, the coupling of ATP hydrolysis to movements of the precursor through the translocon is much less efficient than the coupling by the SecA2:SecB4 complex. Here we used heterotetrameric species of SecB to understand the source of the asymmetry in the contacts and its role in the functioning of the complex. The model of interactions presented suggests a way that binding between SecA and SecB might decrease the affinity of precursor polypeptides for SecB and facilitate the transfer to SecA.
Introduction
The general secretory, Sec, system of Escherichia coli captures precursor proteins before they acquire stably folded structure and exports them from the cytoplasm to their final destination in the periplasm or in the outer membrane. SecB, a homotetrameric chaperone of 69 kDa, organized as a dimer of dimers, binds the unstructured precursors. SecA, the ATPase of the Sec system specifically binds the SecB:precursor complex and delivers the complex to the translocon, SecYEG. It is not known when the precursor is transferred from SecB to SecA; whether the transfer occurs within the complex before it binds to SecYEG or after, and whether the transfer requires ATP binding and hydrolysis. Efficient export of precursors through the translocon is mediated by a complex between tetrameric SecB and two protomers of SecA (A2:B4 complex) [1]. The complex is stabilized by two areas of contact. Each SecA protomer binds through its zinc-containing C-terminal 21 aminoacyl residues to a site on the flat side of SecB. The flat sides are formed by two eight-stranded β sheets, composed of four strands from each of the two protomers in a dimer. We refer to those sites on SecB as the side sites. There are additional areas of contact between the C-terminal 13 residues of SecB and both the amino-terminal ten residues of SecA and a stretch of aminoacyl residues 600–610 that form a linker helix between the nucleotide binding fold II (NBDII) and the helix scaffold domain [2, 3]. These sites of interaction on SecB are referred to as tail sites.
This symmetric complex between two protomers of SecA and tetrameric SecB is unusual in that the interactions that stabilize it are distributed asymmetrically. Breaking the contact of SecA with side sites on SecB results in only one protomer of SecA bound, yielding a complex with stoichiometry of A1:B4 [4]. Although A1:B4 complexes do function to mediate translocation of precursors they display much less efficient coupling of the hydrolysis of ATP to the movement of precursors through the translocon [1]. Further evidence for asymmetry is provided by electron microscopy of the complex between SecA and SecB. In the presence of proOmpA one of the SecA protomers predominantly binds to SecB:proOmpA [5]. Another unusual property of SecA is that it interacts with its numerous binding partners, SecB, precursor proteins, SecY and lipids, on the same surface through overlapping, but not identical binding sites [6]. Based on our understanding of these interactions we have proposed that during the process of passage of the precursor from SecB to SecA and on to SecYEG there is a dynamic interplay among the components that involves the making and breaking of subsites of interaction between SecA and SecB. This would allow the transfer of precursors from SecB to SecA and further to the translocon without complete dissociation of SecA and SecB.
Further elucidation of the transfer of precursor along the pathway of export requires understanding of the asymmetric interactions between SecA and SecB. Here we use hybrid species of SecB to determine the relative contribution of the multiple sites of interaction to the stability of the A2:B4 complex.
Results and Discussion
Strategy to investigate the asymmetry of the SecB tetramer
It was necessary to break the symmetry of the SecB dimer of dimers to identify the region that is the basis for the asymmetric interactions and to determine contributions to overall stability by the different regions of contact. SecB undergoes a spontaneous dimer-tetramer equilibrium [7]. Therefore, we generated hybrid species that carry different binding sites on each dimer of the tetramer by mixing two species of SecB. To test the role of the side sites and the tail sites we generated six hybrid heterotetramers using the following species of SecB: wild-type SecB, all sites present; SecBE77K, nonfunctional side sites [8]; SecB142, a truncation lacking the C-terminal 13 aminoacyl residues, thus, no tail sites; SecB142,E77K, a truncation that also has a mutation in the side site, thus, no tails and defective sides. Figure 1 shows these parent species as well as the heterotetramers that are generated. Cartoons and nomenclature for each species are given and will be used throughout to aid in following the data presented.
Figure 1. Parent and hybrid species of SecB.

The four parent species and each of the hybrid tetramers generated are shown in cartoon form. The hybrids are designated by letters which denote the two species of parents that were mixed. The hybrids were stabilized by introduction of cysteines and formation of disulfide bonds between the dimers. The first letter is upper case and indicates the parent species that carried the Q12C substitution whereas the lower case letter in the second position indicates the parent that carried the A129C substitution. For example, the parents of hybrid Ba are SecBQ12C E77K (B) and SecBA129C (A, otherwise wild-type). Thus, the only difference between Dc and Cd is the Cys residue present in the parent.
The interface between monomers in a dimer of SecB is formed by two of the eight β strands in the β sheet on the flat side (Fig. 2). This β sheet does not spontaneously dissociate; therefore, we are not able to generate heterodimers. Thus we were limited in this study to using heterotetramers (Fig. 1) comprising dimers that either have two tails (from SecB species A and B) or none (from SecB species C and D). Because of this limitation the dimers are discussed as either with or without a functional side site, the two tails on a dimer are considered one tail site and within the tetramer are referred to as either on the same dimer as the side site or on the opposite dimer. The reader is encouraged to use the cartoons shown in Figure 1 to understand the discussion of results.
Figure 2. Structures of SecB and SecA.

(a) Structure of tetrameric SecB in ribbon representation from E. coli (Protein Data Bank [PDB] code 1QYN). The image on the right corresponds to a 90° rotation counterclockwise around the y-axis. SecB is structurally a dimer of dimers. One dimer, blue and blue-green protomers forming an eight-stranded β sheet, associates with the other dimer of purple and gray protomers via α helices on the interface. The residues altered to Cys to use in the creation of stable heterotetramers through introduction of disulfide bonds are shown as CPK models, Q12 (red) on the purple:gray dimer and A129 (yellow) on the blue:blue-green dimer. (b) Structure of SecA protomer in CPK models from E. coli protein. PDB code 2FSF with the PBD modeled in by A. Economou based on B. subtilis SecA PDB code ITF5. The separate domains are colored: Nucleotide Binding Domain (NBD) I (yellow), NBD2 (brown), 600 region (green), Helix Scaffold Domain (blue), Precursor Binding Domain (PBD, pink), two helix finger (orange), Helical Wing Domain (HWD, lavender). The location of the N terminus, the first residue resolved is Val 9, is indicated by the red CPK models. The final 65 residues from the C terminus were not resolved but would emerge from residue Glu 836 (cyan).
Preparation of hybrid heterotetramers of SecB
After a mixture of two parent species of SecB reaches equilibrium, there will be three species, the two homotetrameric parent species and the desired heterotetramer [7]. The heterotetramers were stabilized to stop further exchange by introducing disulfide bonds between single cysteine substitutions at residue Q12 in one parent species and at A129 in the other (Fig. 2). In the heterotetramer these residues lie directly across the interface of the dimers and can be oxidized, whereas they lie diagonally across the interface in homotetramers at a distance that does not allow formation of a disulfide bond. The homotetramers Aa (wild-type SecB) and Cc (SecB142) were also subjected to oxidative crosslinking so that we could control for any effects of the nonnative disulfide bond in our studies of the hybrids. After oxidation, catalyzed by copper phenanthroline, the cross-linked tetramers were purified by ion-exchange chromatography and subjected to electrophoresis on denaturing sodium dodecyl sulfate (SDS) polyacrylamide gels (Fig. 3). When denatured and reduced the hybrid species migrate to positions corresponding to those of the parent monomeric species. Species A and B are full length, mass 17,146 Da, whereas C and D are truncated (mass 15,765 Da) and migrate faster. If the disulfide bonds are not reduced each denatured hybrid species migrates as a dimer that is covalently-linked across the interface of the heterotetramer and displays a mass corresponding to the sum of the two parents: Ba having the highest mass and Dc, the lowest.
Figure 3. The six hybrid species.

Each of the hybrids (see cartoons in Fig. 1 for nomenclature) was analyzed by sodium dodecyl (14%) polyacrylamide gel electrophoresis. The samples in the first 6 lanes (reduced) were boiled 5 min. in sample buffer containing 1% SDS and 20 mM DTT before loading. Those in the 6 lanes on the right were boiled in sample buffer containing 1% SDS without DTT, but with 1 mM NEM (N-ethylmaleimide), which reacts with any free sulfhydryls, to avoid oxidative crosslinking.
Stoichiometry of complexes between hybrid SecB species and SecA
The difference in stoichiometry between complexes having maximal efficiency in mediating protein export and those having low activity was originally demonstrated [1, 4] by determining the molar mass of the complexes using size-exclusion chromatography with the proteins applied at concentrations well above the dissociation constants, previously determined by calorimetry [9]. The molar mass determined for the highly efficient wild-type complex (272 kDa) was consistent with two protomers of SecA (204 kDa) bound to a tetramer of SecB (69 kDa) whereas complexes with defective interaction at either the side sites or the tail sites of SecB had masses consistent with A1:B4 (171 kDa) [4]. Position of elution during size-exclusion chromatography cannot be used to give a true estimate of mass since separation of particles involves both size and shape. Therefore the absolute molar mass of each complex was determined by passing the eluent through a detector of refractive index, which provides a measure of concentration, in series with a multi-angle static light scatter detector. The intensity of the light scattered is proportional to the product of the concentration and the weight average molar mass of the particles in solution. The same approach was applied in this work to investigate the complexes formed between SecA and the hybrid SecB species.
Each of the hybrid SecB species was mixed with SecA and subjected to column chromatography (Fig. 4) coupled to light scatter and analysis of the protein content of the eluted fractions by gel electrophoresis. Only three species formed an A2:B4 complex: the parent species Aa and Cc and the heterotetramer Ca (Fig. 4a). Although the complex comprising SecA and wild-type SecB (Aa) elutes ahead of the complexes containing Cc or Ca, this difference does not arise from a difference in stoichiometry. Analyses using static light scatter show both complexes have molar masses of approximately 270 kDa, consistent with A2:B4. Since the molar masses are the same, the difference in elution is likely to result from a difference in shape. Complexes with five of the hybrids, Ba, Da, Cd, Bc and Db elute close to the position of a complex with SecBL75Q, a species of SecB that has defective side sites and makes an A1:B4 complex (Fig. 4b, Fig. 5, data not shown for Db). The slight differences in peak position among the A1:B4 complexes is also attributed to differences in shape (see further evidence below). Figure 5 shows analyses of the protein content in fractions eluted during chromatography of SecB hybrid Bc with and without SecA as a representative example of all characterizations carried out for all analyses by column chromatography. When applied separately, SecA and SecB elute at resolved positions (SecA at 9.3 mL, fraction 25, and SecB at 9.7 mL, fraction 27, Fig. 5a – 5c). The observed co-elution of SecA and SecB at 9 mL (fraction 23) when applied as a mixture (Fig. 5a, 5d) indicates formation of a complex. Since only Aa, Ca and Cc bind two protomers of SecA, we conclude that two functional side sites are necessary to form a stable A2:B4 complex (see Fig. 1).
Figure 4. Stoichiometry of SecA:SecB complexes.

Protein mixtures were subjected to size exclusion chromatography and the eluent was monitored to determine protein concentration by change in refractive index and molar mass by static light scatter. The traces represent concentration and the symbols molar mass. Only every tenth data point is shown for mass determination. The traces were normalized to the maximal peak height for each of the chromatograms for ease of comparing the positions of the peaks. The samples applied in 100 μL contained both SecA and the SecB species indicated at 20 μM each. (a) SecA with: SecB wild-type (red), SecB142 (Cc, green), SecB Ca (blue); (b) SecA with: SecB142(Cc, green) repeated for reference, SecB Ba (cyan), SecBL75Q (magenta), SecB Da (gold), and SecB Cd (brown).
Figure 5. Representative analysis of SecA:SecB complex by column chromatography and gel electrophoresis.

(a) Absorbance profiles of size exclusion chromatography of SecA only applied at 20 μM (green), SecB hybrid Bc only applied at 20 μM (red) and a mixture of SecA and SecB hybrid at 20 μM each (blue). Analyses of the protein content of the fractions eluted by gel electrophoresis: (b) SecA only, (c) SecB hybrid Bc only, and (d) a mixture of SecA and SecB hybrid Bc. Similar analyses were carried out for each of the complexes of SecA with hybrid SecB species. SecA and SecB stain with different efficiency. Therefore, it was necessary to load four times the quantity of SecB relative to the quantity of SecA for analyses by gel electrophoresis.
Differences in shape among the complexes with hybrid SecB species
In addition to analyses using size exclusion chromatography and static light scatter, we characterized the complexes by sedimentation velocity centrifugation. While this technique does not directly determine molar mass it is more sensitive to differences in shape than is chromatography. The parameter determined, the sedimentation coefficient (s value), is directly proportional to molar mass and inversely proportional to the frictional coefficient, which is itself proportional to the radius of hydration (Rh). When protein is loaded into a sample cell and subjected to centrifugation the boundary of protein (Fig. 6a) moves away from the meniscus as it sediments with time and also spreads because of diffusion. Analyses of interactions between SecA and SecB are complicated because the system contains multiple species that are in equilibrium. Therefore, we use the method of van Holde and Weischet [10, 11] (See Fig. 6a and 6b for explanation of analysis) to extract s values that are independent of the diffusional spreading. The distribution of s values for SecA loaded into a cell at 4 μM dimer shows a smooth increase from 4S up to 6.5 S as a function of concentration (Fig. 6c, gray circles); sedimentation behavior that is consistent with the equilibrium between SecA monomer and dimer characterized by an equilibrium constant of 1μM [12]. The SecB tetramer does not undergo dissociation and displays a vertical distribution at approximately 4 S (Fig. 6c, blue circles). The distribution of s values of an equimolar mixture of SecA and SecB increases as a function of concentration from values near the sedimentation coefficients of the individual species. Complexes of stoichiometry A1:B4 and A2:B4 each display characteristic distributions of s values. The distribution plot of the A2:B4 wild-type complex (Fig. 6C, red squares) has a maximal value of approximately 12 S at the highest concentrations, whereas the distribution of complexes between SecA and SecBL75Q, the variant with the defective side site, has a maximal value of 7 S (A1:B4; Fig. 6C open squares). The distribution of s values for the complex with hybrid SecB Ca (green squares), which showed a mass of 273 kDa by light scatter (Fig. 4), is very similar to that of the wild-type complex. We have shown previously that a complex with stoichiometry A2:B4 can be converted to A1:B4 by addition of a zinc-containing synthetic peptide that competes with the C terminus of SecA and disrupts the interactions at the side sites [9]. As expected, addition of the peptide to the cell containing the wild-type complex (red triangles) converted it to a species with the same s value distribution as that of SecA:SecBL75Q (open squares). Addition of the peptide to the SecA:SecB Ca complex moved the distribution back to lower s values (green triangles) consistent with the release of a protomer of SecA; however, the resulting A1:B4 complex sediments faster than does the disrupted wild-type complex. This can be explained if the A1:B4 complex derived from SecB Ca is more compact (smaller Rh) than is A1:B4 derived from wild-type complex. This difference in Rh may result from the different number of tails present in the complexes. In the slower sedimenting complex both dimers of wild-type SecB have tails whereas in the case of the complex with SecB Ca tails are present on only one of the two dimers.
Figure 6. Sedimentation velocity centrifugation.

(a) Raw data. The centrifuge cell contained SecA (4 μM dimer) and SecBL75Q (8 μM tetramer) in 10 mM Hepes-KOH, 300 mM KOAc, 5 mM Mg(OAc)2, 1 mM TCEP, pH 7.3. Thirty-five successive scans were analyzed as shown in (b). (b) van Holde & Weischet analysis. The sedimenting boundaries shown in (a) were divided into 50 equal segments along the concentration axis. Each point is the apparent sedimentation coefficient (s*) of each fraction of the boundary, and each vertical array of 50 points represents one sedimenting boundary. The s* values are extrapolated to the y-axis (infinite time) and plotted to give distribution plots as shown in (c). (c) Distribution plots of s values. The samples were: SecB wild-type (blue circles); SecA (gray circles); SecA and SecBL75Q (open squares); SecA and SecB wild-type (red squares); SecA, SecB wild-type and zinc-peptide (red triangles); SecA and SecB hybrid Ca (green squares); SecA, SecB hybrid Ca and zinc-peptide (green triangles). In all cases SecA was 4 μM dimer and SecB was 4 μM tetramer, except SecBL75Q which was 8 μM, and zinc-containing peptide was 25 μM. The peptide was added to the centrifuge cells after the first centrifugation to gather data for the complexes. The cells are shaken to suspend pelleted material and centrifugation repeated to assess the effect of the peptide.
Differences in shape are also revealed among the complexes having A1:B4 stoichiometry that are formed by interaction of SecA with hybrid species of SecB (Fig. 7). Hybrid Db (gold squares) makes a complex that has an Rh similar to that of the A1:B4 complex derived by peptide-induced dissociation of a SecA protomer from the wild-type complex (Fig. 6d; red triangles). The complex with hybrid Da (blue squares) sediments faster (smaller Rh) and complexes with hybrids Ba (not shown) and Bc (magenta squares) have similar distributions and are the most compact. The differences in shape probably have their origins in the presence or absence of different interactions among the possible sites of binding between SecA and SecB.
Figure 7. Distribution plots for SecA1:SecB4 species.
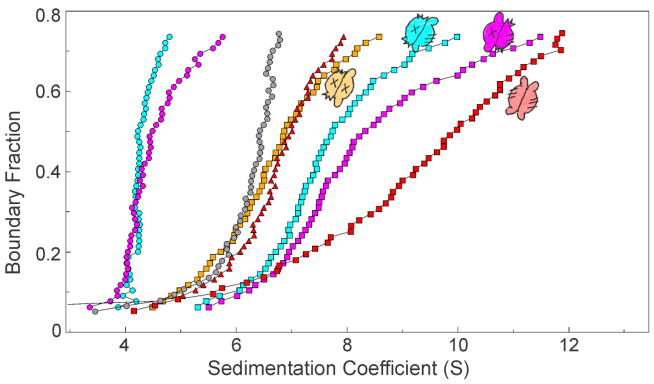
For reference distributions from (6c) are shown: SecA (gray circles); SecA with SecB wild-type (red squares) and with zinc peptide (red triangles). SecA and SecB hybrids were 4 μM each: SecB hybrid Da only (blue circles) and with SecA (blue squares); SecB hybrid Bc only (magenta circles) and with SecA (magenta squares); SecB hybrid Db with SecA (gold squares).
Relative strength of binding between SecB hybrids and SecA
To elucidate the relative contribution of the different sites of interaction to the overall stability of binding, the ability of the hybrids to compete with wild-type SecB for binding to SecA was assessed. Competitions were carried out with each of the heterotetramers as well as with the homotetramers Aa, Bb and Cc. Each of the nonradioactive species of SecB to be tested for competition was mixed with 14C-SecB (6 μM) at molar ratios of 1:1, 2:1 and 3:1, and SecA was added at 4 μM. A competing species of SecB that has the same affinity (dissociation constant, Kd) for SecA as does wild-type SecB would be expected to displace 50% of the 14C-SecB if added to the complex at the same molarity as the wild type 14C-SecB. The experimental design and the effect of introduction of the disulfide bonds to stabilize the hybrids were tested by comparing the efficiency of competition between the 14C-SecB and the nonradioactive wild-type SecB (Fig. 8a) with the efficiency of competition by the cross-linked version of wild-type SecB (hybrid Aa, Fig. 8b). In the presence of the competitors, 14C –SecB was released from the complex (Fig. 8a, 8b black, apex at fraction 32) and appeared at the position of free SecB (gray, apex at fraction 36). Figure 8c shows that both competitors displaced the same fraction of SecB. The data points lie slightly above a theoretical curve calculated using the same affinity of each of the species of SecB for the SecA (Fig. 8c). A fit of the data to the equation used to generate the curve yields a relative affinity of the competing species to that of the radiolabeled SecB of 0.82 +/− 0.03. This difference in the affinity between the species, indicating slightly tighter binding of the competitors (the affinity is expressed in terms equivalent to Kd, i.e., the lower number indicates tighter binding), is likely the result of small errors in determination of the concentrations of the three proteins. These results demonstrate the validity of the experimental design and that the disulfide bonds in SecB do not significantly affect binding to SecA.
Figure 8. Validation of competition experiment to determine relative affinity.
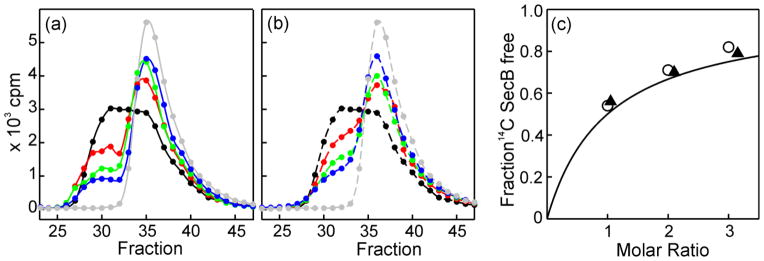
SecA (4 μM dimer) was added either to 14C-SecB (6 μM wild-type tetramer) alone or to 14C-SecB mixed with a nonradioactive competing species of SecB and subjected to size exclusion chromatography. The amount of radioactive SecB in each fraction of eluent was determined. The fractions displayed (23–47) correspond to 7.4 mL to 10.2 mL of eluent. (a) Competition by wild-type SecB. Samples applied to the column contained: SecA mixed with 14C-SecB (6 μM) alone (black); or with competing nonradioactive SecB wild-type at molar ratios of 1(red), 2 (green) or 3 (blue); 14C-SecB (6 μM) without SecA (gray). (b) Competition as described in (a) except that the cross-linked SecB hybrid Aa was the competing SecB species at molar ratios of 1.05 (red), 2.1 (green) and 3.15 (blue). (c) Determination of relative affinity. The fraction of 14C-SecB free was determined by deconvolution of the chromatograms (PeakFit, Systat Software, Inc., San Jose, CA) to resolve the amount of radioactive SecB that was free from that in complex. The continuous line is a theoretical curve generated from the equation, F = R/(A+R) where F is the fraction of SecB free, R the molar ratio of the SecB species, and A, the relative strength of binding, is taken as 1, i.e., equal affinities. Data from competition with wild-type SecB (○) or disulfide cross-linked SecB Aa (▲).
Comparison of the ability of each hybrid SecB species to compete with wild-type SecB allows us to assess the relative contribution to the stability of the SecA:SecB complex by the different sites of interaction. When mixed with the 14C wild-type SecB and added to SecA each hybrid species caused a release of 14C-SecB that increased incrementally at one, two, and threefold molar excess of competitor. The results of the assays carried out with each of the competing hybrid species added at a molar ratio of 1 hybrid:1 wild-type allows one to rank the affinities by visual inspection. Relative to wild type SecB (Aa) (Fig. 9a, red), hybrid Cc (green) binds more tightly and has the highest affinity, followed by hybrid Ca (blue). The remaining hybrids bind more weakly than does Aa (Aa is repeated in Figure 9b as a reference). Hybrid Ad (cyan) displays the weakest binding. Hybrids Cb (orange) and Dc (purple) bind more strongly than does hybrid Ad and appear to have almost identical affinities. The competition experiments carried out at the three molar ratios for each hybrid, including hybrids Bb, Ba and Db, which are not shown in Fig. 9, were subjected to the quantitative analysis described above (Fig. 8c) and the affinities relative to the radiolabeled SecB are given in Figure 10a.
Figure 9. Relative strength of binding to SecA of heterotetrameric SecB species.
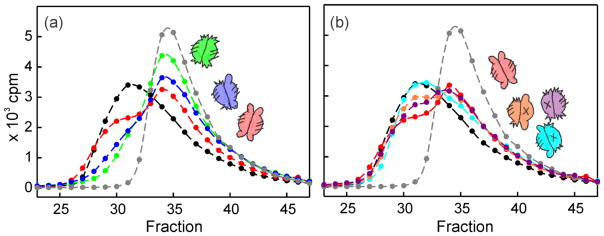
The experimental design was as described in Fig. 8. To facilitate visual comparison only the competitions done at a molar ratio of 1 are shown. The competing species are shown as cartoons in a color that matches the symbol. (a) Hybrid Cc (green); Hybrid Ca (blue) and Hybrid Aa (red). (b) Hybrid Aa (red) is included for comparison; Hybrid Dc (purple), Hybrid Cb (orange) and Hybrid Ad (cyan).
Figure 10. Relative Strength of interaction between Hybrid SecB species and SecA.

(a) The affinities are unitless, expressed relative to that of wild-type SecB without crosslink. Relative affinity determined from fit to F = R/(A+R). See legend to Fig. 8c. The affinity of Db for SecA relative to that of wild-type was determined by competition of hybrid Bb with 14C-SecBE77K. Hybrid Db had an affinity relative to that of Bb of 3.6; the relative affinity of Bb compared to 14C SecB wild type is 2.7. Thus, (3.6)(2.7) = 9.7. (b) Relative ΔG was calculated from the equation ΔG = −RTln (Kd−1) using the values of relative affinity in Table 1 as the relative Kd. The dot in the cartoon is plotted at the value of % ΔG using the energy of Cc as 100%.
The relative strength of binding of the complexes can be expressed as relative free energy (ΔG) of stabilization of the complex using the relationship: ΔG = −RTlnKa, Ka = 1/Kd. Figure 10b illustrates the ranking of the energy of stabilization taking the ΔG of Cc as 100% (titration calorimetry at 8 °C indicates that ΔG for binding of SecA to SecB142 is approximately 8 kcal/mol [13]). The cartoons of each hybrid species are positioned at the corresponding energy level to help in deciphering the features which underlie the ranking. It should be noted that the largest difference in energy, corresponding to a 24-fold difference in relative affinity between Cc and Db, represents a loss of only 22% of binding energy. The three species having the highest affinities, Cc, Ca and Aa have two side sites and make A2:B4 complexes, but differ in the distribution of tails. The tightest binding occurs in the complete absence of tails, consistent with calorimetric studies [13] showing that the tails make a negative contribution to the binding energy when the side sites are involved in stabilizing the complex. Addition of the tail site on one dimer (Ca) results in a loss of 4% of the energy and addition of the tail site to the second dimer (Aa) results in an additional loss of 1%. All other species have only one side site and bind more weakly to SecA than does wild type SecB. Comparison of affinities of Ad and Cb (Fig. 10, orange cartoons) allows us to conclude that a negative energy contribution comes from the tails on the same dimer that carries the active side site; Ad binds more weakly than does Cb (corresponding to a decrease of 7% of the stabilizing energy). Furthermore, we can conclude that tails on the dimer opposite that with the active side site do not contribute to binding energy since the tails on hybrid Cb had no significant effect on the affinity as compared to that of Dc which has no tails.
Models of Binding
Based on the data presented here, in combination with previously published observations, we propose a model of interactions that accounts for this asymmetry. To facilitate discussion of the model we will assume that the dominant contributors to binding energy fall within the sites referred to as side sites and tail sites. Indeed, we know that this is an oversimplification. Our previous mapping of contacts by electron paramagnetic resonance (EPR) spectroscopy [14] and by crosslinking [3] as well as studies using titration calorimetry [13] indicate that there are areas of contact outside these limits (See Fig. 4 in Patel et al.).
The construction of a model begins with the observation that each dimer of the SecB tetramer must have a functional side site to allow two protomers of SecA to bind SecB. The side sites in the absence of the SecB tails (SecB142 and Hybrid Cc) provide the highest energy of stabilization for the A2:B4 complex. When SecA interacts with the side sites of SecB, resulting in binding of two protomers, the presence of the tails on SecB decreases the binding energy (i.e., makes a negative contribution to the binding energy). If only one side site is involved, as occurs in A1:B4 complexes, then the tails on SecB either decrease or increase the stability of the complex depending on whether the tails are on the same dimer that makes contact at the side (decreased stability) or on the opposite dimer (increased stability, see Fig. 10). Two regions of SecA provide binding partners for the tails: one, the first 10 aminoacyl residues of SecA [2, 3] and the second, a stretch of aminoacyl residues in the region 600 to 610 [3]. Both interactions can be shown by crosslinking [3] and by EPR (data not shown). The interaction of the N terminus of SecA seems to be the stronger of the two since a peptide mimic of residues 2 through 11 of SecA was shown by titration calorimetry to bind SecB with a Kd of 30 μM [2], whereas no interaction could be detected by calorimetry with an isolated peptide mimic of residues 600–609. Thus, when comparing binding affinities of the isolated peptides, the N-terminal peptide binds more strongly; however, in the context of the intact SecA the affinity of the 600 region might increase.
In proposing a model to account for this asymmetry, we assume that the same side of SecB cannot simultaneously make contact with the zinc-binding sites of both SecA protomers. This assumption is supported by the crystal structure of the C-terminal 24 aminoacyl residues of SecA, complexed with zinc, bound to the flat β-sheet of SecB [15]. We propose that when the C-terminal zinc-region of the first protomer of SecA to encounter SecB binds to the side of SecB, the N terminus of the SecA binds to the SecB tail on the opposite dimer. We have shown that the complex is more stable if the interacting tail is on the dimer opposite the side contact (compare the stabilizing energy of SecA bound to Ad versus to Cb, Fig. 10). To impose asymmetry in binding we suggest that the 600 region of SecA binds a SecB tail on the dimer across the interface in one of two arrangements: either directly across the dimer interface from the binding site for the N terminus (Fig. 2, blue and purple protomers) or diagonally opposite it (Fig. 2, purple and blue-green protomers). Both orientations are consistent with the crosslinking data in Suo et al [3].
First consider the asymmetry in the case in which the tails involved in binding lie directly across the interface of the SecB tetramer. The figures are schematic cartoons intended to give an approximation and are not meant to convey precise orientations. Following the illustrations in Figure 11 allows the reader to see the relative orientation of the two SecA protomers and the basis of the asymmetry of contacts. There is only one SecB tetramer in the complex, however Figures 11a and 11b each show a SecB positioned so that the tails make contact with the N terminus (indicated by N) and the 600 region (indicated by a green area, and also shown as green CPK models in Figure 2). Let us assume that SecB has actually docked on the black protomer (Fig. 11a). The second (red) protomer joins the complex by a rotation of 180° around the x-axis so that the image of SecB is on the lower face (Fig. 11c). Movement of the red protomer over the black and rotation clockwise until the images of SecB overlay results in a complex with asymmetric contacts. It is clear in the overlay (Fig. 11d) that the 600 region of the upper (red) protomer, which is shown in dark green, cannot interact with the tail to which it is aligned because that tail is bound to the N-terminal region (black N) of the lower (black) protomer. In a similar way the tail to which the N terminus of the upper protomer (red N) is positioned to bind is occupied by the 600 region (light green) of the first protomer that bound.
Figure 11.

Model for asymmetric binding: tails directly across the dimer interface.
Two X-ray structures provide data to help in determining the approach of the C-terminal region of SecA to the side binding site of SecB. One is the crystal structure of a peptide corresponding to the C-terminal 27 aminoacyl residues of SecA bound to SecB. Twenty-four aminoacyl residues in the peptide are structured around a zinc atom in a manner to position the side chains to interact with an acidic patch on the β sheet that forms the side contact site on SecB. The structure shows that the N terminus of the peptide lies over an edge β-strand and the loop containing the zinc extends ~17 Å to reach the β-strand at the interface with the adjoining protomer in the dimer [15]. The structured zinc binding site is attached to the body of SecA by a stretch of 41 disordered aminoacyl residues (not resolved in the crystal structure). If extended, this tether would reach more than 120 Å. An idea of the pathway it might take to reach the side of SecB is provided by the structure of SecA from Bacillus subtilis (PDB 1M6N) [16] which has a greater length of the C-terminal region, referred to as the carboxyl-terminal linker (CTL) resolved then does the E. coli SecA structure. The linker passes below SecA relative to the surface to which SecB binds. As it traverses the PBD it forms the third strand of a short β sheet. The other two β strands are the linkers between the PBD and NBFI. The final residue of the CTL, which is attached to the 22 aminoacyl residues, containing the bound zinc atom, emerges through the cleft between the PBD and NBFII, ideally situated to reach the side of SecB as it is positioned in Figures 11 and 12 (see asterisks in Fig. 2b).
Figure 12.

Model for symmetric binding: tails diagonally across the dimer interface.
The second arrangement in which the active protomers lie diagonally across the interface permits generation of complexes both with symmetric and with asymmetric contacts. First consider the symmetric case illustrated in Figure 12. Here rotation of one protomer (red) 180° around the y-axis followed by positioning over the black protomer and rotation clockwise approximately 45° allows contacts with all four SecB tails. This arrangement is not consistent with the demonstration that only one SecA protomer is bound by SecB tails. It is possible to generate a complex in which only one protomer makes contacts with the tails (Fig. 13); however, this scenario seems less likely than the asymmetric case discussed above (Fig. 11) for two reasons. The first reason is that the two SecA protomers would be required to interact with SecB in different orientations. The interface of the SecB dimer of dimers relative to the SecA protomer bound is rotated (compare Figs. 13a and 13b). The angle of rotation could vary over a wide range. We have chosen to illustrate a 90° difference between the relative position of the SecB tetramer to each SecA protomer. Rotation of the red protomer 180° about the x-axis (Fig. 13b to c), movement over the black protomer followed by rotation clockwise to align the SecB (Fig. 13c to d) again generates asymmetry in the contacts. The second protomer of SecA to bind cannot make interactions with the SecB tails. Figure 13 shows an alternative asymmetric arrangement that is achieved by rotation of the red protomer 180° about the y-axis, movement over the black protomer and alignment of the SecB (Fig. 13b to e). The second reason that leads us to favor the model (Fig. 11) in which the active tails are directly across the interface of the SecB tetramer as opposed to diagonally across the interface is that the C terminus of only one of the SecA protomers (Fig. 13, black) would have the access to the side of SecB as described for Figure 11. The other C terminus (Fig. 13, red protomer) would have a greater distance to traverse, and the side of SecB would not be as accessible.
Figure 13.

Model for asymmetric binding: tails diagonally across the dimer interface.
Concluding remarks
In conclusion, considering all the evidence, we favor a model in which contacts between SecA and tails of SecB involve SecB protomers that lie directly across the dimer interface. This model displays an overall two-fold symmetry of the complex between SecA and SecB, but imposes a functional asymmetry in the stabilizing contacts.
During export SecB carrying a precursor ligand binds SecA and the ternary complex interacts with a membrane-bound translocon to transfer the precursor through the SecY channel. The binding of precursor to SecB is of high affinity (Kd for pGBP and pMBP ~35–55 nM) [17], so it is likely that to achieve transfer from SecB to SecA, the binding of precursor to SecB is weakened by interaction of SecB with SecA.
Mapping of the contact sites between SecB and a ligand using site-directed spin labelling combined with EPR spectroscopy shows that the polypeptide is wrapped around the surface of SecB. It occupies a channel at the interface of the dimer of dimers and could cross over the ends or the sides of SecB to bind in the symmetrically placed channel on the opposite side [18–20]. A study by Haimann and colleagues using pulse EPR techniques indicated that the distance between spin labels situated at the edges of the channel across the dimer interface of the tetramer undergoes a change upon binding a ligand [21]. The model of interactions presented here suggests a way that binding between SecA and SecB might reverse that change at the interface and thereby decrease the affinity of SecB for a bound precursor, allowing it to be passed to SecA. The binding of the N terminus of SecA and the 600 region on opposite dimers of a SecB tetramer would allow the binding of SecA to exert a strain on the interface and move the dimers relative to each other. Additional energy to effect a conformational change in SecB might come from interaction of the complex with SecYEG at the membrane as well as from binding or hydrolysis of ATP by SecA. Studies are underway to elucidate the mechanism in further detail.
Materials and Methods
Mutagenesis and protein purification
Variants of each of the four species of SecB used here as parents of the heterotetramers, wild-type SecB, SecB142, SecBE77K and SecB142,E77K, were created to substitute one of the four natural cysteines C102 with leucine. For each of the variants carrying C102L an additional substitution of cysteine was made separately at Q12 and at A129. Thus we had eight variants, each parent carrying either Q12C or A129C for use in crosslinking heterotetramers. The substitutions were made by standard recombinant DNA techniques (QuikChange, Stratagene). SecB and SecA were purified as described [14, 22]. 14C –SecB was purified from a culture radiolabelled by growth in minimal medium (M9, vitamin B1, 0.4% glycerol). At an optical density of 0.6 at 560 nm, IPTG (0.1 mM) was added to induce the synthesis of SecB. After 90 min., 0.5 μCi of a 14C-amino acid mixture (Amersham Biosciences) was added per milliliter of culture. Growth was continued for 10 min., the cells were harvested, and SecB was purified. All purified proteins were stored at −80°C in 10 mM Hepes (KOH), 300 mM potassium acetate (KOAc), 2 mM tris(2-carboxyethyl)-phosphine (TCEP), pH 7.6. Concentrations of uncrosslinked proteins were determined spectrophotometrically at 280 nm using coefficients of extinction as follows: SecB tetramer 47,600 M−1cm−1, SecA 157,800 M−1cm−1. Concentrations are given in terms of tetrameric SecB and dimeric SecA.
Crosslinking of SecB heterotetramers
To form heterotetramers the SecB wild-type and SecB variants, SecBE77K, SecB142, and SecBE77K,142 were incubated together pairwise at 0.5 mg/mL each in 10 mM Hepes (KOH), 300 mM KOAc, pH 7.6, and 1 mM dithiothreitol (DTT) overnight on ice to allow spontaneous formation of mixed heterotetramers. In each pair one species contained a Cys substitution at aminoacyl residue Q12 and the other at residue A129. The heterotetramers were stabilized by oxidative crosslinking as follows. The reducing agent, DTT, was removed and the buffer changed to 20 mM Tris (HCl), pH 7.6, 300 mM NaCl using a NAP-10 desalting column (GE Healthcare). The eluted protein was collected directly into an Amicon Ultra-15 centrifugal filter device (molecular weight cutoff, 30K, Millipore), copper phenanthroline was added at 0.1 mM and the mixture was incubated on ice. After 5 min., the reaction was stopped by addition of 10 mM EDTA and the buffer was exchanged by repeated dilution and concentration in 20 mM Tris, pH 7.6, until the NaCl concentration was below 50 mM. The cross-linked hybrid was separated from the homotetramers using an ion exchange column (IEC QA-825, 8.0 mm i.d. × 15 cm, Shodex) with a gradient from 270 to 540 mM NaCl. The fractions containing the hybrids were pooled, concentrated and dialyzed against 10 mM Hepes (KOH), 300 mM KOAc, 5 mM Mg(OAc)2, pH 7.6 and stored at −80 °C. Concentration of each cross-linked heterotetramer was determined by electrophoresis using 10 mM TCEP as the reducing agent. At least three quantities of each species were run on the same SDS (13% w/v) polyacrylamide gel as a standard generated by electrophoresis of three quantities of both SecB wild-type and SecB142 of known concentration. The amount of protein in each lane was determined using a Kodak EDAS 290 digital camera to capture the images and TotalLab software (Nonlinear Dynamics, Ltd.) to quantify the intensities. Each cross-linked species was analyzed by MALDI mass spectrometry and the determined masses were all within 0.1% of the calculated masses.
Activity of hybrid species of SecB
Each of the cross-linked SecB heterotetramers was shown to bind precursor galactose-binding protein by size exclusion chromatography using a TSKG3000SWXL (Tosoh Corp., Japan) column as described below except the buffer contained 1 mM ethylene glycol tetraacetic acid to keep the precursor in an unfolded state. The relative affinities for the precursors were not quantitatively determined and might vary as much as four-fold.
Each hybrid except SecBDb, which has the lowest affinity for SecA (at least 10-fold lower than does wild-type SecB), was assayed for in vitro translocation using inverted membrane vesicles and precursor galactose-binding protein as described in Mao et al. [22]. Species that populate A2:B4 complexes showed approximately 1.7-fold higher activity as compared to those that form A1:B4 complexes as expected [1] (see Supplemental Data).
Size exclusion column chromatography and molar mass determination
High performance liquid chromatography was performed as previously described [4] on a Biosep-SEC-S4000 (7.8 mm i.d. × 30 cm, Phenomenex) with a flow rate of 0.7 mL/min. at 7°C in 10 mM Hepes (KOH), 300 mM KOAc, 2 mM Mg(OAc)2, pH 7.0. The absolute molar mass of proteins was determined directly using static light scatter by passing the eluent through a multi-angle laser light scatter detector followed by a differential refractometer (DAWN-HELEOS-II and Optilab rEX, respectively; Wyatt Technology Corporation, Santa Barbara, CA). The molar mass was determined using a specific refractive index increment (dn/dc) of 0.195 mL/gm and the Debye plotting formalism of the Astra software supplied with the instrument. The relationship between the weight average molar mass (Mw) and the excess Rayleigh ratio R(θ) at the low protein concentrations used here is given by:
Where R(θ) is the light scattered by the solution at angle θ in excess of that scattered by pure solvent divided by the incident light intensity, c is the concentration of protein, P(θ) is the form factor that describes the angular dependence of the scatter, and K* is a constant dependent on the parameters of the system used in the study.
SDS-polyacrylamide gel electrophoresis
SDS gel electrophoresis was performed using 14% acrylamide (w/v) gels. DTT (20 mM) was present or absent in the samples applied as indicated. The protein in the fractions eluted from the size exclusion columns was precipitated by addition of trichloroacetic acid to 10%; the precipitates were collected by centrifugation, washed with acetone and suspended in sample buffer.
Analytical Ultracentrifugation
Solutions containing mixtures of proteins at indicated concentrations in 10 mM Hepes (KOH), 300 mM KOAc, 5 mM Mg(OAc)2, pH 7.0 were subjected to centrifugation at 6° or 10°C using an XL-I ultracentrifuge (Beckman Coulter). Samples were loaded into cells with two-sector centerpieces either in the An60Ti rotor and centrifuged at 48,000 rpm for between 2 and 3 h or in the An50 rotor centrifuged at 45,000 rpm for 4 h. Absorbance at 280 nm was measured as a function of radial position. The data were analyzed by the method of van Holde and Weischet using the UltraScan Data Analysis Program version 9.9 from Borries Demeler (University of Texas Science Center, San Antonio, Texas). This analysis extracts s values independent of diffusional spreading. For a detailed description see Demeler et al. (1997). The values used for density and viscosity of the solution relative to water were 1.014 and 1.063, respectively. The s values reported are corrected to water at 20°C.
Determination of relative strength of binding to SecA
Competition between 14C wild-type SecB and nonradioactive SecB or SecB hybrids was carried out as follows. SecA (4 μM dimer) was added to 14C SecB (6 μM tetramer) or to 14C-SecB (6μM tetramer) which was mixed with a competing nonradioactive SecB species at 6, 12 or 18 μM. The protein mixtures were subjected to size exclusion chromatography on a TSK G3000 SWXL column (7.8 mm i.d. × 30 cm) at 7 °C with a flow rate of 0.6 mL/min. in 10 mM Hepes, 350 mM NH4OAc, 5 mM Mg(OAc)2, pH 7.6. Fractions (115 μL) were collected and 80 μL from each fraction was added to 3 mL ScintiSafe 30% (Fisher) and the radioactivity measured using a liquid scintillation counter (Beckman Coulter, LS6500). These data were plotted against volume of eluate so that by deconvolution (PeakFit, 4.11 Auto Fit Peaks II) of the profile we were able to determine the proportion of 14C SecB that was in complex and that which was free. This allowed us to determine the affinities of the competitors for SecA relative to that of wild-type SecB using the equation
where F is the fraction of 14C SecB that is free; R is the molar ratio of the competing SecB species to the 14C SecB and A is the ratio of the affinity of 14C SecB for SecA to the affinity of the competing species. Thus, the numbers we report are lower than one if the competitor binds more tightly than does the wild-type SecB and higher than one if the competitor binds more weakly. In other words, the relative affinity we report would correspond to the ratio of the Kds.
Supplementary Material
Highlights.
An active protein export complex contains tetrameric SecB and two SecA protomers
In the symmetric complex the stabilization energy is distributed asymmetrically
Hybrid SecB species were used to determine the relative strength of the contacts
We show one SecA is bound only to SecB sides and the other also binds SecB tails
A model suggests a mechanism for transfer of precursors from SecB to SecA
Acknowledgments
We thank Angela A. Lilly for construction of the SecB variants, Beverly DaGue (The Proteomics Center, University of Missouri) for MALDI mass spectrometry, Chunfeng Mao for translocation assays, Virginia F. Smith for suggestions on the manuscript, and Jennine M. Crane for critical discussion and preparation of the manuscript. This work was supported by an endowment from the Hugo Wurdack Trust at the University of Missouri and National Institutes of Health Grant GM29798 (to L.L.R.).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Mao C, Hardy SJS, Randall LL. Maximal efficiency of coupling between ATP hydrolysis and translocation of polypeptides mediated by SecB requires two protomers of SecA. J Bacteriol. 2009;191:978–984. doi: 10.1128/JB.01321-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Randall LL, Henzl MT. Direct identification of the site of binding on the chaperone SecB for the amino terminus of the translocon motor SecA. Protein Science. 2010;19:1173–1179. doi: 10.1002/pro.392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Suo Y, Hardy SJS, Randall LL. Orientation of SecA and SecB in complex, derived from disulfide cross-linking. J Bacteriol. 2011;193:190–196. doi: 10.1128/JB.00975-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Randall LL, Crane JM, Lilly AA, Liu G, Mao C, Patel CN, et al. Asymmetric binding between SecA and SecB two symmetric proteins: implications for function in export. J Mol Biol. 2005;348:479–489. doi: 10.1016/j.jmb.2005.02.036. [DOI] [PubMed] [Google Scholar]
- 5.Tang Y, Pan X, Chen Y, Tai PC, Sui SF. Dimeric SecA couples the preprotein translocation in an asymmetric manner. PLoS One. 2011;6:e16498. doi: 10.1371/journal.pone.0016498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cooper DB, Smith VF, Crane JM, Roth HC, Lilly AA, Randall LL. SecA, the motor of the secretion machine, binds diverse partners on one interactive surface. J Mol Biol. 2008;382:74–87. doi: 10.1016/j.jmb.2008.06.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Topping TB, Woodbury RL, Diamond DL, Hardy SJS, Randall LL. Direct demonstration that homotetrameric chaperone SecB undergoes a dynamic dimer-tetramer equilibrium. J Biol Chem. 2001;276:7437–7441. doi: 10.1074/jbc.M009584200. [DOI] [PubMed] [Google Scholar]
- 8.Gannon PM, Kumamoto CA. Mutations of the molecular chaperone protein SecB which alter the interaction between SecB and maltose-binding protein.[erratum appears in J Biol Chem 1993 Jul 15;268(20):15324] J Biol Chem. 1993;268:1590–1595. [PubMed] [Google Scholar]
- 9.Randall LL, Crane JM, Liu G, Hardy SJS. Sites of interaction between SecA and the chaperone SecB, two proteins involved in export. Protein Sci. 2004;13:1124–1133. doi: 10.1110/ps.03410104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.van Holde KE, Weischet WO. Boundary analysis of sedimentation velocity experiments with monodisperse and paucidisperse solutes. Biopolymers. 1978;17:1387–1403. [Google Scholar]
- 11.Demeler B, Saber H, Hansen JC. Identification and interpretation of complexity in sedimentation velocity boundaries. Biophys J. 1997;72:397–407. doi: 10.1016/S0006-3495(97)78680-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Woodbury RL, Hardy SJS, Randall LL. Complex behavior in solution of homodimeric SecA. Protein Sci. 2002;11:875–882. doi: 10.1110/ps.4090102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Patel CN, Smith VF, Randall LL. Characterization of three areas of interactions stabilizing complexes between SecA and SecB, two proteins involved in protein export. Protein Sci. 2006;15:1379–1386. doi: 10.1110/ps.062141006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Crane JM, Mao C, Lilly AA, Smith VF, Suo Y, Hubbell WL, et al. Mapping of the docking of SecA onto the chaperone SecB by site-directed spin labeling: insight into the mechanism of ligand transfer during protein export. J Mol Biol. 2005;353:295–307. doi: 10.1016/j.jmb.2005.08.022. [DOI] [PubMed] [Google Scholar]
- 15.Zhou J, Xu Z. Structural determinants of SecB recognition by SecA in bacterial protein translocation. Nat Struct Biol. 2003;10:942–947. doi: 10.1038/nsb980. [DOI] [PubMed] [Google Scholar]
- 16.Hunt JF, Weinkauf S, Henry L, Fak JJ, McNicholas P, Oliver DB, et al. Nucleotide control of interdomain interactions in the conformational reaction cycle of SecA. Science. 2002;297:2018–2026. doi: 10.1126/science.1074424. [DOI] [PubMed] [Google Scholar]
- 17.Randall LL, Topping TB, Suciu D, Hardy SJS. Calorimetric analyses of the interaction between SecB and its ligands. Protein Sci. 1998;7:1195–1200. doi: 10.1002/pro.5560070514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Xu Z, Knafels JD, Yoshino K. Crystal structure of the bacterial protein export chaperone secB. Nat Struct Biol. 2000;7:1172–1177. doi: 10.1038/82040. [DOI] [PubMed] [Google Scholar]
- 19.Crane JM, Suo Y, Lilly AA, Mao C, Hubbell WL, Randall LL. Sites of interaction of a precursor polypeptide on the export chaperone SecB mapped by site-directed spin labeling. J Mol Biol. 2006;363:63–74. doi: 10.1016/j.jmb.2006.07.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lilly AA, Crane JM, Randall LL. Export chaperone SecB uses one surface of interaction for diverse unfolded polypeptide ligands. Protein Science. 2009;18:1860–1868. doi: 10.1002/pro.197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Haimann MM, Akdogan Y, Philipp R, Varadarajan R, Hinderberger D, Trommer WE. Conformational changes of the chaperone SecB upon binding to a model substrate--bovine pancreatic trypsin inhibitor (BPTI) Biological chemistry. 2011;392:849–858. doi: 10.1515/BC.2011.151. [DOI] [PubMed] [Google Scholar]
- 22.Mao C, Cheadle CE, Hardy SJS, Lilly AA, Suo Y, Gari RRS, et al. Stoichiometry of SecYEG in the active translocase of escherichia coli varies with precursor species. Proceedings of the National Academy of Sciences of the United States of America. 2013;110:11815–11820. doi: 10.1073/pnas.1303289110. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.