

Abstract
Helicobacter pylori is a common pathogen correlated with several severe digestive diseases. It has been reported that isolates associated with different geographic areas, different diseases and different individuals might have variable genomic features. Here, we describe draft genomic sequences of H. pylori strains YN4-84 and YN1-91 isolated from patients with gastritis from the Naxi and Han populations of Yunnan, China, respectively. The draft sequences were compared to 45 other publically available genomes, and a total of 1059 core genes were identified. Genes involved in restriction modification systems, type four secretion system three (TFS3) and type four secretion system four (TFS4), were identified as highly divergent. Both YN4-84 and YN1-91 harbor intact cag pathogenicity island (cagPAI) and have EPIYA-A/B/D type at the carboxyl terminal of cagA. The vacA gene type is s1m2i1. Another major finding was a 32.5-kb prophage integrated in the YN4-84 genome. The prophage shares most of its genes (30/33) with Helicobacter pylori prophage KHP30. Moreover, a 1,886 bp transposable sequence (IS605) was found in the prophage. Our results imply that the Naxi ethnic minority isolate YN4-84 and Han isolate YN1-91 belong to the hspEAsia subgroup and have diverse genome structure. The genome has been extensively modified in several regions involved in horizontal DNA transfer. The important roles played by phages in the ecology and microevolution of H. pylori were further emphasized. The current data will provide valuable information regarding the H. pylori genome based on historic human migrations and population structure.
Introduction
Helicobater pylori (H. pylori) is a well-defined pathogen that may be correlated with several digestive diseases such as gastritis, ulcers and gastric cancer [1–3]. It is well known that Helicobacter pylori can be divided into seven subpopulations with distinct geographical distributions based on the sequence diversity of seven house-keeping genes. Traces of human migrations could be well reflected and integrated with historical stories or events [4–6] by the gene pools of the different populations. Based on several population-level or individual analyses, it was noted that H. pylori has a high genetic diversity characterized by horizontal transfer and recombination [7–15]. DNA delivery is affected by several type four secretion systems, such as the comB system, TFS3 and TFS4. Cryptic plasmids and prophages were also considered as potential vectors for the transfer of DNA across different strains. However, the mechanisms of these transfers are not well understood [16–21].
Since strain 26695 was first sequenced in 1999, exploration of the comprehensive mechanisms contributing to the flexibility of pathogenesis at the genomic level have been ongoing. Especially during the last three years, there has been a dramatic increase of genomic sequences of Helicobacter pylori in the GenBank genomics database (45 complete sequences up to May 2013, when the analysis for this study began). As the rapid development of next generation sequencing technology has generated many genome sequences [22–30], more studies are being focused on the investigation of potential molecular mechanisms. However, only one of these complete sequences originated from China. We have reported draft genome sequences for three H. pylori strains isolated from Heilongjiang province, which has quite a high incidence of gastric disease [31, 32]. Additionally, we recently completed draft genomes of two other isolates recovered from gastritis patients from Yunnan province. Yunnan, a province located in southwest China, borders Myanmar, Laos, and Vietnam and has many local ethnic minorities. Lijiang is located on the Northwest Yunnan Plateau adjacent to the southeast side of the Tibetan Plateau, which is considered to be the "Roof of the World". One of the largest populations in Yunnan is the Naxi population, numbering approximately 300,000, who mainly inhabit the Lijiang Naxi Autonomous County. The Naxi inhabit a relatively limited area and have a particular lifestyle quite different from that of other ethnic groups. Moreover, reports indicate that this area has a high H. pylori infection rate. Another large population, the Han, mainly inhabit the eastern areas of Yunnan, Kunming. In this study, we performed a global overview of genomic features for the isolates from Yunnan and other publicly available genomes worldwide. The results provide useful information on the genomic diversity of H. pylori among different locations and ethnicities.
Materials and Methods
H. pylori strains for genome sequencing
Two isolates from Yunnan province were sequenced in a comprehensive study. YN4–84 was isolated from a Naxi gastritis patient. YN1–91 was isolated from a Han gastritis patient. The strains were cultured on a Columbia agar base supplemented with 5% sheep blood, and DNA was extracted as previously described [31, 32].
Ethics Statement
All patients involved gave informed consent for the use of the samples in studies in writing, and ethical approval was obtained from the ethics committee of the Chinese Center for Disease Control and Prevention (China CDC) and the academic committee of the National Institute for Communicable Disease Control and Prevention, China CDC.
Genome sequencing
Whole-genome sequencing was performed for each strain using the Illumina HiSeq 2000 by generating paired-end libraries (500 bp and 2 kb) following the manufacturer’s instructions. The read lengths were 90 bp and 50 bp for each library, from which more than 100 Mb of high-quality data were generated. Next, the paired-end reads from the two libraries were assembled de novo into scaffolds. Gene prediction was performed using Glimmer. The tRNA genes were identified with tRNAScan-SE2. rRNA genes were identified with RNAmmer3. The best result for each BLAST search was imported as the gene annotation.
Genomic data deposition
This whole-genome shotgun project was deposited at the DDBJ/EMBL/GenBank under the accession numbers JPXD00000000 (YN4–84), and JPXC01000000 (YN1–91).
Gene annotation and comparative genomics
The sequences were uploaded to the RAST server for gene identification and automatic annotation [33]. A total of 47 genome sequences were further analyzed. These sequences were designated on a world map according to their geographic location (Fig. 1A). Detailed background information for these sequenced strains is shown in Table 1. To identify specific regions in YN4–84, YN4–84 was compared with YN1–91 using MAUVE [34]. GVIEW was used to determine the core genomes of the analyzed strains. BLASTATLAS was used to identify variable genome regions based on a comparison of YN4–84 with an additional forty-six genomes [35]. BLAST parameters were set as follows: Expected cutoff value 1e-10, Alignment length cutoff 100bp, Percent identity cutoff 85%. To identify genomic variable regions among all analyzed genomes, P12 was used as a seed genome for pan-genome construction. P12 has a relatively large genome size and dramatic genome diversity. Therefore using P12 as a reference genome might reflect the variation more comprehensively, although it is not the most phylogenetically close strain to YN4–84. The most diverse genomic regions among the 47 sequences were labeled in an arc line. YN4–84 was in the outermost circle. Neighboring YN4–84 were XZ274 and YN1–91.
Fig 1. A. Global distribution of sequenced H. pylori strains from different populations B. phylogenetic comparison based on seven house-keeping genes (Left) and core genomes (Right).

Table 1. Genomic features and backgrounds of the 47 H. pylori strains used in this study.
| strain | ACCESSION | length | origin | Clinical diagnosis |
|---|---|---|---|---|
| 26695 | AE000511 | 1667892 | UK | Gastritis |
| J99 | AE001439 | 1643831 | USA | Duodenal ulcer |
| HPAG1 | CP000241 | 1596366 | Sweden | Atrophic gastritis |
| P12 | CP001217 | 1673813 | German | Duodenal ulcer |
| G27 | CP001173 | 1652982 | Italy | NA |
| Shi470 | CP001072 | 1608548 | Peru | Gastritis |
| Shi169 | NC_017740 | 1616909 | Peru | NA |
| Shi417 | NC_017739 | 1665719 | Peru | NA |
| Shi112 | NC_017741 | 1663456 | Peru | NA |
| 35A | CP002096 | 1566655 | Japan | NA |
| 51 | CP000012 | 1589954 | Korea | Duodenal ulcer |
| 52 | CP001680 | 1568826 | Korea | NA |
| 908 | CP002184 | 1549666 | France | Duodenal ulcer |
| B38 | FM991728 | 1576758 | France | MALT lymphoma |
| B8 | FN598874 | 1673997 | unknown | Gastric ulcer |
| Cuz20 | CP002076 | 1635449 | Peru | NA |
| Gambia94/24 | CP002332 | 1709911 | The Gambia | NA |
| India7 | CP002331 | 1675918 | India | Peptic ulcer |
| Lithuania75 | CP002334 | 1624644 | Lithuania | NA |
| PeCan4 | CP002074 | 1629557 | Peru | Peruvian gastric cancer |
| PeCan18 | NC_017742 | 1660685 | Peru | Peruvian gastric cancer |
| SJM180 | CP002073 | 1658051 | Peru | Gastritis |
| Sat464 | CP002071 | 1560342 | Peru | NA |
| SouthAfrica7 | CP002336 | 1501960 | South Africa | NA |
| v225d | CP001582 | 1588278 | Venezuela | Gastritis |
| 2017 | CP002571 | 1548238 | France | Duodenal ulcer |
| 2018 | CP002572 | 1562832 | France | Duodenal ulcer |
| F16 | AP011940 | 1575399 | Japan | Gastritis |
| F30 | AP011941 | 1570564 | Japan | Duodenal ulcer |
| F32 | AP011943 | 1578824 | Japan | Gastric cancer |
| F57 | AP011945 | 1609006 | Japan | Gastric cancer |
| 83 | CP002605 | 1617426 | unknown | NA |
| XZ274 | NC_017926 | 1634138 | China | Gastric cancer |
| Rif1 | CP003905 | 1667883 | German | NA |
| Rif2 | CP003906 | 1667890 | German | NA |
| Puno120 | NC_017378 | 1624979 | Peru | Gastritis |
| Puno135 | NC_017379 | 1646139 | Peru | Gastritis |
| HUP-B14 | NC_017733 | 1599280 | Spain | NA |
| ELS37 | NC_017063 | 1664587 | El Salvador | Gastric cancer |
| SNT49 | NC_017376 | 1607577 | India | Asymptomatic |
| Aklavik86 | CP003476 | 1494183 | Canada | NA |
| Aklavik117 | CP003483 | 1614447 | Canada | NA |
| HLJ193 | ALJI00000000 | 1552322 | China | Atrophic gastritis |
| HLJ256 | ALKA00000000 | 1576324 | China | Atrophic gastritis |
| HLJ271 | ALKB00000000 | 1588141 | China | Gastric ulcer |
| YN1–91 | JPXC01000000 | 1609835 | China | Gastritis |
| YN4–84 | JPXD00000000 | 1633405 | China | Gastritis |
Phage prediction
Possible phage sequences were predicted using PHAST [36]. Information on H. pylori phages from other strains were imported from the database in PHAST (http://phast.wishartlab.com/Download.html, update: Jan 1 2014). PHAST can predict phage completeness based on a score calculation system. The criteria for scoring prophage regions as intact, questionable, or incomplete was described in detail in the website above. If a region's total score is less than 70, it is designated incomplete; if between 70 to 90, it is designated questionable; if greater than 90, it is designated intact.
Phylogenetic analysis
Seven house-keeping genes were extracted from these genome sequences and aligned with concatenation to construct a phylogenetic tree using MEGA5 [37]. MUMMER3 was used to perform genome comparison to identify core genome SNPs [38]. Based on a core genome SNP analysis of 47 H. pylori strains distributed in various worldwide regions, a phylogenetic tree was generated to show the YN4–84 and YN1–91 subtype. Groups of the 47 sequenced strains were identified with different colors based on the phylogenetic analysis of the house-keeping gene sequences and core genome SNPs.
Virulence gene analysis for YN4–84 and YN1–91
To characterize the virulence of Yunnan isolates, we extracted gene clusters of the cag pathogenetic island from the 34 sequenced strains that harbored the intact island and aligned the sequences. We built a neighbor-joining tree based on the sequence diversity of both cagPAI and cagA. We compared the EPIYA motif among the 34 sequences. We also searched for other virulence genes, including sabA, babA, vacA, iceA1, iceA2, oipA, dupA and homB.
Results
General genomic features of the two Yunnan isolates
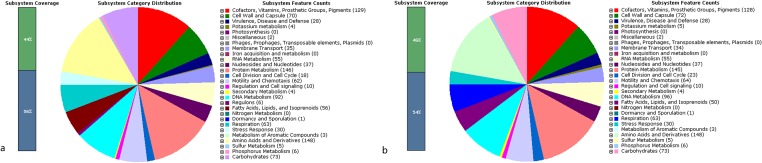
We obtained 11 scaffolds with a total length of 1,609,835 bp for the draft genome of strain YN1–91. For strain YN4–84, we obtained 9 scaffolds with a total length of 1,633,405 bp. The average genomic GC content for each strain was 38.5%. The subsystem distribution and general information about the potential functional distribution of YN4–84 and YN1–91are shown in Fig. 2.
Fig 2. a. Subsystem distribution statistics of Helicobacter pylori strain YN4–84 generated by the rapid annotation using a subsystem technology server. b. Subsystem distribution statistics of Helicobacter pylori strain YN1–91 generated by the rapid annotation using a subsystem technology server.
Comparative genomic and phylogenetic analysis
We extracted seven house-keeping genes from these genome sequences and concatenated them to a length of 3,404 bp and used them to construct a neighbor-joining tree. We found a total of 8,644 core SNPs among the 47 analyzed genome sequences. Based on the phylogenetic analysis of the house-keeping gene sequences and core genome SNPs, the groups of the 47 sequenced strains are identified with different colors in Fig. 1B (left) and Fig. 1B (right).
Fig. 3 shows a global overview of genomic diversity of YN4–84 with other 46 H. pylori genome sequences. The regions showing high diversity were quite consistent with our previous study, from which twelve variable genomic regions were identified based on a high density genome tiling microarray technology. In a picture from BLASTATLAS, four regions with the greatest genome diversity were labeled with a brown arc line. The gene clusters from these regions mainly encoded the type IV secretion system 4, the type IV secretion system 3, the cag pathogenicity island and a serine/threonine kinase C-like protein. Genes coding for restriction modification system enzymes were also quite different in these genomes. Both YN4–84 and YN1–91 have an intact cag pathogenetic island. YN4–84 has an intact TFS3 and partial TFS4 system, while YN1–91 has incomplete TFS3 and TFS4 systems. For the core genome analysis, we found 1059 core genes in total present in all of the sequenced strains.
Fig 3. Global overview of genomic diversity of YN4–84 and YN1–91 with other 46 H. pylori genome sequences.

The four regions with most divergence were labeled with brown arcs along the chromosome.
Phage sequence analysis
By comparing YN4–84 with the isolate recovered from the Han patient (YN1–91), we found a large insertion fragment 32,517 bp in length in YN4–84 (Fig. 4). This fragment was also determined as a strain specific region for YN4–84 compared to the other 45 genome sequences available in GenBank. Further analysis of this fragment in Genbank showed it had a high sequence homology with a reported H. pylori phage (KHP30). This was also confirmed preliminarily by the results of a PHAST analysis. PHAST indicated that the strain specific region was 32,517 bp in length and had 39 predicted CDSs (Table 2). We designated this new phage as YN4–84P. According to the criteria for scoring prophage regions [36], YN4–84P was identified as an intact phage with a high score of 140. The G+C percentage was 35.62%, which was lower than that for the YN4–84 genome (38.43%). The left flanking region was a gene encoding RNA polymerase sigma-54 factor, a transcription factor required for the expression of several flagellar genes. The right flanking region was a homB gene that is reported to have a high positive ratio in gastric cancer isolates. Most of the predicted genes (30/33) were homologous with genes from KHP30. Unlike KHP30, YN4–84P had a 1886 bp transposable sequence (IS605) inserted into the prophage sequence (Fig. 5).
Fig 4. Global alignment of YN4–84 and YN1–91.
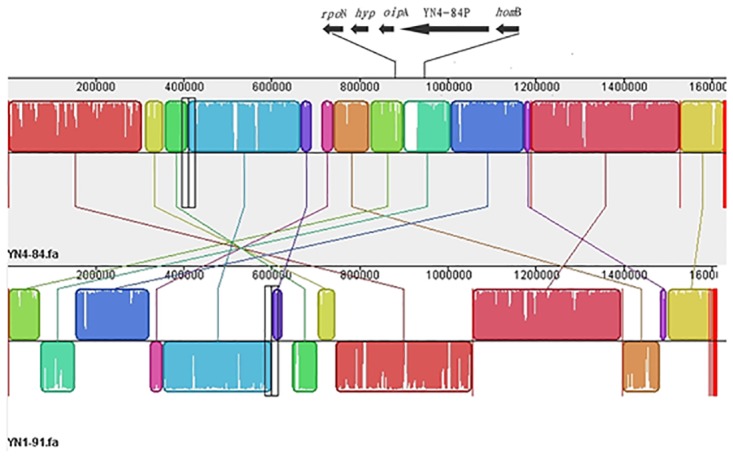
The black arrows above YN4–84 show the insertion of YN4–84P and its flanking gene fragments.
Table 2. Gene contents of the predicted phage YN4–84P.
| code | CDS_POSITION | BLAST_HIT | E-VALUE |
|---|---|---|---|
| 1 | 901213..901485 | PHAGE_Helico_KHP30_NC_019928: hypothetical protein; PP_00960; phage(gi431810541) | 1.00E-25 |
| 2 | 901416..901427 | attL TCAAAAAACCAC | 0 |
| 3 | 901478..902599 | PHAGE_Helico_KHP30_NC_019928: putative phage integrase; PP_00961; phage(gi431810542) | 7.00E-148 |
| 4 | 902626..902811 | PHAGE_Helico_KHP30_NC_019928: hypothetical protein; PP_00962; phage(gi431810543) | 3.00E-22 |
| 5 | 902813..903121 | PHAGE_Helico_KHP30_NC_019928: hypothetical protein; PP_00963; phage(gi431810544) | 1.00E-46 |
| 6 | 903118..904113 | PHAGE_Helico_KHP30_NC_019928: hypothetical protein; PP_00964; phage(gi431810545) | 2.00E-167 |
| 7 | 904311..904577 | PHAGE_Helico_KHP30_NC_019928: DNA helicase; PP_00965; phage(gi431810547) | 2.00E-38 |
| 8 | 904588..905757 | PHAGE_Helico_KHP30_NC_019928: putative DNA helicase, putative DNA repair protein;PP_00966; phage(gi431810548) | |
| 9 | 905768..907327 | PHAGE_Helico_KHP30_NC_019928: putative primase; PP_00967; phage(gi431810549) | 0 |
| 10 | 907324..908997 | PHAGE_Helico_KHP30_NC_019928: hypothetical protein; PP_00968; phage(gi431810550) | 0 |
| 11 | 909058..914190 | PHAGE_Helico_KHP30_NC_019928: hypothetical protein; PP_00969; phage(gi431810551) | 0 |
| 12 | 914563..915150 | PHAGE_Helico_KHP30_NC_019928: hypothetical protein; PP_00970; phage(gi431810552) | 1.00E-97 |
| 13 | 915385..915945 | PHAGE_Helico_KHP30_NC_019928: hypothetical protein; PP_00971; phage(gi431810553) | 5.00E-98 |
| 14 | 915956..917101 | PHAGE_Helico_KHP30_NC_019928: structural protein; PP_00972; phage(gi431810554) | 0 |
| 15 | 917115..917477 | PHAGE_Helico_KHP30_NC_019928: hypothetical protein; PP_00973; phage(gi431810555) | 9.00E-43 |
| 16 | 917528..917959 | PHAGE_Helico_KHP30_NC_019928: hypothetical protein; PP_00974; phage(gi431810556) | 6.00E-70 |
| 17 | 918030..919418 | PHAGE_Helico_KHP30_NC_019928: putative portal protein; PP_00975; phage(gi431810557) | 0 |
| 18 | complement(919375..920658) | PHAGE_Clostr_c_st_NC_007581: putative IS transposase (OrfB); PP_00976; phage(gi80159731) | 9.00E-57 |
| 19 | 920727..921155 | PHAGE_Clostr_c_st_NC_007581: putative transposase; PP_00977; phage(gi80159828) | 3.00E-18 |
| 20 | 921347..921733 | PHAGE_Helico_KHP30_NC_019928: putative portal protein; PP_00978; phage(gi431810557) | 3.00E-60 |
| 21 | 921694..921840 | hypothetical; PP_00979 | 0 |
| 22 | 921833..922228 | PHAGE_Helico_KHP30_NC_019928: putative terminase; PP_00980; phage(gi431810558) | 2.00E-67 |
| 23 | 922234..923388 | PHAGE_Helico_KHP30_NC_019928: putative terminase; PP_00981; phage(gi431810558) | 0 |
| 24 | 923443..923652 | PHAGE_Helico_KHP30_NC_019928: hypothetical protein; PP_00982; phage(gi431810559) | 4.00E-27 |
| 25 | 923664..923879 | PHAGE_Helico_KHP30_NC_019928: hypothetical protein; PP_00983; phage(gi431810560) | 4.00E-08 |
| 26 | 923879..924205 | PHAGE_Helico_KHP30_NC_019928: putative holin; PP_00984; phage(gi431810561) | 7.00E-42 |
| 27 | 924272..924592 | PHAGE_Helico_KHP30_NC_019928: hypothetical protein; PP_00985; phage(gi431810563) | 4.00E-33 |
| 28 | 924640..925185 | PHAGE_Helico_KHP30_NC_019928: hypothetical protein; PP_00986; phage(gi431810564) | 2.00E-97 |
| 29 | 925187..925984 | PHAGE_Helico_KHP30_NC_019928: hypothetical protein; PP_00987; phage(gi431810565) | 4.00E-134 |
| 30 | 925984..926556 | PHAGE_Helico_KHP30_NC_019928: hypothetical protein; PP_00988; phage(gi431810566) | 3.00E-88 |
| 31 | 926595..926882 | PHAGE_Helico_KHP30_NC_019928: hypothetical protein; PP_00989; phage(gi431810567) | 6.00E-39 |
| 32 | 926935..927267 | PHAGE_Helico_KHP30_NC_019928: hypothetical protein; PP_00990; phage(gi431810568) | 2.00E-12 |
| 33 | 927288..928121 | PHAGE_Helico_KHP30_NC_019928: hypothetical protein; PP_00991; phage(gi431810569) | 7.00E-125 |
| 34 | 928121..928903 | PHAGE_Helico_KHP30_NC_019928: hypothetical protein; PP_00992; phage(gi431810570) | 4.00E-133 |
| 35 | 933718..933729 | attR TCAAAAAACCAC | 0 |
Fig 5. Gene content of YN4–84P.

Virulence genes in Yunnan isolates
CagPAI from YN4–84 is 37,296 bp in length and has 26 cag protein encoding genes. The cagA gene is 3,549 bp in length. CagPAI from YN1–91 is 37,492 bp in length and has 26 cag protein encoding genes. The cagA gene is 3,525 bp in length. For the 47 genome sequences, only 34 had intact cagPAI genes with an identical sequence length. From the NJ tree constructed based on cagA and cagPAI, quite a similar phylogenetic relationship was observed, except for minor differences in the bootstrap value of some branches (Fig. 6). YN4–84 was related to another strain isolated from Tibet in China, which was consistent with the results of the phylogenetic analysis based on the core genome SNPs. YN1–91 was related to other hspEAsia strains. Analysis of the EPIYA motif showed that both YN4–84 and YN1–91 belonged to the EPIYA-A/B/D type. Unexpectedly, we found a conserved motif LFGNS in the left flanking regions of EPIYA-A in all of the analyzed hspEAsia subgroup strains (Fig. 7). Other previously reported possible virulence genes like sabA (887355–889637), babA (1528133–1526690), vacA (1179426–1183640) and iceA1 (1386799–1384980) were also found in YN4–84, while dupA and iceA2 were absent.
Fig 6. Neighbor-joining tree based on the sequence diversity of cagA (Left) and cagPAI (Right).
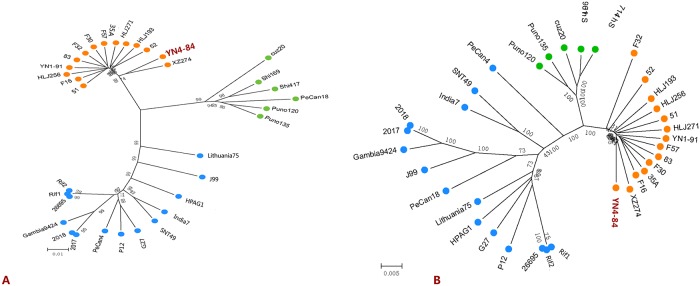
YN4–84 is highlighted in red.
Fig 7. EPIYA motif analysis of the CagA C-terminal region.

Discussion
Genomic features of H. pylori strains recovered from various geographic regions and ethnic groups is a topic of considerable interest. Even though dozens of strains have complete genome sequences, few reports of isolates recovered from high altitude areas exist. Our previous analysis indicated that Yunnan isolates have significantly different genetic features compared to isolates from other areas, especially for the Naxi population, one of the largest ethnic minorities in Yunnan. To enhance our knowledge of the genomic features of the Naxi isolates, we initiated this genome sequencing project.
One of our most striking findings is a putative intact phage sequence in the Naxi isolate YN4–84. Helicobacter pylori was considered devoid of prophages until 2012 when the presence of an incomplete prophage sequence in strain B38 and a complete prophage sequence in strain B45 were reported. Since then, several reports described prophage sequences found in H. pylori isolates. For example, the phage KHP30 and KHP40 were isolated from culture supernatants of East Asian-type isolates from Japanese patients living in distinct geographic regions and the temperate bacteriophage 1961P was found in a lysate of a clinical strain of H. pylori isolated in Taiwan [39–42]. In this study, we report an intact phage sequence found in a Chinese H. pylori isolate for the first time. YN4–84P was inserted between two putative virulence genes, oipA and homB (Fig. 4). The HomB protein was expressed in the H. pylori outer membrane and was antigenic in humans. H. pylori homB knockout mutant strains had a reduced ability to induce interleukin-8 secretion in human gastric epithelial cells, as well as a reduced capacity to bind to the cells, which suggests that HomB is involved in the inflammatory response and in H. pylori adherence [43–45]. OipA, another member of the H. pylori outer membrane protein family similar to HomB, has been called the “outer inflammatory protein” because of its association with increased interleukin (IL)–8 secretion from epithelial cells in vitro and heightened gastric inflammation in vivo [46,47]. Both HomB and OipA are surface-exposed adherence factors that can mediate the interactions of H. pylori with the host microenvironment. The presence of a prophage inserted into this region suggests that HomB and OipA may act as a receptor for YN4–84P. According to the results from PHAST, YN4–84 has a high sequence homology with the other three previously reported H. pylori phages KHP30, KHP40 and 1961P. These phages were all isolated from H. pylori isolates of the hspEAsia subgroup. This phenomenon preliminarily indicates that the host bacteria infected by phage may be population specific for H. pylori species and that the subgroup of H. pylori hspEAsia may only be sensitive to specific phages like YN4–84P or KHP30, etc. We also found a transformable fragment (IS605) inserted into a putative phage portal protein sequence (Fig. 5), which is rare and different from a previous report that described an IS605 fragment inserted between a hypothetical protein HPF16_0942 and a putative site-specific integrase-resolvase HPF16_0945 in an incomplete, questionable phage in F16 [41]. The distribution of IS605 in H. pylori phages should be further investigated and characterized. This study reported a phage in a Chinese H. pylori isolate and its complete genome sequence, however there are still several interesting questions to clarify. Can the phage be released during bacterial growth, or can it be induced by other physical or chemical methods? What is the role of phage in the microevolution or pathogenesis of the H. pylori hspEAsia subgroup? We are currently undertaking a series of further comprehensive experiments to clarify these issues.
We primarily used four sets of SNPs data for the phylogenetic analyses in this study, including clustering based on the sequence diversity of seven commonly used house-keeping genes, the core genome, the cagA gene and the cagPAI. All the results showed a similar phylogenetic profile for the tested strains; however, core genome SNP analysis had higher bootstrap values for some branches compared to cluster results based on house-keeping genes, which illustrated that phylogenetic analysis based on global genomic diversity might be more reliable than local alignment. There was also a minor difference for the genetic relationship of YN4–84. Fig. 1B (left) shows that YN4–84 is related to F32 (a Japanese isolate), and YN1–91 is related to 51(a Korean isolate). While in Fig. 1B (right), YN4–84 is more closely related to XZ274 (a Chinese Tibetan isolate) and YN1–91 is more closely related to HLJ271(a Han isolate). It is noteworthy that geographically, Yunnan neighbors Tibet. We are more confident in the results based on core genome SNPs, which were confirmed by the relationships among the other three Chinese Heilongjiang isolates. In the core genome SNPs tree, they are related to 51 (a Korean isolate). Heilongjiang is located in the northeast of China and neighbors Korea. Therefore, it seems that these isolates can be grouped more precisely according to core genome SNPs though they all belong to hspEAsia subgroup. Both YN1–91 and HLJ271 are Han isolates, from the core genome SNPs phylogenetic tree, YN1–91 is close to HLJ271. While from the seven house-keeping genes SNPs tree, YN1–91 is close 51. The results further emphasize that it is more accurate to construct phylogenetic relationship using core genome SNPs.
The core gene number found in this study was 1059, which is a bit lower than that in previous reports [25, 26]. It is unquestionable that, given the increasing number of sequenced H. pylori isolates based on next-generation sequencing technology, the number of core genes will slightly but gradually decrease. However, according to several estimates from previous studies, the extreme core gene contents cannot be less than 1000 if the genome structure stability for bacterial survival is to be maintained [27–29].
From the global overview of genomic diversity among YN4–84, YN1–91 and the other 45 H. pylori genome sequences, three genomic regions were found to have dramatic genetic divergence in YN4–84. These regions mainly encode type four secretion system three (TFS3), type four secretion system four (TFS4), and a serine threonine kinase protein. YN4–84 has an intact TFS3 and partial TFS4 system, while YN1–91 has incomplete TFS3 and TFS4 systems. The three Heilongjiang isolates all lack these two systems. Although the core genome SNPs tree reflects the close phylogenetic relationship of YN1–91 and HLJ271 for they all belong to Han population, the climate, geographic environment and habitant lifestyle are quite different for these areas. Whether the absence of genomic TFS3 and TFS4 systems are due to the geographic and environmental distinctions needs to be further explored from the genomic features of more isolates from these area. Some other relatively short variable regions that were not labeled along the chromosome circle mainly included genes encoding type I, type II and type III restriction modification systems. These results are consistent with previous genomic studies and further emphasize the contribution of these ‘plasticity zones’ to external niche adaption and pathogenesis for H. pylori [32,48,49]. However, there are still few reports that indicate correlation of these regions with geographic differences or disease clinical outcomes except for cagPAI. Further studies should focus on the functional analysis of these gene clusters to explore potential mechanisms.
From the phylogenetic tree based on cagA and cagPAI sequence diversity, 34 strains were divided into three groups correlating with hspEAsia (orange dots), hspAmerind (green dots) and hpEurope (blue dots) (Fig. 6). YN4–84 was shown to be related to XZ274, which was consistent with the results from the core genome SNPs and suggested that the genetic diversity of cagA could be used to construct a phylogenetic tree to define strain relationships instead of cagPAI, house-keeping genes or core genomes in cagA positive isolates. Further comparison of the EPIYA motifs of cagA shows that YN4–84 has a EPIYA-A/B/D profile, which is a characteristic of eastern isolates. We also found a conserved LFGNS motif in the left flanking regions of EPIYA-A present in all analyzed hspEAsia strains (Fig. 7). Therefore, we think that this motif is a potential marker to identify H. pylori isolates that belong to the hspEAsia subgroup, though more strains should be screened to confirm this. We also briefly investigated other important virulence genes in YN4–84, such as sabA, babA, vacA and iceA1, which were present in YN4–84. Sequence analysis of vacA revealed that it belonged to the s1m2 gene type, which is a predominant vacA gene type in the hspEAsia subgroup and is considered characteristic of highly-virulent strains [50, 51]. YN4–84 did not contain dupA and iceA2. To further investigate the correlation between virulence gene diversity and disease status, more Yunnan isolates are necessary.
In summary, the genome sequencing of Naxi isolate YN4–84 and Han isolate YN1–91 provided useful information for a deep exploration of genetic variations among different H. pylori populations. High genomic diversity and presence of a phage sequence were found in YN4–84 compared to YN1–91 and genomes from strains found worldwide. In future studies, additional Naxi isolates should be sequenced to explore the potential microevolution mechanism for this unique population.
Data Availability
This whole-genome shotgun project has been deposited at DDBJ/EMBL/GenBank under the accession numbers JPXD00000000 (YN4-84), and JPXC01000000 (YN1-91).
Funding Statement
The study was supported by a fund for China Mega-Project for Infectious Disease (2011ZX10004-001) and a grant from the National Technology R & D Program in the 12th Five-Year Plan of China (2012BAI06B02). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Marshall B. Helicobacter pylori. Am J Gastroenterol. 1994;89: S116–S128. [PubMed] [Google Scholar]
- 2. Gerhard M, Rad R, Prinz C, Naumann M. Pathogenesis of Helicobacter pylori infection. Helicobacter 7(Suppl 1). 2002; 17–23. [DOI] [PubMed] [Google Scholar]
- 3. Suerbaum S, Michetti P. Helicobacter pylori infection. N Engl J Med. 2002; 347: 1175–1186. [DOI] [PubMed] [Google Scholar]
- 4. Linz B, Balloux F, Moodley Y, Manica A, Liu H, Roumagnac P, et al. An African origin for the intimate association between humans and Helicobacter pylori . Nature. 2007;445: 915–918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Falush D, Wirth T, Linz B, Pritchard JK, Stephens M. Traces of human migrations in Helicobacter pylori populations. Science. 2003;299: 1582–1585. [DOI] [PubMed] [Google Scholar]
- 6. Moodley Y, Linz B, Yamaoka Y, Windsor HM, Breurec S. The peopling of the Pacific from a bacterial perspective. Science. 2009;323: 527–530. 10.1126/science.1166083 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Cao Q, Didelot X, Wu Z, Li Z, He L, Li Y, et al. Progressive genomic convergence of two Helicobacter pylori strains during mixed infection of a patient with chronic gastritis. Gut. 2014; pii: gutjnl-2014–307345. [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Avasthi TS, Devi SH, Taylor TD, Kumar N, Baddam R, Kondo S, et al. Genomes of Two chronological isolates (Helicobacter pylori 2017 and 2018) of the West African Helicobacter pylori strain 908 obtained from a single patient. J Bacteriol. 2011;193: 3385–3386. 10.1128/JB.05006-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Gustavsson A, Unemo M, Blomberg B, Danielsson D. Genotypic and phenotypic stability of Helicobacter pylori markers in a nine-year follow-up study of patients with noneradicated infection. Dig Dis Sci. 2005; 50: 375–380. [DOI] [PubMed] [Google Scholar]
- 10. Israel DA, Salama N, Krishna U, Rieger UM, Atherton JC, Falkow S, et al. Helicobacter pylori genetic diversity within the gastric niche of a single human host. Proc Natl Acad Sci USA. 2001;98: 14625–14630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Blanchard TG, Czinn SJ, Correa P, Nakazawa T, Keelan M, Morningstar L, et al. Genome sequences of 65 Helicobacter pylori strains isolated from asymptomatic individuals and patients with gastric cancer, peptic ulcer disease, or gastritis. Pathog Dis. 2013;68: 39–43. 10.1111/2049-632X.12045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Baltrus DA, Blaser MJ, Guillemin K. Helicobacter pylori genome plasticity. Genome Dyn. 2009;6: 75–90. 10.1159/000235764 [DOI] [PubMed] [Google Scholar]
- 13. Olbermann P, Josenhans C, Moodley Y, Uhr M, Stamer C, Vauterin M, et al. A global overview of the genetic and functional diversity in the Helicobacter pylori cag pathogenicity island. PLoS Genet. 2010;6: e1001069 10.1371/journal.pgen.1001069 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Duncan SS, Valk PL, McClain MS, Shaffer CL, Metcalf JA, Bordenstein SR, et al. Comparative genomic analysis of East Asian and non-Asian Helicobacter pylori strains identifies rapidly evolving genes. PLoS One. 2013;8: e55120 10.1371/journal.pone.0055120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Mikihiko K, Yoshikazu F, Koji Y, Takeshi T, Kenshiro O. Evolution in an oncogenic bacterial species with extreme genome plasticity: Helicobacter pylori East Asian genomes. BMC Microbiology. 2011;11: 104 10.1186/1471-2180-11-104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Yamaoka Y. Roles of the plasticity regions of Helicobacter pylori in gastroduodenal pathogenesis. J Med Microbiol. 2008;57: 545–553. 10.1099/jmm.0.2008/000570-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Karnholz A, Hoefler C, Odenbreit S, Fischer W, Hofreuter D. Functional and topological characterization of novel components of the comB DNA transformation competence system in Helicobacter pylori . J Bacteriol. 2006; 188: 882–893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Janssen P, Audit B, Ouzounis C. Strain-specific genes of Helicobacter pylori: distribution, function and dynamics. Nucleic Acids Res. 2001;29: 4395–4404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Dangeruta K, Billie V, Asish K, Lizbeth C, Alejandro B. Cluster of type IV secretion genes in Helicobacter pylori’s plasticity zone. J Bacteriol. 2003; 185:3764–3772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Hoffmann R, Zimmer R, Haas R. Strain-specific genes of Helicobacter pylori: genome evolution driven by a novel type IV secretion system and genomic island transfer. Nucleic Acids Res. 2010;38: 6089–6101. 10.1093/nar/gkq378 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Kersulyte D, Lee W, Subramaniam D, Anant S, Herrera P, Cabrera L, et al. Helicobacter pylori’s plasticity zones are novel transposable elements. PLoS ONE. 2009;4: e6859 10.1371/journal.pone.0006859 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Tomb JF, White O, Kerlavage AR, Clayton RA, Sutton GG, Fleischmann RD, et al. The complete genome sequence of the gastric pathogen Helicobacter pylori . Nature.1997;388: 539–547. [DOI] [PubMed] [Google Scholar]
- 23. Alm RA, Ling LS, Moir DT, King BL, Brown ED, Doig PC, et al. Genomic-sequence comparison of two unrelated isolates of the human gastric pathogen Helicobacter pylori . Nature.1999;397: 176–180. [DOI] [PubMed] [Google Scholar]
- 24. Oh JD, Kling-Bäckhed H, Giannakis M, Xu J, Fulton RS, Fulton LA, et al. The complete genome sequence of a chronic atrophic gastritis Helicobacter pylori strain: evolution during disease progression. Proc Natl Acad Sci USA.2006;103: 9999–10004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Gressmann H, Linz B, Ghai R, Pleissner KP, Schlapbach R, Yamaoka Y, et al. Gain and loss of multiple genes during the evolution of Helicobacter pylori . PLoS Genet. 2005;1: e43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. McClain MS, Shaffer CL, Israel DA, Peek RM, Cover TL. Genome sequence analysis of Helicobacter pylori strains associated with gastric ulceration and gastric cancer. BMC Genomics. 2009;10: 3 10.1186/1471-2164-10-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Farnbacher M, Jahns T, Willrodt D, Daniel R, Haas R, Goesmann A, et al. Sequencing, annotation and comparative genome analysis of the gerbil-adapted Helicobacter pylori strain B8. BMC Genomics. 2010; 11: 335 10.1186/1471-2164-11-335 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Ahmed N, Loke MF, Kumar N, Vadivelu J: Helicobacter pylori in 2013: multiplying genomes, emerging insights. Helicobacter 2013;18(Suppl 1): 1–4. 10.1111/hel.12069 [DOI] [PubMed] [Google Scholar]
- 29. Lu W, Wise MJ, Tay CY, Windsor HM, Marshall BJ, Peacock C, et al. Comparative analysis of the full genome of Helicobacter pylori isolate Sahul64 identifies genes of high divergence. J Bacteriol. 2014;196: 1073–1083. 10.1128/JB.01021-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Kumar N, Mukhopadhyay AK, Patra R, De R, Baddam R, Shaik S, et al. Next-generation sequencing and de novo assembly, genome organization, and comparative genomic analyses of the genomes of two Helicobacter pylori isolates from duodenal ulcer patients in India. J Bacteriol. 2012;194: 5963–5964. 10.1128/JB.01371-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. You Y, Liu L, Zhang M, Han X, He L, Zhu Y, et al. Genome sequences of three Helicobacter pylori strains isolated from atrophic gastritis and gastric ulcer patients in China. J Bacteriol. 2012;194: 6314–6315. 10.1128/JB.01399-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. You Y, He L, Zhang M, Fu J, Gu Y, Tao X, et al. Comparative genomics of Helicobacter pylori strains of China associated with different clinical outcome. PLoS ONE. 2012;7: e38528 10.1371/journal.pone.0038528 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Aziz RK, Bartels D, Best AA, DeJongh M, Disz T, Edwards RA, et al. The RAST Server: rapid annotations using subsystems technology. BMC Genomics. 2008;9: 75 10.1186/1471-2164-9-75 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Darling AC, Mau B, Blattner FR, Perna NT. Mauve: multiple alignment of conserved genomic sequence with rearrangements. Genome Res. 2004; 14:1394–1403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Petkau A, Stuart-Edwards M, Stothard P, and Van Domselaar G. Interactive microbial genome visualization with GView. Bioinformatics. 2010;26: 3125–3126. 10.1093/bioinformatics/btq588 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Zhou Y, Liang Y, Lynch KH, Dennis JJ, Wishart DS. “PHAST: A Fast Phage Search Tool”. Nucl. Acids Res. 2011;39: W347–W352 10.1093/nar/gkr485 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Tamura K, Peterson D, Peterson N, Stecher G, Nei M, Kumar S, et al. MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol Biol Evol. 2011; 28: 2731–2739. 10.1093/molbev/msr121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Kurtz S, Phillippy A, Delcher AL, Smoot M, Shumway M, Antonescu C, et al. Versatile and open software for comparing large genomes. Genome Biol. 2004;5: R12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Lehours P, Vale FF, Bjursell MK, Melefors O, Advani R, Glavas S, et al. Genome sequencing reveals a phage in Helicobacter pylori . mBio. 2011; 2: e00239–11. 10.1128/mBio.00239-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Uchiyama J1, Takeuchi H, Kato S, Gamoh K, Takemura-Uchiyama I, Ujihara T, et al. Characterization of Helicobacter pylori Bacteriophage KHP30. Appl. Environ. Microbiol. 2013;79: 3176–3184 10.1128/AEM.03530-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Luo CH, Chiou PY, Yang CY, Lin NT. Genome, integration, and transduction of a novel temperate phage of Helicobacter pylori . J. Virol. 2012; 86: 8781–8792 10.1128/JVI.00446-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Uchiyama J, Takeuchi H, Kato S, Takemura-Uchiyama I, Ujihara T, Daibata M, et al. Complete genome sequences of two Helicobacter pylori bacteriophages isolated from Japanese patients.J Virol. 2012;86: 11400–11401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Oleastro M, Cordeiro R, Ferrand J, Nunes B, Lehours P, Carvalho-Oliveira I et al. Evaluation of the clinical significance of homB, a novel candidate marker of Helicobacter pylori strains associated with peptic ulcer disease. J Infect Dis.2008; 198: 1379–1387. 10.1086/592166 [DOI] [PubMed] [Google Scholar]
- 44. Talebi B, Abadi A, Rafiei A, Ajami A, Hosseini V, Jones KR, et al. Helicobacter pylori homB, but not cagA, is associated with gastric cancer in Iran. J Clin Microbiol. 2011;49: 3191–3197. 10.1128/JCM.00947-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Jung SW, Sugimoto M, Graham DY, Yamaoka Y. homB status of Helicobacter pylori as a novel marker to distinguish gastric cancer from duodenal ulcer.J Clin Microbiol. 2009;47: 3241–3245. 10.1128/JCM.00293-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Yamaoka Y. Mechanisms of disease: Helicobacter pylori virulence factors. Nat. Rev. Gastroenterol. Hepatol. 2010;7: 629–641. 10.1038/nrgastro.2010.154 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Matteo MJ, Armitano RI, Granados G, Wonaga AD, Sanches C, Olmos M, et al. Helicobacter pylori oipA, vacA and dupA genetic diversity in individual hosts. J. Med. Microbiol. 2010;59: 89–95. 10.1099/jmm.0.011684-0 [DOI] [PubMed] [Google Scholar]
- 48. Baltrus DA, Blaser MJ, Guillemin K. Helicobacter pylori genome plasticity. Genome Dyn. 2009;6: 75–90. 10.1159/000235764 [DOI] [PubMed] [Google Scholar]
- 49. Kersulyte D, Lee W, Subramaniam D, Anant S, Herrera P, Cabrera L, et al. Helicobacter Pylori’s plasticity zones are novel transposable elements. PLoS One. 20094:e6859 10.1371/journal.pone.0006859 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Atherton JC, Peek RM, Tham KT, Cover TL, Blaser MJ. Clinical and pathological importance of heterogeneity in vacA, the vacuolating cytotoxin gene of Helicobacter pylori . Gastroenterology. 1997;112: 92–99. [DOI] [PubMed] [Google Scholar]
- 51. Gangwer KA, Shaffer CL, Suerbaum S, Lacy DB, Cover TL, Bordenstein SR, et al. Molecular evolution of the Helicobacter pylori vacuolating toxin gene vacA . J. Bacteriol. 2010;192: 6126–6135. 10.1128/JB.01081-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
This whole-genome shotgun project has been deposited at DDBJ/EMBL/GenBank under the accession numbers JPXD00000000 (YN4-84), and JPXC01000000 (YN1-91).