

Abstract
Molecular taxonomy and evolution of nematodes have been recently the focus of several studies. Mitochondrial sequences were proposed as an alternative for precise identification of Meloidogyne species, to study intraspecific variability and to follow maternal lineages. We characterized the mitochondrial genomes (mtDNAs) of the root knot nematodes M. floridensis, M. hapla and M. incognita. These were AT rich (81–83%) and highly compact, encoding 12 proteins, 2 rRNAs, and 22 tRNAs. Comparisons with published mtDNAs of M. chitwoodi, M. incognita (another strain) and M. graminicola revealed that they share protein and rRNA gene order but differ in the order of tRNAs. The mtDNAs of M. floridensis and M. incognita were strikingly similar (97–100% identity for all coding regions). In contrast, M. floridensis, M. chitwoodi, M. hapla and M. graminicola showed 65–84% nucleotide identity for coding regions. Variable mitochondrial sequences are potentially useful for evolutionary and taxonomic studies. We developed a molecular taxonomic marker by sequencing a highly-variable ~2 kb mitochondrial region, nad5-cox1, from 36 populations of root-knot nematodes to elucidate relationships within the genus Meloidogyne. Isolates of five species formed monophyletic groups and showed little intraspecific variability. We also present a thorough analysis of the mitochondrial region cox2-rrnS. Phylogenies based on either mitochondrial region had good discrimination power but could not discriminate between M. arenaria, M. incognita and M. floridensis.
Introduction
Mitochondria are subcellular organelles derived from an alpha-proteobacterial endosymbiont, wherein oxidative phosphorylation and other important biochemical functions take place [1]. Most of the genes of the original bacterial chromosome were lost or transferred to the nucleus, with a handful of genes remaining in animal mitochondrial genomes (mtDNAs) [2]. Animal mtDNAs share several features, including relatively constant gene content and order, maternal inheritance, reduced recombination rate, and high evolutionary rate. This molecule is central in disease, apoptosis and ageing studies, in genetic and genomic analyses, and in developing genetic markers for molecular systematics [3]. Animal mtDNAs are generally small and circular molecules containing 12–13 protein-coding genes for enzymes of the oxidative phosphorylation pathway, 22 transfer RNA-coding genes (tRNAs), and 2 ribosomal RNA-coding genes [2]. This gene content is variable only in a few groups of metazoans, such as some nematodes that lack the gene atp8 [3], a bivalve that also lacks atp8 and has an extra tRNA gene [4], and cnidarians that have lost nearly all tRNAs genes [5]. Usually, no introns are present in animal mtDNAs and intergenic sequences are small, although one large non-coding region with control elements is found in some metazoan species [6]. To date, more than 100 complete mtDNA sequences of the phylum Nematoda have been published. A phylum-wide comparison revealed that nematode mtDNAs are characterized by a very rapid rate of mitochondrial gene rearrangement and are smaller in size than those of other metazoan groups, varying in size from 13 kb to 15 kb with some exceptions as Romanomermis culicivorax (26 kb), Pratylenchus vulnus (21 kb) Hexamermis agrotis (24 kb) [2,3,7,8,9].
Root-knot nematodes (genus Meloidogyne, infraorder Tylenchomorpha, suborder Tylenchina, order Rhabditida, class Chromadorea) [10] are plant parasites, comprising more than 100 species, in addition to several described races [11] and rank first based on a list of scientifically and economically important plant parasitic nematodes [12]. Their common name refers to typical galls on the roots of host plants that reduce the uptake of water and nutrients resulting in lower crop productivity [13]. The genus Meloidogyne includes the most widespread and economically damaging nematodes worldwide: M. incognita, M. javanica, M. arenaria, M. hapla, M. chitwoodi and M. enterolobii [14]. Accurate identification of Meloidogyne species is critical for effective crop management. Traditionally, species diagnosis was done based on morphological characters, host range and esterase isozyme electrophoresis and were later combined with DNA-based methods [15]. These molecular techniques were initially based on analyses of DNA fragments and sequencing of the nuclear rDNA operon and became obsolete or less effective with the increasing description of new species [16]. Mitochondrial sequences were proposed as an alternative for precise identification of Meloidogyne species, to study intraspecific variability and to follow maternal lineages [17]. Mitochondrial markers have been used in the past, but with a different fragment than in this current study [17,18,19,20].
To gain insight into the evolutionary patterns, sequence diversity and potential taxonomic use of the mitochondrial genome of the genus Meloidogyne, we characterized the mtDNAs of three root-knot nematodes: a facultative meiotic parthenogenetic strain of M. hapla and M. floridensis and a strain of the obligate mitotic parthenogenetic M. incognita, taking advantage of genomic projects that focused on these species. We undertook detailed genomic comparisons among them and with mtDNAs of Meloidogyne spp. recently published [21,22,23]. Based on genomic alignments of Meloidogyne, we identified a 2 kb variable mitochondrial marker and performed phylogenetic analyses to aid in Meloidogyne species identification and evolution and to elucidate relationships within the genus.
Materials and Methods
Mitochondrial genome annotation
The nuclear genomes of three species of root-knot nematodes, M. floridensis [24], M. hapla [25], and M. incognita [26] have been reported and mitochondrial sequences were made available in public sequence databases as part of these genome sequencing projects but have not been yet assembled or analyzed. Sequence similarity searches were performed with BLASTn [27] against the GenBank databases nr, WGS (whole-genome shotgun), and EST (expressed sequence tag), restricted to the genus Meloidogyne without filtering low complexity regions and using the mtDNA of several Nematoda as the query sequence. A total of 17 contigs of the root-knot nematodes M. floridensis, M. hapla and M. incognita had similarity to mitochondrial genes (S1 Table). Mitochondrial fragments of each species were assembled based on overlapping sequences and by amplification and sequencing of one joining fragment. We designed primers to link two mitochondrial contigs of M. hapla ABLG01002800 and ABLG01002664 (S1 Table): Mh32800R (5`-AGAGTGAATTGGTAAGAGG-3`) and Mh12664F (5`-CGGTAACCAAAAACCTCCAAGC-3`). We amplified and sequenced a product of 1500 bp using DNA of M. hapla 7J2 (S2 Table). It is not possible to rule out the possibility that some of these mitochondrial sequences belong to NUMTs in the nuclear genome. However, the lack of nuclear sequences in the fragments containing mitochondrial genes suggests that those DNA fragments reside in the mitochondria.
Open reading frames of the mitochondrial sequences of M. floridensis, M. hapla and M. incognita were recognized with Sequencher 5.2.2 (Genes Codes Corporation) using the invertebrate mitochondrial genetic code. Each putative protein-coding gene was used as query for BLASTx searches against GenBank protein databases to identify them. Ribosomal RNA genes (rRNAs) were recognized by comparison to other rRNAs of nematodes using BLASTn [27]. The tRNA genes were identified using tRNAscan-SE search server [28] or by similarity to those reported in other Meloidogyne species. Secondary structures of tRNAs were predicted with tRNAscan-SE.
Gene boundaries, i.e. start and stop codons, are generally predicted based on the presence of a canonical start codon followed by a standard stop codon. When non-universal genetic codes or non-canonical stop codons are involved, as reported for animal mtDNAs [3], gene boundaries are less obvious and several start or stop codons may be available. A disparity in the criterion for establishing gene boundaries was observed for several genes in Meloidogyne mtDNA, even between two strains of the same species [21,23]. In this study, start codons were set at the first start codon available (including non-universal ones) that had the minimum overlap with an upstream gene. Complete universal stop codons (TAA or TAG) were selected even when they overlapped with a downstream gene.
Analyses of mitochondrial sequences
Pairwise comparisons of protein, rRNA, and tRNA genes of M. chitwoodi, M. floridensis, M. graminicola, M. hapla and M. incognita were done in MEGA6 [29]. Codon usage of protein-coding genes and nucleotide composition were calculated with the software SMSv2 (Sequence Manipulation Suite) [30]. Whole-genome alignments of Meloidogyne spp. mitochondrial genomes were performed with the VISTA pipeline infrastructure [31]. Tandem repetitive elements were identified using Tandem Repeat Finder software [32].
PCR amplification and sequencing of a mitochondrial region
DNA samples of 28 populations of 5 Meloidogyne species were kindly provided by Andrea Skantar (USDA) (S2 Table). DNA of a M. arenaria isolate from Argentina was extracted as reported previously [33]. We amplified and sequenced the mitochondrial region between the genes nad5 and cox1 from different Meloidogyne species and isolates using the following primers: Mmt5 (5`-GGTTTAATTGGTGGTTTTGG-3`), Mmt2rc (5`-TGTCCTCAAACTAAACAACC-3`), Mmt9 (5`-TTGGTTGATTGGTGAAAGC-3`), and Mmt4rc (5`-AAACCACCAATTAAACCAGG-3`). Primers were designed with the software Primaclade [34] based on a sequence alignment of available Meloidogyne species. PCR conditions included an initial denaturing step at 94°C for 2 min, followed by 35 cycles of 94°C for 30 seconds, 45°C for 30 seconds and 72°C for 1 min; followed by a final extension step at 72°C for 8 min. Amplified fragments were sequenced by Sanger sequencing with Applied Biosystems 3730XL. Sequences were deposited in GenBank (accession numbers KM491188-KM491216).
Phylogenetic analyses
Three mitochondrial nucleotide data sets were constructed and analyzed. One consisted of 8 protein-coding genes (cox1, cox2, cox3, cob, nad2, nad3, nad4 and nad5) from 43 diverse nematode species that were retrieved from GenBank. The 8 protein-coding genes were concatenated in a single nucleotide data set of 4,920 bp. The second data set included sequences of the nad5-cox1 region from 36 populations of 8 species of Meloidogyne (S2 Table). The third data set consisted of mitochondrial sequences of the cox2-rrnS region from 151 isolates of 20 Meloidogyne species that were retrieved from GenBank. Nucleotide sequences were manually aligned with MacClade 4.07 [35]. Alignment regions for which positional homology could not be determined with confidence by visual inspection were excluded. Alignments and trees have been deposited in Treebase (study S17102).
Maximum Likelihood (ML) phylogenetic analyses were performed with Garli 0.951 [36] under the General Time Reversible model with parameters for invariable sites and gamma-distributed rate heterogeneity. The GTR substitution model was chosen by using Modeltest for each data set [37]. Ten independent runs were conducted using either the automated stopping criterion or for up to 5,000,000 generations to ensure convergence to a similar topology and likelihood score. A hundred ML bootstrap replicates were performed. Maximum Parsimony (MP) analyses were conducted using PAUP* 4.0 [38]. The search strategy involved tree-bisection reconnection branch swapping, MulTrees option in effect. To assess node support, MP bootstrap analyses were performed using 1,000-replicate heuristic search.
Results and Discussion
Characterization of the mitochondrial genomes of M. incognita, M. floridensis, and M. hapla
The mtDNA of M. incognita assembled as a circular molecule of 17,985–18,332 bp, depending on the number of 63-bp repeats (shown linearized in Fig. 1). The incomplete mitochondrial genomes of Meloidogyne hapla and M. floridensis assembled as single molecules of 17,355 bp and 15,811 bp respectively (Fig. 1, Table 1). The total length of the latter genomes is unknown due to missing data in the long intergenic region between the genes nad4 and atp6 (Fig. 1). Complete mitochondrial genomes of three Meloidogyne spp. have been described with similar genome length (Table 1, Fig. 1). Also, the size of the mtDNA of M. javanica (20.5 kb) has been reported [39]. A comparison to other animal mtDNAs revealed that Meloidogyne mtDNAs fall within the size range of Metazoa (8 kb to 48 kb)[6,40].
Fig 1. Linear maps of the mitochondrial genomes of five Meloidogyne species.
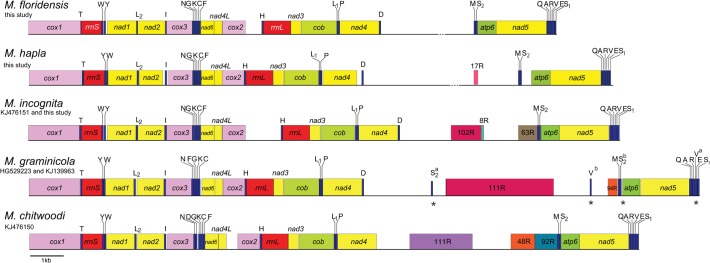
The maps were drawn to scale. Dots interrupting the main genomic line indicate unknown sequences. All genes are encoded on the same strand of the mtDNA. Protein and rRNA genes have standard nomenclature. tRNA genes are designated by single-letter abbreviation. Two tRNAs for Leucine (L) and Serine (S) are present: L1 (anticodon UAG), L2 (UAA), S1 (UCN) and S2 (AGN). Sa, Va, Sb, and Vb correspond to tRNA genes annotated differently in two previous studies (Sa and Va in GenBank KJ139963 [23]; Sb and Vb in GenBank HG529223 [21]). Tandem repeats are represented by boxes labeled by the repeat length in kb followed by “R”.
Table 1. Comparisons between sequenced mitochondrial genomes of the genus Meloidogyne.
| M. floridensis (isolate 5) | M. hapla (isolate VW9) | M. incognita (isolate Morelos) | M. incognita (isolate NCMI4) | M. chitwoodi (isolate CAMC2) | M. graminicola (isolate from Batangas) | M. graminicola (isolate from Hainan) | |
|---|---|---|---|---|---|---|---|
| Genome size | >15,811 bp | >17,355 bp | 17,985–18,332 bp b | 17,662–19,100 bp | 18,201 bp | 20,030 bp | 19,589 bp |
| AT content (%) | 83.5% | 81.4% | 83.3% | 83.0% | 85.0% | 84.3% | 83.5% |
| Protein-coding sequence length (%) | nd c | nd c | 53–54% | 51–54% | 49.4% | 49.0% | 50.6% |
| Repeats a | no repeats found | 17 bp (7.6) | 102 bp (12.8) | 102 bp (19–24) | 111 bp (16.9) | 111 bp (29) | 111 bp (25) |
| 8 bp (9.4) | 8 bp (10.4) | 48 bp (15.6) | 94 bp (3) | 94 bp (3) | |||
| 63 bp (7.9–13.4) | 63 bp (9) | 92 bp (8.1) | |||||
| References | this study | [15] | [14] | [16] | |||
a Numbers in parentheses indicate the copy-number of each repeat.
b The genome size depends on the number of copies of the R63 repeat, which varies from 7 to 13 in the genomic assembly.
c Not determined because the total genome length is unknown.
The mitochondrial genomes of the five Meloidogyne spp. available, M. hapla, M. incognita, M. graminicola, M. chitwoodi and M. floridensis, shared several features (Fig. 1). They contained 12 protein-coding genes (atp8 was missing), 2 rRNA-coding genes (rrnS and rrnL), and 22 tRNA-coding genes (Fig. 1). Loss of the gene atp8 has been related to the parasitic lifestyle in other organisms [41]; however, both free-living and parasitic chromadorean nematodes lack this gene [3].
We identified 22 discrete sequences (ranging from 49 to 59 bp) predicted to fold into secondary structures of tRNAs in the mtDNAs of M. floridensis, M. hapla, and M. incognita, respectively. In each of the species, 20 tRNAs had the predicted T-arm-lacking structure [5] and two putative tRNA genes folded into different secondary structures, such as the genes tRNA-S1(UCN) and tRNA-S2(AGN) that had a TyC arm and loop but not the DHU arm.
All genes in Meloidogyne mtDNAs were encoded on the same strand, with very few and often short intergenic sequences (Fig. 1; S3 Table). This asymmetry in transcriptional architecture is typical of chromadoreans, while enopleans and most animal species have circular mitochondrial genomes with coding genes on both strands [42,43]. The nucleotide composition of Meloidogyne spp. mtDNAs showed a large bias towards AT (Table 1), similar to that of their nuclear genomes [24]. In general, mitochondrial genomes of the phylum Nematoda showed a trend for AT richness, ranging from 66% to 85%. In addition, a bias towards T and G content in the coding strand was observed in the mtDNAs of M. floridensis, M. hapla and M. incognita, as reported for other nematodes [23,44,45,46].
Start and stop codons of protein-coding genes in Meloidogyne mtDNAs were often non-universal. Putative start codons for the 12 mitochondrial protein-coding genes in M. hapla, M. incognita and M. floridensis were ATA, ATG, ATT, TTA, TTG; stop codons were TAA and TAG (S3 Table). Some of these start codons are non-universal and have been previously reported for nematode mitochondrial genomes [21,22,23,47,48,49]. In addition, the termination codon TGA encoded tryptophan, among other deviations from the universal genetic code in nematode mitochondria [50].
Phylogeny of Nematoda based on mitochondrial sequences
We performed a Maximum Likelihood phylogenetic analysis based on nucleotide sequences of eight mitochondrial protein-coding genes (4,920 bp) from 43 species of the phylum Nematoda, including 5 Meloidogyne spp. and 8 taxa of the class Enoplea as outgroups (S1 Fig.). Mitochondrial genes could resolve most phylogenetic relationships within the phylum Nematoda and these were consistent with previous studies based on mitochondrial [7,8,22] and nuclear genes [51,52]. Meloidogyne spp. grouped together as sister to Pratylenchus vulnus within the infraorder Tylenchomorpha with high bootstrap support (S1 Fig.). Within the clade of Meloidogyne, M. incognita + M. floridensis were sister to M. hapla with great bootstrap support (BS = 100%), and these were sister to M. chitwoodi + M. graminicola. These findings agree with previous studies based on the nuclear gene 18S rRNA [53].
Gene order in nematode mitochondrial genomes
Protein and rRNA gene order was identical within the genus Meloidogyne (Fig. 1), but different to other members of the infraorder Tylenchomorpha and any other nematode (S1 Fig.). Furthermore, the location of the tRNA genes was unique for four species of Meloidogyne, except for M. incognita and M. floridensis that were identical in gene order (Fig. 1). Differences occurred mainly in two blocks of tRNAs; those located between rrnS and nad1 and the group found between the genes cox3 and nad6.
Identification of tRNA genes is controversial in nematode mitochondrial genomes given that tRNAs are short, divergent and have atypical structures [54]. As a result, we noticed discrepancies in the prediction of tRNA genes in published Meloidogyne mtDNAs. For example, there was no consensus between the tRNA annotations of the highly similar (see below) mitochondrial genomes of two strains of M. graminicola (asterisks in Fig. 1) using two different tRNAs prediction programs [21,23].
Nematode mtDNAs, including those of root-knot nematodes, showed mitochondrial evolutionary dynamics that differ from most metazoan [55]. For example, mitochondrial genomic rearrangements within the phylum Nematoda were significantly higher than those of other animal groups [3]. A previous study found 25 different gene rearrangements (GA) in 62 mitochondrial genomes of nematodes, considering the position of tRNAs; of them, the most common was GA3, which was shared by 32 nematode species [8]. Within the infraorder Tylenchomorpha, several mtDNAs have recently become available. Heterodera glycines and Pratylenchus vulnus [7,46] showed two different gene arrangements to those already described (indicated as GA26 and GA27 in S1 Fig.). The mtDNAs of Meloidogyne spp. added four novel gene rearrangements to the list of described GA in Nematoda (GA28-GA31; S1 Fig.). The genus Meloidogyne (and the infraorder Tylenchomorpha as a whole) are particularly variable in gene order in comparison to other lineages of Nematoda, along with the highly re-arranged enopleans [43,56], and the genus Onchocerca [49,57].
Non-coding regions in Meloidogyne mtDNAs
The mtDNA of M. hapla was highly compact and presented 3 intergenic regions larger than 100 bp (Fig. 1): nad4-trnD (144 bp), trnS-atp6 (290 bp), and trnD-trnM (>4,596 bp, the major non-coding region). In the latter, a tandem repeat of 17 bp (17R) was found (Table 1). Previously, three tandem repeats of 102, 8 and 63 bp were reported for M. hapla [39], but subsequent reports [19] and this study did not find such sequences in M. hapla.
The mtDNA of M. floridensis had only 2 intergenic regions >100 bp: cox2-trnH (535 bp) and trnD-trnM (>2,774 bp) and no tandem repeats were identified (Fig. 1). M. incognita mtDNA also had 2 intergenic regions in the same locations as in M. floridensis: cox2-trnH (1,063 bp) and trnD-trnM (4,420–5,910 bp, depending on the number of repeats). Within the latter, 102-bp (102R) and 63-bp (63R) tandem repeats were found in M. incognita (Fig. 1). The 63R had also been reported for M. arenaria, M. javanica [39], and M. enterolobii [19]. Different copy-numbers of the 63R were described for a single individual (heteroplasmy) or among populations of the same species for M. javanica, M. arenaria and M. incognita [19,39]. The 63R region has been explored as a molecular marker but the results were difficult to interpret and its use was questionable [19,20].
The cox2-rrnS region has been largely studied and used in molecular systematics [17,18,19,58,59] because it has indels and polymorphisms useful to differentiate species of the genus Meloidogyne. Until recently, it was possible to identify the major species of Meloidogyne by PCR amplification and digestion of this fragment, or by DNA sequencing of this region. Lately, the analysis of the cox2-rrnS in additional Meloidogyne species uncovered limitations in the taxonomic use of this marker (see below).
The largest intergenic region in all Meloidogyne spp. mtDNAs was found between the protein-coding genes nad4 and atp6 and included 2–5 tRNA-coding genes (Fig. 1). This region also contained tandem repeats in most species, but the repeats were not homologous with a few exceptions (Fig. 1). M. graminicola had a 111-bp (111R) tandem repeat that was similar to the 102-bp (102R) tandem repeat of M. incognita, but different to the 111-bp (111R) tandem repeat of M. chitwoodi. Also, M. chitwoodi had a 48-bp (48R) tandem repeat that aligned with the 94-bp (94R) repeat of M. graminicola (Fig. 1).
Variability of mitochondrial genomes within a single Meloidogyne species
The mtDNAs of 2 strains of M. incognita (this study and [15]) and 2 strains of M. graminicola [21,23] are now available for intraspecific comparisons. Variation in the copy-number of tandem repeats and the presence of indels in intergenic regions are responsible for the differences in genome length within each species (Table 1). A pairwise comparison between the mtDNA of the 2 strains of M. incognita and M. graminicola, respectively, indicated that they shared the same gene content and order (not considering the discrepancy in tRNA annotation of the M. graminicola mtDNAs shown in Fig. 1 given that the putative tRNAs were present in both strains).
Mitochondrial sequences between the strains of M. incognita were extremely similar showing an identity of 99.2% (excluding gaps) with only a few substitutions and indels across coding and non-coding regions (S2-A Fig.). A pairwise comparison between the mtDNAs of both strains of M. graminicola showed that they were almost identical (99.9% identity, excluding gaps) with very few polymorphisms in the coding regions and most substitutions clustered within the trnD-trnV non-coding region (S2-B Fig.). The closer geographic distance between the M. graminicola strains (China and Philippines) in comparison to the two isolates of M. incognita (Mexico and France) could relate to the higher mtDNA similarity observed within M. graminicola. It is also possible that the mitochondrial sequence of M. incognita strain Morelos were contaminated with NUMTs, showing more substitutions than the actual mtDNA residing in the mitochondria [60,61].
Variability of mitochondrial genomes among Meloidogyne spp.
The mtDNA of M. floridensis was compared to that of other Meloidogyne spp. with the program VISTA (Fig. 2). M. floridensis and M. incognita mtDNAs were strikingly similar across the genome with an identity of 97–100% for all coding regions. The genes cox3, nad6 and nad3, along with 19 tRNAs, were identical in both species while the other genes showed 1–7 substitutions/gene. This result is in agreement with recent evidence showing that M. incognita is a double hybrid of M. floridensis and another species [24]. Comparisons to M. hapla, M. graminicola and M. chitwoodi revealed the presence of conserved genic regions and highly divergent (including some non-homologous) intergenic sequences (Fig. 2). The same is true for pairwise comparisons between the other Meloidogyne species.
Fig 2. Pairwise comparisons between the mitochondrial genome of M. floridensis (this study) and that of M. incognita (KJ47615), M. hapla (this study), M. chitwoodi (KJ476150) and M. graminicola (KJ139963) using the program VISTA.

The Y-axis represents the sequence identity (50–100%). Protein (in violet) and rRNA (in yellow) genes are indicated in the standard nomenclature. tRNA genes (in red) are designated by a single-letter abbreviation. The range of identity (%) of protein-coding genes among Meloidogyne spp. is shown at the top.
The alignments of each mitochondrial gene from M. floridensis, M. incognita, M. hapla, M. graminicola and M. chitwoodi showed >65% nucleotide identity for most coding regions (Fig. 2; S3 Table). Overall comparisons within the genus Meloidogyne indicated that the most conserved protein-coding gene was cox1 (81–99% identity; Fig. 2). Genes encoding rRNAs had an identity of 78–99% (rrnS) and 76–99% (rrnL). In contrast, tRNA genes had a wider range of identity (47–100%) within the genus, being trnS and trnV the most divergent tRNAs (S3 Table).
Analysis of mitochondrial regions for taxonomic use
Variable intergenic mitochondrial sequences are potentially useful for phylogenetic studies and species identification. Based on mitochondrial genomic comparisons (i.e. Fig. 2), we identified a highly variable ~2 kb mitochondrial segment starting on the gene nad5 through the middle of the gene cox1, including 6 tRNAs and intergenic regions. We selected this region because it showed interspecific variability and it was easy to amplify by PCR from a wide range of species given the presence of highly conserved flanking sequences. The nad5-cox1 region was sequenced and analyzed in 36 populations of 8 Meloidogyne spp. (S2 Table). The overall topologies of the ML and MP phylogenetic trees were almost identical (Fig. 3).
Fig 3. Maximum Likelihood phylogenetic tree of Meloidogyne spp. based on the nad5-cox1 mitochondrial region.

The tree was rooted according to Holterman et al. [53]. Numbers above and below branches represent ML and MP bootstrap support values >50%, respectively. Species names in red and green indicate mitotic and meiotic parthenogenetic species, respectively.
Isolates of each species formed monophyletic groups with high bootstrap support (95–100%), except for populations of M. arenaria, M. incognita and M. floridensis that grouped in a single clade but were not separated by species (Fig. 3). The few polymorphisms observed in this heterogeneous group fell within the range of intraspecific variability. Within each of the monophyletic species in the tree, sequences were highly conserved with very few or no polymorphisms. This limited intraspecific variability (Fig. 3) has been reported for parthenogenetic nematodes [17,62] and it is now extended to sexual species (Fig. 3). The mitochondrial genomic comparisons between the 2 strains of M. incognita and M. graminicola (S2 Fig.) also agreed with this statement. Phylogenetic relationships observed among the sampled species (Fig. 3) were congruent with previous phylogenies based on the nuclear 18S rRNA [63] or the mitochondrial region cox2-rrnS.
Besides the mitochondrial marker nad5-cox1, a single mitochondrial region, cox2-rrnS, has been extensively analyzed within the genus Meloidogyne [17,18,19,20]. Phylogenetic trees based on this marker had good discriminating power but relationships among several species of root-knot nematodes were unresolved and several isolates could not be identified at the species level [58,64,65,66]. A phylogeny including all cox2-rrnS sequences available in public databases for Meloidogyne species (S3 Fig.) showed that M. graminis, M. marylandi, M. hapla, M. partityla, M. haplanaria, M. enterolobii, M. naasi and M. graminicola could be separated based on a number of shared substitutions that took place since each species diverged. In contrast, some species had highly similar cox2-rrnS sequences and could not be differentiated. For example, M. fallax was paraphyletic with respect to M. chitwoodi with almost identical sequences (S3 Fig.). The greatest unresolved clade included the major and most economically damaging Meloidogyne spp, along with other minor root-knot nematodes (S3 Fig.). These 10 species formed a monophyletic clade with highly similar cox2-rrnS sequences and indels shared by different taxa, which prevented a clear distinction of species based on this marker.
These and previous studies based on nuclear or mitochondrial data [53,58,63,67] found that mitotic parthenogenetic Meloidogyne species (shown in red in Fig. 3 and S3 Fig.) were evolutionary distinct from the meiotic parthenogenetic ones (shown in green in Fig. 3 and S3 Fig.), with a few exceptions such as M. floridensis that is meiotic parthenogenetic and clusters within the first group (Fig. 3; S3 Fig.).
In addition to the results found with both mitochondrial markers, other studies also indicated that mitotic parthenogenetic species and M. floridensis are hard to distinguish using the nuclear regions ITS and 18S rDNA [33,53,65,68] and occasionally showed contradictory results between nuclear and mitochondrial phylogenies [17,58]. Species boundaries, especially for mitotic parthenogenetic nematodes, have been discussed [17] and interspecific hybridizations prior to the recent establishment of parthenogenesis were proposed [17,69]. A comparative analysis of gene families based on the complete nuclear genomes of M. hapla [25], M. incognita [26] and M. floridensis [24] suggested that M. incognita is a double-hybrid between M. floridensis and an unidentified species [24].
It remains challenging to distinguish most mitotic parthenogenetic Meloidogyne isolates and to identify the species, if they, indeed, belong to different phylogenetic species. Extensive studies that include a wide range of populations and species of Meloidogyne while comparing mitochondrial and nuclear sequences are needed to elucidate the complex evolution of this group. Single copy nuclear genes may aid in the identification of mitotic parthenogenetic species.
Supporting Information
Numbers above branches represent bootstrap support values >50%. The mtDNA gene arrangement (GA) according to Liu et al. [8] is shown next to each species name.
(PDF)
The Y-axis represents the sequence identity (50–100%). Protein (in violet) and rRNA (in yellow) genes are indicated in the standard nomenclature. tRNA genes (in red) are designated by a single-letter abbreviation.
(PDF)
Numbers above branches represent ML bootstrap values >50%. Species names in red, green or black indicate species with mitotic parthenogenesis, meiotic parthenogenesis or unknown reproductive strategy, respectively.
(PDF)
(PDF)
(PDF)
(PDF)
Acknowledgments
We would like to thank A. Skantar for sharing nematode DNA samples. M.V.S.P. is a researcher of CONICET.
Data Availability
Sequences were deposited in GenBank (accession numbers KM491188-KM491216). Alignments and trees have been deposited in Treebase (study S17102).
Funding Statement
This work was supported by Agencia Nacional de Promoción Científica y Tecnológica, www.agencia.mincyt.gob.ar, PICT-2008-277 to MVSP and by Universidad Nacional de Cuyo, www.uncu.edu.ar, Sectyp 06/M053 to MVSP. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Koonin EV. The origin and early evolution of eukaryotes in the light of phylogenomics. Genome Biol. 2010; 11: 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Boore JL. Animal mitochondrial genomes. Nucleic Acids Res. 1999; 27: 1767–1780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Hu M, Gasser RB. Mitochondrial genomes of parasitic nematodes-progress and perspectives. Trends parasitol. 2006; 22: 78–84. [DOI] [PubMed] [Google Scholar]
- 4. Hoffmann RJ, Boore JL, Brown WM. A novel mitochondrial genome organization for the blue mussel, Mytilus edulis. Genetics. 1992; 131: 397–412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Wolstenholme DR. Animal mitochondrial DNA estructure and evolution. Int Rev Cytol. 1992; 141: 173–216. [DOI] [PubMed] [Google Scholar]
- 6. Lavrov D. Mitochondrial Genomes in Invertebrate Animals In: Bell E, editor. Molecular Life Sciences: Springer New York; 2014. pp. 1–8. [Google Scholar]
- 7. Sultana T, Kim J, Lee SH, Han H, Kim S, Min GS, et al. Comparative analysis of complete mitochondrial genome sequences confirms independent origins of plant-parasitic nematodes. BMC Evol Biol. 2013; 13: 1471–2148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Liu HG, Shao R, Li JY, Zhou DH, Li H, Zhu XQ. The complete mitochondrial genomes of three parasitic nematodes of birds: a unique gene order and insights into nematode phylogeny. BMC Genomics. 2013; 14: 414 10.1186/1471-2164-14-414 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Azevedo JL, Hyman BC. Molecular characterization of lengthy mitochondrial DNA duplications from the parasitic nematode Romanomermis culicivorax. Genetics. 1993; 133: 933–942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. De Ley P, Blaxter ML. A new system for Nematoda: combining morphological characters with molecular trees, and translating clades into ranks and taxa. Nematol Monogr Persp. 2004; 2: 633–653. [Google Scholar]
- 11. Karssen G. The plant parasitic nematode genus Meloidogyne Goldi, 1892 (Tylenchida) in Europe Leiden, Netherlands: Brill; 2002. [Google Scholar]
- 12. Jones JT, Haegeman A, Danchin EG, Gaur HS, Helder J, Jones MG, et al. Top 10 plant-parasitic nematodes in molecular plant pathology. Mol Plant Pathol. 2013; 14: 946–961. 10.1111/mpp.12057 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Abad P, Favery B, Rosso MN, Castagnone-Sereno P. Root-knot nematode parasitism and host response: molecular basis of a sophisticated interaction. Mol Plant Pathol. 2003; 4: 217–224. 10.1046/j.1364-3703.2003.00170.x [DOI] [PubMed] [Google Scholar]
- 14. Sasser JN, Eisenback JD, Carter CC, Triantaphyllou AC. The international Meloidogyne project—its goals and accomplishments. Ann Rev Phytopathol. 1983; 21: 271–288. [Google Scholar]
- 15. Hunt D, Handoo Z. Taxonomy, identification and principal species In: Perry RN, Moens M, Starr JL, editors. Root- knot nematodes. CABI, Wallingford; 2009. pp. 55–97. [Google Scholar]
- 16. Carneiro R, Correa V, Almeida MR, Gomes AC, Mohammad Deimi A, Castagnone-Sereno P, et al. Meloidogyne luci n. sp. (Nematoda: Meloidogynidae), a root-knot nematode parasitising different crops in Brazil, Chile and Iran. Nematology. 2014; 16: 289–301. [Google Scholar]
- 17. Fargette M, Berthier K, Richaud M, Lollier V, Franck P, Hernandez A, et al. Crosses prior to parthenogenesis explain the current genetic diversity of tropical plant parasitic Meloidogyne species (Nematoda: Tylenchida). Infect Genet Evol. 2010; 10: 807–814. 10.1016/j.meegid.2009.04.013 [DOI] [PubMed] [Google Scholar]
- 18. Powers TO, Harris TS. A polymerase chain reaction method for identification of five major Meloidogyne species. J Nematol. 1993; 25: 1–6. [PMC free article] [PubMed] [Google Scholar]
- 19. Blok VC, Wishart J, Fargette M, Berthier K, Phillips M. Mitochondrial DNA differences distinguishing Meloidogyne mayaguensis from the major species of tropical root-knot nematodes. Nematology. 2002; 4: 773–781. [Google Scholar]
- 20. Lunt DH, Whipple LE, Hyman BC. Mitochondrial DNA variable number tandem repeats (VNTR): utility and problems in molecular ecology. Mol Ecol. 1998; 7: 1441–1455. [DOI] [PubMed] [Google Scholar]
- 21. Besnard G, Juhling F, Chapius E, Zedane L, Lhuillier E, Mateille T, et al. Fast assembly of the mitochondrial genome of a plant parastic nematode (Meloidogyne graminicola) using next generation sequencing. C R Biol. 2014; 337: 295–301. 10.1016/j.crvi.2014.03.003 [DOI] [PubMed] [Google Scholar]
- 22. Humphreys- Pereira DA, Elling A. Mitochondrial genomes of Meloidogyne chitwoodi and Meloidogyne incognita (Nematoda: Tylenchina): Comparative analysis, gene order and phylogenetics relationship with other nematodes. Mol Biochem Parasit. 2014; 194: 20–32. 10.1016/j.molbiopara.2014.04.003 [DOI] [PubMed] [Google Scholar]
- 23. Sun L, Zhuo K, Lin B, Wang H, Liao J. The complete mitochondrial genome of Meloidogyne graminicola (Tylenchina): a unique gene arrangement and its phylogenetics implications. Plos one. 2014; 9: e98558 10.1371/journal.pone.0098558 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Lunt DH, Kumar S, Koutsovoulos G, Blaxter ML. The complex hybrid origins of the root knot nematodes revealed through comparative genomics. PeerJ. 2014; 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Opperman C, Birda D, Williamson V, Rokhsare D, Burkea M, Cohna J, et al. Sequence and genetic map of Meloidogyne hapla: a compact nematode genome for plant parasitism. Proc Natl Acad Sci. 2008; 105: 14802–14807. 10.1073/pnas.0805946105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Abad P, Gouzy J, Aury JM, Castagnone-Sereno P, Danchin E, Deleury E, et al. Genome sequence of the metazoan plant—parasitic nematode Meloidogyne incognita . Nat Biotechnol. 2008; 26: 909–915. 10.1038/nbt.1482 [DOI] [PubMed] [Google Scholar]
- 27. Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. Basic local alignment search tool. J Mol Biol. 1990; 215: 403–410. [DOI] [PubMed] [Google Scholar]
- 28. Schattner P, Brooks AN, Lowe TM. The tRNAscan-SE, snoscan and snoGPS web servers for the detection of tRNAs and snoRNAs. Nucleic Acids Research. 2005; 33: W686–W689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Tamura K, Stecher G, Peterson D, Filipski A, Kumar S. MEGA6: Molecular Evolutionary Genetics Analysis version 6.0 Mol Biol Evol. 2013; 30 2725–2729. 10.1093/molbev/mst197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Stothard P. The sequence Manipulation Suite: JavaScript programs for analyzing and formating protein and DNA sequences. Biotechniques. 2000; 28: 1102–1104. [DOI] [PubMed] [Google Scholar]
- 31. Frazer KA, Pachter L, Poliakov A, Rubin EM, Dubchak I. VISTA: Computational tool for comparative genomics. Nucleid Acid Res. 2004; 1: 273–279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Benson G. Tandem repeat finder: a program to analyze DNA sequences. Nucleic Acid Res. 1999; 27: 573–580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. García LE, Sanchez-Puerta MV. Characterization of a root-knot nematode population of Meloidogyne arenaria from Tupungato (Mendoza, Argentina). J Nematol. 2012; 44: 291–301. [PMC free article] [PubMed] [Google Scholar]
- 34. Gadberry MD, Malcomber ST, Doust AN, Kellog EA. Primaclade-a flexible tool for find conserved PCR primers across multiple species. Bioinformatics. 2005; 21: 1263–1264. [DOI] [PubMed] [Google Scholar]
- 35. Madison DR, Madison WP. Macclade 4: Analysis of Phylogeny and Character solution. Sinauer Associates, Sunderland, M A: 2000. [Google Scholar]
- 36. Zwickl DJ. Genetic algorithm approaches for the phylogenetic analysis of large biological sequence dataset under the maximun likelihood criterion University of Texas, Austin: 2006. [Google Scholar]
- 37. Posada D, Crandall KA. ModelTest. Testing the modal of DNA substitution. Bioinformatics. 1998; 14: 817–818. [DOI] [PubMed] [Google Scholar]
- 38.Swofford DL (1998) PAUP*. Phylogenetic Analysis Using Parsimony (*and Other Methods). In: Associates S, editor. Suderland, Massachusetts.
- 39. Okimoto R, Chamberline HM, Macfarlane JL, Wolstenholme DR. Repeat sequences sets in mitochondrial DNA molecules of root knot nematodes (Meloidogyne) nucleotide sequences, genome locations and potencial for host race identification Nucleic Acid Res. 1991; 19: 1619–1626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Cameron SL, Yoshizawa K, Mizukoshi A, Whiting MF, Johnson KP. Mitochondrial genome deletions and minicircles are common in lice (Insecta: Phthiraptera). BMC Genomics. 2011; 12: 1471–2164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Hancock L, Goff L, Lane C. Red algae lose key mitochondrial genes in response to becoming parasitic. Genome Biol Evol. 2010; 2: 897–910. 10.1093/gbe/evq075 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Bernt M, Bleidorn C, Braband A, Dambach J, Donath A, Fritzch G, et al. A comprehensive analysis of bilaterian mitochondrial genomes and phylogeny. Mol Phylogenet Evol. 2013; 69: 352–354. 10.1016/j.ympev.2013.05.002 [DOI] [PubMed] [Google Scholar]
- 43. Hyman BC, Lewis S, Tang S, Wu Z. Rampant gene rearrangement and haplotype hypervariation among nematode mitochondrial genomes. Genetica. 2011; 139: 611–615. 10.1007/s10709-010-9531-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Jacob JE, Vanholme B, Van Leeuwen T, Gheysen G. A unique genetic code change in the mitochondrial genome of the parasitic nematode Radopholus similis BMC Res Notes. 2009. [DOI] [PMC free article] [PubMed]
- 45. Hu M, Chilton NB, Gasser RB. The mitochondrial genome of Strongyloides stercoralis (Nematoda)—idiosyncratic gene order and evolutionary implications. Int J Parasitol. 2003; 33: 1393–1408. [DOI] [PubMed] [Google Scholar]
- 46. Gibson T, Farrugia D, Barret J, Chitwood DJ, Rowe J, Subottin S, et al. The mitochondrial genome of the soybean cyst nematode, Heterodera glycines . Genome Biol. 2010; 54: 565–564. [DOI] [PubMed] [Google Scholar]
- 47.Li MW, Lin RQ, Song H, Q., Wu XY, Zhu Q, X. The complete mitochondrial genomes of three Toxocara species of human and animal health significance. BMC Genomics. 2008: e224. [DOI] [PMC free article] [PubMed]
- 48. Kim K-H, Eom KS, Park J-K. The complete mitochondrial genome of Anisakis simplex (Ascaridida: Nematoda) and phylogenetic implications. Int J Parasitol. 2006; 36: 319–328. [DOI] [PubMed] [Google Scholar]
- 49. McNulty SN, Mullin AS, Vaughan JA, Tkach VV, Weil GJ, Fischer PU. Comparing the mitochondrial genomes of Wolbachia-dependent and independent filarial nematode species. BMC Genomics. 2012; 13: 1471–2164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Watanabe K. Unique features of animal mitochondrial translation system. Prc Jpn Acad Ser B Phys Biol Sci. 2010; 86: 11–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Blaxter M, De Ley P, Garey JR, Liu LX, Scheldeman P, Vierstraete A, et al. A molecular evolutionary framework for the Phylum Nematoda. Nature. 1998; 392: 71–75. [DOI] [PubMed] [Google Scholar]
- 52. Holterman M, van der Wurff A, van den Elsen S, van Megen H, Bongers T, Holovachov O, et al. Phylum-wide analysis of ssu rDNA reveals deep phylogenetic relationships among nematodes and accelerated evolution toward crown clades. Mol Biol Evol. 2006; 23: 1792–1800. [DOI] [PubMed] [Google Scholar]
- 53. Holterman M, Karssen G, van den Elsen S, van Megen H, Bakker J, Helder J. Small subunit rDNA- based phylogeny of the Tylenchida sheds light on relationship among some high-impact plant parasitic nematodes and the evolution of plant feeding. Phytopathology. 2009; 99: 227–235. 10.1094/PHYTO-99-3-0227 [DOI] [PubMed] [Google Scholar]
- 54. Wende S, Platzer EG, Juhling F, Putz J, Florentz C, Stadler PF, et al. Biological evidence for the world's smallest tRNAs. Biochimie. 2014; 100: 151–158. 10.1016/j.biochi.2013.07.034 [DOI] [PubMed] [Google Scholar]
- 55. Gissi C, Iannelli F, Pesole G. Evolution of the mitochondrial genome of Metazoa as exemplified by comparison of congeneric species. Heredity. 2008; 101: 301–320. 10.1038/hdy.2008.62 [DOI] [PubMed] [Google Scholar]
- 56. Powers TO, Harris TS, Hyman BC. Mitochondrial DNA Sequence Divergence among Meloidogyne incognita, Romanomermis culicivorax, Ascaris suum, and Caenorhabditis elegans . J Nematol. 1993; 25: 564–572. [PMC free article] [PubMed] [Google Scholar]
- 57. Keddie EM, Higazi T, Unnasch TR. The mitochondrial genome of Onchocerca volvulus: sequence, structure and phylogenetic analysis. Mol Biochem Parasitol. 1998; 95: 111–127. [DOI] [PubMed] [Google Scholar]
- 58. Tigano MS, Carneiro R, Jeyaprakash A, Dickson DW, Adams BJ. Phylogeny of Meloidogyne spp. based on 18s rDNA and the intergenic region of mitochondrial DNA sequences. Nematology. 2005; 7: 851–862. [Google Scholar]
- 59. Onkendi EM, Moleleki LN. Detection of Meloidogyne enterolobii in potatoes in South Africa and phylogenetic analysis based on intergenic region and the mitochondrial DNA sequences. Eu J Plant Pathol. 2013; 136: 1–5. [Google Scholar]
- 60. Hazkani-Covo E, Zeller RM, Martin W. Molecular poltergeists: mitochondrial DNA copies (numts) in sequenced nuclear genomes. PLoS Genet. 2010; 6: 1000834 10.1371/journal.pgen.1000834 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Parr RL, Maki J, Reguly B, Dakubo GD, Aguirre A, Wittock R, et al. The pseudo-mitochondrial genome influences mistakes in heteroplasmy interpretation. BMC Genomics. 2006; 7: 185 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Hugall A, Moritz C, Stanton J, Wolstenholme DR. Low but strongly structured mitochondrial DNA diversity in root knot nematodes (Meloidogyne). Genetics. 1994; 136: 903–912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Castagnone-Sereno P, Danchin EGJ. Parasitic success without sex—the nematode experience. J Evol Biol. 2014; in press. [DOI] [PubMed]
- 64. Skantar AM, Carta LK, Handoo ZA. Molecular and morphological characterization of an unusual Meloidogyne arenaria population from Traveler´s tree Ravenala madagascariensis . J Nematol. 2008; 40: 179–189. [PMC free article] [PubMed] [Google Scholar]
- 65. McClure MA, Nischwits C, Skantar AM, Schmitt ME, Subbotin SA. Root-Knot Nematodes in Golf course greens of the western United States. Plant Dis. 2012; 96: 635–347. [DOI] [PubMed] [Google Scholar]
- 66. Maleita CM, Simoes MJ, Egas C, Curtis RHC, de O. Abrantes I. Biometrical, biochemical and molecular diagnosis of portuguese Meloidogyne hispanica isolates. Plant Dis. 2012; 96: 865–874. [DOI] [PubMed] [Google Scholar]
- 67. Tandingan De Ley I, De Ley P, Vierstraete A, Karssen G, Moens M, Vanfleteren J. Phylogenetic analyses of Meloidogyne small subunit rDNA. J Nematol. 2002; 34: 319–327. [PMC free article] [PubMed] [Google Scholar]
- 68.Blok V, Powers T. Biochemical and molecular identification. In: Perry RN, Moens M, Starr J, editors. Root-knot nematodes. Lincoln; 2009. pp. 98–112.
- 69. Lunt DH. Genetic test of ancient asexuality in root knot nematodes reveal recent hybrid origins. BMC Evol Biol. 2008; 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Numbers above branches represent bootstrap support values >50%. The mtDNA gene arrangement (GA) according to Liu et al. [8] is shown next to each species name.
(PDF)
The Y-axis represents the sequence identity (50–100%). Protein (in violet) and rRNA (in yellow) genes are indicated in the standard nomenclature. tRNA genes (in red) are designated by a single-letter abbreviation.
(PDF)
Numbers above branches represent ML bootstrap values >50%. Species names in red, green or black indicate species with mitotic parthenogenesis, meiotic parthenogenesis or unknown reproductive strategy, respectively.
(PDF)
(PDF)
(PDF)
(PDF)
Data Availability Statement
Sequences were deposited in GenBank (accession numbers KM491188-KM491216). Alignments and trees have been deposited in Treebase (study S17102).