

Abstract
Acid challenge of the gastric mucosa is signalled to the brainstem. This study examined whether mild gastritis due to dextrane sulfate sodium (DSS) or iodoacetamide (IAA) enhances gastric acid-evoked input to the brainstem and whether this effect is related to gastric myeloperoxidase activity, gastric histology, gastric volume retention or cyclooxygenase stimulation. The stomach of conscious mice was challenged with NaCl (0.15 M) or HCl (0.15 and 0.25 M) administered via gastric gavage. Two hours later, activation of neurons in the nucleus tractus solitarii (NTS) was visualized by c-Fos immunocytochemistry. Gastritis was induced by DSS (molecular weight 8,000; 5 %) or IAA (0.1 %) added to the drinking water for 7 days. Relative to NaCl, intragastric HCl increased the number of c-Fos protein-expressing cells in the NTS. Pretreatment with DSS or IAA for 1 week did not alter the c-Fos response to NaCl but significantly enhanced the response to HCl by 54 and 74 %, respectively. Either pretreatment elevated gastric myeloperoxidase activity and induced histological injury of the mucosal surface. In addition, DSS caused dilatation of the gastric glands and damage to the parietal cells. HCl-induced gastric volume retention was not altered by IAA but attenuated by DSS pretreatment. Indomethacin (5 mg/kg) failed to significantly alter HCl-evoked expression of c-Fos in the NTS of control, DSS-pretreated and IAA-pretreated mice. We conclude that the gastritis-evoked increase in the gastric acid-evoked c-Fos expression in the NTS is related to disruption of the gastric mucosal barrier, mucosal inflammation, mucosal acid influx and enhanced activation of the afferent stomach – NTS axis.
Keywords: Gastric acid-induced expression of c-Fos, nucleus of the solitary tract, myeloperoxidase activity, superficial injury of the gastric mucosa, cyclooxygenase
Gastric hydrochloric acid (HCl) is essential for the digestion of food. Apart from its physiological function, HCl is also a potentially noxious stimulus if it penetrates the mucosal barrier. This dual role of HCl requires acid surveillance mechanisms in order to regulate its physiological functions and keep its noxious potential under control. We have previously reported that vagal afferent neurons make an important contribution to the acid-sensing mechanisms within the rat stomach (Schuligoi et al., 1998; Michl et al., 2001; Danzer et al., 2004). Intramucosal acidosis, induced by exposure of the gastric lumen to supraphysiological HCl concentrations that create a transmucosal H+ gradient high enough to drive H+ ions into the lamina propria, is signalled to the nucleus tractus solitarii (NTS) of the brainstem, the central projection area of vagal afferent neurons. In addition, intragastric (IG) administration of excess HCl elicits a visceromotor response indicative of pain, which is suppressed by vagotomy (Lamb et al., 2003).
There is experimental evidence that gastritis and gastric ulceration are associated with gastric hypersensitivity (Ozaki et al., 2002; Lamb et al., 2003). Iodoacetamide (IAA)-induced gastritis enhances the visceromotor response to gastric distension and HCl challenge (Ozaki et al., 2002; Lamb et al., 2003), and systemic administration of the proinflammatory interleukin-1β causes a prolonged increase in the medullary c-Fos response to gastric HCl challenge (Holzer et al., 2004). Gastric ulceration alters the kinetics of tetrodotoxin-resistant sodium currents as well as acid-induced currents in nodose ganglion neurons (Bielefeldt et al., 2002; Sugiura et al., 2005), and acute exposure of the oesophagus to HCl plus pepsin sensitizes brainstem neurons to oesophageal distension (Medda et al., 2005). The current study was undertaken to address the question as to how mucosal inflammation and injury enhance the responsiveness of the afferent stomach – NTS axis to gastric acid challenge. Afferent activity arriving in the brainstem was visualized by the induction of c-Fos, which is widely used to trace central neurons activated by excitatory input from the periphery (Hughes and Dragunow, 1995; Munglani and Hunt, 1995).
The major hypothesis pursued in this study was that mild gastritis enhances the acid sensitivity of the afferent stomach – NTS system via damage to the gastric mucosal surface, which is likely to facilitate the influx of excess luminal acid into the lamina propria. In testing this hypothesis, the first aim was to establish two models of mild gastritis induced by dextrane sulfate sodium (DSS) and IAA. While IAA added to the drinking water for several days is known to induce gastritis in rats and mice (Ozaki et al., 2002; Lamb et al., 2003; Piqueras et al., 2003; Larauche et al., 2004; Takeeda et al., 2004), DSS is primarily used to induce chronic colitis (Okayasu et al., 1990; Kitajima et al., 2000). We were able to demonstrate that, like IAA, DSS added to the drinking water for 1 week causes gastritis and enhances gastric HCl-evoked expression of c-Fos in the brainstem. The two gastritis models were used to clarify whether gastritis per se, independent of the underlying mechanisms, leads to enhanced acid responsiveness of the afferent stomach – NTS system.
The second aim was to characterize the DSS- and IAA-induced gastritis models in terms of gastric myeloperoxidase (MPO) activity, gastric histology and gastric volume retention. These experiments addressed the question whether the acid hyperresponsiveness of the afferent stomach – NTS system seen in DSS- and IAA-pretreated animals is related to distinct changes in biochemical, histological and functional indices of gastritis.
The third aim of this study was to explore whether cyclooxygenase products generated during DSS- and IAA-induced gastritis contribute to the hyperresponsiveness of the afferent stomach – NTS system to gastric acid challenge, given that both IAA (Barnett et al., 2000; Piqueras et al., 2003; Takeeda et al., 2004) and DSS (Kabashima et al., 2002; Singh et al., 2004) have been found to affect the prostaglandin system.
EXPERIMENTAL PROCEDURES
Experimental animals
This study was carried out with adult female mice of the outbred strain Him:OF1 (Division of Laboratory Animal Science and Genetics, Department of Biomedical Research, Medical University of Vienna, Himberg, Austria) weighing 25 - 35 g. The animals were housed in groups of 3 - 4 per cage under controlled temperature (21 °C) and a 12 h light/dark cycle (lights on at 6:00 AM, lights off at 6:00 PM). All experiments were approved by an Ethical Committee at the Federal Ministry of Education, Science and Culture of the Republic of Austria and conducted according to the Directive of the European Communities Council of 24 November 1986 (86/609/EEC). The experiments were designed in such a way that the number of animals used and their suffering was minimized.
Experimental gastritis
Mild gastritis was induced by adding either DSS or IAA to the drinking water (tap water) for a period of seven days. IAA (Sigma, Vienna, Austria) was added to the drinking water at a concentration of 0.1 % (w/v) (14,20,23), while DSS (molecular weight: 8,000; Sigma, Vienna, Austria) was administered at a concentration of 5 % (w/v) in the drinking water. The control animals received normal tap water. Since IAA is light-sensitive, the IAA-containing drinking water was made up fresh every day. For protocol consistency, the drinking bottles containing normal tap water and DSS-containing water were also renewed daily.
Experimental protocols
Ten specific studies were performed. Study 1 was carried out to establish that, relative to intake of normal tap water, pretreatment of mice with oral DSS (5 %) for 1 week affects the expression of c-Fos in the brainstem following IG administration of NaCl (0.15 M) or HCl (0.25 M). This test was made on the 8th day of DSS or control pretreatment, when the NaCl and HCl solutions were administered IG at a volume of 0.02 ml/g 2 h before collection of the brainstem. Study 2 examined whether, relative to intake of normal tap water, pretreatment of mice with oral DSS for 1 week alters the activity of MPO in the gastric wall and causes gastric mucosal damage at the macroscopical and histological level. To this end, the stomachs were analyzed on the 8th day of DSS or control pretreatment without any prior exposure to IG NaCl or HCl. IG pH was also determined in some experiments.
Study 3 was carried out to determine gastric fluid retention and gross gastric lesion formation following IG administration of HCl (0.25 M given 30 min before collection of the stomach) to mice that had drunk normal tap water or water containing DSS for 1 week. In study 4 it was investigated whether the DSS-induced enhancement of gastric HCl-evoked expression of c-Fos in the brainstem was maintained for 1 week after cessation of the pretreatment. For this purpose, the experimental mice drank tap water containing DSS for 1 week followed by normal tap water for another week; the control animals received normal tap water throughout the 2 week period. On the 15th day of the pretreatment protocol, the animals were exposed to IG HCl (0.25 M ) administered 2 h before collection of the brainstem for immunocytochemical visualization of c-Fos.
Study 5 was performed to examine the effect of indomethacin on the DSS-induced enhancement of gastric HCl-evoked expression of c-Fos in the brainstem. Mice who had drunk normal tap water (control animals) or water containing DSS for 1 week received an IP injection of indomethacin (5 mg/kg; Sigma, Vienna, Austria) or its vehicle (2 ml/kg; 0.15 M NaOH adjusted to pH 8.5 with 1 M HCl ) on the 8th pretreatment day. Thirty minutes later HCl (0.25 M) was administered IG; the brainstem was collected for determination of c-Fos expression 2 h post-HCl administration.
The experimental protocols of studies 6 - 10 were identical to those of studies 1 - 5, respectively, except that DSS was replaced by IAA (0.1 %).
c-Fos immunocytochemistry
c-Fos-like immunoreactivity was visualized as described previously (Wultsch et al., 2005). In brief, the mice were fasted for 16 h to ensure that the stomach was empty, but had free access to water, before HCl (0.15 or 0.25 M) or physiological saline (0.15 M NaCl) was administered IG at a volume of 0.02 ml/g through a soft infant feeding tube (outer diameter 1.5 mm; Rüsch, Montevideo, Uruguay). After the IG treatment the animals were no longer allowed to drink until tissue collection 2 h later. For this purpose, the mice were euthanized by intraperitoneal injection of an overdose of pentobarbital (100 mg/kg). Following euthanasia, the mice were transcardially perfused with 0.1 M phosphate-buffered saline (PBS) of pH 7.4 (25 ml), followed by 4 % buffered paraformaldehyde (25 ml). The brainstems were removed and postfixed overnight in 4 % buffered paraformaldehyde at 4 °C. Then the tissues were cryoprotected for 48 h in 20 % sucrose at 4 °C, frozen on dry ice and stored at −70 °C until use. Serial coronal sections of 40 μm thickness were cut from the brainstem over the whole length of the area postrema (AP) with a cryostat. The coordinates of the brainstem region under study were interaural −3.96 to −3.40 and bregma −7.76 to −7.20 according to Paxinos and Franklin (2001). Only every second section was used. Immunocytochemistry was performed with free-floating sections which first were washed once in 0.1 M PBS, then washed twice in washing buffer (WB; 0.1 M PBS with 0.01 % Triton X 100), and incubated in 0.3 % H2O2 for 30 min. After three further washes (each for 10 min in WB), the tissues were incubated for 1.5 h with a blocking serum (2.5 % goat serum) at room temperature and then with the primary antibody (rabbit polyclonal anti-c-Fos, 1:20,000, Santa Cruz Biotech, Santa Cruz, California, USA) for 40 h at 4 °C. Both the blocking serum and primary antibody were dissolved in 0.1 M PBS containing 0.3 % Triton X 100 and 1 % bovine serum albumin. Afterwards the sections were washed three times in WB and incubated for 1.5 h in a solution containing the biotinylated secondary antibody (goat anti-rabbit IgG, Vectastain Elite Kit, Vector Laboratories, Burlingame, California, USA). After three other washes in WB they were incubated for 1 h in avidin-biotin complex (Vectastain Elite Kit). The tissues were rinsed afterwards and developed with 3,3-diaminobenzidine (DAB) substrate (Vectastain Elite Kit) intensified with nickel sulfate for 135 s. Subsequently the sections were mounted on gelatin-covered slides, air-dried and dehydrated through an alcohol series (50 - 70 - 100 % ethanol followed by 100 % xylol). The slides were coverslipped with Entellan (Merck, Darmstadt, Germany). To control for the specificity of the anti-c-Fos antibody signal, a c-Fos blocking peptide (Santa Cruz Biotech) was added to the primary antibody dilution.
The immunocytochemically processed brainstem sections were examined with a light microscope (Axiophot, Zeiss, Oberkochen, Germany) coupled to a computerized image analysis system (MCID-M2, version 3.0, Rev 1.1, Imaging Research Inc., Brock University, St. Catharines, Ontario, Canada). The sections were coded such that the examiner did not know which treatment group they came from. As described previously (Wultsch et al., 200%), eight sections per animal were analyzed, and all c-Fos-positive cells were randomly counted on one side of the NTS and AP. In order to avoid that the same cells were counted twice, only every second section was taken for analysis. All counts in each section of each animal were averaged to give the number of c-Fos-positive cells in the NTS and AP of that animal. These average values from each animal were then used to calculate the mean number of c-Fos-positive cells per section in the unilateral NTS and AP of each experimental group.
Myeloperoxidase activity
The enzyme activity of MPO (donor:H2O2 oxidoreductase, EC 1.11.1.7) was determined according to previously described spectrophotometric techniques (Suzuki et al., 1983; Krawisz et al., 1984; Graff et al., 1998). At autopsy, full-thickness pieces of the gastric corpus (120-160 mg) were excised, shock-frozen in liquid nitrogen and stored at −70 °C until assay. After weighing, the frozen tissues were placed, at a ratio of 100 mg : 1 ml, in potassium phosphate buffer (0.05 M) of pH 7.4 and homogenized on ice with an Ultraturrax (IKA, Staufen, Germany). The homogenates were then subjected to 3 cycles of freezing on solid carbon dioxide and thawing, followed by centrifugation at 40,000 g and 4 °C for 20 min. The supernatants were discarded, while the pellets were taken up, at a ratio of 100 mg : 1 ml, in potassium phosphate buffer (0.05 M) of pH = 6.0 containing 5 g/l hexadecyltrimethylammonium bromide (HETAB, Sigma, Vienna, Austria) and 3.72 g/l ethylenediaminetetraacetic acid (Roth, Karlsruhe, Germany). HETAB is a detergent that releases MPO from the primary granules of the neutrophils (Krawisz et al., 1984). The suspensions were homogenized on ice with an Ultraturrax and immediately used for the assay of MPO.
The assay is based on the MPO-catalyzed oxidation of tetramethylbenzidine (Merck, Darmstadt, Germany) in the presence of H2O2 (Suzuki et al., 1983). To this end 0.1 ml aliquots of the homogenates were combined with 0.39 ml of potassium phosphate buffer (0.08 M) of pH = 5.4 containing HETAB (5 g/l) and with 0.05 ml tetramethylbenzidine solution (16 mM in dimethyl formamide). These assay samples were incubated for 5 min in a water bath at 37 °C. The spectrophotometric reaction was initiated by addition of 0.01 ml hydrogen peroxide solution (0.04 % in water) to the samples, 3 min later followed by the addition of 0.01 ml catalase (Fluka, Buchs, Switzerland) solution (13,300 units/ml in water) to remove excess hydrogen peroxide. After another 3 min the reaction was stopped by the addition of 2 ml ice-cold sodium acetate (0.2 M) of pH = 3.0. Immediately afterwards, the samples were placed on ice, mixed and centrifuged at 9,500 g and 4 °C for 3 min. Aliquots of the supernatants (1.0 ml) were transferred to cuvettes, and the absorbance of the samples at 655 nm was measured with a spectrophotometer (Spectronic Genesys 5; Milton Roy, Ivyland, PA, USA). Blank assay samples were run in the same way except that the homogenate aliquots were replaced by 0.1 ml aliquots of potassium phosphate buffer (0.08 M) of pH = 5.4 containing HETAB (5 g/l).
MPO activity was expressed relative to the MPO activity of human neutrophils. To this end, a sample of human neutrophils (5 million cells in 2 ml of 0.05 M potassium phosphate buffer of pH = 7.4) was extracted in an identical manner as the tissue samples and assayed as described above. A standard curve was constructed by measuring the absorbance of the reaction mixture with various dilutions of the neutrophil extract. Since the MPO activity in the gastric tissue samples was determined as an index of neutrophil infiltration, the MPO activity of the tissue extracts was expressed as human neutrophil equivalents/mg wet tissue.
Gastric fluid recovery, gastric lesion formation and intragastric pH
The mice were fasted for 16 h to ensure that the stomach was empty, but had free access to water, before HCl (0.25 M) was administered IG at a volume of 0.02 ml/g through a soft infant feeding tube as described above. After the IG treatment the animals were no longer allowed to drink until tissue collection 0.5 h later. For this purpose, the mice were euthanized by intraperitoneal injection of an overdose of pentobarbital (100 mg/kg). After euthanasia the abdomen of the animals was quickly opened by a midline incision, and the stomach clamped at the lower oesophageal sphincter and pylorus. Then the fluid-filled stomach was weighed, opened along the greater curvature, blotted dry on tissue paper and re-weighed to determine the volume of the fluid present in the stomach. Gastric fluid recovery was calculated by expressing the weight of the fluid present in the stomach as a percentage of the weight of the fluid administered into the stomach (Wultsch et al., 2005).
The integrity of the gastric mucosa was examined at the macroscopical, light and electron microscopical level. To quantify macroscopically visible damage, the stomach was pinned flat on a silicon elastomer-coated plate and covered with PBS. The stomach was photographed, the image transferred to a personal computer, and macroscopical gastric injury assessed with computerized planimetry by an observer who was unaware of the experimental treatment (Schuligoi et al., 1998). The mucosal area covered by visible hemorrhagic damage was expressed as a percentage of the total area of the glandular mucosa (Wultsch et al., 2005).
For histological examination, specimens of the gastric corpus were fixed in a medium containing 2 % paraformaldehyde, 2.5 % glutaraldehyde and 0.1 M cacodylate buffer of pH 7.4 and embedded in Technovit 7100 (Heraeus Kulzer, Wehrheim, Germany). Then 2.5 μm sections were cut and stained with a mixture of methylene blue - azure II and basic fuchsin (Schuligoi et al., 1998). The sections were taken randomly from the gastric corpus and included areas of hemorrhagic damage, if present. Histological injury was assessed in qualitative terms, special attention being drawn to the appearance of the surface epithelium. Some tissues were also processed for transmission electron microscopy. To this end, the tissues were postfixed for 2 h in 2 % OsO4 in 0.1 M cacodylate buffer at room temperature and then dehydrated in a series of ascending ethanol concentrations and propylene oxide (Pabst et al., 1996). Thereafter, the tissue specimens were embedded in Taab epoxy resin (Taab, Berkshire, UK) and blocks cut with a Leica Ultracut T microtome. Ultrathin sections were stained with uranyl acetate and lead citrate and examined on a Zeiss EM 902 electron microscope at 50 kV.
The pH of the gastric contents was determined with a pH meter that was fitted with a Micro Line pH electrode (ThermoOrion, New Hyde Park, NY, USA) and calibrated with standard buffers of pH 1, 4 and 7.
Statistics
Statistical evaluation of the results was performed on SPSS 11.5 (SPSS Inc., Chicago, IL, USA) with Student’s t-test (two treatments groups), one-way analysis of variance (ANOVA) or two-way ANOVA (more than two treatment groups), as appropriate, followed by the Tukey test. All data are presented as means ± SEM, n referring to the number of mice in each group. Probability values of P < 0.05 were regarded as significant.
RESULTS
Effect of IG administration of NaCl and HCl to induce c-Fos in the brainstem of mice pretreated with oral IAA or DSS
Pretreatment of mice with oral IAA (0.1 %) or DSS (5 %) for 1 week did not significantly enhance the expression of c-Fos in the NTS seen after IG administration of 0.15 M NaCl, as compared with the c-Fos expression occurring in mice that had drunk normal tap water (Figure 1). The expression of c-Fos in the AP observed after IG administration of 0.15 M NaCl also remained unaltered after 1 week pretreatment with IAA but was significantly enhanced after 1 week pretreatment with DSS (Figure 1).
Figure 1.
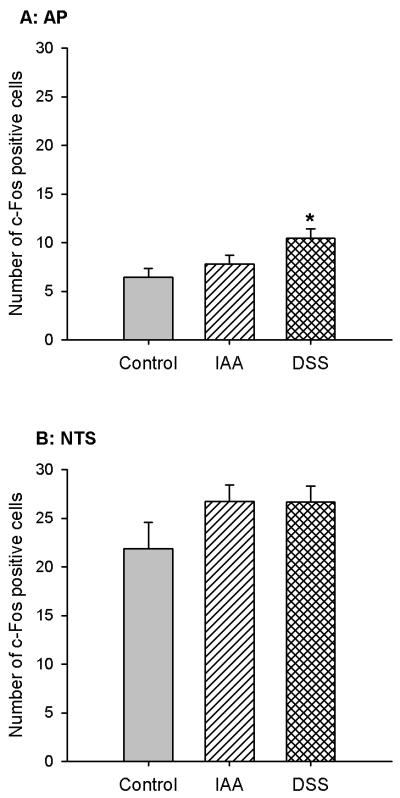
Expression of c-Fos in the AP and NTS of control mice and mice pretreated with IAA or DSS. The animals either drank normal tap water (control) or received water containing IAA (0.1 %) or DSS (5 %) for 1 week. On the 8th pretreatment day the animals were given IG NaCl (0. 15 M) 2 h before collection of the brainstem for immunocytochemical visualization of c-Fos. The values, given as means ± SEM (n = 7), reflect the number of c-Fos positive cells per section in the unilateral AP and NTS. * P < 0.05 versus control (one-way ANOVA followed by the Tukey test).
As reported previously (Wultsch et al., 2005), IG administration of HCl (0.15 and 0.25 M) caused a concentration-related increase in the expression of c-Fos in the NTS relative to that measured after IG administration of 0.15 M NaCl (Figures 2A,B and 3A,B). The largest number of c-Fos positive cells was encountered in the medial and subpostremal nuclei of the NTS (Figure 3B,C). Pretreatment of mice with IAA or DSS for 1 week significantly enhanced the expression of c-Fos in the NTS seen after IG administration of 0.15 M and 0.25 M HCl, as compared with the c-Fos expression determined in mice that had drunk normal tap water (Figures 2A,B,C and 3B,C). Although the effect of 0.25 M HCl to induce c-Fos in the AP also tended to be enhanced after 1 week pretreatment with IAA and DSS, this tendency did not reach statistical significance (Figure 2B,C).
Figure 2.

Gastric HCl-evoked expression of c-Fos in the AP and NTS of control mice and mice pretreated with IAA or DSS. The animals either drank normal tap water (controls) or received water containing IAA (0.1 %) or DSS (5 %) for 1 week. On the 8th pretreatment day the animals were given IG 0.15 M HCl (A) or 0.25 M HCl (B,C) 2 h before collection of the brainstem for immunocytochemical visualization of c-Fos. The values, given as means ± SEM (n as indicated), reflect the number of c-Fos positive cells per section in the unilateral AP and NTS. * P < 0.05, ** P < 0.01 versus control (Student’s t test).
Figure 3.

Photomicrographs showing expression of c-Fos in the AP and NTS taken from control mice (A,B) and mice pretreated with DSS (C). The animals either drank normal tap water (controls) or received water containing DSS (5 % ) for 1 week. On the 8th pretreatment day the animals were exposed IG to 0.15 M NaCl (A) or 0.25 M HCl (B,C) administered 2 h before collection of the brainstem for immunocytochemical visualization of c-Fos. HCl induced many cells in the medial and subpostremal nuclei of the NTS and some cells in the AP to express c-Fos. CC, central canal.
The specificity of c-Fos immunoreactivity was proved by the absence of any immunoreactive signal when the c-Fos blocking peptide had been added to the primary antibody dilution.
Effect of pretreatment with oral IAA or DSS on body weight, gastric myeloperoxidase activity and gastric mucosal integrity
Mice pretreated with oral IAA or DSS for 1 week appeared healthy and did not exhibit any overt sign of sickness. In addition, the body weight of mice pretreated with IAA did not significantly differ from that of mice that had drunk normal tap water for 1 week. Accordingly, the minor weight loss during the 1 week period of pretreatment with IAA (− 1.53 ± 0.52 g, n = 9) was not different from that seen in animals drinking normal tap water (− 1.90 ± 0.49 g, n = 9). Similar observations were made in mice pretreated with DSS in which only the total weight of the animals per cage was determined. The animals receiving DSS lost on average 1.68 g in weight during the 1 week pretreatment, compared with a loss of 1.90 g seen in control animals. In contrast, the MPO activity in the gastric wall was moderately, but significantly, enhanced after 1 week pretreatment with IAA or DSS, as compared with the MPO activity measured in the control mice (Figure 4).
Figure 4.
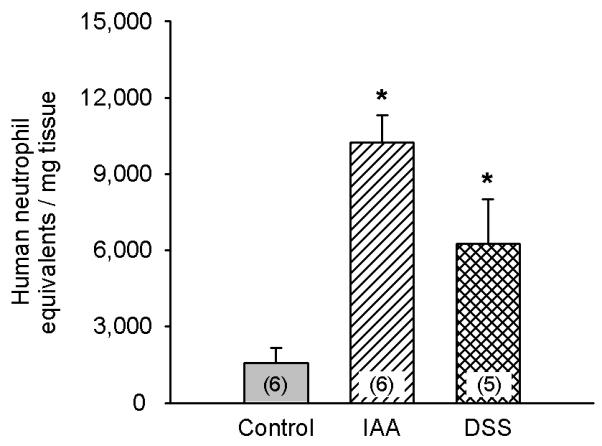
Myeloperoxidase (MPO) activity in the gastric corpus wall of mice that had drunk normal tap water (controls), water containing IAA (0.1 %) or water containing DSS (5 %) for 1 week. MPO activity in the stomach was determined on the 8th day of pretreatment and expressed as neutrophil equivalents per mg tissue. The values represent means ± SEM, n as indicated. * P < 0.05 versus control (one-way ANOVA followed by the Tukey test).
The macroscopical appearance of the gastric mucosa did not differ between mice that had been pretreated with IAA or DSS or which had drunk normal tap water. Apart from some petechiae, the gastric mucosa was considered macroscopically normal. Histological analysis revealed, however, that the stomachs taken from mice pretreated with IAA or DSS for 1 week (n = 5 in each group) exhibited more superficial damage of the gastric surface epithelium than stomachs taken from control mice (Figure 5A,B,C). While only 2 of the 5 control stomachs displayed extensive damage to the superficial mucosa, all 5 stomachs taken from IAA-pretreated mice exhibited considerable surface injury (Figure 5B) and two stomachs occasionally showed abnormalities that went deeper than 10 % of the mucosal height. In many instances the surface mucosa was covered by loose mucosal cells that were shed from the superficial epithelium (Figure 5B). Likewise, all 5 stomachs taken from DSS-pretreated mice exhibited damage of the surface mucosa which occasionally was covered by loose epithelial cells (Figure 5C). In addition, the gastric glands were conspicuously dilated in all specimens taken from DSS-pretreated mice, and many parietal cells in DSS-exposed stomachs exhibited vacuolization (Figure 5C).
Figure 5.

Light microscopical appearance of the gastric corpus mucosa taken from a control mouse (A), a mouse pretreated with IAA (B) and a mouse pretreated with DSS (C). The animals either drank normal tap water (controls) or received water containing IAA (0.1 %) or DSS (5 %) for 1 week. On the 8th pretreatment day the gastric corpus mucosa was collected for histological examination. The photographs show morphological details characteristic of a normal mucosa (A), a mucosa with some superficial damage (B) and a mucosa exhibiting dilated gastric glands and vacuolized parietal cells (C). Parietal cells are indicated by arrows.
The appearance of the parietal cells in DSS-pretreated mice was also examined by electron microscopy. As shown in Figure 6B-D, several degrees of cellular abnormalities were seen when compared with parietal cells from control animals (Figure 6A). The nuclei of the parietal cells showed various degrees of disintegration (Figure 6B,D), and most parietal cells contained vacuoles of various sizes (Figure 6B,C,D,F), whereas the canaliculi appeared rather normal (Figure 6B,C,E). A few parietal cells seemed to be expelled from the continuity of the gastric gland epithelium (Figure 6E). Examination of the material found in the dilated gastric glands revealed the presence of dead cells (Figure 6F).
Figure 6.

Electron microscopical appearance of parietal cells from a control mouse (A) and mice pretreated with DSS (B-F). The animals either drank normal tap water (controls) or received water containing DSS (5 %) for 1 week. On the 8th pretreatment day the gastric corpus mucosa was collected for histological examination. The photographs show details of a normal parietal cell (A), parietal cell damage involving disintegration of the nuclear envelope (D), formation of vacuoles (B,C,D), cell expulsion from the continuity of the gland epithelium (E), and a dead cell in the material present in a dilated gland. CA, canalicular apparatus; NU, nucleus; VA, vacuole.
DSS pretreatment tended to decrease IG pH (pH = 2.14 ± 0.14, n = 5) relative to that measured in control animals (pH = 2.99 ± 0.63, n = 5), but this tendency did not reach statistical significance.
Effect of pretreatment with oral IAA or DSS on gastric volume recovery and mucosal damage caused by IG administration of HCl
Gastric fluid recovery and gross mucosal damage were examined 30 min after IG administration of HCl (0.25 M), at a time point when these parameters have been shown to be maximal (Wultsch et al., 2005). In control mice, 114 % of the IG administered volume was regained 30 min post-HCl (Table 1). This rate of gastric volume recovery did not differ in mice that had been pretreated with IAA for 1 week, whereas in DSS-pretreated mice gastric volume retention was significantly attenuated relative to that measured in control animals (Table 1).
Table 1.
Gastric volume recovery and gross mucosal lesion formation after gastric HCl challenge in control as well as IAA- and DSS-pretreated mice
| Test parameter | Control (IAA) | IAA | Control (DSS) | DSS |
|---|---|---|---|---|
| Volume recovery (%) |
111 ± 10.7 (6) | 112 ± 10.1 (7) | 116 ± 7.64 (7) | 91.1 ± 6.24 (6) * |
| Gastric lesions (%) | 0.49 ± 0.07 (6) | 0.44 ± 0.06 (6) | 0.53 ± 0.25 (6) | 0.63 ± 0.07 (6) |
Gastric volume recovery and gross gastric lesion formation were quantified 30 min after IG administration of HCl (0.25 M) to mice that had drunk normal tap water (controls), water containing IAA (0.1 %) or water containing DSS (5 %) for 1 week. The gastric volume recovery is expressed as a percentage of the volume administered into the stomach, and the extent of the gastric lesions is given as a percentage of the area of the total glandular mucosa. The values represent means ± SEM, n as indicated in brackets.
P < 0.05 versus respective control (Student’s t test).
The macroscopical injury of the gastric corpus mucosa observed after IG administration of HCl (0.25 M) was minor and in a preceding study (Wultsch et al., 2005) was found to be indistinguishable from that observed after IG treatment with NaCl (0.15 M). The visible lesions consisted of petechiae and occasionally a few small streaks of hemorrhage. As shown in Table 1, the formation of gastric hemorrhagic damage following gastric HCl challenge did not differ between control mice and mice pretreated with oral IAA or DSS for 1 week.
Duration of the effect of 1 week pretreatment with oral IAA or DSS on the gastric HCl-evoked expression of c-Fos in the brainstem
Mice were pretreated with oral IAA or DSS for 1 week, followed by normal tap water for another week; the control animals received normal tap water throughout the 2 week period. On the 15th day of the pretreatment protocol, the animals were exposed to IG HCl (0.25 M ) administered 2 h before collection of the brainstem. As shown in Table 2, the HCl-evoked expression of c-Fos in the NTS and AP of the IAA- and DSS-pretreated mice did no longer significantly differ from the respective values measured in control mice.
Table 2.
Gastric HCl-evoked expression of c-Fos in the brainstem of control mice and mice pretreated with IAA or DSS for 7 days followed by 1 week recovery
| Brainstem area | Control | IAA | DSS |
|---|---|---|---|
| NTS | 80.8 ± 8.24 (13) | 79.1 ± 11.5 (9) | 82.7 ± 10.4 (8) |
| AP | 15.8 ± 2.37 (13) | 12.0 ± 2.78 (9) | 10.9 ± 1.69 (8) |
The animals received water containing IAA (0.1 %) or DSS (5 %) for 1 week followed by normal tap water for another week; the control animals received normal tap water throughout the 2 week period. On the 15th pretreatment day the animals were exposed to IG HCl (0.25 M) administered 2 h before collection of the brainstem for immunocytochemical visualization of c-Fos. The values, given as means ± SEM (n as indicated), reflect the number of c-Fos positive cells per section in the unilateral NTS and AP. There were no statistically significant differences (one-way ANOVA).
Effect of indomethacin on the gastric HCl-evoked expression of c-Fos in the brainstem of mice pretreated with oral IAA or DSS
Mice who had drunk normal tap water or water containing IAA or DSS for 1 week received an IP injection of indomethacin (5 mg/kg) or its vehicle on the 8th pretreatment day. Thirty minutes later HCl (0.25 M) was administered IG and the brainstem examined for c-Fos 2 h post-HCl. Pretreatment with IAA enhanced the HCl-evoked expression of c-Fos in the NTS but not AP, whereas pretreatment with DSS increased the effect of HCl to induce c-Fos both in the NTS and AP (Figure 7A,B). Indomethacin did not alter the gastric HCl-evoked expression of c-Fos in the NTS of control and DSS-pretreated mice and failed to significantly reduce it in IAA-pretreated animals (Figure 7B). The gastric HCl-associated expression of c-Fos in the AP of IAA-pretreated mice was significantly diminished by indomethacin whereas that of control and DSS-pretreated animals was not significantly altered (Figure 7A).
Figure 7.
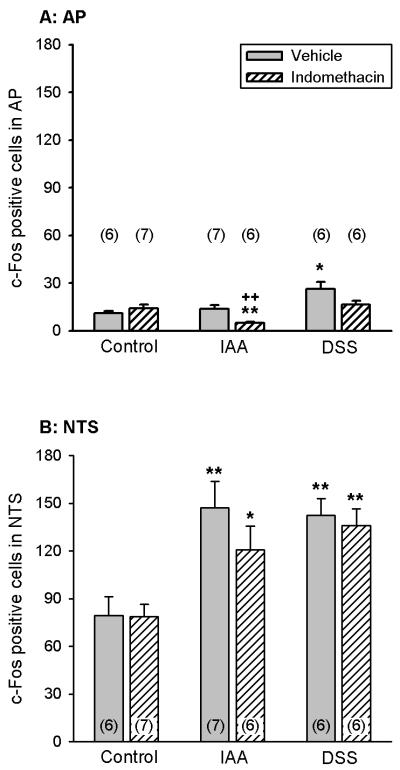
Effect of indomethacin on the gastric HCl-evoked expression of c-Fos in the AP (A) and NTS (B) of control mice and mice pretreated with IAA or DSS. The animals either drank normal tap water (controls) or received water containing IAA (0.1 %) or DSS (5 %) for 1 week. On the 8th pretreatment day the animals received an IP injection of indomethacin (5 mg/kg) or its vehicle. Thirty minutes later HCl (0.25 M) was administered IG; the brainstem was collected for determination of c-Fos expression 2 h post-HCl. The values, given as means ± SEM (n as indicated), reflect the number of c-Fos positive cells per section in the unilateral AP and NTS. * P < 0.05, ** P < 0.01 versus control, ++ P < 0.01 versus vehicle (two-way ANOVA followed by the Tukey test).
DISCUSSION
The current study shows that expression of c-Fos can be used to visualize an inflammation-induced increase in the afferent input from the acid-threatened stomach to the mouse brainstem. By using two different experimental models of gastritis, DSS- and IAA-induced inflammation, the question was addressed whether gastritis per se, independent of the underlying mechanisms, leads to gastric acid hyperresponsiveness of the afferent stomach – NTS axis. Further insight into this gastritis-induced alteration was sought by a comparison of the two gastritis models in terms of gastric MPO activity, gastric histology and gastric volume retention and by examining the effect of indomethacin.
Our data demonstrate that challenge of the mouse gastric mucosa with excess HCl signals NTS neurons to express c-Fos and that this afferent input is significantly enhanced when the stomach is inflamed. In contrast, the medullary c-Fos response to IG administration of saline was left unaffected by gastritis, which indicates that the response to a nonnoxious chemical and the distension associated with the administration of this stimulus (Wultsch et al., 2005) is not modified by gastric inflammation. It can, in addition, be inferred that baseline activity in the afferent stomach – brainstem system is not altered by gastritis. The gastritis-induced amplification of c-Fos protein-expressing cells in the brainstem thus parallels the gastritis-induced enforcement of the behavioural pain response to gastric acid (Lamb et al., 2003). The medullary c-Fos response and visceromotor pain reaction to gastric acid challenge in the rat are largely mediated by vagal afferent neurons since they are suppressed by chronic bilateral subdiaphragmatic vagotomy (Schuligoi et al., 1998; Michl et al., 2001; Lamb et al., 2003). In analogy to the situation in rats, we suppose that the major pathway which in mice signals a gastric acid insult to the brainstem is vagal afferent in nature. It need be considered, however, that the NTS receives visceral input not only via vagal afferent neurons (Altschuler et al., 1989) but also via a spinosolitary pathway (Menetrey and Basbaum, 1987; Gamboa-Esteves et al., 2001).
The gastric acid-evoked expression of c-Fos in the rat and mouse brainstem has previously been characterized with regard to its concentration-effect relationship and time course (Danzer et al., 2004; Wultsch et al., 2005). Consequently, expression of c-Fos in the brainstem was assessed 2 h post-stimulus, at a time when the response is maximal (Wultsch et al., 2005). Given that the concentrations of IG HCl (0.15 - 0.25 M) required to induce c-Fos in the brainstem are supraphysiological, it has been inferred that the afferent input from the acid-threatened stomach is determined by intramucosal acidosis (Danzer et al., 2004; Wultsch et al., 2005). The excess transmucosal H+ gradient is likely to drive luminal H+ ions across the gastric mucosal barrier into the lamina propria (Danzer et al., 2004; Wultsch et al., 2005) where they excite afferent nerve fibres either directly (Clarke and Davison, 1978; Hillsley and Grundy, 1998; Sugiura et al., 2005) or indirectly via neuroactive factors released in the tissue. In contrast, gastric injury (Schuligoi et al., 1998; Danzer et al., 2004; Wultsch et al., 2005) and hyperosmolarity of the HCl stimulus (Michl et al., 2001) have been ruled out as major factors determining the gastric HCl-evoked afferent input to the NTS.
While IAA-induced gastritis has been found to increase the visceromotor pain response to gastric acid and distension (Lamb et al., 2003), there is no report that DSS induces gastritis and that this type of gastritis is associated with hyperresponsiveness of the afferent stomach – NTS system to gastric acid. The observation that DSS causes gastric inflammation was not totally unexpected because this chemical has been widely used to induce chronic colitis in a region- and molecular weight-dependent manner (Okayasu et al., 1990; Kitajima et al., 2000). We used DSS of 8,000 molecular weight because DSS of low molecular weight (5,000) had previously been found to cause inflammation, particularly in the proximal part of the colon (Kitajima et al., 2000), and we therefore reasoned that it may also adversely affect more proximal parts of the digestive tract such as the stomach. Both DSS and IAA added to the drinking water for one week led to a moderate increase of MPO activity in the gastric wall, as has been observed for IAA in another study (Ozaki et al., 2002). This parameter reflects inflammation-associated infiltration of neutrophils and monocytes into the tissue (Suzuki et al., 1983; Krawisz et al., 1984; Graff et al., 1998). While inflammatory cells could be seen in the mucosa and submucosa of IAA-pretreated Swiss CD-1 mice (Piqueras et al., 2003), the rise of MPO activity which IAA and DSS pretreatment caused in the stomach of Him:OF1 mice was apparently too moderate to be seen as an appreciable accumulation of immune cells by routine histology.
It is known that the proinflammatory activity of DSS in the gastrointestinal tract depends on the molecular weight of the compound (Kitajima et al., 2000), the mouse strain (Mähler et al., 1998) and the treatment protocol (Okayasu et al., 1990; Kitajima et al., 2000 ). Preliminary data (Maria A. Pabst, personal communication) indicate that the DSS (molecular weight: 8,000, 5 %) used here failed to cause overt colitis as examined by routine histology. We assume, therefore, that afferent inputs to the brainstem from the distal part of the GI tract are unlikely to be affected. In addition, the gastric acid stimulus is unlikely to reach the distal gut because of the acid-induced closure of the pylorus (Forster et al., 1990; Raybould and Hölzer, 1993; Holzer et al., 2003; Danzer et al., 2004; Wultsch et al., 2005). The absence of severe gastrointestinal inflammation is in keeping with the failure of DSS pretreatment to affect the weight of the animals.
Close examination of the superficial epithelium revealed that both DSS and IAA caused some injury to the mucosal surface. An injurious effect of chronic peroral intake of 0.1 % IAA was also reported by Takeeda et al. (2004) but went unnoticed in other studies (Ozaki et al., 2002; Lamb et al., 2003; Piqueras et al., 2003). The histology of stomachs exposed to DSS was strikingly different from that of IAA-pretreated stomachs inasmuch as many gastric glands were prominently dilated and many parietal cells were vacuolized. Despite this conspicuous damage to the parietal cells as seen by light and electron microscopy, DSS pretreatment tended to enhance basal acid secretion, which is consistent with the relatively normal appearance of the canalicular apparatus. IAA pretreatment had previously been found to increase basal acid secretion as well as acid secretion stimulated by pentagastrin or histamine (Piqueras et al., 2003) and for this reason was not re-examined with regard to its influence on gastric acidity.
In spite of the profound histological differences, both DSS- and IAA-induced gastritis caused a similar enhancement of the medullary c-Fos response to gastric HCl challenge. Since damage to the superficial mucosa and a moderate increase in gastric MPO activity are common features of both gastritis models, we infer that the HCl hyperresponsiveness of the afferent stomach – NTS axis is related to gastric inflammation and mucosal surface injury. The gastritis-induced increase in the NTS response to gastric acid challenge cannot be explained by HCl-evoked exaggeration of mucosal injury because the effect of HCl to cause macroscopical gastric lesions was not altered by DSS and IAA pretreatment.
While the c-Fos response of the NTS to gastric HCl was similarly enhanced in mice with DSS- and IAA-induced gastritis, the c-Fos response of the AP to gastric HCl challenge was enhanced in the DSS gastritis model only. This finding suggests that pretreatment of mice with DSS facilitates the transmission of a gastric acid insult to the AP either by a neural or hormonal pathway. It is unlikely, however, that the expression of c-Fos in the AP and NTS was caused by systemic acidosis because, in the rat, exposure of the stomach to 0.5 M HCl (double the concentration used here) failed to affect blood pH (Schuligoi et al., 1998). In addition, it need be emphasized that the increase in c-Fos expression within the AP was small and its functional significance need be interpreted cautiously, given that the AP receives input from vagal afferents and has reciprocal connections with the NTS (Altschuler et al., 1989; Cai et al., 1996). Another difference concerns the HCl-evoked retention of gastric fluid. Unlike IAA, DSS pretreatment led to a significant decrease in gastric volume recovery. It has been shown before that HCl-induced retention of gastric fluid reflects inhibition of gastric emptying and secretion of gastric fluid (Forster et al., 1990; Raybould and Hölzer, 1993; Holzer et al., 2003; Danzer et al., 2004; Wultsch et al., 2005). The ability of DSS to reduce HCl-induced gastric fluid retention could thus point to a disturbed regulation of gastric emptying or fluid secretion in the presence of HCl and may be related to the differential effect of DSS and IAA on gastric acid secretion. Gastric fluid retention was assessed to rule out that afferent signalling of gastric HCl challenge was enhanced because gastritis increased gastric volume or prolonged stimulus exposure (Danzer et al., 2004, Wultsch et al., 2005). The observation that both IAA- and DSS-induced gastritis led to a similar increase in the HCl-evoked stimulation of the NTS indicates that the different rates of gastric volume retention in the two gastritis models were of minor relevance in this respect.
Indomethacin was used to explore whether proinflammatory products of the cyclooxygenase pathway (e.g., prostaglandins) contribute to the gastritis-associated hyperresponsiveness of the afferent stomach – NTS axis to excess gastric HCl. This possibility was envisaged because IAA-induced gastritis is associated with enhanced prostaglandin production in the gastric mucosa (Takeeda et al., 2004) and the nonselective cyclooxygenase inhibitor indomethacin modifies the effect of IAA to cause gastric mucosal damage and alter gastric acid secretion (Barnett et al., 2000; Piqueras et al., 2003; Takeeda et al., 2004). In addition, it has been reported that prostaglandins acting via the EP1 receptor mediate acid-induced pain hypersensitivity in the human oesophagus (Sarkar et al., 2003) and contribute to protection from acid/ethanol-induced gastric lesions in mice (Takeuchi et al., 2002). The ability of DSS to induce colitis is likewise modified by experimental manipulation of the prostaglandin system (Kabashima et al., 2002; Singh et al., 2004). At a dose (5 mg/kg) that has been shown to effectively augment pentagastrin- and histamine-stimulated gastric acid secretion in IAA-pretreated mice for more than 2 hours (Piqueras et al., 2003), indomethacin failed to significantly diminish the gastric HCl-evoked expression of c-Fos in the NTS of control, IAA- and DSS-pretreated mice. It would appear, therefore, that cyclooxygenase products do not participate in the gastritis-associated hyperresponsiveness of the afferent stomach – NTS axis to HCl. In view of the limited expression of c-Fos in the AP it would be premature to attribute any functional significance to the indomethacin-induced alterations of input from the stomach to this brainstem region.
In summary, the present findings show that experimental gastritis induced by two different proinflammatory chemicals, DSS and IAA, enhances the afferent signalling of a gastric acid insult to the NTS, although the two gastritis models differ in their structural and functional characteristics. Although we cannot deduce from the current data whether the increase in the medullary c-Fos response reflects enhanced sensory gain by primary afferent neurons or enhanced responsiveness of NTS neurons to the afferent input, several lines of evidence favour the notion that gastritis-associated hyperresponsiveness takes place at the level of primary afferent neurons. Firstly, it need be considered that the superficial mucosal injury associated with DSS- and IAA-induced gastritis facilitates the influx of excess luminal HCl into the lamina propria, which in turn will enhance the sensory gain of primary afferent neurons. Secondly, gastric injury has been found to enhance the excitability of nodose ganglion neurons and their responsiveness to acidosis (Bielefeldt et al., 2002; Sugiura et al., 2005). Thirdly, systemic administration of the proinflammatory cytokine interleukin-1β in the absence of overt gastritis has also been found to augment the medullary c-Fos response to a gastric HCl insult, most probably by a peripheral site of action on vagal nerve fibres (Holzer et al., 2004). In a wider perspective, our observations have two important implications. Firstly, it is possible to demonstrate by histological c-Fos imaging that gastritis is associated with hyperresponsiveness of the afferent stomach – NTS system to gastric acid. This method of functional neuroanatomy can also be used to trace the pathways within the brain that are activated by afferent input from the normal and inflamed stomach (Michl et al., 2001). Secondly, the two gastritis-associated hyperresponsiveness paradigms may represent valuable models in the identification and development of novel medicines targeting afferent pathways for the treatment of gastric pain.
ACKNOWLEDGEMENTS
This study was supported by the Jubilee Funds of the Austrian National Bank (grant 9858), the Zukunftsfonds Steiermark (grant 262) and the Austrian Scientific Research Funds (FWF grant L25-B05). The authors thank Rudolf Schmied for his expert help in image data processing.
Abbreviations
- ANOVA
analysis of variance
- AP
area postrema
- DSS
dextrane sulfate sodium
- HCl
hydrochloric acid
- HETAB
hexadecyltrimethylammonium bromide
- IAA
iodoacetamide
- IG
intragastric
- MPO
myeloperoxidase
- NTS
nucleus tractus solitarii
- PBS
phosphate-buffered saline
- WB
washing buffer
REFERENCES
- Altschuler SM, Bao XM, Bieger D, Hopkins DA, Miselis RR. Viscerotopic representation of the upper alimentary tract in the rat: sensory ganglia and nuclei of the solitary and spinal trigeminal tracts. J Comp Neurol. 1989;283:248–268. doi: 10.1002/cne.902830207. [DOI] [PubMed] [Google Scholar]
- Barnett K, Bell CJ, McKnight W, Dicay M, Sharkey KA, Wallace JL. Role of cyclooxygenase-2 in modulating gastric acid secretion in the normal and inflamed rat stomach. Am J Physiol. 2000;279:G1292–G1297. doi: 10.1152/ajpgi.2000.279.6.G1292. [DOI] [PubMed] [Google Scholar]
- Bielefeldt K, Ozaki N, Gebhart GF. Experimental ulcers alter voltage-sensitive sodium currents in rat gastric sensory neurons. Gastroenterology. 2002;122:394–405. doi: 10.1053/gast.2002.31026. [DOI] [PubMed] [Google Scholar]
- Cai Y, Hay M, Bishop VS. Synaptic connections and interactions between area postrema and nucleus tractus solitarius. Brain Res. 1996;724:121–124. doi: 10.1016/0006-8993(96)00282-x. [DOI] [PubMed] [Google Scholar]
- Clarke GD, Davison JS. Mucosal receptors in the gastric antrum and small intestine of the rat with afferent fibres in the cervical vagus. J Physiol. 1978;284:55–67. doi: 10.1113/jphysiol.1978.sp012527. (London) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danzer M, Jocic M, Samberger C, Painsipp E, Bock E, Pabst MA, Crailsheim K, Schicho R, Lippe IT, Holzer P. Stomach-brain communication by vagal afferents in response to luminal acid backdiffusion, gastrin, and gastric acid secretion. Am J Physiol. 2004;286:G403–G411. doi: 10.1152/ajpgi.00308.2003. [DOI] [PubMed] [Google Scholar]
- Forster ER, Green T, Elliot M, Bremner A, Dockray GJ. Gastric emptying in rats: role of afferent neurons and cholecystokinin. Am J Physiol. 1990;258:G552–G556. doi: 10.1152/ajpgi.1990.258.4.G552. [DOI] [PubMed] [Google Scholar]
- Gamboa-Esteves FO, Kaye JC, McWilliam PN, Lima D, Batten TF. Immunohistochemical profiles of spinal lamina I neurones retrogradely labelled from the nucleus tractus solitarii in rat suggest excitatory projections. Neuroscience. 2001;104:523–538. doi: 10.1016/s0306-4522(01)00071-9. [DOI] [PubMed] [Google Scholar]
- Graff G, Gamache DA, Brady MT, Spellman JM, Yanni JM. Improved myeloperoxidase assay for quantitation of neutrophil influx in a rat model of endotoxin-induced uveitis. J Pharmacol Toxicol Methods. 1998;39:169–178. doi: 10.1016/s1056-8719(98)00023-9. [DOI] [PubMed] [Google Scholar]
- Hillsley K, Grundy D. Sensitivity to 5-hydroxytryptamine in different afferent subpopulations within mesenteric nerves supplying the rat jejunum. J Physiol. 1998;509:717–727. doi: 10.1111/j.1469-7793.1998.717bm.x. (London) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holzer P, Danzer M, Schicho R, Samberger C, Painsipp E, Lippe IT. Vagal afferent input from the acid-challenged rat stomach to the brainstem: enhancement by interleukin-1β. Neuroscience. 2004;129:439–445. doi: 10.1016/j.neuroscience.2004.07.040. [DOI] [PubMed] [Google Scholar]
- Holzer P, Painsipp E, Jocic M, Heinemann A. Acid challenge delays gastric pressure adaptation, blocks gastric emptying and stimulates gastric fluid secretion in the rat. Neurogastroenterol Motil. 2003;15:45–55. doi: 10.1046/j.1365-2982.2003.00382.x. [DOI] [PubMed] [Google Scholar]
- Hughes P, Dragunow M. Induction of immediate-early genes and the control of neurotransmitter-regulated gene expression within the nervous system. Pharmacol Rev. 1995;47:133–178. [PubMed] [Google Scholar]
- Kabashima K, Saji T, Murata T, Nagamachi M, Matsuoka T, Segi E, Tsuboi K, Sugimoto Y, Kobayashi T, Miyachi Y, Ichikawa A, Narumiya S. The prostaglandin receptor EP4 suppresses colitis, mucosal damage and CD4 cell activation in the gut. J Clin Invest. 2002;109:883–893. doi: 10.1172/JCI14459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitajima S, Takuma S, Morimoto M. Histological analysis of murine colitis induced by dextran sulfate sodium of different molecular weights. Exp Anim. 2000;49:9–15. doi: 10.1538/expanim.49.9. [DOI] [PubMed] [Google Scholar]
- Krawisz JE, Sharon P, Stenson WF. Quantitative assay for acute intestinal inflammation based on myeloperoxidase activity. Assessment of inflammation in rat and hamster models. Gastroenterology. 1984;87:1344–1350. [PubMed] [Google Scholar]
- Lamb K, Kang YM, Gebhart GF, Bielefeldt K. Gastric inflammation triggers hypersensitivity to acid in awake rats. Gastroenterology. 2003;125:1410–1418. doi: 10.1016/j.gastro.2003.07.010. [DOI] [PubMed] [Google Scholar]
- Larauche M, Anton PM, Peiro G, Eutamene H, Bueno L, Fioramonti J. Role of capsaicin-sensitive afferent nerves in different models of gastric inflammation in rats. Auton Neurosci. 2004;110:89–97. doi: 10.1016/j.autneu.2003.11.003. [DOI] [PubMed] [Google Scholar]
- Mähler M, Bristol IJ, Leiter EH, Workman AE, Birkenmeier EH, Elson CO, Sundberg JP. Differential susceptibility of inbred mouse strains to dextran sulfate sodium-induced colitis. Am J Physiol. 1998;274:G544–G551. doi: 10.1152/ajpgi.1998.274.3.G544. [DOI] [PubMed] [Google Scholar]
- Medda BK, Sengupta JN, Lang IM, Shaker R. Response properties of the brainstem neurons of the cat following intra-esophageal acid-pepsin infusion. Neuroscience. 2005;135:1285–1294. doi: 10.1016/j.neuroscience.2005.07.016. [DOI] [PubMed] [Google Scholar]
- Menetrey D, Basbaum AI. Spinal and trigeminal projections to the nucleus of the solitary tract: a possible substrate for somatovisceral and viscerovisceral reflex activation. J Comp Neurol. 1987;255:439–450. doi: 10.1002/cne.902550310. [DOI] [PubMed] [Google Scholar]
- Michl T, Jocic M, Heinemann A, Schuligoi R, Holzer P. Vagal afferent signaling of a gastric mucosal acid insult to medullary, pontine, thalamic, hypothalamic and limbic, but not cortical, nuclei of the rat brain. Pain. 2001;92:19–27. doi: 10.1016/s0304-3959(00)00467-x. [DOI] [PubMed] [Google Scholar]
- Munglani R, Hunt SP. Proto-oncogenes: basic concepts and stimulation induced changes in the spinal cord. Prog Brain Res. 1995;104:283–298. doi: 10.1016/s0079-6123(08)61796-3. [DOI] [PubMed] [Google Scholar]
- Okayasu I, Hatakeyama S, Yamada M, Ohkusa T, Inagaki Y, Nakaya R. A novel method in the induction of reliable experimental acute and chronic ulcerative colitis in mice. Gastroenterology. 1990;98:694–702. doi: 10.1016/0016-5085(90)90290-h. [DOI] [PubMed] [Google Scholar]
- Ozaki N, Bielefeldt K, Sengupta JN, Gebhart GF. Models of gastric hyperalgesia in the rat. Am J Physiol. 2002;283:G666–G676. doi: 10.1152/ajpgi.00001.2002. [DOI] [PubMed] [Google Scholar]
- Pabst MA, Wachter C, Holzer P. Morphologic basis of the functional gastric acid barrier. Lab Invest. 1996;74:78–85. [PubMed] [Google Scholar]
- Paxinos G, Franklin KBJ. The Mouse Brain in Stereotaxic Coordinates. Academic Press; San Diego: 2001. [Google Scholar]
- Piqueras L, Corpa JM, Martinez J, Martinez V. Gastric hypersecretion associated to iodoacetamide-induced mild gastritis in mice. Naunyn-Schmiedeberg’s Arch Pharmacol. 2003;367:140–150. doi: 10.1007/s00210-002-0670-7. [DOI] [PubMed] [Google Scholar]
- Raybould HE, Hölzer H. Duodenal acid-induced inhibition of gastric motility and emptying in rats. Am J Physiol. 1993;265:G540–G546. doi: 10.1152/ajpgi.1993.265.3.G540. [DOI] [PubMed] [Google Scholar]
- Sarkar S, Hobson AR, Hughes A, Growcott J, Woolf CJ, Thompson DG, Aziz Q. The prostaglandin E2 receptor-1 (EP-1) mediates acid-induced visceral pain hypersensitivity in humans. Gastroenterology. 2003;124:18–25. doi: 10.1053/gast.2003.50022. [DOI] [PubMed] [Google Scholar]
- Schuligoi R, Jocic M, Heinemann A, Schöninkle E, Pabst MA, Holzer P. Gastric acid-evoked c-fos messenger RNA expression in rat brainstem is signaled by capsaicin-resistant vagal afferents. Gastroenterology. 1998;115:649–660. doi: 10.1016/s0016-5085(98)70144-1. [DOI] [PubMed] [Google Scholar]
- Singh VP, Patil CS, Jain NK, Kulkarni SK. Aggravation of inflammatory bowel disease by cyclooxygenase-2 inhibitors in rats. Pharmacology. 2004;72:77–84. doi: 10.1159/000079135. [DOI] [PubMed] [Google Scholar]
- Sugiura T, Dang K, Lamb K, Bielefeldt K, Gebhart GF. Acid-sensing properties in rat gastric sensory neurons from normal and ulcerated stomach. J Neurosci. 2005;25:2617–2627. doi: 10.1523/JNEUROSCI.2894-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki K, Ota H, Sasagawa S, Sakatani T, Fujikura T. Assay method for myeloperoxidase in human polymorphonuclear leukocytes. Anal Biochem. 1983;132:345–352. doi: 10.1016/0003-2697(83)90019-2. [DOI] [PubMed] [Google Scholar]
- Takeeda M, Hayashi Y, Yamato M, Murakami M, Takeuchi K. Roles of endogenous prostaglandins and cyclooxygenase isoenzymes in mucosal defense of inflamed rat stomach. J Physiol Pharmacol. 2004;55:193–205. [PubMed] [Google Scholar]
- Takeuchi K, Araki H, Umeda M, Komoike Y, Suzuki K. Adaptive gastric cytoprotection is mediated by prostaglandin EP1 receptors: a study using rats and knockout mice. J Pharmacol Exp Ther. 2001;297:1160–1165. [PubMed] [Google Scholar]
- Wultsch T, Painsipp E, Thoeringer CK, Herzog H, Sperk G, Holzer P. Endogenous neuropeptide Y depresses the afferent signaling of gastric acid challenge to the mouse brainstem via neuropeptide Y type Y2 and Y4 receptors. Neuroscience. 2005;136:1097–1107. doi: 10.1016/j.neuroscience.2005.08.038. [DOI] [PMC free article] [PubMed] [Google Scholar]