

Abstract
Real-time quantitative polymerase chain reaction (qPCR) has become widely used as a method to compare gene transcript levels across different conditions. However, selection of suitable reference genes to normalize qPCR data is required for accurate transcript level analysis. Recently, Marchantia polymorpha has been adopted as a model for the study of liverwort development and land plant evolution. Identification of appropriate reference genes has therefore become a necessity for gene expression studies. In this study, transcript levels of eleven candidate reference genes have been analyzed across a range of biological contexts that encompass abiotic stress, hormone treatment and different developmental stages. The consistency of transcript levels was assessed using both geNorm and NormFinder algorithms, and a consensus ranking of the different candidate genes was then obtained. MpAPT and MpACT showed relatively constant transcript levels across all conditions tested whereas the transcript levels of other candidate genes were clearly influenced by experimental conditions. By analyzing transcript levels of phosphate and nitrate starvation reporter genes, we confirmed that MpAPT and MpACT are suitable reference genes in M. polymorpha and also demonstrated that normalization with an inappropriate gene can lead to erroneous analysis of qPCR data.
Introduction
Real-time quantitative polymerase chain reaction (qPCR) is a sensitive and reproducible technique that is used to measure and compare transcript accumulation levels for genes in many organisms. Correct interpretation of qPCR results, however, is subject to an understanding of several parameters (e.g. the amount and integrity of initial RNA, variations between batches of reagents, and intrinsic stochastic variability in the biochemical reactions). In an attempt to standardize procedures and thus allow comparability between experiments, guidelines establishing a checklist for qPCR experiments were published several years ago [1–3]. Notably, the use of an appropriate normalization strategy is perhaps the most important point to consider for accurate transcript level comparison, because normalization can compensate for experimental variations between different samples. Normalization in qPCR experiments is most commonly carried out by comparing transcript levels of the gene of interest with transcript levels of a ‘constitutively expressed’ reference gene. As such, qPCR experiments can only accurately report transcript levels for a gene of interest, if there are suitable reference genes available for use with the biological samples under investigation [4,5].
Marchantia polymorpha is a liverwort that belongs to the early divergent bryophyte grade of land plants [6,7]. Although the phylogenetic relationship between the bryophyte lineages of liverworts, mosses and hornworts is still debated, the position of liverworts as one of (if not) the earliest land plant lineage(s) is undisputed [8]. As such, studies of liverwort biology are important for understanding the evolution of development processes in land plants. Over the last few years, M. polymorpha has become the liverwort species of choice, not least because it is easy to propagate, mutate and genetically transform [9–11]. The adoption of M. polymorpha as a model [12–14], has led to the need to develop quantitative methods for analyzing mRNA levels. As such, reference genes for which transcript levels are relatively constant across a range of routine conditions must now be identified. The identification of suitable genes for normalization of qPCR analyses in M. polymorpha will facilitate gene expression studies in a representative of one of the earliest land plant lineages.
Specialized tools have been developed over the years to help identify good reference genes for qPCR analyses [15–18], and a number of studies reporting suitable genes have been published in a diverse range of organisms [4,5,18–28]. Notably, studies suggested that commonly used reference genes such as ACTIN, TUBULIN, UBIQUITIN or GLYCERALDEHYDE-3-PHOSPHATE DEHYDROGENASE are not the most appropriate [4,5,20,24,25], and a number of studies have suggested that genes such as SAND [20], EUKARYOTIC ELONGATION FACTOR 5 [28], or CULLIN [24] are better alternatives. Here, we report the analysis of transcript levels for eleven genes in M. polymorpha across different growth conditions, a range of developmental stages, and under a variety of abiotic stress and hormone treatments. We identify MpAPT and MpACT as a good pair of reference genes for normalization of qPCR assays in M. polymorpha and demonstrate that MpGAPC1, MpTUB8 and MpUBQ10 are unsuitable. Finally, using phosphate and nitrate starvation reporter genes, we exemplify how choosing suitable reference genes is crucial for the accurate analysis of qPCR data.
Materials and Methods
Plant material and culture conditions
Gemmae from female Marchantia polymorpha (accession Takaragaike-2) [11] were propagated on Petri dishes filled with ½ Gamborg’s medium pH 5.6 (# G0210, Duchefa) supplemented with 1% agar, or in pots filled with a soil-based compost (Levingston M2) under a 16h light:8h dark photoperiod, respectively at 23°C and a light intensity of 56 μE.m−2.s−1, or at 18°C and a light intensity of 150 μE.m−2.s−1. Male and female sexual organs were obtained from 2-month-old plants (accession Takaragaike-1 and Takaragaike-2 respectively) cultivated in soil conditions supplemented with far-red light [29,30].
For cold treatment, 17 day old plants were transferred to 4°C for 24 hours. Hormone treatments were carried out on ½ Gamborg’s supplemented with, abscisic acid (ABA), 1-naphthaleneacetic acid (NAA) or GR24 at a final concentration of 2 μM, 750 nM and 1 μM respectively. Hormones were initially dissolved in 100% acetone then dilutions to 1/10000th were prepared to supplement the medium. Medium for starvation experiments was prepared by modulating phosphate or nitrate concentration in ½ Johnson’s medium [31]. Low phosphate medium was prepared by adding only 1 mM of (NH4)2SO4 instead of 1 mM of NH4H2PO4. For both low and high nitrate media, 2 mM of CaCl2 was added to the medium instead of 2 mM of Ca(NO3). Low nitrate medium was prepared by adding 1 mM KNO3 instead of 3 mM KNO3, and 2 mM of KH2PO4 as phosphate source. High nitrate medium was prepared by adding 5 mM of NH4NO3, and 2 mM of NH4H2PO4 as phosphate source. For 24 hours treatments (with the exception of cold treatment), plants were grown on cellophane disks (AA Package Ltd.) placed on top of untreated medium. Cellophane disks were then transferred onto untreated (control) or treated (induction) medium.
RNA extraction, cDNA synthesis and qPCR
Total RNA was extracted from ground frozen tissue with the RNeasy plant mini kit (#74904, Qiagen). On-column DNase I treatment was performed using RNase-free DNase I (#79254, Qiagen) according to the manufacturer’s recommendations. Total RNA samples were quantified using a Nanodrop ND-1000 spectrophotometer. RNA integrity was checked using a 2100 BioAnalyzer (Agilent Technologies) and RNA-Nano chips (#5067-1511, Agilent Technologies). The RIN (RNA integrity number) provided by the BioAnalyzer software ranges from 1 (corresponding to a very degraded sample) to 10 (for a mostly intact sample).
cDNA was synthesized from 1 μg of total RNA. Reverse transcription was performed in a 20 μl volume with 2 μM of poly-dT17 primer and 5 U of superscript III reverse transcriptase (#18064, Fermentas) according to the manufacturer’s protocol. cDNA was then diluted 10 times by adding 180 μl nuclease-free water.
Amplification experiments were carried out using a 7300 Applied Biosystem thermocycler. Reactions were performed in 10 μL volumes containing 5 μL 2X SYBR-green mastermix (#4309155, Applied Biosystem), 500 nM forward and reverse primers, and 4 μl diluted cDNA (equivalent to 20 ng of reverse transcribed total RNA). A two-step cycle composed of denaturation at 94°C for 15 seconds followed by hybridization/elongation at 60°C for 60 seconds, was repeated 40 times and then followed by a dissociation step. Three technical replicates were performed for each reaction. Genomic contamination was determined by direct amplification of 20 ng total RNA for 50 cycles.
Data analysis
Fluorescence data were first manually checked for any substantial variation between technical replicates and any outlying values were discarded. Fluorescence data were then exported and transformed using a Perl script into a format suitable for analysis using LinRegPCR v2012.0 [32]. Briefly, LinRegPCR determines mean reaction efficiency (Em) for any specific primer pair, by linearly regressing fluorescence data from several amplification reactions. Because mean efficiencies are calculated in this way, where possible, reactions were grouped in qPCR plates by amplicon rather than by treatment/growth condition. Using the mean efficiency and the window of linearity of the reaction, LinRegPCR computes a N0 value which reflects the initial amount of template in the reaction mix. The computed N0 values from the technical replicates were then averaged. Genomic contamination was taken into account by subtracting N0 values obtained from MpEF1α amplified from total RNA. Contamination was insignificant in all cases, with N0 values from genomic amplifications at least two orders of magnitude lower than N0 from cDNA amplifications.
The extent of variation in transcript levels across samples was determined using the software GenEx version 6 (http://www.biomcc.com/genex-software.html), including geNorm [16] and NormFinder [15] algorithms. N0 values from three biological replicates were first averaged and then normalized by the highest averaged N0 value for a given gene. Normalized N0 were thus rendered equivalent to relative quantification data classically obtained by the ∆Ct method and suitable to be used in GenEx (see Equation 1 adapted from Ruijeter et al. [32]).
| Equation1 |
Stability values were produced by geNorm and NormFinder algorithms (the lower the value, the more stable the gene transcript level). For a given gene, geNorm computes the standard deviation of transcript level ratios between the gene and another gene in all tested conditions. The stability value is then equal to the average of the standard deviations computed for every pairwise comparison of the gene. In contrast, NormFinder relies on a mathematical model to describe gene transcript level variations for a given tested condition and between all tested conditions. The sum of both terms produces a stability value.
Stability values were transformed into z-scores (z) using Equation 2.
| Equation2 |
with Sn the stability value of a given gene n, and δS the mean and the standard deviation of the stability values respectively.
Results
Identification of candidate reference genes
To select putative reference genes for qPCR in M. polymorpha, we conducted a survey of genes used in other plant species [4,5,17,19–28]. 11 candidate genes were selected and orthologous sequences were identified in a M. polymorpha thallus transcriptome (Giulia Morieri, Hélène Proust & Steve Kelly, unpublished) using Arabidopsis gene sequences and reciprocal best BLAST [33] (Table 1). Proteins encoded by the selected genes play roles in genome organization and expression (EF1α, ELONGATION FACTOR 1α; ELF5, EUKARYOTIC ELONGATION FACTOR 5A-1; APT, ADENINE PHOSPHORIBOSYL TRANSFERASE; H3, HISTONE 3), cytoskeleton formation (TUB8, TUBULIN BETA CHAIN 8; ACT, ACTIN 7), protein modification and degradation (CUL, CULLIN 1; UBQ10, POLYUBIQUITIN 10; PEX, PEROXIN 4), glycolysis (GAPC1, GLYCERALDEHYDE-3-PHOSPHATE DEHYDROGENASE C SUBUNIT 1) and vesicle trafficking (SAND).
Table 1. qPCR candidate reference genes in M. polymorpha.
| Gene name | Best hit in uniprot library | Primer sequences (Forward / Reverse) | PCR efficiency | Amplicon size (bp) |
|---|---|---|---|---|
| MpAPT | ADENINE PHOSPHORIBOSYL TRANSFERASE 3 | CGAAAGCCCAAGAAGCTACC / GTACCCCCGGTTGCAATAAG | 1.870 | 146 |
| MpACT | ACTIN 7 | AGGCATCTGGTATCCACGAG / ACATGGTCGTTCCTCCAGAC | 1.908 | 108 |
| MpPEX | PEROXIN 4 | CAGTCAGTTTGCCGTGCTG / GATTGTCCCCCGATCGTAAC | 1.929 | 100 |
| MpUBQ10 | POLYUBIQUITIN 10 | TGAAGGCCAAGATTCAGGAC / ACGAAGCACCAAATGGAGAG | 1.883 | 140 |
| MpCUL | CULLIN 1 | AGGATGTGGACAAGGATAGACG / GTTGATGTGGCAACACCTTG | 1.905 | 84 |
| MpELF5 | EUKARYOTIC ELONGATION FACTOR 5A-1 | AGGTTTCCACCTCCAAGACC / AACGACCTCAGGGACATCAC | 1.918 | 131 |
| MpEF1α | ELONGATION FACTOR 1-ALPHA | CCGAGATCCTGACCAAGG / GAGGTGGGTACTCAGCGAAG | 1.921 | 144 |
| MpTUB8 | TUBULIN BETA 8 | ATCCCGACAGAATGATGCTC / ATTCATCGGCGTTCTCTACG | 1.888 | 120 |
| MpH3 | HISTONE 3 | ACTGATTTGCGGTTCCAGAG / CATAATCGTCACACGCTTGG | 1.921 | 123 |
| MpGAPC1 | GLYCERALDEHYDE-3-PHOSPHATE DEHYDROGENASE C SUBUNIT 1 | GTTCACCACCAAGGACAAGG / CTCGTTCACTCCCATGCAG | 1.930 | 109 |
| MpSAND | SAND protein (At2g28390) | GTTGATGTGTGGCACAAAGG / CAGGCATACGGGAGAAAATG | 1.969 | 142 |
Quantification of transcript levels in different biological contexts
To obtain a set of reference genes that is suitable for use across a broad range of conditions, transcript levels were quantified in 22 biological contexts regrouping different tissue types and plants subjected to different treatments (Table 2). The range of developmental stages included whole plants of different ages (17 days and 24 days old) grown in sterile (Gamborg’s) or soil conditions, plus gemmae (vegetative propagules), antheridiophores (male reproductive structures), and archegoniophores (female reproductive structures). Treatments included abiotic stresses such as nitrogen and phosphate starvation for 17 days or 24 hours (nitrogen only) or 24 hours cold exposure; and hormone treatments where plants were either grown on NAA and ABA-containing media or exposed to NAA, ABA and GR24 (a synthetic strigolactone) for 24 hours.
Table 2. Tissue-types, growth conditions, and exogenous treatments tested (biological contexts).
| Subgroup | Tissue | Condition | Age (days) |
|---|---|---|---|
| Development | |||
| Whole plant | ½ Gamborg’s | 17 | |
| Whole plant | ½ Gamborg’s | 24 | |
| Whole plant | Soil | 17 | |
| Whole plant | Soil | 24 | |
| Gemmae | Soil | 0 | |
| Antheridiophore | Soil | 30 | |
| Archegoniophore | Soil | 30 | |
| Abiotic Stress | |||
| Whole plant | Johnson’s HP | 17 | |
| Whole plant | Johnson’s LP | 17 | |
| Whole plant | Johnson’s HN | 17 | |
| Whole plant | Johnson’s LN | 17 | |
| Whole plant | Johnson’s HN + 24h Johnson’s HN | 18 | |
| Whole plant | Johnson’s HN + 24h Johnson’s LN | 18 | |
| Whole plant | ½ Gamborg’s + 24h cold | 18 | |
| Hormom treatments | |||
| Whole plant | ½ Gamborg’s mock* | 17 | |
| Whole plant | ½ Gamborg’s ABA 2uM | 17 | |
| Whole plant | ½ Gamborg’s mock* | 17 | |
| Whole plant | ½ Gamborg’s NAA 750nM | 17 | |
| Whole plant | ½ Gamborg’s + 24h mock* | 18 | |
| Whole plant | ½ Gamborg’s + 24h ABA 2uM | 18 | |
| Whole plant | ½ Gamborg’s + 24h NAA 2uM | 18 | |
| Whole plant | ½ Gamborg’s + 24h GR24 1uM | 18 | |
*acetone 1/10000e
Where applicable, phenotypic differences between normal and treated plants were recorded (S1 Fig.). Plants cultivated for 17 days on ABA (S1B Fig.) showed a notable size reduction compared to untreated plants (S1A Fig.). Similarly, when cultivated on NAA (S1D Fig.), plant growth was drastically impaired compared to untreated controls (S1C Fig.). The observed ABA– and NAA–induced phenotypes are consistent with published reports [13,34]. Minimal differences were observed between growth on low (S1F Fig.) versus high (S1E Fig.) phosphate levels for 17 days, but nitrogen starvation (S1H Fig.) caused a severe reduction in growth compared to growth on high nitrate levels (S1G Fig.). Nitrogen starvation for just 24 hours did not lead to visible growth perturbations (not shown).
qPCR amplifications were carried out using RNA extracted from plants from the 22 biological contexts (Table 2) and the entire experiment was replicated three times. All RNA samples showed a RIN > 7 (S1 Table), which is sufficient integrity for qPCR experiments [35]. cDNA was synthesized from total RNA extracts to perform qPCR reactions. Primers were designed for the 11 candidate reference genes using the Primer3Plus software [36]. To minimize amplification bias, amplicons were designed to be 80 to 150 nucleotides in length and were located in the 3’ part of the gene. Primer specificity was first checked by PCR and subsequent gel electrophoresis (not shown). The specificity of each qPCR reaction was then checked by performing a dissociation step. For each pair of primers, the dissociation curve showed a unique peak of fluorescence, indicating that a single DNA fragment was amplified during the PCR reaction (S2 Fig.—a few reactions are shown superimposed).
To determine transcript levels for each candidate reference gene in each of the 22 biological contexts, LinRegPCR software [32] was used to compute mean efficiencies (Em) and initial cDNA amount (N0) values for every reaction. All Em values were above 1.85 (Table 1), a value representative of an efficient amplification reaction [37]. Fig. 1 shows a box plot of the log transformed N0/N0min ratio calculated per gene across all conditions. The data reveal that MpEF1α transcript levels were the highest over all conditions tested whereas MpSAND levels were the lowest. Furthermore, MpPEX and MpAPT transcript levels varied least across the whole experiment whereas MpGAPC1 and MpUBQ10 transcript levels varied greatly depending on biological context. Importantly, however, transcript levels were not normalized by known reference genes and thus values are subject to biological and technical perturbations. Therefore, whilst this preliminary analysis showed that the amplification procedure was successful, further refinement was needed to identify reference genes.
Fig 1. Range of transcript levels detected for each of the eleven candidate genes across all 22 conditions.

Average N0 values per gene and condition (n = 3) were normalized on the smallest value then log transformed. Data were then represented for each gene as a box-plot.
Assessment of variation in transcript levels for each candidate reference gene
To assess variation in transcript levels across samples, qPCR data were analyzed in groups using geNorm [16] and NormFinder [15] algorithms. Groups were defined as ‘all’, ‘development’, ‘abiotic stress’ and ‘hormone’, which encompassed all biological contexts, developmental stages and organs, nutrient starvation and cold exposure, and hormone treatments respectively. Both approaches produced a ranked list of genes with the lowest values representing the most consistent transcript levels. Both geNorm and NormFinder ranked similar genes in the top five but not necessarily in the same order (S2 and S3 Tables) (see for example MpACT, MpAPT, MpCUL and MpEF1 when considering all biological contexts). However, in some cases important discrepancies existed between rankings produced by the two algorithms: for example MpELF5 is ranked 1st by geNorm but 7th by NormFinder for the abiotic stress group. In an attempt to reconcile the discrepancies observed, we averaged the values obtained from the two algorithms. Values could not be compared directly as geNorm and NormFinder do not rely on the same calculations. Consequently, we averaged z-scores derived from the data produced by each algorithm for each gene, and for each group (Fig. 2). As for rankings produced by the two algorithms, the lower the z-score for a gene, the more consistent transcript levels are across samples.
Fig 2. Average variation in transcript levels for each candidate gene.

All 22 conditions tested (A, highlighted in blue), development group (B), abiotic stress group (C), and hormone group (D). Variation is represented as the average of z-scores derived from geNorm and NormFinder results. The lower the z-score, the more invariant the transcript levels between samples.
When using z-scores, a different gene ranking was revealed for each group. Transcript levels of MpGAPC1, MpUBQ10 and MpTUB8 varied most across the four groups (Fig. 2). As such, these three genes are unsuitable for use as reference genes in qPCR assays. MpEF1α and MpSAND appeared to be suitable for developmental comparisons (Fig. 2B), MpACT and MpSAND for abiotic stress (Fig. 2C), and MpCUL and MpAPT for hormone treatments (Fig. 2D). Taking into account all biological contexts, MpAPT and MpACT had the lowest z-scores. In addition, MpAPT was ranked third by geNorm and second by NormFinder, and MpACT was ranked second and fifth—in this last case with a value almost identical to the value of the genes ranked third and fourth. Consequently, MpAPT and MpACT are the most suitable reference genes for generic studies. Notably, some genes appeared to have relatively constant transcript levels in some contexts but very variable levels in others. For example, MpSAND had a very low z-score when assayed in the development and abiotic stress groups (Fig. 2B and C) but the highest score in the hormone group (Fig. 2D). In contrast, MpH3 had a low score in the hormone group but a very high score in all other cases.
Estimation of the number of reference genes required for normalization
To further improve normalization strategies, several reference genes can be used simultaneously. Vandesompele et al. [16] described a method to estimate the optimum number of genes needed that computes the variation Vn/n+1 induced by the inclusion of the (n+1)th best ranked gene in the calculation of the normalization factor. A variation greater than 0.15 was assumed to be significant [16]. We implemented the method as a Perl script (available as a supplemental file) and, for each group of biological contexts, computed the stepwise variation obtained when a new gene was added to the normalization factor, starting from the best to the worse ranked gene. When adding the second ranked gene to the normalization factor (V1/2), a variation greater than 0.15 was obtained for every group except the hormone group (Fig. 3). Using the third ranked gene in addition to the first two decreased the variation again but did not improve any of the normalization factors calculated (V2/3 < 0.15 in all cases). Adding more genes tend to make the stepwise variation rise again for three of the four groups, an observation previously reported (see for example Vandesompele et al. [16]). Since the variation when adding the third best-ranked gene was below 0.15, we concluded that using the two most highly ranked genes to compute the normalization factor was sufficient.
Fig 3. Estimation of the optimal number of reference genes required for accurate qPCR data normalization.
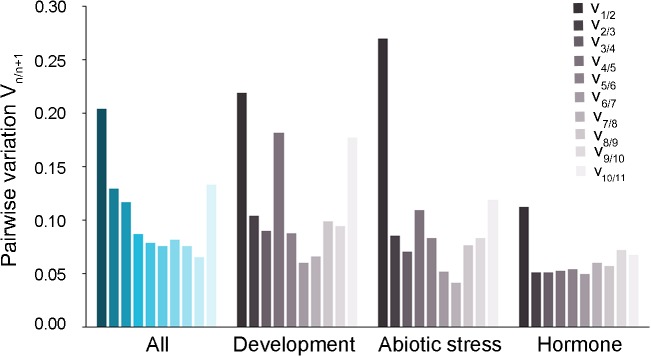
Estimation was done by calculating the pairwise variation (Vn/n+1) of the normalization factors NFn and NFn+1 as described in Vandesomple et al., [16].
Validation of reference genes for qPCR analysis during nitrate and phosphate starvation
In Arabidopsis, AtPHT1-1 (PHOSPHATE TRANSPORTER 1) that encodes a phosphate transporter and AtNRT2-1 (NITRATE TRANSPORTER 2) that encodes a nitrate transporter involved in the High Affinity Transport System are induced during phosphate and nitrate starvation, respectively [38,39]. We identified a similar sequence for each gene in our M. polymorpha transcriptome dataset and measured transcript levels after nutrient starvation (Fig. 4). Non-normalized qPCR data showed a ∼4-fold increase in MpPHT1 transcript levels after phosphate starvation, and a ∼27-fold or ∼14-fold increase in MpNRT2 transcript levels after long term or short term nitrate starvation (Fig. 4A and B, ‘no ref’). The induction of both genes transcription is consistent with previously published results in Arabidopsis [38,39]. Given the dramatic differences detected in non-normalized qPCR between transcript levels before and after starvation, these two genes were deemed appropriate for validation of our candidate reference genes in M. polymorpha.
Fig 4. Transcript levels of MpPHT1-1 and MpNRT2-1 after phosphate and nitrogen starvation.

Histograms represent the fold-change of the average transcript levels (n = 3) compared to the long-term control condition of MpPHT1-1 (A) and MpNRT2-1 (B). Data were non-normalized (‘no ref’) or normalized by a factor consisting of the geometric mean of the transcript level of MpAPT and MpACT (highlighted in blue), of MpACT and MpSAND, of MpACT, MpSAND and MpELF5, and only by the transcript level of MpGAPC1. Biological replicates are depicted superimposed with crosses, diamonds and squares, respectively for replicate one, two and three. NP stands for normal phosphate, LP for low phosphate, NN for normal nitrate, and LN for low nitrate media respectively. A ‘s’ indicates a short (24 h) treatment, a ‘l’ a long (17 days) treatment.
Fig. 4 illustrates different strategies of normalization. Using MpAPT and MpACT, which we showed to be the best candidate reference genes for generic studies, MpPHT1 and MpNRT2 transcript levels exhibited a profile reminiscent of non-normalized data, with fold-change values in the same range: ∼5-fold increase for MpPHT1, and ∼25-fold or ∼17-fold increase (long or short term starvation) for MpNRT2 (Fig. 4A and B, highlighted in blue). The same profile was observed when the data were normalized with the two best candidate genes for the abiotic stress group (MpACT and MpSAND), with a small change, if any, in the fold-change increase (∼5-fold, ∼25-fold and ∼14-fold). Addition of the third ranked gene of the abiotic stress group (MpELF5) to normalize the data only marginally changed the obtained fold-change increase for both MpPHT1 and MpNRT2 after starvation (Fig. 4A and B). This result confirmed that two reference genes are sufficient to normalize qPCR data in M. polymorpha. In combination, these data demonstrate that the two generic reference genes, as well as the two genes specific to the abiotic stress group can be used to reliably normalize qPCR data across abiotic stress contexts.
Data normalization was also carried out using the most variable gene, MpGAPC1. Drastically different (and obviously inaccurate) transcript levels were obtained for both MpPHT1 and MpNRT2. For example, MpPHT1 transcript levels appeared to be equivalent in high and low phosphate contexts, as opposed to being higher in low phosphate. Similarly, whereas in our experiment MpNRT2 transcript levels are higher after long-term nitrate starvation than after 24 hours (non-normalized data and data normalized with the best candidate reference genes), normalization with MpGAPC1 reversed the trend.
These results demonstrate that the methods used here for the identification of reference genes are robust, and also that improper normalization can lead to erroneous analysis of qPCR raw data.
Discussion
Normalization of qPCR results is needed to correct for biological and experimentally introduced variability. A common method of normalization is to compare transcript levels of the gene of interest to those of a reference gene that, through either transcriptional or post-transcriptional mechanisms, is characterized by relatively constant transcript levels across the conditions and/or tissue types being analyzed. Selection of suitable reference genes is not a trivial task and requires careful analysis of non-normalized transcript levels. To facilitate this task, several algorithms such as geNorm and NormFinder are available [15,16]. Here we applied both algorithms, and averaged their results, to identify a set of suitable reference genes for qPCR studies in the model liverwort species M. polymorpha.
We tested eleven candidate genes in a broad range of experimental conditions. We show that genes such as GAPC1, TUB, or UBQ that are commonly used in other organisms [4,5,20] are poor reference genes in M. polymorpha. Instead, our results indicate that for generic applications, normalization using MpAPT and MpACT is optimal. Orthologs of both genes have been shown to be good reference genes in previous studies with other species [4,5,23,25].
The suitability of any particular gene as a reference for normalization can be surprisingly variable depending on biological context. For example, MpSAND transcript levels are elevated after hormone treatments, rendering the gene unsuitable for normalization in these conditions. However, MpSAND transcript levels are relatively constant under different growth conditions, after nutrient starvation and cold response and in different developmental contexts. This constancy has also been observed in other organisms such as Eucalyptus or Arabidopsis [20,25]. This example demonstrates that where possible, reference genes should be determined for each subset of conditions, as restricted as possible; a conclusion also drawn previously by others [5,18].
An important question that arises when testing for reference genes is the number of genes necessary for optimal normalization. The authors of geNorm recommend the use of at least three reference genes (and up to five in some cases) in order to compensate for individual reference gene variation in different study conditions [16]. In contrast, NormFinder authors argue that the use of several reference genes does not necessarily improve normalization accuracy [15]. The use of multiple reference genes increases the number of qPCR reactions required for any particular experiment. This can become problematic for large-scale projects or when working with material from which only small amounts of RNA can be extracted. Here we have shown that two reference genes are sufficient to produce a reliable normalization factor.
Conclusion
We report the identification of a reference gene set suitable for normalizing transcript level data obtained by qPCR in Marchantia polymorpha. We showed that two reference genes were necessary and sufficient to normalize qPCR data and determined the best gene pairs for different experimental conditions such as hormone treatments and abiotic stress, and across a variety of developmental stages. While different pairs were found to be the most appropriate for these biological contexts, we observed that the MpAPT and MpACT combination was the best for generic studies. Therefore, we recommend the use of MpAPT and MpACT for Marchantia polymorpha qPCR analysis.
Supporting Information
17 days mock (A) and 1 μM ABA (B) treatments, mock (C) and 750 nm NAA (D) treatments, high (E) and low (F) phosphate, high (G) and low (H) nitrate. Scale bars equals to 1 cm (A, B, G, H) or 2 cm (C, D, E, F).
(TIF)
Pictures were taken using the qPCR instrument’s software: MpAPT (A), MpACT (B), MpPEX (C), MpUBQ10 (D), MpCUL (E), MpELF5 (F), MpEF1α (G), MpTUB8 (H), MpH3 (I), MpGAPC1 (J), MpSAND (K), MpPHT1 (L), MpNRT2 (M).
(TIF)
(DOCX)
(DOCX)
(DOCX)
(XLSX)
(DOCX)
(TXT)
Acknowledgments
We are grateful to Giulia Morieri and Steve Kelly (Department of Plant Sciences, University of Oxford) for the generation and analysis of M. polymorpha transcriptomes, Francois Didier Boyer (INRA Versailles Grignon) for providing GR24, Cécile Proust-Lima (INSERM Bordeaux) for providing advice on mathematical analysis, and Julie Bull, Mary Saxton, Helen Prescott and Lida Chen for technical assistance.
Data Availability
All relevant data are within the paper and its Supporting Information files.
Funding Statement
This work was funded by ERC Advanced Investigator grants to JAL (EDIP) and LD (EVO500). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Bustin SA. Why the need for qPCR publication guidelines?—The case for MIQE. Methods. 2010;50: 217–226. 10.1016/j.ymeth.2009.12.006 [DOI] [PubMed] [Google Scholar]
- 2. Bustin SA, Benes V, Garson JA, Hellemans J, Huggett J, Kubista M, et al. The MIQE guidelines: minimum information for publication of quantitative real-time PCR experiments. Clin Chem. 2009;55: 611–622. 10.1373/clinchem.2008.112797 [DOI] [PubMed] [Google Scholar]
- 3. Derveaux S, Vandesompele J, Hellemans J. How to do successful gene expression analysis using real-time PCR. Methods. 2010;50: 227–230. 10.1016/j.ymeth.2009.11.001 [DOI] [PubMed] [Google Scholar]
- 4. Gutierrez L, Mauriat M, Guenin S, Pelloux J, Lefebvre JF, Louvet R, et al. The lack of a systematic validation of reference genes: a serious pitfall undervalued in reverse transcription-polymerase chain reaction (RT-PCR) analysis in plants. Plant Biotechnol J. 2008;6: 609–618. 10.1111/j.1467-7652.2008.00346.x [DOI] [PubMed] [Google Scholar]
- 5. Borowski JM, Galli V, Messias Rda S, Perin EC, Buss JH, dos Anjos e Silva SD, et al. Selection of candidate reference genes for real-time PCR studies in lettuce under abiotic stresses. Planta. 2014;239: 1187–1200. 10.1007/s00425-014-2041-2 [DOI] [PubMed] [Google Scholar]
- 6. Bowman JL, Floyd SK, Sakakibara K. Green genes-comparative genomics of the green branch of life. Cell. 2007;129: 229–234. [DOI] [PubMed] [Google Scholar]
- 7. Qiu YL, Li L, Wang B, Chen Z, Knoop V, Groth-Malonek M, et al. The deepest divergences in land plants inferred from phylogenomic evidence. Proc Natl Acad Sci U S A. 2006;103: 15511–15516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Wickett NJ, Mirarab S, Nguyen N, Warnow T, Carpenter E, Matasci N, et al. Phylotranscriptomic analysis of the origin and early diversification of land plants. Proc Natl Acad Sci U S A. 2014;111: E4859–4868. 10.1073/pnas.1323926111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Tàren N. Factors regulating the initial development of gemmae in Marchantia polymorpha. The Bryologist. 1958;61: 191–204. [Google Scholar]
- 10. Ishizaki K, Johzuka-Hisatomi Y, Ishida S, Iida S, Kohchi T. Homologous recombination-mediated gene targeting in the liverwort Marchantia polymorpha L. Sci Rep. 2013;3: 1532 10.1038/srep01532 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Ishizaki K, Chiyoda S, Yamato KT, Kohchi T. Agrobacterium-mediated transformation of the haploid liverwort Marchantia polymorpha L., an emerging model for plant biology. Plant Cell Physiol. 2008;49: 1084–1091. 10.1093/pcp/pcn085 [DOI] [PubMed] [Google Scholar]
- 12. Ishizaki K, Mizutani M, Shimamura M, Masuda A, Nishihama R, Kohchi T. Essential role of the E3 ubiquitin ligase nopperabo1 in schizogenous intercellular space formation in the liverwort Marchantia polymorpha. Plant Cell. 2013;25: 4075–4084. 10.1105/tpc.113.117051 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Tougane K, Komatsu K, Bhyan SB, Sakata Y, Ishizaki K, Yamato KT, et al. Evolutionarily conserved regulatory mechanisms of abscisic acid signaling in land plants: characterization of ABSCISIC ACID INSENSITIVE1-like type 2C protein phosphatase in the liverwort Marchantia polymorpha. Plant Physiol. 2010;152: 1529–1543. 10.1104/pp.110.153387 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Kubota A, Ishizaki K, Hosaka M, Kohchi T. Efficient Agrobacterium-mediated transformation of the liverwort Marchantia polymorpha using regenerating thalli. Biosci Biotechnol Biochem. 2013;77: 167–172. [DOI] [PubMed] [Google Scholar]
- 15. Andersen CL, Jensen JL, Orntoft TF. Normalization of real-time quantitative reverse transcription-PCR data: a model-based variance estimation approach to identify genes suited for normalization, applied to bladder and colon cancer data sets. Cancer Res. 2004;64: 5245–5250. [DOI] [PubMed] [Google Scholar]
- 16. Vandesompele J, De Preter K, Pattyn F, Poppe B, Van Roy N, De Paepe A, et al. Accurate normalization of real-time quantitative RT-PCR data by geometric averaging of multiple internal control genes. Genome Biol. 2002;3: RESEARCH0034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Pfaffl MW, Tichopad A, Prgomet C, Neuvians TP. Determination of stable housekeeping genes, differentially regulated target genes and sample integrity: BestKeeper—Excel-based tool using pair-wise correlations. Biotechnol Lett. 2004;26: 509–515. [DOI] [PubMed] [Google Scholar]
- 18. Hruz T, Wyss M, Docquier M, Pfaffl MW, Masanetz S, Borghi L, et al. RefGenes: identification of reliable and condition specific reference genes for RT-qPCR data normalization. BMC Genomics. 2011;12: 156 10.1186/1471-2164-12-156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Amil-Ruiz F, Garrido-Gala J, Blanco-Portales R, Folta KM, Munoz-Blanco J, Caballero JL. Identification and validation of reference genes for transcript normalization in strawberry (Fragaria x ananassa) defense responses. PLoS One. 2013;8: e70603 10.1371/journal.pone.0070603 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Czechowski T, Stitt M, Altmann T, Udvardi MK, Scheible WR. Genome-wide identification and testing of superior reference genes for transcript normalization in Arabidopsis. Plant Physiol. 2005;139: 5–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Hong SY, Seo PJ, Yang MS, Xiang F, Park CM. Exploring valid reference genes for gene expression studies in Brachypodium distachyon by real-time PCR. BMC Plant Biol. 2008;8: 112 10.1186/1471-2229-8-112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Le Bail A, Dittami SM, de Franco PO, Rousvoal S, Cock MJ, Tonon T, et al. Normalisation genes for expression analyses in the brown alga model Ectocarpus siliculosus. BMC Mol Biol. 2008;9: 75 10.1186/1471-2199-9-75 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Le Bail A, Scholz S, Kost B. Evaluation of reference genes for RT qPCR analyses of structure-specific and hormone regulated gene expression in Physcomitrella patens gametophytes. PLoS One. 2013;8: e70998 10.1371/journal.pone.0070998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Manoli A, Sturaro A, Trevisan S, Quaggiotti S, Nonis A. Evaluation of candidate reference genes for qPCR in maize. J Plant Physiol. 2012;169: 807–815. 10.1016/j.jplph.2012.01.019 [DOI] [PubMed] [Google Scholar]
- 25. Moura JC, Araujo P, Brito Mdos S, Souza UR, Viana Jde O, Mazzafera P. Validation of reference genes from Eucalyptus spp. under different stress conditions. BMC Res Notes. 2012;5: 634 10.1186/1756-0500-5-634 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Wang HL, Chen J, Tian Q, Wang S, Xia X, Yin W. Identification and validation of reference genes for Populus euphratica gene expression analysis during abiotic stresses by quantitative real-time PCR. Physiol Plant. 2014;152: 529–545. 10.1111/ppl.12206 [DOI] [PubMed] [Google Scholar]
- 27. Wang Z, Chen Y, Fang H, Shi H, Chen K, Zhang Z, et al. Selection of reference genes for quantitative reverse-transcription polymerase chain reaction normalization in Brassica napus under various stress conditions. Mol Genet Genomics. 2014;289: 1023–1035. 10.1007/s00438-014-0853-1 [DOI] [PubMed] [Google Scholar]
- 28. Yang H, Liu J, Huang S, Guo T, Deng L, Hua W. Selection and evaluation of novel reference genes for quantitative reverse transcription PCR (qRT-PCR) based on genome and transcriptome data in Brassica napus L. Gene. 2014;538: 113–122. 10.1016/j.gene.2013.12.057 [DOI] [PubMed] [Google Scholar]
- 29. Fredericq H, De Greef J. Red (R), far-red (FR) photoreversible control of growth and chlorophyll content in light-grown thalli of Marchantia polymorpha L. Naturwissenschaften. 1966;53: 337 [DOI] [PubMed] [Google Scholar]
- 30. Wann F. Some of the factors involved in the sexual reproduction of Marchantia polymorpha. Am J Bot. 1925;12: 307–318. [Google Scholar]
- 31. Johnson CM, Stout PR, Broyer TC, Carlton AB. Comparative chlorine requirements of different plant species. Plant and Soil. 1957;8: 337–353. [Google Scholar]
- 32. Ruijter JM, Ramakers C, Hoogaars WM, Karlen Y, Bakker O, van den Hoff MJ, et al. Amplification efficiency: linking baseline and bias in the analysis of quantitative PCR data. Nucleic Acids Res. 2009;37: e45 10.1093/nar/gkp045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. Basic local alignment search tool. J Mol Biol. 1990;215: 403–410. [DOI] [PubMed] [Google Scholar]
- 34. Ishizaki K, Nonomura M, Kato H, Yamato KT, Kohchi T. Visualization of auxin-mediated transcriptional activation using a common auxin-responsive reporter system in the liverwort Marchantia polymorpha. J Plant Res. 2012;125: 643–651. 10.1007/s10265-012-0477-7 [DOI] [PubMed] [Google Scholar]
- 35. Schroeder A, Mueller O, Stocker S, Salowsky R, Leiber M, Gassmann M, et al. The RIN: an RNA integrity number for assigning integrity values to RNA measurements. BMC Mol Biol. 2006;7: 3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Untergasser A, Nijveen H, Rao X, Bisseling T, Geurts R, Leunissen JA. Primer3Plus, an enhanced web interface to Primer3. Nucleic Acids Res. 2007;35: W71–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Remans T, Keunen E, Bex GJ, Smeets K, Vangronsveld J, Cuypers A. Reliable Gene Expression Analysis by Reverse Transcription-Quantitative PCR: Reporting and Minimizing the Uncertainty in Data Accuracy. Plant Cell. 2014; 10.1105/tpc.114.130641 [DOI] [PMC free article] [PubMed]
- 38. Orsel M, Krapp A, Daniel-Vedele F. Analysis of the NRT2 nitrate transporter family in Arabidopsis. Structure and gene expression. Plant Physiol. 2002;129: 886–896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Shin H, Shin HS, Dewbre GR, Harrison MJ. Phosphate transport in Arabidopsis: Pht1;1 and Pht1;4 play a major role in phosphate acquisition from both low- and high-phosphate environments. Plant J. 2004;39: 629–642. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
17 days mock (A) and 1 μM ABA (B) treatments, mock (C) and 750 nm NAA (D) treatments, high (E) and low (F) phosphate, high (G) and low (H) nitrate. Scale bars equals to 1 cm (A, B, G, H) or 2 cm (C, D, E, F).
(TIF)
Pictures were taken using the qPCR instrument’s software: MpAPT (A), MpACT (B), MpPEX (C), MpUBQ10 (D), MpCUL (E), MpELF5 (F), MpEF1α (G), MpTUB8 (H), MpH3 (I), MpGAPC1 (J), MpSAND (K), MpPHT1 (L), MpNRT2 (M).
(TIF)
(DOCX)
(DOCX)
(DOCX)
(XLSX)
(DOCX)
(TXT)
Data Availability Statement
All relevant data are within the paper and its Supporting Information files.