

Abstract
Rationale
Dilated cardiomyopathy is a leading cause of congestive heart failure and a debilitating complication of anti-neoplastic therapies. Despite disparate causes for dilated cardiomyopathy, maladaptive cardiac remodeling and decreased systolic function are common clinical consequences, begging an investigation of in vivo contractile dynamics in development and disease, one that has been impossible to date.
Objective
Imaging in vivo myocardial contractile filament dynamics and assess potential causes of dilated cardiomyopathy in anti-neoplastic therapies targeting Erbb2.
Methods and Results
We generated a transgenic zebrafish line expressing an actin-binding GFP in cardiomyocytes, allowing in vivo imaging of myofilaments. Analysis of this line revealed architectural differences in myofibrils of the distinct cardiomyocyte subtypes. We used this model to investigate the effects of Erbb2 signaling on myofibrillar organization, since drugs targeting ERBB2 (HER2/NEU) signaling, a mainstay of breast cancer chemotherapy, cause dilated cardiomyopathy in many patients. High-resolution in vivo imaging revealed that Erbb2 signaling regulates a switch between a dense apical network of filamentous myofibrils and the assembly of basally localized myofibrils in ventricular cardiomyocytes.
Conclusions
Using this novel line, we compiled a reference for myofibrillar microarchitecture among myocardial subtypes in vivo and at different developmental stages, establishing this model as a tool to analyze in vivo cardiomyocyte contractility and remodeling for a broad range of cardiovascular questions. Further, we applied this model to study Erbb2 signaling in cardiomyopathy. We show a direct link between Erbb2 activity and remodeling of myofibrils, revealing an unexpected mechanism with potentially important implications for prevention and treatment of cardiomyopathy.
Keywords: Imaging, cardiomyopathy, contractility, myofibrils, Erbb2, development, cardiac remodeling, myocardial contraction, myofilament protein, sarcomere
INTRODUCTION
Heritable and acquired dilated cardiomyopathies (DCM) are the third most common cause of heart failure and the leading cause for cardiac transplant1. Recent genomic and pharmacological studies have identified genes involved in the development of DCM; most of these genes encode components of the contractile machinery, but some encode cell surface receptors such as members of the ERBB2/HER2 family2–5. Studies validating these genes in animal models have started to elucidate their role in DCM6,7. Unfortunately, models used to date are static — relying on fixation-based protocols including immunohistochemistry and electron microscopy, or artificial cell culture — while contractility and remodeling are dynamic. Studying contractile physiology and remodeling in a live heart is therefore key to understanding cardiomyopathy.
While these limitations apply to all recent models, the zebrafish offers the potential to image contractile structures in vivo due to the optical transparency of its embryos and larvae and its genetic tractability8. However, this potential has not been exploited to date, in part due to a historical lack of the appropriate transgenic reporter to image contractile structures and the lack of a clinical model for dilated cardiomyopathy. To address these limitations, we developed a novel transgenic line, Tg(myl7:LifeAct-GFP), in which filamentous actin (F-actin), a major component of sarcomeres, is labeled with GFP. LifeAct consists of a fluorescent protein fused to a low-affinity actin binding domain derived from Abp140, a yeast protein that lacks close homologs outside fungi. The 17 amino acids domain binds F-actin with a dissociation constant of 2.2 μM, a 30 fold higher affinity for F-actin than for G-actin9. We designed a zebrafish codon-optimized version based on the zebrafish acta1b, which according to our data is the most highly expressed actin gene in the adult zebrafish heart. One major advantage of the LifeAct system is that as a fungal derivative, it does not sequester endogenous actin-binding proteins as do the utrophin and moesin fusions, amongst others. Using Tg(myl7:LifeAct-GFP), we were able to image myofibrils, which principally contain F-actin in vivo and in high-resolution for the first time. We generated a reference dataset of native myofibril genesis and architecture in the atrium and ventricle at different stages of development. We identified three myocardial subtypes according to their myofibrillar architecture. We further showed that this line, together with state of the art confocal microscopy, allows high-resolution study of myofibrillar dynamics in the beating heart in four-dimensions (4D) of space and time. We then used this tool to uncover the role of Erbb2 signaling in the development of DCM.
The four vertebrate members of the epidermal growth factor receptor (Egfr/ErbB) family of receptor tyrosine kinases regulate many essential processes in development and disease, including cell differentiation, proliferation, survival, adhesion and migration10–12. ErbB2, although incapable of binding ligands itself, plays a particularly important role as the preferred dimerization partner for the three other ligand-binding ErbB receptors13. In disease, ERBB2/HER2 overexpression is evident in more than 30 percent of all breast cancer patients and serves as a marker for poor prognosis and survival14. Consequently, several breast cancer therapies target ERBB2/HER2 function15. However, the great success of anti-ERBB2/HER2 drugs has been tempered by the unexpected and unfortunate fact that they can cause DCM, thus the need to better understand the role of ERBB2/HER2 signaling in cardiomyocyte development and function.
Loss of ErbB signaling causes widespread defects in cardiac development in mammals and zebrafish, including an absence of trabeculation16,17. Advanced microscopy in zebrafish further revealed that Erbb2 signaling is required both formitosis within the early myocardium and for stabilizing the basal processes that cardiomyocytes of the compact wall use to delaminate during trabeculation18,19. ErbB signaling is also essential for adult cardiac function. Perinatal removal of ErbB2 from murine myocardium causes DCM7. Similarly, and unfortunately, ERBB2/HER2 antagonism in the adult human heart, as occurs with anti-ERBB2/HER2 cancer therapies, also causes DCM. Seven percent of patients treated with Trastuzumab, a monoclonal antibody targeting ERBB2/HER2, develop DCM; this incidence rises to 27 percent when combined with the anthracycline doxorubicin20.
Inherited DCM is linked to genes encoding cytoskeletal and contractile proteins, such as cardiac actin (ACTC), troponin T type 2a (TNNT2a), α-tropomyosin (TPM1), myosin heavy chain 7 (MYH7) and others3. Susceptibility to acquired DCM from anti-ERBB2/HER2 agents may depend on more subtle genetic differences in those same sarcomeric proteins. However, exploring a possible link between ERBB2/HER2 signaling and myofibrillar architecture in vivo has been impossible to date. To test the effects of Erbb2 signaling on cardiomyocyte myofibrils, we used our Tg(myl7:LifeAct-GFP) line to perform high-resolution in vivo imaging of myofibrils in 3D and 4D. We used erbb2 mutants, Erbb2 inhibitors and cardiomyocyte-specific dominant-negative Erbb2 overexpression (dnErbb2) to obtain a detailed picture of the role of Erbb2 signaling on the actin cytoskeleton and myofibrils during development and in the adult.
METHODS
Zebrafish lines and DNA constructs
LifeAct-GFP was cloned by PCR primer extension, with the forward primer including a zebrafish codon-optimized version of the 17aa actin-binding domain of S. cerevisiae Abp1409. Subsequently, LifeAct-GFP was cloned into a miniTol2 vector under the control of a myl7 or acta1b promoter element (Figure 1). Transgenesis was performed in TL background as described21, resulting in the establishment of Tg(myl7:LifeAct-GFP)s974. A zebrafish version of a dominant-negative Erbb2 receptor (Erbb2-CD533) based on previous work 22 was cloned into a dual myl7 promoter plasmid allowing the simultaneous expression of RFP and dnErbB2 in cardiomyocytes. Transgenic animals were produced. We used erbb2 (st61)23 and Tg(myl7:nDsRed2)f2Tg24. Zebrafish were maintained under standard conditions.
Figure 1. Myofilament components are associated with inherited dilated cardiomyopathy and can be visualized using LifeAct-GFP expression in vivo.

(A) Schematic representation of sarcomere units and components associated with familial cases of dilated cardiomyopathy (DCM).(B) Schematic representation of the constructs and primary sequence of the LifeAct-GFP used in this study.(C, D) Comparative imaging of skeletal muscle myofibrils fixed and stained with phalloidin (C) vs. in vivo LifeAct-GFP(D). Note that high-affinity phalloidin binds actin within the Z-disc while low-affinity LifeAct-GFP cannot access actin within the Z-disc. (E–H) Transgenic expression of LifeAct-GFP under the myocardial myl7 promoter does not cause any developmental defects. Scale bars indicate equal magnification (E,F;G,H).
Microscopy and image analysis
Zebrafish larvae were mounted in low-melting agarose (GeneMate) under 2 g/LHEPES-buffered tricaine (pH 7.4) to stop cardiac contractions or 75 mg/L tricaine for movie acquisition as published25. Stills were scanned using a Zeiss LSM 5 or 7 series confocal microscope with a W Plan-Apochromat 40x/1.0 DIC dipping objective or LD C-Apochromat 63x/1.1 W Corr objective. Movies were acquired with a Zeiss SPIM (Lightsheet Z.1) or a Zeiss spinning disc confocal system (Z1 Cell Observer SD, CSU-X1). Movies were processed using the open source program VirtualDub and encoded in H.264 in Apple QuickTime format.3D reconstruction was achieved using the Zeiss microscopy ZEN software. Systolic and diastolic area of wild-type and erbb2 mutant larvae was measured to estimate the contractility of the heart as . For 4D rendering, a sequence of non-gated two-dimensional fluorescence time-series (100 frames/s) for every 1 μm slice of beating heart (embedded in 1.2% agarose) was obtained using a Cell Observer SD (Zeiss, CSU-X1, LD C-Apochromat 40x/1.1 W Corr). The acquired non-gated time-series at various z-planes were post-synchronized using a published MATLAB program26. An isosurface rendering (Imaris, Bitplane) was performed on selected regions of the 3D beating hearts for getting a perspective of the inner contours.
Chemical Erbb2 inhibition and Doxorubicin treatments
Erbb2 inhibition was achieved with PD168393 (Millipore) as previously described27. 100 mmol/L Doxorubicin stock solution was prepared in 0.1% DMSO and diluted in H20 prior to injection. Zebrafish larvae were transiently anesthetized in 133 mg/L tricaine during the procedure.
Expression analysis
Expression analysis was performed using the Eco Real-Time PCR System (Illumina). RNA was isolated using Trizol (Sigma). cDNA was synthesized using a random-primed First Strand cDNA Synthesis Kit (Maxima). Data shown were compiled on two specimens and one technical replicate (total of three). Primers were designed using PerlPrimer28 (span intron, overlap intron-exon boundary by 7 bp). gapdh and kcnj3a were used for normalization (ΔΔC(t) method).
Cell culture experiments
Isolation and culture of primary cardiomyocytes was done as published29. Hearts were isolated from one year old fish. Cell culture dishes were coated with 1% gelatin.
RESULTS
Novel transgenic LifeAct-GFP line allows in vivo imaging of myofibrillar architecture
Together with microtubules and intermediate filaments, actin filaments represent a major component of the eukaryotic cytoskeleton. Actin is also a main component of sarcomeres, where it localizes to the I-bands and the overlapping parts of the A-bands (Figure 1A). While myofibrils of skeletal muscles are very large and relatively resistant to fixation in zebrafish, the filamentous actin of the thinner myocardial myofibrils is difficult to image by fixation-based protocols. To circumvent the limitations of established protocols, we tested the recently developed LifeAct-GFP for actin imaging in vivo.
In skeletal muscle, the high actin-binding affinity of phalloidin clearly reveals the Z-, I+A and M-bands (Figure 1C). To test LifeAct-GFP, we first expressed it clonally in skeletal muscle under the acta1b promoter (Figures 1B, 1D) and imaged at 72 hours post fertilization (hpf). As in phalloidin stainings on fixed tissue, LifeAct-GFP expression in skeletal muscle nicely reveals sarcomeric organization (Figure 1D). LifeAct-GFP was then cloned under the pan-myocardial myl7 promoter. To test whether LifeAct-GFP expression reveals structures other than sarcomeres in cardiomyocytes, we performed transient clonal expression in a tnnt2a mutant background in which no sarcomeres are assembled30. Confocal scans of these mutants revealed that no structures other than initially formed Z-bodies could be detected at 4 days post fertilization (dpf) even after overexposure (Online Figure I). Using the transposase-based TOL2 system, we then generated the transgenic line Tg(myl7:LifeAct-GFP). Transgenic animals express high levels of LifeAct-GFP in cardiomyocytes from about 19 hpf to adulthood. No phenotype induced by LifeAct-GFP expression was apparent at any stage. Adult transgenic zebrafish hearts show no morphological abnormalities or signs of insufficiency (Figures 1E–1H).
Myocardial filament organization changes dramatically during cardiac development
In zebrafish, the primitive heart tube has formed by 20 hpf and becomes contractile shortly thereafter31. At 2.5 dpf, some ventricular cardiomyocytes start to delaminate to form trabeculae. To obtain a detailed perspective on actin dynamics during cardiac development, we imaged the Tg(myl7:LifeAct-GFP) line starting at 20 hpf. Between 20 and 27 hpf, high-resolution imaging was restricted to the atrium because during this time the ventricle is positioned deep within the body (Figure 2).
Figure 2. Myofibrillar architecture changes during development.

Myocardial expression of LifeAct-GFP allows the detailed in vivo analysis of myofibril architectural features at highresolution.(A–A‴) Early cardiac tube at 20 hpf, before the initiation of contractions. (A′–A‴) Optical sections through the cardiac tube reveal no signs of myofilament assembly. (B–B‴) Early cardiac tube at 22 hpf, after the initiation of contractions (individual cells outlined). (B′–B‴) Optical sections through the cardiac tube show the establishment of myofilaments starting from actin rich I-Z-I bodies (thin filament/Z-band precursors) in a highly branched fashion where I-Z-I bodies act as branch points (red arrowheads). This early contractile network strictly localizes to the basal (luminal) side of cardiomyocytes. (C) Overview of the heart during cardiac looping (36 hpf). Cardiomyocytes of the ventricular chamber (C′) exhibit cortical localization of actin-rich structures but no myofilaments are present within the center of the cell. This localization is in contrast to that observed in atrial cardiomyocytes (C″), where myofibrils span the cell in a highly organized fashion along the transverse axis of the heart. (D) 3-D reconstruction of the ventricular chamber at 4.5 dpf, i.e., after the onset of trabeculation (luminal view). See Online Movie I for entire 3-D reconstruction. (E) 3-D reconstruction of the atrial chamber (abluminal view) at 4.5 dpf. (F) Schematic of imaging procedure and mounting.
Before the onset of cardiac contractions, LifeAct-GFP localizes to the cytoplasm and appears not to be spatially restricted within the cell. The round shape of the cardiomyocytes indicates a low level of differentiation. Sagittal confocal sections through the heart tube at 20 hpf reveal minor cortical LifeAct-GFP localization at intercellular membrane compartments, suggesting the enrichment of actin at cellular junctions but no sign of filamentous actin or sarcomere assembly (Figure 2A).
Only two hours later, after the initiation of cardiac contractions, LifeAct-GFP localization reflects the formation of myofilaments within cardiomyocytes of the atrial chamber, progressively functionalizing the cardiac tube. Intriguingly, initial myofilament assembly appears in a branched pattern forming a cell-spanning network (Figures 2B––2B′; red arrows, individual cell outlined), whereas more mature cardiomyocytes align the vast majority of their myofibrils orthogonal to the direction of blood flow to sustain the function of the organ as a peristaltic pump. Sagittal confocal sections reveal the progressive flattening of cardiomyocytes at this stage and highly polarized LifeAct-GFP localization (Figures 2B″; 2B‴), showing a strict localization of myofibrils to the luminal (basal) side of atrial cardiomyocytes. Cardiac looping progresses through 48 hpf, moving the ventricle to its final position. Imaging of ventricular cardiomyocytes at this stage revealed a fundamentally different mode of myofilament formation and localization. Ventricular cardiomyocytes now show high LifeAct-GFP expression exclusively at their cortical membranes (Figure 2C–2C′, individual cells outlined). In contrast to atrial cardiomyocytes, myofilaments spanning the cytoplasm could not be observed at this stage. Imaging of both chambers at 4.5 dpf revealed their mature myofibrillar architecture with a densely trabeculated ventricle and a highly organized and parallel alignment of myofibrils in the atrium (Figures 2D–2E; Online Movie I). Together, these data suggest a divergent mode of development and function of the cardiomyocytes in the individual chambers. Atrial cardiomyocytes assemble contractile structures from an immature yet highly polarized branched network of myofilaments. In contrast, in early ventricular cardiomyocytes, filaments are mainly cortical. At later stages, the myofibrillar structures of the ventricular myocardium become more prominent but increasingly difficult to observe in detail as trabeculation expands.
Single cell analysis of cardiomyocytes reveals subtype-specific myofibrillar organization
In order to analyze each subpopulation of cardiomyocytes, we examined mosaic transgenics at 4.5 dpf and found that each subtype exhibits unique features with regards to myofilament organization, assembly and function. A comparison of the key architectural features of cardiomyocyte subtypes is illustrated in Figure 3.
Figure 3. Clonal analysis of myofibril organization in individual myocardial subtypes at 4.5 dpf.

The larval zebrafish heart contains three clearly distinguishable myocardial subtypes. (A) Clone of three atrial cardiomyocytes. (B) Abluminal surface scan of a single cardiomyocyte in the ventricular compact wall. (C) Cross-section through the ventricular lumen showing trabecular myofibers. (D) Cardiomyocytes of the ventricular wall during the process of trabeculation. While the apical (abluminal) side of ventricular wall cardiomyocytes is filled with a dense network of myofibrils (B, D′), the basal side contains thicker, cortically localized myofibrils which are clearly observable in a maximum intensity projection of the whole cell (D, white arrowhead) but not in a scan along the apical surface (D′, red arrowhead). (D″) Optical cross-sectioning through the cortical region of cardiomyocytes of the compact wall also show this polarized localization (dashed lines indicate cell-cell boundaries). Cellular protrusions (pseudocolored in red) which extend into the ventricular lumen appear entirely free of thin myofibrils (D). (E) Trabecular cardiomyocytes are tubular cells of variable length with cortically localized cell-spanning myofibrils and a central nucleus (red). At their respective ends, the myofibrils branch and connect to intercellular membranes of compact layer (CL) cardiomyocytes or adjacent trabecular cells. (F) Clonal analysis of ventricular wall cardiomyocytes, one of which (boxed) is in the process of leaving the ventricular wall. (F′) Closeup of the cardiomyocyte from (F); luminal protrusions (pseudocolored in red), the nucleus containing cell body residing in the wall (green). See Online Movie II for 3D reconstruction. (G–H) Schematic representation of individual myocardial subtypes and their typical myofibril organization and localization (nucleus in blue). (G) Atrial cardiomyocyte. (H) Ventricular wall cardiomyocyte. (I) Trabecular cardiomyocyte. These architectural features can also be observed in Online Movies IIIa and IIIb in beating hearts.
Atrium
The atrial cardiomyocytes exhibit exclusively linear myofibrils, strictly localized to their basal side. The majority of myofibrils align orthogonally to the direction of blood flow. Intriguingly, the crossing points of myofibrils within single cardiomyocytes show almost exclusively a 90° angle (Figures 2E, 3A), with little signs of the branched morphology that was dominant during early developmental stages (Figure 2B′). Further, the atrial myofibrils appear to span across intercellular membranes, an illusion which is probably a result of their tight attachment to fascia adherens on both sides of intercellular membranes.
Ventricular wall
The ventricular chamber at 4.5 dpf consists of two clearly distinct populations of cardiomyocytes: compact wall cardiomyocytes and trabecular cardiomyocytes. Compact wall cardiomyocytes have a flat, round or polygonal, epithelial-like shape18. Intriguingly, sequential imaging along the Z-axis revealed two distinct actin-rich structures within these cells. Abluminal (apically), they are filled with a dense, highly branched filamentous myofibrillar network, similar to the network seen earlier in development. Basolaterally, myofibrils localize to the cell cortex, often forming a non-continuous ring. At about 3 dpf, compact wall cardiomyocytes start extending protrusions on their basal side32. These protrusions are filled with cortical myofibrils but show no sign of the filamentous network (Figure 3D).
Ventricular trabeculae
After their formation, trabeculae mature into long, tubular structures, often branched at their ends and preferentially attached to other trabeculae, forming intraventricular rings orthogonal to blood flow (Online Figure II). Numerous cortically localized myofibrils span the length of trabecular cardiomyocytes, often showing branches distally. This cortical localization leads to the encompassing of the nucleus which is localized centrally as seen in Figure 3E.
To complement this in vivo analysis, we cultured primary cardiomyocytes and observed significant differences in terms of the density and localization of myofibrils (see Online Data Supplement and Online Figure III).
Loss of ErbB2 signaling causes sarcomere remodeling in ventricular cardiomyocytes
Genome-wide association studies suggest a causal relationship between defects in sarcomere assembly and function, and the development of DCM. To investigate the potential effects of ErbB signaling on myocardial myofilaments, we crossed anerbb2 loss-of-function allele into the Tg(myl7:LifeAct-GFP) background and performed imaging studies on ventricular wall cardiomyocytes from 3 to 4.5 dpf. Trabeculae form in wild-type but not in erbb2-deficient larvae (Figure 4, Online Movies IVc–IVd), hence we imaged actin localization in the ventricular wall. At 3 dpf, as described previously, ventricular wall cardiomyocytes form two distinct types of myofibrils, a highly branched network of thin myofibrils on their apical side and a cortical and discontinuous ring of thicker myofibrils on their basolateral side. In erbb2 mutants at 3 dpf, the apical network appears less prominent than in wild-type siblings; however, at the same time, mutants show an excess of myofibrils basolaterally (Figures 4A – A″). This observation is consistent with imaging data acquired from the atrium, where in mutants the density of myofibrils appears higher and the branch points present in wild-type siblings are mostly absent (data not shown), suggesting a general effect of Erbb2-mediated signaling on the architecture and localization of myofibrils in cardiomyocytes. In 4.5 dpf mutants, myofibril reorganization becomes even more prominent. The apically localized filamentous myofibrils are strongly reduced in abundance (Figures 4B′, 4B‴ - red arrowheads) while basolaterally localized myofibrils gain substantialsize (Figures 4B″, 4B‴ - white arrowheads) and represent the dominant type of myofibrils in ventricular wall cardiomyocytes as strikingly revealed in a maximum intensity projection of the ventricular chamber (Figure 4B) or in 4D rendered confocal movies (Online Movies IVa+IVb, IVc+IVd and single plane high magnification IVe+IVf). To assess how this reorganization impairs cardiac performance, we measured ventricular fractional shortening in three wild-type and mutant animals and found a 25% decrease in mutants (Figure 4E). Together, these results indicate that Erbb2 signaling regulates myofilament dynamics with a direct impact on cardiac performance. Wild-type ventricular wall cardiomyocytes are dominated by myofibrils of narrow diameter. Erbb2-deficient cardiomyocytes show very few filamentous myofibrils, particularly at their apical domain; in contrast, they form massive myofibrils mostly at their basolateral cortex.
Figure 4. Myofibril organization inerbb2−/− cardiomyocytes at different developmental stages.

Confocal scans of erbb2−/− and wild-type siblings reveal differences between density and size of myofibrils of compact wall cardiomyocytes. As described above, cardiomyocytes of the ventricular wall exhibit thin filamentous myofibrils on their apical side and more prominent myofibrils forming a cortical ring on their basal side. In erbb2−/−, ventricular wall cardiomyocytes exhibit a reduction of thin filamentous myofibrils on their apical side while their basolateral myofibrils dramatically increase in size as can be seen in (A) and (B) at 3.5 and 4.5 dpf respectively. Surface scans at higher magnification reveal the reduction of the apical filamentous myofibrils (A′ and B′+B‴ red arrowheads) and thickening of the basolaterally located myofibrils (A″ and B″+B‴ white arrowheads) 5 μm below. (B‴) Sagital cross-sections through cells of the ventricular wall highlight this effect. (C, D) Schematic representation of myofibril localization and architecture in cardiomyocytes of the ventricular compact wall in (C) wild-type and (D) erbb2−/−animals. Note: a comparison of a 4.5 dpf erbb2−/− and a wild-type sibling is also available as full 4D reconstruction, 4D luminal view surface rendering and single plane imaging in Online Movies IVa+IVb, IVc+IVd and IVe+IVf respectively. (E) Measurements of ventricular fractional shortening in erbb2−/− and sibling animals reveal a 25% decrease in cardiac performance in erbb2 mutants at 4.5 dpf.
Chemical ErbB2 inhibition later in development causes myofibrillar reorganization
To further investigate the effects of Erbb2 signaling on sarcomere dynamics, we used the compound PD168393, an alkylating agent that specifically inhibits the kinase domain of Erbb receptors19,27. We used this compound laterin development, after the ventricle had already undergone trabeculation, to eliminate potential indirect effects caused by a lack of trabeculae. Tg(myl7:LifeAct-GFP); Tg(myl7:nDsRed2) larvae were treated with 10 μM PD168393 from 4.5 to 6.5 dpf and subsequently imaged (Figure 5A).
Figure 5. Late-stage inhibition of Erbb2 signaling causes myofibril reorganization and bundling.
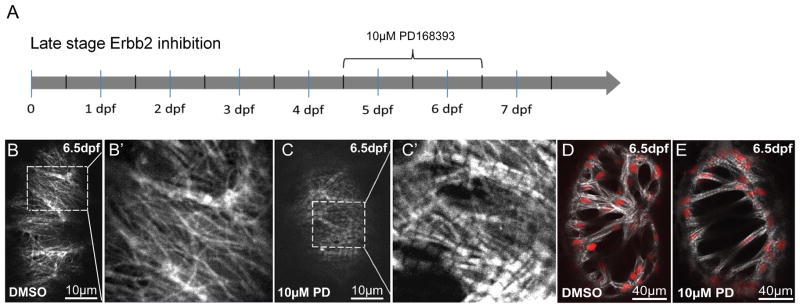
(A) Tg(myl7:LifeAct-GFP);Tg(myl7:nDsRed) larvae where exposed to 10μM PD168393 from 4.5 to 6.5 dpf. (B–B′, C–C′) In ventricular wall cardiomyocytes, Erbb2 inhibition caused a dramatic increase inmyofibril diameter on their apical side. (D, E) Trabecular cardiomyocytes showed increased thickness of myofilament bundles and overall myofibril content.
Whereas no effect could be detected in vehicle-treated larvae, inhibition of Erbb signaling had a dramatic effect on the myofibrils of both ventricular wall and trabecular cardiomyocytes. The apical filamentous network in ventricular wall cardiomyocytes (Figures 5B–B′) was completely lost, replaced by solid linear myofibrils of wide diameter (Figures 5C–C′). Trabecular cardiomyocytes from control animals showed extensively branched myofibrils (Figure 5D). Inhibition of Erbb signaling resulted in a significant overrepresentation of single, non-branched myofibrils which filled the entire cell and caused the centered, usually round shaped nucleus to deform into an ellipsoid shape (Figure 5E). Taken together, these results indicate that Erbb2 signaling prevents the bundling and maturation of ventricular myofibrils, possibly by destabilizing the formation of myofilament bundles and increasing actin turnover.
Long-term Erbb2 suppression causes a DCM-like phenotype in zebrafish and dysregulation of sarcomeric components
As mentioned previously, the analysis of familial DCM cases suggests that defects in sarcomere function can cause DCM. Analysis of Erbb2 function in the adult heart however is challenging, as erbb2−/− zebrafish, like Erbb2−/− mice, die from a variety of developmental defects at early stages. We therefore created a transgenic zebrafish line with cardiac-specific expression of dominant-negative Erbb2 receptor (Tg(myl7:dnerbb2)) to impair Erbb2 signaling specifically in the heart. At first, we tested the effects of dnErbb2 expression on myofibrillar architecture by mosaic expression using DNA injections into fertilized eggs. Identification of cardiomyocytes expressing dnErbb2 was achieved by using a plasmid with two myl7 promoters, one driving dnErbb2 and the other driving RFP. We found that RFP+ cardiomyocytes showed a significant reduction of their apical thin myofibrils and an over-representation of basolateral thick myofibrils, similar to our observation in erbb2 mutants (Figures 6A–A″ – arrowheads point to cell-cell boundaries). These results also suggest a cell-autonomous function of Erbb2 in myofibril remodeling. We next analyzed transgenic fish which we found to develop pericardial edema by about one month of age (Figures 6B–C), exhibiting high lethality afterwards. We were able to raise three F1 fish with stable cardiac expression through two months of age. After euthanizing these fish, we found that their hearts exhibited a significantly dilated ventricle with sparse trabeculae (Figures 6D–G). Using quantitative rtPCR, we tested the expression level of DCM-associated genes and found several of them to be consistently and severely misregulated in transgenic vs. control hearts (Figure 6H). Intriguingly, most of the genes were upregulated rather than downregulated. These genes include acta2, tcap, pdlim3a and vmhc, the zebrafish orthologue of MYH7, which was highly upregulated. This upregulation is consistent with the larval erbb2−/− phenotype, which includes thicker myofibrils.
Figure 6. Erbb2 acts cell-autonomously on myofibrillar architecture, and long-term inhibition of Erbb2 signaling causes DCM and dysregulation of DCM genes.

To reduce Erbb2 signaling exclusively in the myocardium, we developed a cardiomyocyte-specific dominant negative strategy. (A–A″) Expression of the dnErbb2 receptor in clones (with coexpression of RFP) reveals cell-autonomous remodeling of myofibrillar architecture, leading to a reduction of thin myofibrils and an overrepresentation of large diameter myofibrils. This effect is most striking at the boundaries between normal and transgenic cells (arrowheads). We further created transgenic animals expressing dnErbb2 under the pan-myocardial myl7 promoter. The resulting transgenics exhibited severe edema (C; red arrowheads), decreased trabeculation (E, white arrowheads) and dilated cardiac morphology (G) compared to non-transgenic siblings (B, D, F). Using quantitative rtPCR, we tested the expression of genes associated with familial cases of DCM (H), and found a strong and consistent upregulation of several members of this gene set including those encoding components of the contractile machinery. The dashed line indicates the expression level in non-transgenic siblings.
Doxorubicin dismantles myofibrils in a dose-dependent manner, most severely of the filamentous type
Doxorubicin (DOXO), a drug commonly used as an adjuvant chemotherapy for ERBB2/HER2-positive breast cancer patients, has been reported to disrupt myocardial sarcomere structure by a Calpain-dependent mechanism33,34. Also, anti-ERBB2 therapies are much more likely to cause DCM when those therapies are combined withDOXO20. To determine the effects of DOXO on cardiomyocyte myofibrils in vivo, we injected different concentrations into the pericardial cavity and performed confocal imaging one, two and three days post injection (dpi) (Figure 7). We observed a strong, dose-dependent effect of DOXO on overall myofibril integrity and function. Intrapericardial injections of 1 pL (4 mmol/L) DOXO were sufficient to cause pericardial edema and a pronounced decrease of detectable myofilaments in all myocardial subtypes (Figures 7B–B′; Online Movies Va and Vb), most significantly in ventricular wall cardiomyocytes (Figures 7A–B). In these cells, the thin apically localized myofibrils were strongly affected. Since both, the reduction of Erbb2 signaling and DOXO exposure have direct effects on myocardial sarcomeres, these data offer a mechanistic explanation for the increased incidence of acquired DCM in patients treated with both anti-ERBB2 therapy and DOXO compared to anti-ERBB2 therapy alone.
Figure 7. Doxorubicin treatment leads to loss of myofibrils in vivo.

Doxorubicin was injected into the pericardial cavity at 4.5 dpf and the larvae raised for another 24 hours. Subsequent confocal imaging revealed a significant effect on overall myofibril structure and density. While vehicle-injected control larvae showed no detectable alterations (A, A′), injection of 1 pL (4 mmol/L) doxorubicin entirely ablated fine myofibrils in compact wall cardiomyocytes and caused myofibrillar disarray in the thick myofibrils in trabecular cardiomyocytes (B, B′). A′ and B′ are also available as 3-D reconstructions (Online Movies Va and Vb respectively).
DISCUSSION
Current studies of cardiomyopathy are widely based on immunohistochemistry, histology, and electron microscopy from fixed animal or human samples, or cultured cardiomyocytes. A model to investigate myocardial function and remodeling in vivo has been conspicuously lacking. The zebrafish, with its distinct transparency for imaging, has been a useful model for in vivo cardiac development studies, but historically has been less explored to address subcellular events in cardiomyopathy. In this work, we established a novel transgenic zebrafish line that allows detailed analysis of cardiomyocyte myofilament architecture and actin dynamics during development and in disease models in vivo for the first time. The simplicity of the LifeAct system, as well as the reference dataset compiled in this study, will help researchers resolve a variety of questions about cardiac development and pathology. The LifeAct system in this study was used in form of a GFP fusion. From our experience testing LifeAct with mCherry and BFP, we expect it to work with any monomeric fluorescent protein. A limitation of low affinity tags like LifeAct is their unsuitableness for Fluorescence Recovery after Photobleaching experiments as the constant binding and release of the LifeAct-tag to F-Actin leads to unrealistically fast recovery
To characterize this transgenic line, we used the sarcomere-deficient cardiac troponin T mutant to show that LifeAct-GFP expression most clearly labels cytoskeletal actin and myofibrils (Online Figure I). Using high-resolution imaging, we showed for the first time that myofibrils in different myocardial subtypes exhibit a highly distinct architecture that relates to their physiologic function. Atrial myofibrils are exclusively localized to the basal domain. They develop from an initially highly branched network of myofilaments that subsequently gets refined to non-branched linear myofibrils that mostly orient orthogonal to the direction of blood flow. How this refinement and orientation of myofibrils is achieved on a mechanistic level remains unclear and will need further attention in future studies. The orientation of myofibrils is maintained across intercalated discs, literally forming rings of myofibrils that allow the atrium to act as a peristaltic pump pushing the blood into the ventricle. Like the atrium, the ventricle initially forms as a single-layered chamber. The architecture of the myofibrils in compact wall cardiomyocytes, however, differs entirely from that in atrial cardiomyocytes. Compact wall cardiomyocytes start with a ring of thin myofibrils on their basolateral cortex and later develop a highly branched network of filamentous myofibrils in their apical domain that span the entirety of the area. Thicker myofibrils can only be found in the cortex of the basolateral domain, forming ring-like arrangements within each cardiomyocyte. Continuous myofibrillar arrangements across intercalated discs are absent, suggesting an entirely different mode of contraction in the early ventricle compared to the atrium; the ventricle contracts by a reduction in the area of each cardiomyocyte and consequently the contraction of the entire chamber causing a reduction in lumen volume. A third myocardial cell type, trabecular cardiomyocytes, develops from cardiomyocytes of the ventricular wall by first forming basal protrusions which are immediately filled with cortically localized thick myofibrils. Eventually, some cardiomyocytes leave the wall and differentiate into tube-like trabecular cells, which together form ring-like structures (trabeculae) on the inner surface of the ventricle. Each trabecular cardiomyocyte contains several long myofibrils that branch at the ends to connect to myofibrils of neighboring trabecular cells via intercalated discs. The arrangement of the trabecular rings is stereotypically orthogonal to the direction of blood flow from the ventricle. This detailed picture of organ-wide myofibril architecture facilitated the analysis of cardiomyopathies and allowed us to revisit questions about the relationship between anti-ERBB2/HER2 cancer therapies and DCM in previously unachievable resolution.
Since anti-ERBB2/HER2 drugs used to treat breast cancer can cause DCM in humans, we analyzed zebrafish erbb2 mutants in the Tg(myl7:LifeAct-GFP) background as a model for loss of Erbb2 signaling in cardiomyocytes. Our analysis revealed that mutant ventricular wall cardiomyocytes had an increase in cortically located myofibril width and a reduction in apically located filamentous myofibrils. To test whether this effect is a consequence of prolonged absence of Erbb2 signaling, we used a conditional system of chemical Erbb2 inhibition (PD168393) two days after trabeculation had started. Here, Erbb2 inhibition for 48 hours caused an increase in the diameter of the basolateral myofibrils. A similar effect was observed in trabecular cardiomyocytes. The diameter of their myofibrils increased to an extent that the centrally located, usually spheroid nuclei were forced into an ellipsoid shape by the surrounding myofibrils. We next asked whether the observed remodeling was cell-autonomous. We performed clonal analysis of dnErbb2 expression within wild-type hearts and obtained the same results as observed in constitutive loss of Erbb2 signaling. We further found that long-term inhibition of Erbb2 signaling by expressing dnErbb2 under the myl7 promoter caused DCM in zebrafish, a phenotype also reported in mouse7. We performed gene expression analysis in these transgenic hearts and found upregulation of DCM associated genes including acta2, vinculin, tcap and vmhc which encode components of the contractile machinery. Although the sample number was small due to high lethality, we found a clear dysregulation within this set of genes that was consistent with the myofibrillar phenotype found in erbb2 mutants. Together, these data suggest that the link between decreased Erbb2 signaling and DCM is not only about cell survival, as previously reported35, but also about direct action on myofibril remodeling.
Finally, our studies suggest that the increased incidence of acquired DCM in patients treated with both anti-ERBB2 therapies and DOXO may be because both drugs have direct effects on myofibril remodeling. In patients, sequential treatment with DOXO and ERBB2/HER2 antagonists synergistically promotes the development of DCM. Previous studies on cultured cardiomyocytes suggest a direct effect of DOXO on myofibrils assembly and maintenance33,34. We studied the effect of DOXO on cardiomyocytes in vivo and observed a dose-dependent effect on myofibrillar stability. Pericardial injections of 1 pL (4 mmol/L) DOXO completely a blated apical myofibrils of the thin type, the same type positively regulated by Erbb2 signaling. It is interesting to note that DOXO, implicated more often in irreversible Type I chemo-associated DCM, ablated these myofibrils, while loss of Erbb2 signaling, more often seen with reversible Type 2 chemo-associated DCM, reorganized myofibrils of the same type.
Taken together, our data show that both DOXO and Erbb2 inhibition independently affect the integrity and architecture of myofibrils. Intriguingly, ErbB2 signaling acts as a negative regulator of myofilament bundling and a promoter of thinner filamentous myofibrils. We speculate that it is these roles that cause the synergistically negative side effect of sequential DOXO and anti-ERBB2/HER2 therapy in which myofibrillar disarray caused by DOXO cannot be repaired during the phase of ERBB2/HER2 inhibition as myofibril and actin dynamics are reduced. Given our observations, we would like to suggest that breast cancer patients receiving an anti-ERBB2/HER2, doxorubicin, or combination therapy who develop DCM should be tested for variants that are genetically linked to familial cases of DCM so that we can learn more about the potential correlation of these variants with the development of therapy-induced DCM.
Further studies using our model in adults may investigate how Erbb2 acts on myofibril stability and sarcomere dynamics in order to identify mechanisms for other potential DCM therapies, as well as reveal other drugs likely to cause DCM. For example, a pharmacologic screen on Tg(myl7:LifeAct-GFP) zebrafish carrying the dnErbb2 transgene might identify new pharmacologic therapies for DCM. Several assays of adult zebrafish cardiac function, including systolic function, cardiac output, and response to exercise, could complement myofibril assessment using LifeAct-GFP. Additionally, adult Tg(myl7:LifeAct-GFP) zebrafish can be used to test potential drugs for cardiotoxicity and their effects on the cytoskeleton, either as a source for cultured cardiomyocytes or by direct imaging of fixed or freshly explanted hearts.
Supplementary Material
Novelty and Significance.
What Is Known?
The zebrafish model system offers optical transparency and external development combined with genetic tractability to study cardiac development, function and disease in a vertebrate model in vivo.
In contrast to direct tagging, fusions of affinity domains (e.g. LifeAct) to fluorescent proteins allow direct imaging of subcellular localization of the target protein (e.g. F-Actin) without interfering with overall function.
Anti-neoplastic therapies like Doxorubicin and ERBB2/HER2 antagonists induce dilated cardiomyopathy (DCM) with high incidence.
What New Information Does This Article Contribute?
A novel transgenic zebrafish line, which allows direct in vivo imaging of the sarcomere superstructure in high and low magnification and therefore greatly facilitates the analysis of myofibrillar phenotypes in development and disease without fixation artifacts.
A comprehensive reference dataset on myofibril development in different myocardial subtypes based on in vivo imaging.
A detailed analysis of the effect of impaired Erbb2 signaling on myofibrillar architecture and localization as well as the effect of doxorubicin treatment on myofibrillar integrity in vivo.
Myofibrillogenesis, myofibril contractility and myofibril remodeling are dynamic processes and dysfunctions in these processes are associated with cardiac diseases such as dilated cardiomyopathy. As yet, no tools are available for visualizing these processes at high resolution in vivo. The novel zebrafish transgenic line Tg(myl7:LifeAct-GFP) allowed visualization of myofibril organization in developing cardiomyocytes and in larval and adult zebrafish models of DCM. Myofibril architecture and organization were found to change dramatically as the heart developed, and were found to vary by myocardial subtype. Suppression of the ERBB2 signaling pathway caused reorganization of ventricular myofibrils and cardiac dysfunction, while doxorubicin treatment led to the dismantling of myofibrils. These data show, a direct, cell autonomous and highly specific effect of Erbb2 function on myofibril architecture and remodeling and provide a mechanistic explanation for the additive cardiotoxicity of doxorubicin and anti-ERBB2/HER2 inhibitors are used together. The Tg(myl7:LifeAct-GFP) zebrafish line serves as a new tool to investigate changes in contractile dynamics in response to a broad range of genetic mutations and pharmacologic treatments. Clinically, this tool could be adapted to screen for new pharmacologic therapies for DCM, as well as to identify drugs with cardiotoxic side effects.
Acknowledgments
We would like to acknowledge Bilge Reischauer, Oliver Stone and Marion Delous for helpful discussions and critical feedback on the manuscript, and thank Zacharias Kontarakis for help with some of the experiments.
SOURCES OF FUNDING
This work was supported by fellowships from the Deutsche Forschungsgemeinschaft and American Heart Association (S.R.), the NIH (F32HL110489) and Sarnoff Cardiovascular Research Foundation (R.A.), and grants from the NIH (RO1HL54737), the Packard Foundation and the Max Planck Society (D.Y.R.S.)
Nonstandard Abbreviations and Acronyms
- ACTA2
α-actin 2
- ACTC
cardiac actin
- AVC
Atrioventricular canal
- BFP
blue fluorescent protein
- DCM
dilated cardiomyopathy
- DMSO
dimethyl sulfoxide
- DOXO
doxorubicin
- dpf
days post fertilization
- EGFR
epidermal growth factor receptor
- F-actin
filamentous actin
- GAPDH
glyceraldehyde-3-phosphate dehydrogenase
- GFP
green fluorescent protein
- hpf
hours post fertilization
- KCNJ3a
potassium inwardly-rectifying channel, subfamily J, member 3a
- MYH7
myosin heavy chain 7
- MYL7
myosin light chain 7
- PDLIM3A
PDZ and LIM domain 3a
- RFP
red fluorescent protein
- SPIM
selective plane illumination microscopy
- TCAP
titin-cap
- TNNT2a
troponin T type 2a
- TPM1
α-tropomyosin
- VMHC
ventricular myosin heavy chain
Footnotes
DISCLOSURES
S.R., R.A., R.R., D.Y.R.S have no disclosures.
References
- 1.Rakar S, Sinagra G, Di Lenarda A, Poletti A, Bussani R, Silvestri F, Camerini F. Epidemiology of dilated cardiomyopathy. A prospective post-mortem study of 5252 necropsies. The Heart Muscle Disease Study Group. Eur Heart J. 1997;18:117–123. doi: 10.1093/oxfordjournals.eurheartj.a015092. [DOI] [PubMed] [Google Scholar]
- 2.Baselga J. Current and planned clinical trials with trastuzumab (Herceptin) Semin Oncol. 2000;27:27–32. [PubMed] [Google Scholar]
- 3.Taylor MRG, Carniel E, Mestroni L. Cardiomyopathy, familial dilated. Orphanet J Rare Dis. 2006;1:27. doi: 10.1186/1750-1172-1-27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Merlo M, Sinagra G, Carniel E, Slavov D, Zhu X, Barbati G, Spezzacatene A, Ramani F, Salcedo E, Di Lenarda A, Mestroni L, Taylor MRG Familial Cardiomyopathy Registry. Poor prognosis of rare sarcomeric gene variants in patients with dilated cardiomyopathy. Clin Transl Sci. 2013;6:424–428. doi: 10.1111/cts.12116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hershberger RE, Hedges DJ, Morales A. Dilated cardiomyopathy: the complexity of a diverse genetic architecture. Nat Rev Cardiol. 2013;10:531–547. doi: 10.1038/nrcardio.2013.105. [DOI] [PubMed] [Google Scholar]
- 6.Crone SA, Zhao Y-Y, Fan L, Gu Y, Minamisawa S, Liu Y, Peterson KL, Chen J, Kahn R, Condorelli G, Jr JR, Chien KR, Lee K-F. ErbB2 is essential in the prevention of dilated cardiomyopathy. Nat Med. 2002;8:459–465. doi: 10.1038/nm0502-459. [DOI] [PubMed] [Google Scholar]
- 7.Ozcelik C, Erdmann B, Pilz B, Wettschureck N, Britsch S, Hübner N, Chien KR, Birchmeier C, Garratt AN. Conditional mutation of the ErbB2 (HER2) receptor in cardiomyocytes leads to dilated cardiomyopathy. Proc Natl Acad Sci U S A. 2002;99:8880–8885. doi: 10.1073/pnas.122249299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Beis D, Stainier DYR. In vivo cell biology: following the zebrafish trend. Trends Cell Biol. 2006;16:105–112. doi: 10.1016/j.tcb.2005.12.001. [DOI] [PubMed] [Google Scholar]
- 9.Riedl J, Crevenna AH, Kessenbrock K, Yu JH, Neukirchen D, Bista M, Bradke F, Jenne D, Holak TA, Werb Z, Sixt M, Wedlich-Soldner R. Lifeact: a versatile marker to visualize F-actin. Nat Methods. 2008;5:605–607. doi: 10.1038/nmeth.1220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Andrechek ER, Hardy WR, Girgis-Gabardo AA, Perry RLS, Butler R, Graham FL, Kahn RC, Rudnicki MA, Muller WJ. ErbB2 is required for muscle spindle and myoblast cell survival. Mol Cell Biol. 2002;22:4714–4722. doi: 10.1128/MCB.22.13.4714-4722.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Muthuswamy SK, Li D, Lelievre S, Bissell MJ, Brugge JS. ErbB2, but not ErbB1, reinitiates proliferation and induces luminal repopulation in epithelial acini. Nat Cell Biol. 2001;3:785–792. doi: 10.1038/ncb0901-785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Heermann S, Schwab MH. Molecular control of Schwann cell migration along peripheral axons. Cell Adhes Migr. 2013;7:18–22. doi: 10.4161/cam.22123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Citri A, Yarden Y. EGF–ERBB signalling: towards the systems level. Nat Rev Mol Cell Biol. 2006;7:505–516. doi: 10.1038/nrm1962. [DOI] [PubMed] [Google Scholar]
- 14.Slamon DJ, Godolphin W, Jones LA, Holt JA, Wong SG, Keith DE, Levin WJ, Stuart SG, Udove J, Ullrich A. Studies of the HER-2/neu proto-oncogene in human breast and ovarian cancer. Science. 1989;244:707–712. doi: 10.1126/science.2470152. [DOI] [PubMed] [Google Scholar]
- 15.Incorvati JA, Shah S, Mu Y, Lu J. Targeted therapy for HER2 positive breast cancer. J Hematol Oncol J Hematol Oncol. 2013;6:38. doi: 10.1186/1756-8722-6-38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gassmann M, Casagranda F, Orioli D, Simon H, Lai C, Klein R, Lemke G. Aberrant neural and cardiac development in mice lacking the ErbB4 neuregulin receptor. Nature. 1995;378:390–394. doi: 10.1038/378390a0. [DOI] [PubMed] [Google Scholar]
- 17.Lee KF, Simon H, Chen H, Bates B, Hung MC, Hauser C. Requirement for neuregulin receptor erbB2 in neural and cardiac development. Nature. 1995;378:394–398. doi: 10.1038/378394a0. [DOI] [PubMed] [Google Scholar]
- 18.Staudt DW, Liu J, Thorn KS, Stuurman N, Liebling M, Stainier DYR. High-resolution imaging of cardiomyocyte behavior reveals two distinct steps in ventricular trabeculation. Development. 2014;141:585–593. doi: 10.1242/dev.098632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Liu J, Bressan M, Hassel D, Huisken J, Staudt D, Kikuchi K, Poss KD, Mikawa T, Stainier DYR. A dual role for ErbB2 signaling in cardiac trabeculation. Dev Camb Engl. 2010;137:3867–3875. doi: 10.1242/dev.053736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Slamon DJ, Leyland-Jones B, Shak S, Fuchs H, Paton V, Bajamonde A, Fleming T, Eiermann W, Wolter J, Pegram M, Baselga J, Norton L. Use of chemotherapy plus a monoclonal antibody against HER2 for metastatic breast cancer that overexpresses HER2. N Engl J Med. 2001;344:783–792. doi: 10.1056/NEJM200103153441101. [DOI] [PubMed] [Google Scholar]
- 21.Suster ML, Kikuta H, Urasaki A, Asakawa K, Kawakami K. Transgenesis in zebrafish with the tol2 transposon system. Methods Mol Biol Clifton NJ. 2009;561:41–63. doi: 10.1007/978-1-60327-019-9_3. [DOI] [PubMed] [Google Scholar]
- 22.Kashles O, Yarden Y, Fischer R, Ullrich A, Schlessinger J. A dominant negative mutation suppresses the function of normal epidermal growth factor receptors by heterodimerization. Mol Cell Biol. 1991;11:1454–1463. doi: 10.1128/mcb.11.3.1454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lyons DA, Pogoda H-M, Voas MG, Woods IG, Diamond B, Nix R, Arana N, Jacobs J, Talbot WS. erbb3 and erbb2 are essential for schwann cell migration and myelination in zebrafish. Curr Biol CB. 2005;15:513–524. doi: 10.1016/j.cub.2005.02.030. [DOI] [PubMed] [Google Scholar]
- 24.Mably JD, Burns CG, Chen J-N, Fishman MC, Mohideen M-APK. heart of glass Regulates the Concentric Growth of the Heart in Zebrafish. Curr Biol. 2003;13:2138–2147. doi: 10.1016/j.cub.2003.11.055. [DOI] [PubMed] [Google Scholar]
- 25.Kaufmann A, Mickoleit M, Weber M, Huisken J. Multilayer mounting enables long-term imaging of zebrafish development in a light sheet microscope. Development. 2012;139:3242–3247. doi: 10.1242/dev.082586. [DOI] [PubMed] [Google Scholar]
- 26.Liebling M, Forouhar AS, Gharib M, Fraser SE, Dickinson ME. Four-dimensional cardiac imaging in living embryos via postacquisition synchronization of nongated slice sequences. J Biomed Opt. 2005;10:054001. doi: 10.1117/1.2061567. [DOI] [PubMed] [Google Scholar]
- 27.Reischauer S, Levesque MP, Nüsslein-Volhard C, Sonawane M. Lgl2 Executes Its Function as a Tumor Suppressor by Regulating ErbB Signaling in the Zebrafish Epidermis. PLoS Genet. 2009;5:e1000720. doi: 10.1371/journal.pgen.1000720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Marshall OJ. PerlPrimer: cross-platform, graphical primer design for standard, bisulphite and real-time PCR. Bioinforma Oxf Engl. 2004;20:2471–2472. doi: 10.1093/bioinformatics/bth254. [DOI] [PubMed] [Google Scholar]
- 29.Sander V, Suñe G, Jopling C, Morera C, Izpisua Belmonte JC. Isolation and in vitro culture of primary cardiomyocytes from adult zebrafish hearts. Nat Protoc. 2013;8:800–809. doi: 10.1038/nprot.2013.041. [DOI] [PubMed] [Google Scholar]
- 30.Sehnert AJ, Huq A, Weinstein BM, Walker C, Fishman M, Stainier DYR. Cardiac troponin T is essential in sarcomere assembly and cardiac contractility. Nat Genet. 2002;31:106–110. doi: 10.1038/ng875. [DOI] [PubMed] [Google Scholar]
- 31.Stainier DYR, Fishman MC. Patterning the zebrafish heart tube: Acquisition of anteroposterior polarity. Dev Biol. 1992;153:91–101. doi: 10.1016/0012-1606(92)90094-w. [DOI] [PubMed] [Google Scholar]
- 32.Peshkovsky C, Totong R, Yelon D. Dependence of cardiac trabeculation on neuregulin signaling and blood flow in zebrafish. Dev Dyn. 2011;240:446–456. doi: 10.1002/dvdy.22526. [DOI] [PubMed] [Google Scholar]
- 33.Sawyer DB, Zuppinger C, Miller TA, Eppenberger HM, Suter TM. Modulation of Anthracycline-Induced Myofibrillar Disarray in Rat Ventricular Myocytes by Neuregulin-1β and Anti-erbB2: Potential Mechanism for Trastuzumab-Induced Cardiotoxicity. Circulation. 2002;105:1551–1554. doi: 10.1161/01.cir.0000013839.41224.1c. [DOI] [PubMed] [Google Scholar]
- 34.Lim CC, Zuppinger C, Guo X, Kuster GM, Helmes M, Eppenberger HM, Suter TM, Liao R, Sawyer DB. Anthracyclines Induce Calpain-dependent Titin Proteolysis and Necrosis in Cardiomyocytes. J Biol Chem. 2004;279:8290–8299. doi: 10.1074/jbc.M308033200. [DOI] [PubMed] [Google Scholar]
- 35.Fukazawa R, Miller TA, Kuramochi Y, Frantz S, Kim YD, Marchionni MA, Kelly RA, Sawyer DB. Neuregulin-1 protects ventricular myocytes from anthracycline-induced apoptosis via erbB4-dependent activation of PI3-kinase/Akt. J Mol Cell Cardiol. 2003;35:1473–1479. doi: 10.1016/j.yjmcc.2003.09.012. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.