

Abstract
Evidence indicates that broad, nonspecific histone deacetylase (HDAC) inhibition enhances learning and memory, however, the contribution of the various HDACs to specific forms of learning is incompletely understood. Here, we show that the Class I HDAC inhibitor, RGFP963, enhances consolidation of cued fear extinction. However, RGFP966, a strong inhibitor of HDAC3, does not significantly enhance consolidation of cued fear extinction. These data extend previous evidence that demonstrate the Class I HDACs play a role in the consolidation of long-term memory, suggesting that HDAC1 and/or HDAC2, but less likely HDAC3, may function as negative regulators of extinction retention. The development of specific HDAC inhibitors, such as RGFP963, will further illuminate the role of specific HDACs in various types of learning and memory. Moreover, HDAC inhibitors that enhance cued fear extinction may show translational promise for the treatment of fear-related disorders, including post-traumatic stress disorder (PTSD).
Post-traumatic stress disorder (PTSD) and other fear-related disorders are characterized by pathological fear and anxiety. An inability to control fear has led researchers and clinicians to hypothesize that PTSD is a disorder in the inhibition, or extinction, of fear. Evidence suggests that extinction learning establishes a new, inhibitory memory trace that suppresses previously established fear memories (Myers and Davis 2007). Numerous studies implicate the amygdala, hippocampus, and medial prefrontal cortex in the acquisition and extinction of aversive memories (Goosens and Maren 2001; Milad and Quirk 2002; Akirav et al. 2006; Heldt et al. 2007; Herry et al. 2008).
At the molecular level, cued fear and extinction are forms of long-term memory that are consolidated via changes in the expression of specific genes in the amygdala and other associated regions (Josselyn et al. 2001; Ressler et al. 2002; Ploski et al. 2008, 2010). During the consolidation window, generally thought to occur minutes to hours after learning, transcription of specific genes is associated with the activity of various histone deacetylase enzymes (HDACs) (Dudai 2004).
Broad, nonspecific HDAC inhibition enhances emotional learning and cued fear extinction (Bredy et al. 2007; Bredy and Barad 2008). The Class I HDACs, which include HDAC1, HDAC2, HDAC3, and HDAC8, are associated with learning and memory (Bredy et al. 2007; Bredy and Barad 2008; Whittle et al. 2013; Hait et al. 2014; Whittle and Singewald 2014). Specifically, HDAC3 appears to be a negative regulator of long-term memory for spatial learning and extinction of drug-seeking behavior (McQuown et al. 2011; McQuown and Wood 2011; Malvaez et al. 2013). Overexpression of HDAC2 causes deficits in context and cue-dependent fear learning in mice. Conversely, HDAC2 knockout mice exhibit enhanced context and cued fear memory (Guan et al. 2009). Similarly, forebrain-specific knockout of HDAC2 enhances context fear and cued fear extinction (Morris et al. 2013). Furthermore, modulation of HDAC1 expression or activity alters extinction of context fear extinction, where overexpression of HDAC1 enhances extinction and siRNA knockdown or inhibition of HDAC1 blocks extinction (Bahari-Javan et al. 2012). However, others demonstrate little to no effect on memory consolidation by HDAC1 (Guan et al. 2009; Morris et al. 2013).
As the Class I HDACs have been increasingly implicated in learning and memory processes, the need for specific HDAC inhibitor compounds has increased. Two compounds with unique inhibitory properties have been developed—RGFP966 and RGFP963. To elaborate on previous studies which have demonstrated a role for Class I HDACs in memory consolidation and to determine whether RGFP966 and/or RGFP963 show translational potential for the treatment of post-traumatic stress disorder, we administered RGFP966 and RGFP963 after extinction training in a mouse model. The results of this study and others will be crucial to identify and develop HDAC inhibitors that could ameliorate specific symptoms of fear-related disorders.
RGFP966 and RGFP963 were developed by RepliGen Corp. and sent to Reaction Biology Corp. to determine inhibitory potency against all 11 HDAC enzymes. RGFP963 and RGFP966 were prepared in HDAC assay buffer (25 mM Tris/HCl, 137 mM NaCl, 2.7 mM KCl, 1 mM MgCl2, pH 8.0) in 96-well assay plates from DMSO stock solutions. RGFP963 and RGFP966 were preincubated for 2 h at room temperature in the presence of 100 mg/mL BSA and purified recombinant HDAC enzymes at specific concentrations. Compounds were tested in 10-dose IC50 mode in duplicate with threefold serial dilution starting at 20 μM. The half-maximal inhibitory concentration (IC50) was used to measure the inhibitory potency of each compound against all 11 HDAC enzymes. Following preincubation, fluorogenic HDAC substrate was added and plates were incubated for 30 min at room temperature. The enzymatic reaction was stopped by addition of Trichostatin A and trypsin. After a 15-min incubation at room temperature, fluorescence was recorded using a Spectramax M2 fluorometer with excitation at 365 nm and emission at 460 nm. IC50 values were calculated using a sigmoidal dose–response (variable slope) equation in GraphPad Prism 5. RGFP966 and RGFP963 show effective inhibitory potency for the Class I HDAC enzymes (Fig. 1A). RGFP966 exhibited specific inhibition of HDAC3, while RGFP963 broadly inhibited HDAC1, HDAC2, and HDAC3. RGFP963 also showed weak inhibition of HDAC10 with an IC50 value of 10 µM. RGFP963 and RGFP966 did not inhibit any other HDACs besides HDAC1, HDAC2, HDAC3, and HDAC10.
Figure 1.

RGFP963 and RGFP966 compound properties in vitro and in vivo. (A) RGFP966 exhibits specific inhibition of HDAC3, while RGFP963 broadly inhibits HDAC1, HDAC2, and HDAC3 in vitro. (B) Chemical structures of RGFP963 and RGFP966. (C,D) Of note, 10 mg/kg RGFP966 and RGFP963 are detected 0.25, 1, and 2 h post-IP injection (n = 3/group). Of note, 10 mg/kg RGFP966 is capable of inhibiting HDAC3, while minimally inhibiting HDAC1 and HDAC2, at the various time points assessed. Of note, 10 mg/kg RGFP963 is capable of inhibiting HDAC1, HDAC2, and HDAC3 at the various time points assessed. (E–P″) Vehicle, 10 mg/kg RGFP966, and 10 mg/kg RGFP963 were administered IP (n = 2/group). Subjects were perfused 1 h after drug administration and then tissue was processed for immunocytochemistry to determine an effect of drug on global H3 and H4 acetylation levels in the basolateral amygdala (BLA) and dentate gyrus (DG) region of the hippocampus. (E–E″) Photomicrographs showing colocalization of DAPI (E) and global acetyl H3 (E′) in the BLA with vehicle administration (E″ merged). (F–F″) Photomicrographs showing colocalization of DAPI (F) and global acetyl H3 (F′) in the BLA with 10 mg/kg RGFP966 administration (F″ merged). (G–G″) Photomicrographs showing colocalization of DAPI (G) and global acetyl H3 (G′) in the BLA with 10 mg/kg RGFP963 administration (G″ merged). (H–H″) Photomicrographs showing colocalization of DAPI (H) and global acetyl H3 (H′) in the DG with vehicle administration (H″ merged). (I–I″) Photomicrographs showing colocalization of DAPI (I) and global acetyl H3 (I′) in the DG with 10 mg/kg RGFP966 administration (I″ merged). (J–J″) Photomicrographs showing colocalization of DAPI (J) and global acetyl H3 (J′) in the DG with 10 mg/kg RGFP963 administration (J″ merged). (K–K″) Photomicrographs showing colocalization of DAPI (K) and global acetyl H4 (K′) in the BLA with vehicle administration (K″ merged). (L–L″) Photomicrographs showing colocalization of DAPI (L) and global acetyl H4 (L′) in the BLA with 10 mg/kg RGFP966 administration (L″ merged). (M–M″) Photomicrographs showing colocalization of DAPI (M) and global acetyl H4 (M′) in the BLA with 10 mg/kg RGFP963 administration (M″ merged). (N–N″) Photomicrographs showing colocalization of DAPI (N) and global acetyl H4 (N′) in the DG with vehicle administration (N″ merged). (O–O″) Photomicrographs showing colocalization of DAPI (O) and global acetyl H4 (O′) in the DG with 10 mg/kg RGFP966 administration (O″ merged). (P–P″) Photomicrographs showing colocalization of DAPI (P) and global acetyl H4 (P′) in the DG with 10 mg/kg RGFP963 administration (P″ merged).
To better understand how RGFP963 and RGFP966 act in vivo, we performed pharmacokinetic studies. Adult male C57BL/6J mice 8–12 wk old were dosed intraperitoneally (IP) with RGFP963 or RGFP966. RGFP963 and RGFP966 were dissolved in DMSO (calculated to be 5% of the final volume) and diluted with 30% Hydroxypropyl Beta-Cyclodextran, 0.1 M acetate, pH 5.4. Both drugs were administered at a dose of 10 mg/kg. Animals were sacrificed 0.25, 1, or 2 h after drug administration. Blood was collected via cardiac puncture and stored in EDTA tubes. Brains were flash frozen. Samples were shipped to RepliGen for analysis. Brains were homogenized, followed by protein precipitation with a solution of internal standard in acetonitrile. Samples were filtered and analyzed by LC/MS/MS. The LC/MS/MS system consisted of an Agilent 1100 series quaternary pump and micro degasser (Agilent), a CTC PAL autosampler (Leap Technologies), and an API 4000 triple quadrupole mass spectrometer equipped with an ESI TurboV source (AB Sciex). The system is controlled by Analyst 1.5.2 (AB Sciex) running on Windows XP Professional SP3 (Microsoft). Compound concentrations were determined by calibration curves, which consist of MS response ratios of compounds to an internal standard versus concentration, prepared for each compound in each tissue type using the same sample preparation protocol. Brain and plasma concentrations (in nanograms/gram and nanograms/milliliter, respectively) were averaged according to compound, tissue, and time point. An n of 3 animals were used for each time point (per compound).
RGFP966 and RGFP963 were able to cross the blood brain barrier, however, RGFP966 exhibited lower brain penetrance compared with RGFP963. Maximum brain concentrations were observed at early time points (15 min post-drug administration). Concentration of each compound decreased with an estimated elimination half-life of ∼45 min. Central nervous system (CNS) concentration of RGFP963 was 20- to 80-fold more than the IC50 of RGFP963 required to inhibit HDAC1. For RGFP966, CNS concentration was twofold lower than its respective HDAC1 and HDAC2 IC50. However, maximal brain concentrations of RGFP963 and RGFP966 were 160- and 110-fold their respective HDAC3 IC50 values. We conclude that RGFP963 was able to access the brain and reach a concentration capable of inhibiting HDAC1, HDAC2, and HDAC3 when administered systemically at a dose of 10 mg/kg. At the same dose, RGFP966 was able to specifically inhibit HDAC3, while minimally inhibiting HDAC1 and HDAC2 (Fig. 1C,D). These data suggest that 10 mg/kg RGFP963 and RGFP966 would be active during the consolidation window when administered after learning, exhibiting selective inhibition of HDAC1, HDAC2, and HDAC3 activity.
To determine whether 10 mg/kg RGFP966 and 10 mg/kg RGFP963 differentially affect histone acetylation in vivo, subjects were perfused 1 h after drug administration. Tissue was then processed for immunocytochemistry to determine an effect of drug on global histone H3 (anti-acetyl histone H3 antibody Millipore 06-599) and histone H4 (anti-acetyl histone H4 Millipore 06-598) acetylation levels in the basolateral amygdala (BLA) and dentate gyrus (DG) region of the hippocampus, two areas critical for consolidation of emotional long-term memory. Immunofluorescence experiments were performed as described previously (Bowers and Ressler 2015). While 10 mg/kg RGFP966 seems to increase global acetylation of histone H3 in the BLA compared with vehicle and 10 mg/kg RGFP963 (Fig. 1E–G″), both RGFP963 and RGFP966 seem to decrease global acetylation of histone H3 and H4 in the dentate gyrus (Fig. 1H–J″, N–P″). Neither RGFP966 nor RGFP963 appear to alter global H4 acetylation in the BLA (Fig. 1K–M″). While future experiments should more quantitatively examine the effect of these HDACi compounds on global acetylation of histone H3 and H4 (as well as other histones and specific histone residues), these experiments demonstrate differential in vivo effects of RGFP963 and RGFP966 on global histone H3 acetylation in the BLA, which could underlie differences in observed behavioral effects of these drugs.
For behavioral experiments, adult male C57BL/6J mice 8–12 wk old were group-housed 1 wk prior to testing. Subjects were kept in a temperature-controlled (24°C) animal colony, with ad libitum access to food and water, on a 12-h light–dark cycle. All behavioral procedures were performed during the light cycle. Subjects were handled once per day for 2 d. Twenty-four hours after the last day of handling and 1 d prior to fear conditioning, subjects were habituated to the test chambers. Associative fear conditioning occurred over 2 d and consisted of 10 conditioned stimulus (CS) tones (30 sec, 6 kHz, 75 dB) coterminating with unconditioned stimulus (US) footshocks (500 msec, 1 mA) with a 3-min inter-trial interval (ITI) in “Context A” (MED Associates, standard fear conditioning chambers, Product Number VFC-008). As we hypothesized that RGFP966 and RGFP963 would enhance extinction memory consolidation (as measured by lower levels of freezing behavior during extinction retention tests), we trained subjects to 2 d of associative fear conditioning to maximize freezing levels and avoid a floor effect of extinction training on freezing during extinction retention tests (Bredy and Barad 2008).
Twenty-four hours prior to extinction training and 48 h after fear conditioning, subjects were tested to three CS trials (“grouping”) in a novel context, “Context B”. “Context B” consisted of the same experimental chambers as in “Context A” with changes to visual, tactile, and olfactory cues—PLEXIGLAS covered the shock bars and subjects were exposed to red light and a 1% ammonium hydroxide scent. Freezing to the three trials was averaged and used to sort subjects into three groups (n = 12/group) with equivalent freezing levels. Freezing (the absence of movement except for respiration) in response to the CS was measured using FreezeView software (Coulbourn Instruments). Twenty-four hours following the three CS test, subjects were extinction trained. Extinction training consisted of 30 presentations of the CS (30 sec, 6 kHz, 75 dB) separated with a 30 sec ITI. Vehicle or drug was administered intraperitoneally (IP) 5 min after the last CS trial of extinction training, to explicitly examine an effect of drug on consolidation.
Using a repeated-measures ANOVA, we did not observe a significant main effect of group on freezing across 3 CS or 30 CS trials during grouping (F(2,33) = 0.01, NS) or extinction training (F(2,33) = 1.97, NS). Furthermore, we did not observe an interaction between group and CS trial during grouping (F(4,66) = 0.56, NS) or extinction training (F(19.17,316.23) = 0.63, NS), suggesting that freezing behavior of vehicle, RGFP963, and RGFP966 did not significantly differ from one another at any specific CS trial (Fig. 2C). However, we did observe a significant effect of CS trial on freezing (F(9.58,316.23) = 3.95, P < 0.05) during extinction training, indicating that all groups exhibited within-session extinction. Twenty-four and 48 h later, subjects were tested for retention of extinction training with 15 CS trials. Extinction retention was defined as average freezing to the first 5 CS trials of the 15 CS test. Repeated-measures ANOVA revealed a significant main effect of drug on freezing across extinction retention days 1 and 2 (F(2,33) = 4.33, P < 0.05). A post hoc least significant difference (LSD) test revealed significant differences between 10 mg/kg RGFP963 versus vehicle and 10 mg/kg RGFP963 versus 10 mg/kg RGFP966 (P < 0.05) across extinction retention days (Fig. 2B). We did not detect a significant interaction between drug and extinction retention day (F(2,33) = 0.10, NS). Using a repeated-measures ANOVA, and defining drug as a between-subjects factor and CS as a within-subjects factor, we did not observe a significant main effect of drug across all 15 CS trials on extinction retention day 1 or extinction retention day 2 (extinction retention 1: F(2,33) = 2.81, NS; extinction retention 2: F(2,33) = 1.74, NS) (Fig. 2C). Additionally, we did not observe a significant difference between groups (F(2,33) = 2.32, NS), or an interaction between group and day (F(2,33) = 0.1, NS), when comparing average freezing to the last five CS of extinction training and the first five CS of extinction retention test 1 (repeated-measures ANOVA, between-subjects factor: drug group, within-subjects factor: average freezing to the last five CS of extinction training and average freezing to the first five CS of extinction retention test 1). We conclude that RGFP963, and not RGFP966, enhances consolidation of extinction learning at a dose of 10 mg/kg (Tables 1, 2).
Figure 2.
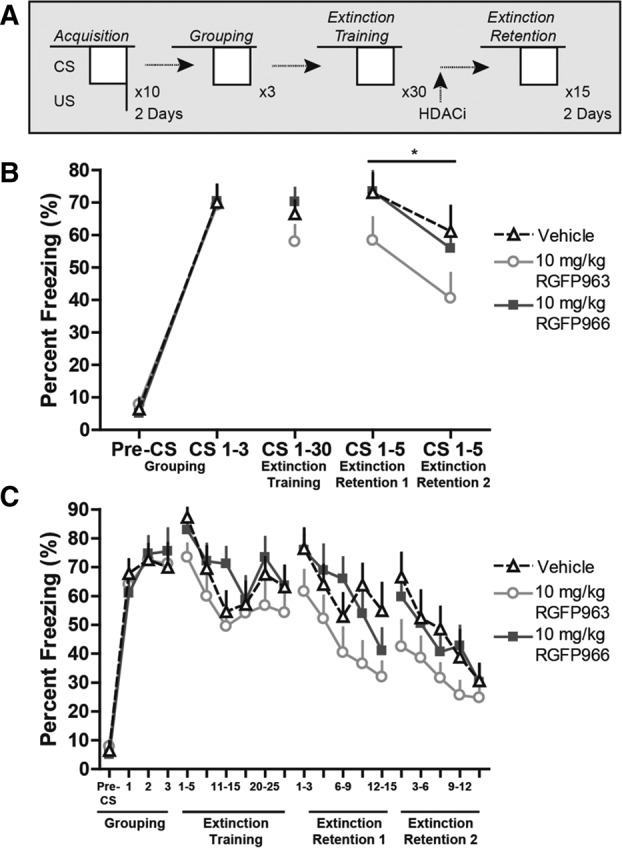
Systemic administration of 10 mg/kg RGFP963 enhances consolidation of cued fear extinction. (A) Fear conditioning and extinction paradigm. No significant differences in freezing behavior between groups prior to drug administration were observed during grouping or extinction training (n = 12/group). Twenty-four and 48 h later, subjects were tested for retention of extinction training with 15 CS trials. Extinction retention was defined as average freezing to the first 5 CS trials of the 15 CS test. (B) Repeated-measures ANOVA revealed a significant main effect of drug on freezing across extinction retention days 1 and 2 (P < 0.05). A post hoc least significant difference (LSD) test revealed significant differences between 10 mg/kg RGFP963 versus vehicle and 10 mg/kg RGFP963 versus 10 mg/kg RGFP966 (P < 0.05). (C) No significant differences in within-session extinction were detected between groups on extinction training, extinction retention 1, or extinction retention 2 test days.
Table 1.
Mean and SEM for data in Figure 2B
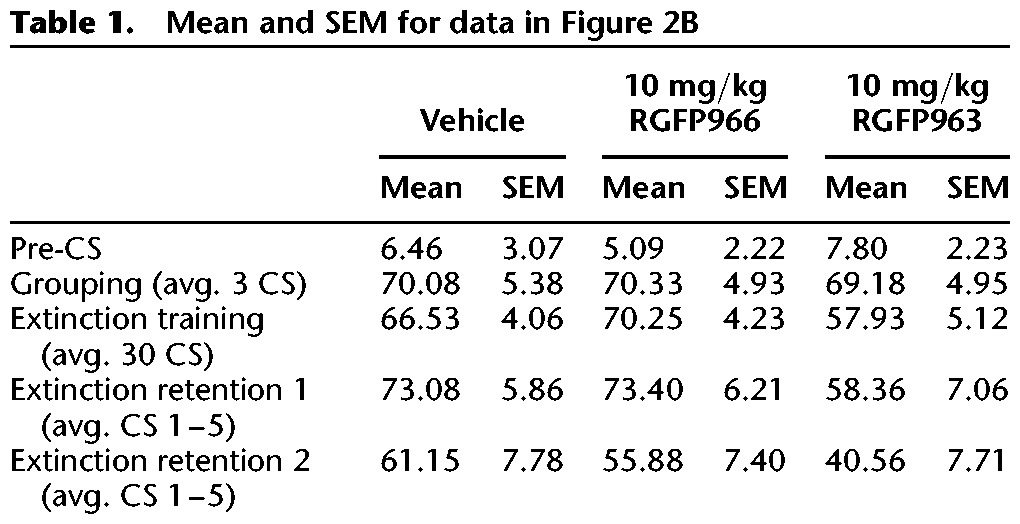
Table 2.
Mean and SEM for data in Figure 2C
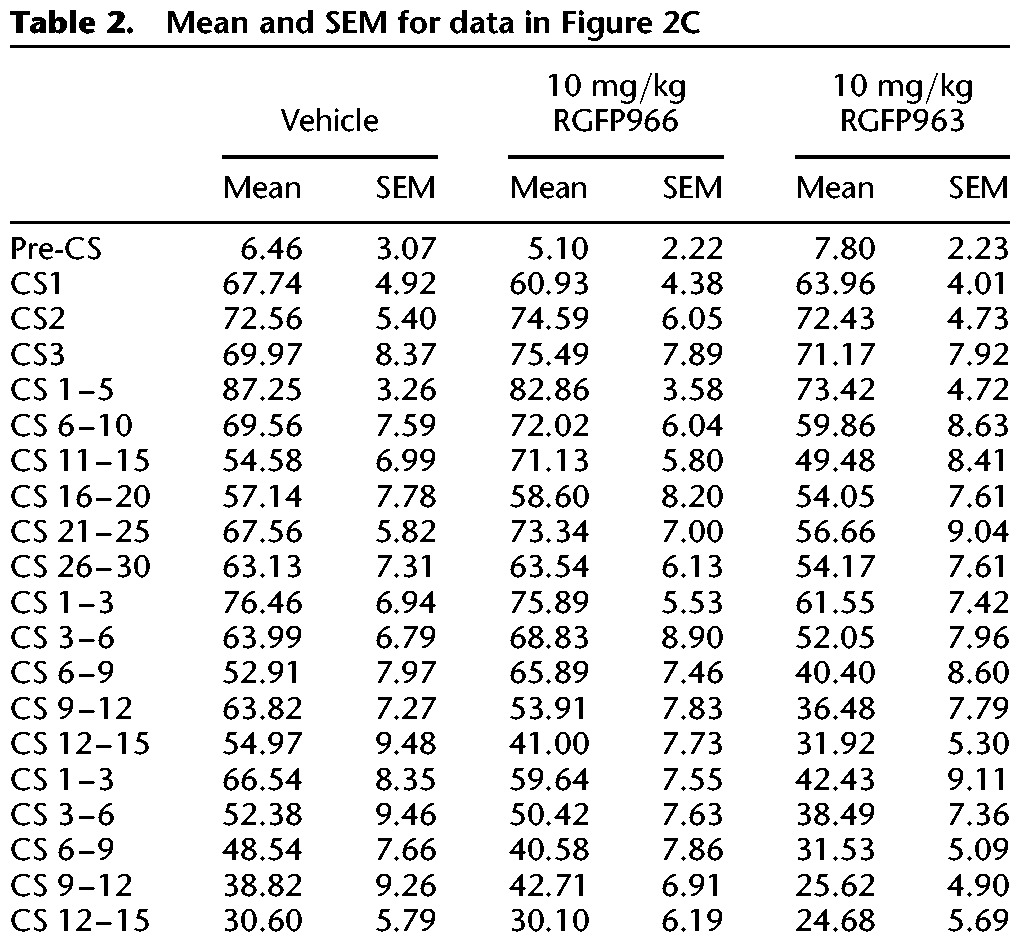
To test whether RGFP963 nonspecifically enhances extinction retention, we: (1) fear conditioned subjects, (2) grouped subjects for equivalent freezing levels 48 h after the last day of fear conditioning, (3) administered drug systemically (IP) without extinction training 24 h after grouping, and (4) tested subjects to extinction retention tests 24 and 48 h after drug administration (Fig. 3). The same training paradigm from the previous experiment was maintained, where subjects received 2 d of 10 CS–US pairings during fear conditioning in “Context A”, 3 CS trials during grouping, and 15 CS trials during extinction retention (all extinction tests were performed in “Context B”), however, only one group, “extinction” underwent a 30 CS trial extinction training between grouping and extinction retention tests. The following groups were tested: “home cage (HC),” “extinction,” “vehicle-no extinction,” “10 mg/kg RGFP963-no extinction,” “10 mg/kg RGFP966-no extinction” (n = 10/group). Interestingly, we did not observe a significant difference between groups on extinction retention day 1 (F(4,45) = 0.47, NS) or extinction retention day 2 (F(4,45) = 1.11, NS) using a repeated-measures ANOVA and defining drug as a between-subjects factor and CS as a within-subjects factor. Furthermore, we did not observe a significant difference (F(4,45) = 1.00, NS) between groups when we define extinction retention as the first 5 CS of the 15 CS test and look across extinction retention days 1 and 2, as in the previous experiment. This suggested that extinction training consisting of 30 CS trials does not significantly diminish freezing after a relatively robust fear conditioning paradigm of 2 d of 10 CS–US pairings, rather than a nonspecific enhancement of extinction retention by RGFP966 or RGFP963 (as we did not detect a significant difference between either HDACi group and vehicle or home cage groups). This conclusion was supported by analysis comparing average freezing to all three CS during grouping to average freezing to the first three CS during extinction retention test 1. We did not observe a significant interaction (F(4,45) = 1.24, NS) between group and day (grouping and extinction retention day 1). Importantly, we did observe an effect of CS on freezing during extinction training in our “extinction group” (F(29,261) = 1.97, P < 0.05), suggesting that within-session extinction was maintained, despite a lack of effect of extinction training on extinction retention (Fig. 3A) (Table 3).
Figure 3.
Systemic administration of 10 mg/kg RGFP963 and 10 mg/kg RGFP966 does not nonspecifically affect extinction retention, motor, or exploratory behavior. (A) To test whether RGFP963 and RGFP966 nonspecifically affect extinction retention, subjects received 2 d of 10 CS–US pairings during fear conditioning in “Context A,” 3 CS trials during grouping, and 15 CS trials during extinction retention (all extinction tests were performed in “Context B”), however, only one group, “extinction” underwent a 30 CS trial extinction training between grouping and extinction retention tests. The following groups were tested: “home cage (HC),” “extinction,” “vehicle-no extinction,” “10 mg/kg RGFP963-no extinction,” “10 mg/kg RGFP966-no extinction” (n = 10/group). No significant differences were observed on extinction retention day 1 or extinction retention day 2. A significant effect of CS on freezing during extinction training in the “extinction group” was observed (P < 0.05), suggesting that within-session extinction was maintained, despite a lack of effect on extinction retention. These data suggest that extinction training consisting of 30 CS trials does not significantly diminish freezing after a relatively robust fear conditioning paradigm of 2 d of 10 CS–US pairings and that 10 mg/kg RGFP963 and RGFP966 does not nonspecifically enhance extinction retention. (B,C) Thirty minutes prior to testing, RGFP963 and RGFP966 were administered IP at a dose of 10 mg/kg. No significant effect of drug on time spent in the center (sec) or distance traveled (cm) was observed (n = 8/group).
Table 3.
Mean and SEM for data in Figure 3A
To test whether RGFP963 or RGFP966 have an effect on exploratory behavior (which can indicate an effect on anxiety-like behavior) and/or locomotion that could confound the observed effect on extinction retention, we performed an open-field test on a separate cohort of C57BL/6J mice (n = 8/group). Subjects were handled once per day for 2 d prior to testing. The open field consisted of an open box (27.9 × 27.9 cm) made of PLEXIGLAS. Subjects were placed in the apparatus to explore for 5 min, and then returned to their home cage. All testing was conducted under standard room lighting. Activity data were analyzed using the Open Field Activity Software (Med Associates Inc.) for locomotor activity (distance traveled in centimeters) and exploratory behavior (time spent in center of chamber in seconds, where center is defined as 6 cm from the perimeter of chamber walls). Thirty minutes prior to testing, RGFP963 and RGFP966 were administered IP at a dose of 10 mg/kg. No significant effect of drug on distance traveled (F(2,23) = 1.06, NS) or time spent in the center (F(2,23) = 0.22, NS) was observed (Fig. 3B,C) (Table 4).
Table 4.
Mean and SEM for data in Figure 3B,C
In this brief report, we describe two Class I HDAC inhibitors, RGFP966 and RGFP963. At a dose of 10 mg/kg, RGFP966 is a potent inhibitor of HDAC3, while 10 mg/kg RGFP963 broadly inhibits HDAC1, HDAC2, and HDAC3. In vivo experiments demonstrate that 10 mg/kg RGFP963 and RGFP966 are able to access the brain. Furthermore, 10 mg/kg RGFP963 reaches a concentration capable of inhibiting brain HDAC1, HDAC2, and HDAC3 15 min after systemic administration. For RGFP966, CNS concentration is sufficient to inhibit HDAC3, but not HDAC1 or HDAC2. Importantly, RGFP966 has been shown to enhance acetylation of lysine 8 on H4—a site regulated by HDAC3, supporting our in vitro and in vivo data suggesting that RGFP966 strongly inhibits HDAC3 (Malvaez et al. 2013).
Immunocytochemistry data demonstrate increased global histone H3 acetylation in the BLA 1 h after IP administration with 10 mg/kg RGFP966. Paradoxically, we observe a decrease in global histone H3 and H4 acetylation in the dentate gyrus 1 h after administration with 10 mg/kg RGFP966 and RGFP963. This result is unexpected given the behavioral effect of RGFP963. We would predict that RGFP963, which enhances extinction consolidation, would increase acetylation. Furthermore, given the lack of a behavioral effect by RGFP996, we would not have expected an increase in acetylation in the amygdala with RGFP966 administration. While these data are unexpected, the results of this experiment might be explained by a complex interaction occurring between RGFP963 and RGFP966 and histone acetyltransferase enzymes (HATs), which acetylate lysine residues on histone proteins. A previous study demonstrates inhibition of HAT activity by trichostatin A (TSA), a Class I and II HDAC inhibitor (Nair et al. 2001). The data presented here should be quantitatively replicated and extended to other brain regions, in particular the infralimbic cortex, as increased histone acetylation in this area is associated with enhanced extinction (Stafford et al. 2012). Future studies will be needed to more directly address, in vivo, RGFP963 and RGFP966 effects on Class I HDAC activity and downstream effects on histone acetylation, particularly over a time course and at a variety of doses.
Behaviorally, 10 mg/kg RGFP963 enhances consolidation of cued fear extinction. RGFP966 has no effect on consolidation of cued fear extinction at the same dose. RGFP966 strongly inhibits HDAC3, while RGFP963 more broadly inhibits HDAC1, HDAC2, and HDAC3. Behavioral dose–response studies with RGFP963 and RGFP966 will need to be conducted to assess the full range of potential extinction effects of these compounds. The IC50 values of RGFP963 and RGFP966 suggest that at higher doses, different behavioral effects might be observed. Additionally, given that RGFP963 and RGFP966 have different pharmacokinetic properties—where RGFP966 exhibits lower brain penetrance compared with RGFP963 at the same systemically administered dose—it would be interesting to determine whether differential behavioral effects than those observed could be achieved with a dose of drug that resulted in equivalent levels of brain penetrance. As RGFP966 strongly inhibits HDAC3, while RGFP963 more broadly inhibits HDAC1, HDAC2, and HDAC3, further data on the behavioral effects of RGFP963 and RGFP966 could be gleaned if both drugs were administered at doses achieving comparable levels of HDAC3 inhibition.
Furthermore, the behavioral effect of RGFP963 may be difficult to interpret based on the current set of experimental controls. Future studies should replicate a similar paradigm as depicted in Figure 3A with “vehicle-extinction,” “10 mg/kg RGFP963-extinction,” “10 mg/kg RGFP966-extinction,” “vehicle-no extinction,” “10 mg/kg RGFP963-no extinction,” and “10 mg/kg RGFP966-no extinction” groups, perhaps with more extensive extinction training, to further clarify an effect of RGFP963 on extinction consolidation.”
Our data suggest that when subjects receive relatively strong fear conditioning (2 d of 10 CS–US pairings), a 30 CS trial extinction training is insufficient to diminish freezing behavior on a subsequent extinction retention test—despite subjects exhibiting within-session extinction—compared with control groups which did not receive extinction training. Future studies should address whether more extensive extinction training (e.g., 50 CS trials) would enhance extinction retention after a similarly robust fear conditioning. Furthermore, as studies demonstrate that the ability of an HDAC inhibitor to modify long-term memory critically depends on the parameters of the learning event and its neural substrates, experiments should address whether RGFP966 would enhance consolidation of extinction learning with alternative training protocols. Interestingly, the data indicate that 10 mg/kg RGFP963 enhances consolidation of extinction memory when paired with a weaker training paradigm that would not normally result in long-term memory formation. This is in line with previous evidence suggesting that HDAC inhibition can transform and strengthen weak learning events to produce long lasting memories (Federman et al. 2009; Stefanko et al. 2009; Stafford et al. 2012). Future studies should determine the strength of RGFP963's effect on long-term extinction memory by assessing measures of fear recovery, including renewal, spontaneous recovery, and reinstatement.
Importantly, previous studies find similar effects for the Class I HDACs HDAC2 and HDAC3 in long-term memory consolidation. Forebrain-specific knockout of HDAC2 enhances contextual fear learning and cued fear extinction (Morris et al. 2013). Systemic HDAC3 inhibition enhances long-term object memory and facilitates extinction of cocaine-conditioned place preference (McQuown et al. 2011; Malvaez et al. 2013). Knockout of HDAC1, however, does not have an effect on measures of contextual or cued fear learning and extinction (Morris et al. 2013), or, conversely, HDAC1 inhibition blocks extinction of contextual fear (Bahari-Javan et al. 2012).
While a number of well-known “positive” regulators (their activity is memory-promoting, e.g., CREB) have been extensively studied in learning and memory, the literature suggests that there is a concomitant increase in the activity of “negative” regulators (i.e., their activity suppresses learning and memory), such as the HDACs. Global and site-specific inhibition or knockdown of specific HDACs indicate that they might be crucial for resisting excessive new learning. This balance in activity between positive and negative regulators likely allows for a more finely graded balance between new learning and memory maintenance (Abel et al. 1998; Akers et al. 2014). This balance might be particularly crucial in fear versus extinction learning, requiring the differential activity of specific HDACs, as fear is quickly learned and long-remembered. Extinction, however, requires extensive training and even then, the fear response often returns. This “fear-promoting” imbalance is thought to be an evolutionarily conserved survival tool.
In summary, the present study suggests that HDACs are involved in long-term amygdala-dependent learning and memory. Our findings contribute to growing evidence that the Class I HDACs play a role in the extinction of cued fear—an analog of exposure therapy in humans. Future studies are necessary to determine the therapeutic potential of specific HDAC inhibitors.
Acknowledgments
This study was supported by NIH (T32 GM 08605, 1F31MH097397-01, R01DA01962), the Center for Behavioral Neuroscience (NSF agreement IBN-987675), the Burroughs Wellcome Fund, and by an NIH/NCRR base grant (P51RR000165) to Yerkes National Primate Research Center.
Footnotes
Article is online at http://www.learnmem.org/cgi/doi/10.1101/lm.036699.114.
References
- Abel T, Martin KC, Bartsch D, Kandel ER 1998. Memory suppressor genes: inhibitory constraints on the storage of long-term memory. Science 279: 338–341. [DOI] [PubMed] [Google Scholar]
- Akers KG, Martinez-Canabal A, Restivo L, Yiu AP, De Cristofaro A, Hsiang HL, Wheeler AL, Guskjolen A, Niibori Y, Shoji H, et al. 2014. Hippocampal neurogenesis regulates forgetting during adulthood and infancy. Science 344: 598–602. [DOI] [PubMed] [Google Scholar]
- Akirav I, Raizel H, Maroun M 2006. Enhancement of conditioned fear extinction by infusion of the GABA(A) agonist muscimol into the rat prefrontal cortex and amygdala. Eur J Neurosci 23: 758–764. [DOI] [PubMed] [Google Scholar]
- Bahari-Javan S, Maddalena A, Kerimoglu C, Wittnam J, Held T, Bähr M, Burkhardt S, Delalle I, Kügler S, Fischer A, et al. 2012. HDAC1 regulates fear extinction in mice. J Neurosci 32: 5062–5073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowers ME, Ressler KJ 2015. Interaction between the cholecystokinin and endogenous cannabinoid systems in cued fear expression and extinction retention. Neuropsychopharmacology 40: 688–700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bredy TW, Barad M 2008. The histone deacetylase inhibitor valproic acid enhances acquisition, extinction, and reconsolidation of conditioned fear. Learn Mem 15: 39–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bredy TW, Wu H, Crego C, Zellhoefer J, Sun YE, Barad M 2007. Histone modifications around individual BDNF gene promoters in prefrontal cortex are associated with extinction of conditioned fear. Learn Mem 14: 268–276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dudai Y 2004. The neurobiology of consolidations, or, how stable is the engram? Annu Rev Psychol 55: 51–86. [DOI] [PubMed] [Google Scholar]
- Federman N, Fustiñana MS, Romano A 2009. Histone acetylation is recruited in consolidation as a molecular feature of stronger memories. Learn Mem 16: 600–606. [DOI] [PubMed] [Google Scholar]
- Goosens KA, Maren S 2001. Contextual and auditory fear conditioning are mediated by the lateral, basal, and central amygdaloid nuclei in rats. Learn Mem 8: 148–155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guan JS, Haggarty SJ, Giacometti E, Dannenberg JH, Joseph N, Gao J, Nieland TJ, Zhou Y, Wang X, Mazitschek R, et al. 2009. HDAC2 negatively regulates memory formation and synaptic plasticity. Nature 459: 55–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hait NC, Wise LE, Allegood JC, O'Brien M, Avni D, Reeves TM, Knapp PE, Lu J, Luo C, Miles MF, et al. 2014. Active, phosphorylated fingolimod inhibits histone deacetylases and facilitates fear extinction memory. Nat Neurosci 17: 971–980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heldt SA, Stanek L, Chhatwal JP, Ressler KJ 2007. Hippocampus-specific deletion of BDNF in adult mice impairs spatial memory and extinction of aversive memories. Mol Psychiatry 12: 656–670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herry C, Ciocchi S, Senn V, Demmou L, Müller C, Lüthi A 2008. Switching on and off fear by distinct neuronal circuits. Nature 454: 600–606. [DOI] [PubMed] [Google Scholar]
- Josselyn SA, Shi C, Carlezon WA Jr, Neve RL, Nestler EJ, Davis M 2001. Long-term memory is facilitated by cAMP response element-binding protein overexpression in the amygdala. J Neurosci 21: 2404–2412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malvaez M, McQuown SC, Rogge GA, Astarabadi M, Jacques V, Carreiro S, Rusche JR, Wood MA 2013. HDAC3-selective inhibitor enhances extinction of cocaine-seeking behavior in a persistent manner. Proc Natl Acad Sci 110: 2647–2652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McQuown SC, Wood MA 2011. HDAC3 and the molecular brake pad hypothesis. Neurobiol Learn Mem 96: 27–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McQuown SC, Barrett RM, Matheos DP, Post RJ, Rogge GA, Alenghat T, Mullican SE, Jones S, Rusche JR, Lazar MA, et al. 2011. HDAC3 is a critical negative regulator of long-term memory formation. J Neurosci 31: 764–774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milad MR, Quirk GJ 2002. Neurons in medial prefrontal cortex signal memory for fear extinction. Nature 420: 70–74. [DOI] [PubMed] [Google Scholar]
- Morris MJ, Mahgoub M, Na ES, Pranav H, Monteggia LM 2013. Loss of histone deacetylase 2 improves working memory and accelerates extinction learning. J Neurosci 33: 6401–6411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Myers KM, Davis M 2007. Mechanisms of fear extinction. Mol Psychiatry 12: 120–150. [DOI] [PubMed] [Google Scholar]
- Nair AR, Boersma LJ, Schiltz L, Chaudhry MA, Muschel RJ 2001. Paradoxical effects of trichostatin A: inhibition of NF-Y-associated histone acetyltransferase activity, phosphorylation of hGCN5 and downregulation of cyclin A and B1 mRNA. Cancer Lett 166: 55–64. [DOI] [PubMed] [Google Scholar]
- Ploski JE, Pierre VJ, Smucny J, Park K, Monsey MS, Overeem KA, Schafe GE 2008. The activity-regulated cytoskeletal-associated protein (Arc/Arg3.1) is required for memory consolidation of Pavlovian fear conditioning in the lateral amygdala. J Neurosci 28: 12383–12395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ploski JE, Park KW, Ping J, Monsey MS, Schafe GE 2010. Identification of plasticity-associated genes regulated by Pavlovian fear conditioning in the lateral amygdala. J Neurochem 112: 636–650. [DOI] [PubMed] [Google Scholar]
- Ressler KJ, Paschall G, Zhou XL, Davis M 2002. Regulation of synaptic plasticity genes during consolidation of fear conditioning. J Neurosci 22: 7892–7902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stafford JM, Raybuck JD, Ryabinin AE, Lattal KM 2012. Increasing histone acetylation in the hippocampus-infralimbic network enhances fear extinction. Biol Psychiatry 72: 25–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stefanko DP, Barrett RM, Ly AR, Reolon GK, Wood MA 2009. Modulation of long-term memory for object recognition via HDAC inhibition. Proc Natl Acad Sci 106: 9447–9452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whittle N, Singewald N 2014. HDAC inhibitors as cognitive enhancers in fear, anxiety and trauma therapy: where do we stand? Biochem Soc Trans 42: 569–581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whittle N, Schmuckermair C, Gunduz Cinar O, Hauschild M, Ferraguti F, Holmes A, Singewald N 2013. Deep brain stimulation, histone deacetylase inhibitors and glutamatergic drugs rescue resistance to fear extinction in a genetic mouse model. Neuropharmacology 64: 414–423. [DOI] [PMC free article] [PubMed] [Google Scholar]