

Abstract
Origins of DNA replication (ORIs) occur at defined regions in the genome. Although DNA sequence defines the position of ORIs in budding yeast, the factors for ORI specification remain elusive in metazoa. Several methods have been used recently to map ORIs in metazoan genomes with the hope that features for ORI specification might emerge. These methods are reviewed here with analysis of their advantages and shortcomings. The various factors that may influence ORI selection for initiation of DNA replication are discussed.
Background
DNA synthesis initiates at multiple replication origins (ORIs) in eukaryotic genomes. When an ORI is used to initiate replication, it is said to have “fired”. Accurate duplication of the genetic material depends on a reliable mechanism that ensures that any given ORI fires at most once per cell cycle by restricting “licensing” to G1 phase and “activation” to S phase. In G1, each ORI that binds an origin recognition complex (ORC) and subsequently Cdc6 and Cdt1/MCM2-7 to form the pre-replication complex (pre-RC) is said to be “licensed” [1–4]. Activation occurs when the pre-RC is converted to the initiation complex (IC) through the exit of Cdc6 and Cdt1 and the entry of Cdc45 [5] and GINS to form the CMG complex (containing Cdc45, MCM2-7, and GINS) [6–8] that ultimately recruits DNA polymerase.
The classic studies of Huberman and Riggs [9,10] using DNA fiber autoradiography demonstrated many key points. DNA replication is bidirectional and starts at ORIs that sometimes fire coordinately in clusters. DNA fiber autoradiography allowed measurement of the rate of DNA replication fork progression. In its original form, this approach was unable to correlate the mapped ORIs with DNA sequence, and it remained a possibility that the ORIs were neither sequence-specific nor site-specific in the population of DNA molecules. In metazoans, the possibility that ORIs were not sequence-specific was supported by the observation that plasmid replication was independent of its sequence content [11,12] and that any DNA sequence could replicate when injected into Xenopus embryos [13]. Nonetheless, the site specificity of ORIs was demonstrated by investigations from the Hamlin laboratory that showed that a 6 kb restriction fragment of the amplified dihydrofolate reductase (DHFR) locus in Chinese hamster ovary cells was always the earliest one labeled by radioactive thymidine after release into S phase [14,15]. This ruled out the possibility that ORIs were totally random in the genome (although this might be the case in early embryos before the mid-blastula transition [16–18]).
Thus, the search for site-specific metazoan ORIs was invigorated by using a variety of methods at a few individual loci [19]. A sequel to the earliest labeled restriction fragment approach was polymerase chain reaction (PCR) mapping of small nascent strands, which was applied to the DHFR locus [20–22] and to other loci to map preferred start sites of DNA replication. In this same era, two-dimensional (2D) gels were developed and revealed a specific restriction fragment on a yeast plasmid where DNA replication starts [23,24]. 2D gels were subsequently used to map ORIs in eukaryotic chromosomes. Neutral-neutral 2D gels took advantage of the differences in gel migration of restriction fragments containing a replication bubble or a replication fork [23]. Neutral-alkaline 2D gels were able to measure the direction of replication fork movement [25]. With all of these pioneering studies showing that preferred regions of the genome were used as ORIs in both yeast and metazoa, the stage was set to elucidate what specifies eukaryotic ORIs. Most ORIs in budding yeast have been confidently mapped by a variety of methods [26] and are relatively well understood. However, metazoan ORIs have remained mysterious with regard to a universal principle, if any, that is shared by all. Initial experiments in metazoans studied a few individual ORIs [27–29]. Recent efforts have been made to map all ORIs in certain metazoan genomes (for example, Drosophila, mouse, and human), hoping to uncover global principles. This article provides an overview of the various current methods used to map ORIs genome-wide. Understanding the methods serves as the foundation to evaluate the conclusions from these studies regarding what features might define metazoan ORIs.
Methods to map origins
DNA combing and single-molecule analysis of replicating DNA
Current approaches to map ORIs tend to be improvements on earlier methods, such as the application of newer technologies to an older technique. For example, labeling with CldU and IdU and subsequent detection by fluorescence microscopy constitute a modern adaptation of DNA fiber autoradiography, which used H3-thymidine labeling. Both the older and newer approaches allow determination of replication fork rate, and pulse-chase labeling allows the ORI to be mapped at the center of the bidirectional fork pattern. The CldU/IdU-labeled DNA can be spread freehand (SMARD: single-molecule analysis of replicating DNA [30]) or with a DNA combing machine that ensures uniform spreading [31–34]. DNA combing of evenly stretched DNA allows accurate genome-wide information to be obtained on replication fork speed, fork asymmetry, and the distance between ORIs that are used in the same S phase on a single DNA molecule [35,36]. When DNA combing or SMARD is coupled with fluorescence in situ hybridization (FISH), it is possible to map the ORI at the locus defined by the hybridization probe. FISH has been combined with DNA combing to analyze replicating DNA in yeast [37,38] and in mammalian samples [39,40]. However, the large size of mammalian genomes and the low frequency of finding replicating DNA molecules with the FISH signal make this a very time-consuming, though feasible, process [39,40]. This problem has been minimized when coupling FISH with SMARD by a prior enrichment step to select the locus of interest by pulsed-field gel electrophoresis [30]. Future developments will be required to allow analysis of spread metazoan DNA with regard to DNA sequence genome-wide rather than just at individual loci and to increase the resolution.
Origin recognition complex chromatin immunoprecipitation
Several approaches have been taken to map ORIs genome-wide [41–43]. Following the same trend as above, these often couple earlier approaches to map ORIs at individual loci with current genomics technologies. Chromatin immunoprecipitation (ChIP) to identify DNA sequences bound to ORC has been used in conjunction with DNA microarrays (ChIP-chip) or genomic sequencing (ChIP-seq) [44] for yeast [45,46] and recently for Drosophila [47] and human samples [48]. Several difficulties face ORC-ChIP, especially for mammalian genomes that are 20 times larger than the Drosophila genome [49]. As for any ChIP experiment, the quality of the data reflects the quality of the antibody; epitope tagged proteins can be used with an antibody against the tag to increase sensitivity and specificity but requires transformation [44]. A challenge is that ORC has low sequence specificity and the ChIP signal is not very high compared with the background control, although CsCl gradient ultracentrifugation was recently used as a pre-enrichment step for ORC-bound DNA in a genome-wide study on human cells [48]. An alternative enrichment approach is to biotinylate the protein of interest and then perform avidin purification of the biotinylated chromatin before doing ChIP [44]. Moreover, since ORC has other roles beyond DNA replication [50–54], such as its localization to heterochromatin, a mixture of ORIs and other sequences might be enriched by ORC-ChIP alone. For higher specificity in identifying sites of pre-RC formation, MCM ChIP-seq can also be done to filter for ORC peaks that coincide with MCM peaks [44,46]. However, it is known that ORC/Cdc6/Cdt1 loads many MCM double-hexamers onto DNA that can move away from the ORC site, raising the possibility of initiation sites that are not coincident with ORC [55,56]. These potential ORC-distal MCM initiation sites would not meet the criterion of overlap with ORC sites; in some cases, the ORC sites loading the MCMs that may be involved in distal initiation might be excluded from the analysis if they do not overlap with MCMs. In addition, pre-RC ChIP studies identify licensed ORIs in the genome but do not indicate the subset that is chosen for activation [57].
5-bromo-2′-deoxyuridine immunoprecipitation
Other methods have been employed to identify those ORIs that are active in metazoan genomes. Immunoprecipitation of nascent DNA strands labeled with 5-bromo-2′-deoxyuridine (BrdU) (BrIP-NS) has been coupled with microarrays (BrIP-NS-chip) to map active ORIs to the ENCODE 1% of the human genome [58,59] or with deep sequencing (BrIP-NS-seq) to map active ORIs genome-wide [60]. In this technique, short, origin-centered BrdU-labeled nascent strands are first enriched by sucrose gradient ultracentrifugation and then further enriched by immunoprecipitation. Sucrose gradient size fractionation is used by some laboratories without further enrichment steps and was the basis for an early microarray study to map ORIs [58]. Another technique, Repli-seq [61], enriches for BrdU-labeled DNA at consecutive time points throughout S-phase to identify the replication timing profile over the genome by tracking where replication forks tend to be at each time point. This information has been used to identify the likely regions where replication forks initiate [48,61–63]. The Repli-seq data are consistent with a computational method of calculating the nucleotide compositional skew profile over the genome, which also predicts the direction of replication forks genome-wide and the likely regions where they initiate ([63,64] and references therein; Olivier Hyrien, personal communication).
Bubble-trap
The bubble-trap method [65,66] is another approach to map active ORIs. It has some similarities to 2D gels since in both methods restriction fragments containing replication bubbles are retarded in their mobility on a gel. However, bubble trapping takes advantage of the circular nature of replication bubbles by allowing the matrix of a gel to form through them, thereby trapping bubble-containing fragments in the gel during electrophoresis. Visualizing blots of 2D gels can interrogate only a single restriction fragment for a bubble arc, whereas trapped bubble-containing restriction fragments are recovered from the gel for library preparation and can be coupled with DNA microarrays (Bubble-chip) [67] or genomic sequencing (Bubble-seq) [68] to identify bubble-containing restriction fragments genome-wide. It is possible that extrachromosomal circular DNAs (eccDNAs) [69–72] that do not contain the specific restriction site will be trapped in the gel matrix too, but owing to their circular nature, they would not be cloned in the subsequent step. If future adaptations of bubble-seq bypass the cloning step by directly fragmenting and sequencing bubble-trapped material, eccDNA may become more of a concern, although it would still be possible to identify and eliminate eccDNA enrichments in downstream analytical steps by using paired-end sequencing and flagging enriched fragments with discordantly mapped reads consistent with circular DNA. In its current form, bubble trapping was shown to have very few false positives as demonstrated by 2D gel analysis [65]. However, the current bubble-trap datasets cannot recover all ORIs in the genome since they interrogate the fragments of only a single restriction enzyme. Any given restriction enzyme will inherently disfavor the identification of certain ORIs that are too near a restriction site or that are in a small restriction fragment. Moreover, the resolution is limited to the sizes of the bubble-containing fragments of the single restriction enzyme. Constructing parallel bubble-seq libraries where each is derived from a different restriction enzyme could increase the sensitivity of this approach for ORI discovery and potentially allow higher-resolution inferences to map ORIs. It is possible, though, that the gain in information from additional restriction enzyme libraries is not enough to justify the increased workload and cost. Because each step in the bubble-trap process favors certain restriction fragment sizes, it is currently unclear whether the enrichment value of a particular fragment can accurately be used to estimate ORI efficiency. Despite the conceptual elegance and high purity of bubble trapping, it has not been widely adopted perhaps because its many steps make it a time-consuming and technically challenging method.
Mapping the transition between leading and lagging nascent strands
There is a transition from leading (continuous) to lagging (discontinuous Okazaki fragment) strand synthesis at each ORI of bidirectional replication (Figure 1). The region of the transition from leading to lagging strand was mapped [73] at the DHFR locus by using emetine, a protein synthesis inhibitor thought at the time to cause nucleosomes to segregate to only the leading strand. Micrococcal nuclease digestion of the assumed naked lagging strand was then used to map the strand-specific transition of the leading strand. However, shortly thereafter, the DePamphilis laboratory showed that emetine actually inhibits Okazaki fragment synthesis and not nucleosome segregation contrary to what was previously assumed [74]. Nonetheless, the conclusion that the emetine approach could map the transition from leading to lagging strand synthesis remained intact. Another study mapped the transition from lagging to leading strand synthesis at the DHFR locus by using strand-specific dot blotting [75], an adaptation of an earlier technique that mapped this transition with single nucleotide resolution in replication origins of animal viruses using sequencing gels [76,77]. The conceptual approach of mapping the transition point is also the basis for the more recent strand-specific sequencing of Okazaki fragments to identify ORIs and estimate ORI efficiencies genome-wide; this mapping technique holds great promise as seen by its application to the budding yeast genome, where isolation of Okazaki fragments was accomplished by DNA ligase I repression in a degron-tagged construct [78,79]. Published data have yet to be released for deep sequencing Okazaki fragments from metazoan genomes, although preliminary results suggest that deep-sequenced Okazaki fragments in the human genome track replication forks and predict initiation regions in agreement with Repli-Seq timing and nucleotide composition skew profiles (Olivier Hyrien, personal communication).
Figure 1. Origin of bidirectional replication.
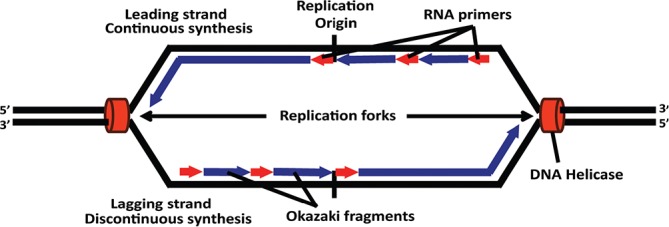
The transition between leading, continuous strand synthesis and lagging, discontinuous strand synthesis marks the origin of bidirectional replication. DNA polymerase can only extend nascent DNA in the 3′ direction, and Okazaki fragments are used to allow net growth of the lagging strand in the 5′ direction.
Lambda exonuclease enrichment of nascent strands
Mapping the transition point from continuous to discontinuous synthesis is also the basis of replication initiation point (RIP) mapping that has been used to map the start site of DNA synthesis to the nucleotide level [80–84] in specific ORIs from budding yeast [81,85], fission yeast [86], fungus gnats [87] and humans [88]. The key to RIP mapping is to obtain enriched preparations of nascent DNA that are not contaminated by broken parental DNA. The inspiration for this approach came from studies in the DePamphilis laboratory that mapped the start site of DNA synthesis with nucleotide resolution in SV40 and polyomavirus [76,77]. Those studies phosphorylated the 5′ end of all DNA fragments (including parental DNA that was broken) and then identified nascent strands by removal of their 5′ primer to expose a 5′-hydroxyl group that was then end-labeled with P32. This approach worked well to map ORIs in viruses but did not have sufficient sensitivity to map ORIs to the nucleotide level in eukaryotic genomes. RIP mapping was able to retain single nucleotide resolution in eukaryotic genomes because of the increased sensitivity gained by employing the enzyme lambda exonuclease (Lexo) that digests parental DNA from its 5′ end [89,90] but leaves intact the nascent DNA containing a 5′ RNA primer. A more recent adaptation of RIP mapping bypasses Lexo digestion and simply performs cell lysis in the well of an alkaline gel to minimize breakage of parental DNA before selection for small nascent strands [91]. Sequencing the primer-extended products from nascent DNA templates derived from an asynchronous population of cells allows the origin of bidirectional replication to be mapped as the transition from continuous to discontinuous synthesis.
The use of Lexo to enrich nascent strands has become a preferred method for nascent strand purification, which has been used with PCR analysis of nascent strand abundance to map individual ORIs at a lower level of resolution than RIP mapping. Lexo-enriched nascent strands (Lexo-NS) also provide the foundation for genome-wide ORI mapping when coupled with DNA microarrays (Lexo-NS-chip) or DNA sequencing (Lexo-NS-seq) [92]. Lexo-NS-chip has been applied to Drosophila [93,94], mouse [93–95], and human [59,96,97] genomes. Lexo-NS-seq has been used to map ORIs in the human genome [60,98–100]. In these approaches, the transition point between continuous and discontinuous synthesis is not determined, but instead ORIs are mapped where short nascent strands are enriched. Nascent strands that are about 500 to 2500 nucleotides long (the size range depends on the laboratory) are used to avoid inclusion of ~200 nt Okazaki fragment sequences that occur throughout the entire genome. Exclusion of Okazaki fragments is usually accomplished by the isolation of short single-stranded DNA through sucrose gradient fractionation. Alternatively, BND-cellulose column chromatography can be used to enrich replicative intermediates [80–84], with a size selection step later in the procedure [100]. Depending on the computational analysis, Lexo-NS-seq has the potential to give nucleotide-level resolution estimates for ORIs that map to fixed locations. It has also been used to gauge ORI efficiency, where an increased number of reads has suggested greater efficiency of early firing ORIs [93,99,101]. However, Lexo has base compositional and other biases that result in enriching various non-origin DNA sequences and influencing these ORI efficiency estimates in favor of GC-rich ORIs as discussed below.
Further insights into genome-wide origin mapping methods
The ORI mapping results from the various methods described above are not in full agreement with one another, likely because most or all methods enrich both origin as well as non-origin DNA with various degrees of sensitivity and specificity. This makes it imperative that we understand the proclivities of each method to fully appreciate their outputs. Overall, it is to be expected that all origin mapping techniques will have some degree of overlap at least where origins are concerned. What are enigmatic are those putative ORIs unique to one method even after reaching saturation. Statistically significant but still incomplete overlap between two or more orthogonal datasets should not be taken as uniform validation of each individual putative ORI in those datasets. Although a significant number of overlaps between two approaches may increase our belief (in the Bayesian sense of the word) in the validity of putative ORIs that are represented in both approaches, it should not necessarily increase our belief in the validity of those ORIs absent from one approach. Conversely, some putative ORIs unique to one approach may be true positives, such as ORIs detected by nascent strand techniques that reside too close to a restriction site for bubble-trap detection. Thus, while studying only ORIs that overlap from several different origin-mapping techniques helps to highlight the most likely putative ORIs, it can also falsely discard true ORIs unique to one method. Therefore, perhaps meta-analyses that consider all datasets could instead use the degree of belief (e.g. posterior probability) of each potential ORI site in the genome to weight downstream analyses in favor of the most probable ORIs rather than allowing all putative ORIs to uniformly influence results and rather than discarding all putative ORIs with lower support. There is a possibility that some putative ORIs that overlap in orthogonal methods are not guaranteed to be true positives if both orthogonal methods contain the same systematic false positives. However, this is likely to be rare and the weighting scheme above would account for this if additional orthogonal methods were included in the analysis. These possibilities highlight the need to better understand the outputs of each method to fine-tune our approaches and analyses to hunt for ORIs in metazoan genomes.
Despite the incomplete agreement between methods, comparing the outputs of orthogonal approaches is the best way to have an estimate on the reliability of a given method in ORI discovery. Bubble-trap was orthogonally validated by 2D gels and can be used to make orthogonal comparisons with nascent strand enrichments. SMARD with FISH, though of lower resolution, is orthogonal to both bubble-trapping and nascent strand techniques. Strand switch detection methods and ORC-ChIP are both orthogonal to SMARD, nascent strand enrichment techniques, bubble trap, and 2D gels, though ORC-ChIP requires making the assumption that ORC binding sites always flank or overlap initiation sites. Other comparisons are supportive but do not orthogonally validate the ORI status of a query selected from a genome-wide dataset. For example, quantitative PCR (qPCR) analysis of the abundance of Lexo-enriched small nascent strands can be used to test the reproducibility of a Lexo-based enrichment and could provide more confidence if the qPCR enrichment is present only in S phase, but since it uses Lexo, is identical in preparation, and differs only at the readout stage, it is not orthogonal to Lexo-NS-seq. Using Lexo-NS and BrIP-NS techniques to validate each other is not strictly orthogonal either because both typically begin with the same sucrose gradient fractionation step to obtain short single-stranded DNA. Nonetheless, since one technique further enriches nascent strands based on enzymatic logic directed at RNA primers and the other based on immunoprecipitation targeted at incorporated BrdU in nascent DNA, their agreement can still be satisfying. Overall, orthogonal comparisons are important for both building confidence in ORI mapping techniques and in quantifying our degree of belief in putative ORI sites across the genome in order to paint a refined picture of the ORI landscape.
Previous Lexo-NS-chip datasets did not have high overlap with other ORI mapping techniques such as Bubble-chip (10-14% of bubbles overlapped 26-35% of Lexo-NS [67]) and BrIP-NS-chip (2.2-12.8% BrIP overlapped 6.4-33.4% Lexo-NS [59,96]). Lexo-NS-chip datasets did not even overlap well with each other (approximately 5-6% [59,96]). Poor overlap was often attributed to a lack of saturation (that is, incomplete sets of ORIs), which was supported by a recent deep-sequencing Lexo-NS-seq peak set that identified more than 350,000 putative ORIs across four different cell lines that encompassed 89-92% of most previous Lexo-NS datasets (25.3% for one outlier) [99]. Nonetheless, even after saturation, overlap of these Lexo-NS-seq peaks with Bubble-chip was not as high (50-65% bubbles) and with BrIP-NS-chip was still low (9-20% BrIP-NS-chip) [99]. More recent analyses of Lexo-NS-seq datasets have shown that 45-46% of Lexo-NS-seq peaks overlap with 36%-37% of the bubble-seq ORI map [101] and that 56.5% of the BrIP-NS-seq peaks overlapped 50.2% of the Lexo-NS-seq peaks from the same study [60]. Preliminary results from Okazaki fragment sequencing in the human genome had better agreement with results from bubble-trap than from Lexo-NS methods (Olivier Hyrien, personal communication). The Okazaki fragment results appear to be supported by their agreement with the bubble-trap method, which was shown to be highly specific [65]. Conversely, more confidence can be given to bubble-trap since the Okazaki fragment sequencing profile was in agreement with Repli-Seq and nucleotide skew profiles. However, the lower concordance between Okazaki fragment sequencing and Lexo-NS-seq supports the idea that non-origin peaks in addition to origins are systematically enriched in Lexo-NS-seq datasets.
Data from single-molecule studies [102–104] and from deep sequencing of Lexo-digested genomic DNA isolated from non-replicating G0 cells [100] demonstrated that Lexo is more efficient in digesting AT-rich DNA than GC-rich DNA and that this enriches GC-rich regions of the genome. Moreover, in vitro experiments and genomic analyses revealed that Lexo digestion is obstructed by G-quadruplexes (G4s) [100], reminiscent of exonuclease 1 digestion [105] and DNA polymerase elongation [106,107], both of which are also impeded by G4s and are used as diagnostics to detect G4 formation in vitro. Therefore, Lexo-NS-seq may also be enriched for G4-protected and GC-rich DNA independent of nascent strands, and efficiency estimates may be highly dependent on base composition. There may also be other nascent strand independent (NSI) Lexo biases. For instance, since parental DNA contains ribonucleotides, with potentially as many as one million ribonucleotide insertions per genome duplication [108], Lexo digestion could be obstructed when it reaches a ribonucleotide in the parental DNA in accordance with its inability to digest RNA-protected DNA. Whether or not in vivo ribonucleotide incorporation events are randomly distributed or occur at hotspots is unknown, but the latter seems to be the case in vitro [108]. Lexo will also not digest eccDNA, such as “microDNA” circles that have been described in mammalian cells and range in size with a peak at 200 to 400 bp [72]. eccDNAs of all sizes appear to be generated from non-random genomic sites such as tandem repeats [69–71]. MicroDNA sequences were enriched in CpG islands (CGIs) and GC-richness, as well as at the 5′ end of genes [72]. In general, non-random genomic sites associated with ribonucleotide insertions, eccDNA, G4s, and GC-rich DNA could pose as ORIs in Lexo-NS-seq datasets and could potentially be more highly reproducible than true ORIs, which are plastic and stochastic.
In contrast to the evidence of pervasive, reproducible NSI Lexo biases [100], other Lexo-NS studies that looked at non-replicating DNA (“mitotic NS”) as well as RNase-treated nascent strands concluded that there were no enrichments independent of nascent strands [92,93]. Why one NS-Lexo study [93], which looked at “mitotic NS” by microarray came to different conclusions on nascent strand independent enrichments of Lexo is somewhat enigmatic. However, finding no enrichment after performing qPCR [92] on sites of known origins in Lexo-treated non-replicating DNA does not qualify as ruling out nascent strand independent enrichments in genome-wide datasets; it only supports the existence of nascent strand dependent enrichments. NSI Lexo biases by definition enrich non-origin sites, which would still be enriched in non-replicating DNA and RNase controls. Despite the inconsistency between these studies, it still seems plausible that NSI Lexo biases affect all previous Lexo-NS studies at least to some extent. Indeed, 47% [100], 72% [96], 74-77% [98] and 70-94% [99] of the Lexo-NS peaks from published datasets overlap peaks from Lexo-enriched non-replicating DNA from G0-synchronized cells ([100] and Gerbi lab, unpublished data]) indicating that NSI Lexo biases may permeate Lexo-NS-seq datasets or that these datasets preferentially enrich origins in regions favorable to Lexo enrichment. The former possibility (non-origin enrichments from NSI Lexo biases in Lexo-NS datasets) might explain why preliminary Okazaki fragment sequencing results in the human genome agreed better with Bubble-seq than Lexo-NS-seq (Olivier Hyrien, personal communication). NSI Lexo biases might also explain the apparent discrepancy between the original Okazaki fragment sequencing studies in yeast [78,79] and a more recent study that used Lexo to enrich Okazaki fragments before sequencing [109].
It is desirable to overcome the aforementioned concerns that can result in Lexo-NS DNA preparations being a mixture of true positives (nascent strands) and systematic false positives (NSI Lexo enrichments). One possibility to overcome G4 protection and GC-rich Lexo biases could be to use very high enzyme-to-DNA ratios to completely eliminate the parental DNA, leaving intact only RNA-protected nascent strands. However, 50 units/μg at pH 9.4 was shown to be insufficient to completely digest G4 structures in plasmid DNA [100], yet higher Lexo-to-DNA ratios have been shown to sacrifice the specificity of Lexo and lead to the digestion of RNA-primed DNA [109]. When 1 μg of DNA was mixed with 50 ng of RNA-primed DNA oligos, the RNA primers conferred protection against Lexo digestion (10 units/μg DNA), but when 100 units/μg DNA was used to digest 50 ng of DNA with 50 ng of RNA-primed DNA, the RNA primers no longer conferred protection [109]. Therefore, starting the digestion with very high enzyme-to-DNA ratios should be avoided in order to preserve the RNA protection of nascent strands. This indicates that other ways are needed to overcome the NSI Lexo biases.
Fortunately, there are several ways to improve Lexo-NS-seq. Most previous NS-Lexo studies have used genomic DNA to account for copy number variation in the genome and to control against biases introduced during library preparation and sequencing, but this does not control for biases introduced by Lexo. Thus, the detected enrichments can be summarized into three categories: (1) enrichments that arise from nascent strands alone (true positives), (2) enrichments that arise solely from NSI Lexo biases (systematic false positives) and (3) enrichments that arise from some combination of both (true positives within NSI Lexo-biased regions) [100]. One needs a way to eliminate category 2 while retaining category 3 enrichments. Many of the NSI Lexo biases can be overcome by using Lexo-digested non-replicating DNA from G0 cells (“Lexo G0”) as a control [100], which simultaneously accounts for nucleotide composition biases, the G4 protection bias, and other possible issues related to Lexo digestion while also controlling for copy number and biases introduced during library preparation and sequencing. Importantly, this approach corrects for the nascent strand independent enrichment components introduced by Lexo across the genome by detecting origins as enrichments of nascent strands over the nascent strand independent Lexo-digested background. Thus, with this approach, regions enriched from NSI Lexo biases alone (category 2) no longer pose a problem, but it is still possible to discover origins in Lexo-biased regions (for example, regions with G4-containing and GC-rich sequences) (category 3). Moreover, if both undigested and Lexo-digested non-replicating genomic DNA controls are performed, it is also possible to compare the two peak sets that arise in Lexo-NS-seq after separately applying each control to analyze what Lexo-based enrichments are lost (or gained) when correcting for NSI Lexo biases. Lexo-digested non-replicating DNA will control against ribonucleotide incorporation and eccDNA hotspots too if the abundance and sites/sequences of these features are similar in S phase and G1/G0. Regardless, if paired-end sequencing is performed, then eccDNA hotspots can be identified in Lexo-NS-seq data by searching for read pairs inside peaks that map in the wrong orientation (the signature of a circle would be outward- instead of inward-facing read pairs) or by removing such discordantly mapped reads before peak calling. Similarly, strand-specific sequencing could aid in identifying ribonucleotide incorporation hotspots and G4-protected DNA enrichments if these features lead to detectable imbalances in the number of reads mapped to Watson and Crick strands since origin-centered nascent strands should contain relatively balanced enrichments. Using emetine in conjunction with strand-specific sequencing could allow identification of leading strand transition points (similar to Okazaki-fragment sequencing that identifies lagging strand transitions), which provides a biological signature to use in addition to enrichment values. Thus, in the future, Lexo-NS-seq experiments could have significant gains in specificity from novel strand-specific, paired-end strategies and analyses. Furthermore, future Lexo-NS-seq studies might be able to circumnavigate the G4 bias altogether by changing the Lexo digestion buffer from glycine-KOH (pH 9.4) to glycine-NaOH (pH 8.8) [100]. This buffer simultaneously replaces K+ with Na+ and lowers the pH from 9.4 to 8.8, both of which were shown to lower the effect of G4s on Lexo digestion [100]. Controlling against NSI biases with the Lexo G0 control, changing the buffer conditions to reduce G4 formation, and using strand-specific, paired-end sequencing strategies will reduce the number of systematic false positives to begin with, aid in identifying and eliminating persistent systematic false positives, and permit ORI efficiency estimates that are less influenced by base composition biases. Given that the potential issues in Lexo-NS-seq can be solved, it is likely to remain a powerful and popular technique in the coming years, although these issues may also motivate the innovation of new ORI mapping methods altogether.
What features define an origin of DNA replication?
In contrast to the punctuate ORIs of budding yeast, metazoan initiation zones contain many potential ORIs, some of which are preferred (reviewed by [27,28,43,110–112]). These observations led to the Jesuit model (“Many are called but few are chosen” [113]) that posited that although there may be multiple potential origins in a local genomic region (an ‘initiation zone’), only one or a few of those origins are activated in any given DNA molecule (Figure 2). If one ORI fires on a given DNA molecule in an initiation zone, its replication forks could inactivate other potential ORIs that are nearby, reflecting the phenomenon of origin interference [114–118]. A well-studied example is the 55 kb intergenic initiation zone of the DHFR locus [119], where 20% of the initiation events occur at two preferred sites called ori β and ori γ [120,121], although other studies concluded that most initiation events occur at ori β [20,21,75]. Even when ori β is deleted, initiation still occurs within the 55 kb initiation zone [122]. Moreover, deletion of 45 kb of the 55 kb intergenic initiation zone still allowed initiation from the remaining 10 kb [123]. These observations reinforce the notion that there are multiple potential initiation sites within the broad initiation zone. Nonetheless, some metazoan ORIs are more tightly circumscribed, such as at the human lamin B2 [88, 124,125] and β-globin [126] loci. Moreover, the width of the initiation zone can change during development, as observed for the 8 to 9 kb initiation zone of the II/9A locus from the fly Sciara that shrinks to 1 to 2 kb during locus-specific re-replication during DNA puff amplification [127].
Figure 2. Multiple potential origins in an initiation zone.
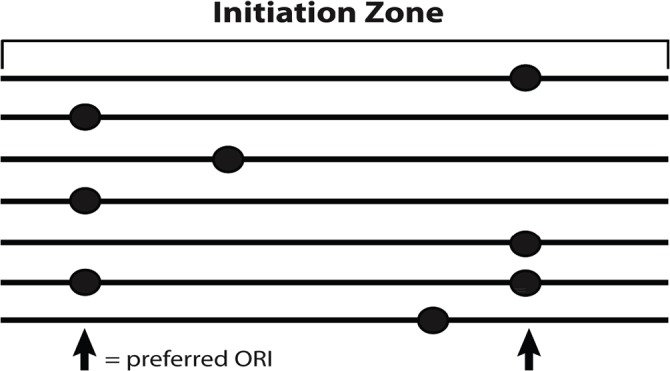
There are many potential origins of DNA replication (ORIs) in an initiation zone for metazoan DNA replication, but only one will be used on a given DNA molecule. Some of the potential ORIs are used more often than others, leading to preferred initiation sites of DNA synthesis in the initiation zone.
Regardless of whether the initiation zone is broad or narrow, the question remains of what features specify the potential ORIs in these regions. Since there are limitations and biases in various methods to map ORIs, the interpretations of results are intimately linked to the methods used. What conclusions have these ORI mapping studies come to about ORI specification? Recurring themes in genome-wide replication origin studies have been proximity to genes and gene promoters, transcription of nearby genes, open versus compact chromatin, GC and AT content, CGIs, and G4 structures.
Metazoan origins, genes, and transcription
ORC binding might be random in early Xenopus embryos, where ORC has a periodic spacing of 9 to 12 kb that appears to be DNA sequence independent and governed by an unknown mechanism [16,17]. However, after the mid-blastula transition and the onset of zygotic transcription, ORIs are found in defined locations [18,128]. In addition, deletion of the DHFR promoter to down-regulate DHFR transcription reduced ORI efficiency in the downstream intergenic initiation zone [129]. These observations suggest a link between transcription and ORI specification and efficiency. ORIs were thought to be absent in transcribed regions [98,130], and previous studies showed that ORC was found in intergenic regions, often in promoters [48,131], suggesting that ORC cannot bind or is displaced by RNA polymerase. However, an initial genomic study identified 28 new putative ORIs (using short nascent strands from sucrose gradient size fractionation), with the majority within genes and not intergenic [58]. Bubble-chip results also suggested that ORIs were equally distributed across genic and intergenic restriction fragments and that many of the genes that overlapped ORIs were actively transcribed [67]. Other results showed that 85% of ORIs were associated with transcription units with nearly half locating to promoters, and they suggested that transcription in early development might be linked to the regulation of ORI efficiency in later development [95]. Subsequent studies also found that ORIs were significantly enriched near transcription starts sites as well as RNA Pol II and transcription factor binding sites [48,59,93,96,97]. Martin and colleagues [98] expanded on this finding showing that ORIs were specifically enriched at moderately expressed genes rather than at lowly or highly expressed ones. Conversely, other studies did not find strong links of ORIs with genes and transcription. Bubble-seq results [68], in contrast to bubble-chip results [67], suggested that ORIs only marginally associated with transcriptionally active genes. Other nascent strand studies reported that relatively few ORIs were located near transcription start sites [60,99] and concluded that the association between ORIs, promoters, and CGIs is independent of transcription [60]. Cadoret and colleagues [96] suggested that there might be no link between ORIs and gene regulation since half the ORIs found were not associated with open chromatin. Moreover, lack of correlation between transcription and ORI selection is evident on the transcriptionally inactive X chromosome [60], where the same ORIs are used as on the active X [132] and only their timing [133] and replication program for the order of ORI firing [134] differ. Earlier plasmid experiments revealed that the presence of the transcription complex is sufficient to specify ORI location even in the absence of active transcription [135]. Overall, in terms of ORI specification, it seems that the replication program interacts with the transcription program but is not dictated by it.
Metazoan origins and chromatin
Histone modifications may play a role in ORI control. ORC binds to regions of open chromatin in Drosophila [47,136], and in mammals the DHFR initiation zone exhibits low nucleosome density [137]. However, genome-wide studies have shown that human ORIs are distributed across regions of both open and closed chromatin [60,68,96,98]. One sign of open chromatin is acetylation. Histones at ORC binding sites are hyperacetylated in Drosophila, and tethering a histone acetyl transferase (HAT) increases ORI activity [138]. Moreover, an increase in histone acetylation correlates with ORI activation [139]. HBO1 is a HAT that has been found to be associated with pre-RC components such as MCM2 and ORC1 [140,141]. Moreover, H4K12ac is dependent on ORC. Overall, histone acetylation is not unique to the active amplicons in Drosophila follicle cells, but there is a quantitative relationship between the level of hyperacetylation and amplicon origin activity [138,142,143].
Methylation of histone H4 at lysine 20 (H4K20me) may play a role in the control of ORIs [144], and ORC recruitment appears to be enhanced by methylation of H4K20 by the histone methyltransferase PR-Set7 [145]. A recent Lexo-NS-seq analysis also found a potential role for H4K20me1 as well as H3K27me3 at ORIs [101]. This is supported by a recent integrative genomics analysis of chromatin marks across the rDNA repeat [146], where these two marks occur over the sites where rDNA replication initiation activity occurs ([100,147] and references therein). There have been reports for and against a significant association of ORIs with H3K4me3 [59,96–98], although most support this association with the caveat that it is not present at all ORIs and perhaps mostly early ones. In addition, different ORIs at the chicken β-globin locus have different patterns of histone modifications [148], suggesting further complexities. In general, conclusions have been that chromatin accessibility appears to be suitable for ORI formation, but is not necessary for it [60], and that no single chromatin mark studied to date can guarantee the presence of an ORI (nor vice versa).
Metazoan origins and DNA sequence elements (G4s, CpG islands, and GC-rich DNA)
Although an AT-rich consensus sequence motif was found in budding yeast ORIs [149–152], the hunt for such a motif in metazoan ORIs has been less successful. Any DNA sequence injected into Xenopus embryos could replicate [13], and plasmid replication was dependent on size rather than sequence [11,12]. In addition, it was shown that ORC binds most DNA equally well and has insufficient DNA sequence binding specificity to be responsible for ORI definition [153–155], although ORC had a slight preference for AT-rich double-stranded DNA consistent with the prediction that metazoan ORIs would likely be AT-rich similar to most studied ORIs across the tree of life [110,156–157]. Conclusions drawn from other experiments suggested that DNA topology rather than a conserved DNA sequence motif may be a more important determinant for ORC binding [154]. The consensus motif possibility was recently revived in the form of G4 motifs [94,99], which can form stable secondary structures. It was shown that ORC preferentially binds G4 structures in RNA and single-stranded DNA [158]. However, it needs to be emphasized that this preference was equivalent to AT-rich double-stranded DNA [158]. Most genome-wide Lexo-NS studies have reported GC-rich ORIs [93,94,96,99], but Karnani and colleagues [59] reported AT-rich ORIs and Foulk and colleagues [100] reported both AT- and GC-rich ORIs with mostly AT-rich ORIs after controlling for NSI Lexo biases. Perhaps the slight preference of ORC binding for both AT-rich double-stranded DNA and G4 motifs as well as the mixed results of AT-rich and GC-rich ORIs reflect the possibility of both AT-rich and GC-rich ORIs in metazoans, as was recently seen for the yeast species Pichia pastoris [159].
In general, a high correlation has been noted between Lexo-NS peak (putative ORI) density and G4 motifs [94,99,101,160], CGIs [93–96,98,99,101], and GC content [93,94,96,99]. In one study, nearly all (91.4%) Lexo-NS-seq peaks overlapped G4 motifs [99]. Moreover, in Lexo-NS studies, G4-associated ORIs appear to be among the most efficient ORIs [99,101,160], as do CGI-associated ORIs [93,95,98,101], and GC-richness tends to peak near ORI centers with G4s and G-richness occurring most frequently within 500 bp 5′ to Lexo-NS enrichments [94,100,160]. In one Lexo-NS study [160], point mutations in origin-associated G4s that lowered the stability of the G4 also lowered the Lexo enrichment level of the associated ORI and switching the strand the G4 was on changed the position of the Lexo-enriched ORI signal to remain 3′ of the G4. It was also seen that areas of higher G4 motif density have higher Lexo-NS enrichments and peak densities [99,101,160] and that 86% of Lexo-NS peaks associated with CGI are constitutive ORIs (present in all or most cell lines studied). At least one Lexo-NS study has claimed that the association with both CGI and genes is actually due to the association with G4s [99], which are correlated with those features. Lexo-NS studies have also highlighted that ORIs with these three features (G4, CGI, and GC richness) are typically early ORIs and that ORI density is correlated with timing (early ORIs being most densely populated and strongest) [93,94,99]. However, a recent study demonstrated that significantly enriched regions of the genome from sequencing Lexo-digested genomic DNA from non-replicating G0 cells were also associated with GC-richness, CGI, and G4s, suggesting that these observations in Lexo-NS studies could be explained, at least to some degree, by NSI Lexo biases [100].
Other techniques have been either supportive of or in conflict with the Lexo-NS-seq results. Bubble-seq did not support the conclusion that G4s are necessary for all ORIs [68]. The majority (59%) of bubble-containing fragments did not overlap G4s, and the number that did was only 1.05-fold enriched over the number expected at random [68]. Nonetheless, it is possible that G4s are important at the subset (41%) of ORIs that do overlap G4s. A small proportion (9.6%) of Bubble-seq enriched fragments [68] overlapped 51.3% of CGIs. BrIP-NS-seq peaks significantly overlapped G4s, although the G4 overlap made up just 37.5% of the peaks [60], and 13.1% BrIP peaks overlapped 35.1% CGI. Approximately a third (34.1%) of ORC binding sites [48] overlapped 2.8% of G4 motifs, and 30.6% of ORC sites overlapped 14.7% CGI. Moreover, it was concluded from nucleotide composition skew analysis that CpG-rich genes are over-represented at skew-predicted ORIs [64], and other Lexo-independent methods have identified ORIs near CGI ([161] and references therein). Given all the results from Lexo-NS, bubble-trap, BrIP-NS, ORC-ChIP, and nucleotide skew studies, it is clear that G4s are likely near 34-41% of ORIs and that CGIs are near approximately 10% to 30% of ORIs (many of which are the same ones associated with G4s). It is also clear that estimates from recent Lexo-NS studies stating that more than 70-90% of ORIs [94,99] are near G4s are inflated relative to other techniques. This inflation of G4 association may arise because Lexo digestion is inefficient for GC-rich DNA [100,102–104] and is impeded by G4 structures [100] and thus may reflect a high abundance of NSI enrichments and/or preferential enrichment of G4-proximal origins in those datasets. Indeed, in a recent study, after controlling for NSI Lexo biases, 35% of Lexo-NS-seq peaks overlapped 6.8% of G4s [100], which is more consistent with the BrIP, bubble, and ORC estimates. Moreover, Bubble-seq results suggested that ORI density was similar in early and late replicating regions [68]. Thus, it is possible that the higher ORI densities in early S phase associated with Lexo-NS studies are due to the combination of higher G4 and CGI occurrences in early replicating regions of the genome and NSI Lexo biases.
Another interpretation for the prevalence and high efficiency of G4 associated ORIs in genome-wide studies is that ORI-proximal G4s might impede replication fork progression in vivo. This would leave the replication bubbles and nascent strands from G4-proximal ORIs over-represented in the population of all ORIs because of their longer half-life, which would inflate the detection sensitivity and apparent efficiency of this subset of G4-proximal ORIs. In fact, higher detection and higher apparent efficiency would be true for ORIs with ORI-proximal pausing of replication forks in vivo for any other reason as well, such as impediments incurred by other structures. Valton and colleagues [160] have presented some compelling arguments against this interpretation of the G4 association with many apparently efficient ORIs, but it remains a formal possibility. Replication fork pauses have been detected 9 to 35 kb away from human ORIs [162], and high abundances of 200 nucleotide pieces of apparently aborted nascent DNA from ORIs have been found [163], but neither of these will be present in nascent strand preparations after size selection (typically 500-2500 bp). For support of this concern, more evidence will be required of origin-proximal pausing of replication forks that affect nascent strands in the target size range.
It is interesting that the observation that 5′ to 3′ Lexo digestion is both impeded by G4 structures and inefficient in digesting GC-rich DNA [100] explains quirks seen in Lexo-NS-based results at least as well as other interpretations. This observation predicts that Lexo will enrich G4-protected and GC-rich DNA. It predicts that Lexo-NS data will correlate with G4 density and that G4s will be 5′ to Lexo-NS enrichments. It predicts that experimentally inverting a G4 to place it on the opposite strand will change the location of Lexo-based enrichment to stay 3′ to the G4. It predicts that the strongest enrichments will be GC-rich. It predicts that GC-rich features such as CGIs will be constitutively enriched in all human cell lines (explaining why constitutive ORIs are near CGIs). It predicts that deleting G4s will result in less or no enrichment and that lowering the stability of G4s will lower the level of enrichment. All in all, given these caveats and others discussed above pertaining to all genome-wide ORI mapping approaches, the conclusions drawn so far about characteristics of metazoan ORIs should be cautiously considered.
Future outlooks
The genetic basis for ORI definition is suggested by many examples where ORI activity is retained by an ORI sequence put into an ectopic location in the genome [125,164–167]. Deletion of certain sequences reduces efficiency for the DHFR ori β [166,168,169], lamin B2 [125], c-myc [165], and β-globin [126] ORIs. Similarly, two elements from the Drosophila chorion gene locus, ACE3 and ori β, together can direct re-replication (amplification) when inserted into other genomic locations [170,171]. These observations raise the question of what features explain the basis for ORIs. As discussed above, the binding of metazoan ORC is more dependent on topology than on DNA sequences [154]. Could chromatin and higher order structure be determinants for ORI specification rather than ORC binding a specific sequence motif in metazoan genomes?
Moving forward, to continue harnessing the power of genomics for studying metazoan ORIs, a goal should be to refine and innovate origin-mapping techniques to hunt for ORIs more specifically. The challenges in genomics studies will be in overcoming systematic errors. Although the level of false positives from random sampling error in a dataset can be controlled by setting a desirable false discovery rate or irreproducible discovery rate ([172] and references therein), these statistical procedures do not control the level of systematic false positives that arise from biases in the method used. There is also a need to use other approaches, in addition to overlap of peak coordinates, to determine if and where datasets agree since the overlap of peak coordinates can be affected by many factors in upstream analytical steps (for example, P value thresholds). With the ever-increasing number of genomic datasets and ORI maps, quantifying our degree of belief in the validity of each individual putative ORI site in the genome on the basis of the accumulation of evidence, rather than maintaining uniform confidence over all putative ORIs in a single dataset, will be of utmost importance. For example, a statistical score such as the Bayesian posterior probability that there is an ORI or ORI potential (given all trusted datasets) could be assigned to each base pair in the genome. With highly authenticated genome-wide maps of replication initiation sites and zones, clearer pictures on what defines different classes of metazoan origins may emerge.
Overall, the field is now poised to address several questions for regulation of DNA replication: (a) what defines where the potential ORIs are located, (b) what determines which potential ORIs will be activated, and (c) what determines when in S phase an ORI will be activated [29]. It is clear that ORIs occur at defined sites in the genome, but their specification could be determined by something other than primary DNA sequence. It also remains possible that we are observing apparent specificity, rather than actual specificity, where ORIs opportunistically occur in reproducibly available sites determined by other features and processes.
Acknowledgments
We gratefully acknowledge support from postdoctoral fellowship DOD W81XWH-11-1-0599 to Cinzia Casella and a fellowship to John M. Urban for work supported by the National Science Foundation Graduate Research Fellowship Program under grant DGE-1058262.
This review article is dedicated to Joyce Hamlin and Mel DePamphilis, two giants in the field of DNA replication, whose years of healthy debate of whether metazoan replication origins are broad initiation zones or punctate sites have enlivened numerous Cold Spring Harbor Laboratory DNA replication meetings.
Abbreviations
- 2D
two-dimensional
- bp
base pairs
- BrdU
5-bromo-2′-deoxyuridine
- BrIP-NS
BrdU-immunoprecipitation enriched nascent strands
- CGI
CpG island
- ChIP
chromatin immunoprecipitation
- DHFR
dihydrofolate reductase
- eccDNA
extrachromosomal circular DNA
- G4
G-quadruplex
- Lexo
lambda exonuclease
- Lexo-NS
Lexo-enriched nascent strands
- NSI
nascent strand-independent
- ORC
origin recognition complex
- ORI
origin of DNA replication
- PCR
polymerase chain reaction
- qPCR
quantitative polymerase chain reaction
- RIP
replication initiation point
Disclosures
The authors declare that they have no disclosures.
The electronic version of this article is the complete one and can be found at: http://f1000.com/prime/reports/b/7/30
References
- 1.Bell SP, Dutta A. DNA replication in eukaryotic cells. Annu Rev Biochem. 2002;71:333–74. doi: 10.1146/annurev.biochem.71.110601.135425. [DOI] [PubMed] [Google Scholar]
- 2.DePamphilis ML. DNA Replication and Human Disease. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press; 2006. [Google Scholar]
- 3.DePamphilis ML, Bell S. Genome Duplication. New York: Garland Science; 2010. [Google Scholar]
- 4.Bell SD, Méchali M, DePamphilis ML. DNA Replication. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press; 2013. [Google Scholar]
- 5.Zou L, Stillman B. Formation of a preinitiation complex by S-phase cyclin CDK-dependent loading of Cdc45p onto chromatin. Science. 1998;280:593–6. doi: 10.1126/science.280.5363.593. [DOI] [PubMed] [Google Scholar]
- 6.Ilves I, Petojevic T, Pesavento JJ, Botchan MR. Activation of the MCM2-7 helicase by association with Cdc45 and GINS proteins. Mol Cell. 2010;37:247–58. doi: 10.1016/j.molcel.2009.12.030. [DOI] [PMC free article] [PubMed] [Google Scholar]; http://f1000.com/prime/1976956
- 7.Costa A, Ilves I, Tamberg N, Petojevic T, Nogales E, Botchan MR, Berger JM. The structural basis for MCM2-7 helicase activation by GINS and Cdc45. Nat Struct Mol Biol. 2011;18:471–7. doi: 10.1038/nsmb.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]; http://f1000.com/prime/9144960
- 8.Gambus A, Khoudoli GA, Jones RC, Blow JJ. MCM2-7 form double hexamers at licensed origins in Xenopus egg extract. J Biol Chem. 2011;286:11855–64. doi: 10.1074/jbc.M110.199521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Huberman JA, Riggs AD. Autoradiography of chromosomal DNA fibers from Chinese hamster cells. Proc Natl Acad Sci USA. 1966;55:599–606. doi: 10.1073/pnas.55.3.599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Huberman JA, Riggs AD. On the mechanism of DNA replication in mammalian chromosomes. J Mol Biol. 1968;32:327–41. doi: 10.1016/0022-2836(68)90013-2. [DOI] [PubMed] [Google Scholar]
- 11.Krysan PJ, Haase SB, Calos MP. Isolation of human sequences that replicate autonomously in human cells. Mol Cell Biol. 1989;9:1026–33. doi: 10.1128/mcb.9.3.1026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Heinzel SS, Krysan PJ, Tran CT, Calos MP. Autonomous DNA replication in human cells is affected by the size and the source of the DNA. Mol Cell Biol. 1991;11:2263–72. doi: 10.1128/mcb.11.4.2263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Harland RM, Laskey RA. Regulated replication of DNA microinjected into eggs of Xenopus laevis. Cell. 1980;21:761–71. doi: 10.1016/0092-8674(80)90439-0. [DOI] [PubMed] [Google Scholar]
- 14.Heintz NH, Hamlin JL. An amplified chromosomal sequence that includes the gene for dihydrofolate reductase initiates replication within specific restriction fragments. Proc Natl Acad Sci USA. 1982;79:4083–7. doi: 10.1073/pnas.79.13.4083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Burhans WC, Selegue JE, Heintz NH. Isolation of the origin of replication associated with the amplified Chinese hamster dihydrofolate reductase domain. Proc Natl Acad Sci USA. 1986;83:7790–4. doi: 10.1073/pnas.83.20.7790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hyrien O, Méchali M. Chromosomal replication initiates and terminates at random sequences but at regular intervals in the ribosomal DNA of Xenopus early embryos. EMBO J. 1993;12:4511–20. doi: 10.1002/j.1460-2075.1993.tb06140.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Blow JJ, Gillespie PJ, Francis D, Jackson DA. Replication origins in Xenopus egg extract Are 5-15 kilobases apart and are activated in clusters that fire at different times. J Cell Biol. 2001;152:15–25. doi: 10.1083/jcb.152.1.15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hyrien O, Maric C, Méchali M. Transition in specification of embryonic metazoan DNA replication origins. Science. 1995;270:994–7. doi: 10.1126/science.270.5238.994. [DOI] [PubMed] [Google Scholar]; http://f1000.com/prime/723296092
- 19.DePamphilis ML. The search for origins of DNA replication. Methods. 1997;13:211–9. doi: 10.1006/meth.1997.0521. [DOI] [PubMed] [Google Scholar]
- 20.Vassilev LT, Burhans WC, DePamphilis ML. Mapping an origin of DNA replication at a single-copy locus in exponentially proliferating mammalian cells. Mol Cell Biol. 1990;10:4685–9. doi: 10.1128/mcb.10.9.4685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Pelizon C, Diviacco S, Falaschi A, Giacca M. High-resolution mapping of the origin of DNA replication in the hamster dihydrofolate reductase gene domain by competitive PCR. Mol Cell Biol. 1996;16:5358–64. doi: 10.1128/mcb.16.10.5358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kobayashi T, Rein T, DePamphilis ML. Identification of primary initiation sites for DNA replication in the hamster dihydrofolate reductase gene initiation zone. Mol Cell Biol. 1998;18:3266–77. doi: 10.1128/mcb.18.6.3266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Brewer BJ, Fangman WL. The localization of replication origins on ARS plasmids in S. cerevisiae. Cell. 1987;51:463–71. doi: 10.1016/0092-8674(87)90642-8. [DOI] [PubMed] [Google Scholar]; http://f1000.com/prime/718701598
- 24.Huberman JA, Spotila LD, Nawotka KA, el-Assouli SM, Davis LR. The in vivo replication origin of the yeast 2 microns plasmid. Cell. 1987;51:473–81. doi: 10.1016/0092-8674(87)90643-X. [DOI] [PubMed] [Google Scholar]
- 25.Nawotka KA, Huberman JA. Two-dimensional gel electrophoretic method for mapping DNA replicons. Mol Cell Biol. 1988;8:1408–13. doi: 10.1128/mcb.8.4.1408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Siow CC, Nieduszynska SR, Müller CA, Nieduszynski CA. OriDB, the DNA replication origin database updated and extended. Nucleic Acids Res. 2012;40:D682–6. doi: 10.1093/nar/gkr1091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Aladjem MI. Replication in context: dynamic regulation of DNA replication patterns in metazoans. Nat Rev Genet. 2007;8:588–600. doi: 10.1038/nrg2143. [DOI] [PubMed] [Google Scholar]
- 28.Hamlin JL, Mesner LD, Lar O, Torres R, Chodaparambil SV, Wang L. A revisionist replicon model for higher eukaryotic genomes. J Cell Biochem. 2008;105:321–9. doi: 10.1002/jcb.21828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Masai H, Matsumoto S, You Z, Yoshizawa-Sugata N, Oda M. Eukaryotic chromosome DNA replication: where, when, and how? Annu Rev Biochem. 2010;79:89–130. doi: 10.1146/annurev.biochem.052308.103205. [DOI] [PubMed] [Google Scholar]
- 30.Norio P, Schildkraut CL. Visualization of DNA replication on individual Epstein-Barr virus episomes. Science. 2001;294:2361–4. doi: 10.1126/science.1064603. [DOI] [PubMed] [Google Scholar]; http://f1000.com/prime/723289596
- 31.Bensimon A, Simon A, Chiffaudel A, Croquette V, Heslot F, Bensimon D. Alignment and sensitive detection of DNA by a moving interface. Science. 1994;265:2096–8. doi: 10.1126/science.7522347. [DOI] [PubMed] [Google Scholar]
- 32.Michalet X, Ekong R, Fougerousse F, Rousseaux S, Schurra C, Hornigold N, van Slegtenhorst M, Wolfe J, Povey S, Beckmann JS, Bensimon A. Dynamic molecular combing: stretching the whole human genome for high-resolution studies. Science. 1997;277:1518–23. doi: 10.1126/science.277.5331.1518. [DOI] [PubMed] [Google Scholar]; http://f1000.com/prime/723294149
- 33.Herrick J, Bensimon A. Single molecule analysis of DNA replication. Biochimie. 1999;81:859–71. doi: 10.1016/S0300-9084(99)00210-2. [DOI] [PubMed] [Google Scholar]
- 34.Herrick J, Bensimon A. Introduction to molecular combing: genomics, DNA replication, and cancer. Methods Mol Biol. 2009;521:71–101. doi: 10.1007/978-1-60327-815-7_5. [DOI] [PubMed] [Google Scholar]
- 35.Bianco JN, Poli J, Saksouk J, Bacal J, Silva MJ, Yoshida K, Lin Y, Tourrière H, Lengronne A, Pasero P. Analysis of DNA replication profiles in budding yeast and mammalian cells using DNA combing. Methods. 2012;57:149–57. doi: 10.1016/j.ymeth.2012.04.007. [DOI] [PubMed] [Google Scholar]; http://f1000.com/prime/720704205
- 36.Técher H, Koundrioukoff S, Azar D, Wilhelm T, Carignon S, Brison O, Debatisse M, Le Tallec B. Replication dynamics: biases and robustness of DNA fiber analysis. J Mol Biol. 2013;425:4845–55. doi: 10.1016/j.jmb.2013.03.040. [DOI] [PubMed] [Google Scholar]; http://f1000.com/prime/717997265
- 37.Pasero P, Bensimon A, Schwob E. Single-molecule analysis reveals clustering and epigenetic regulation of replication origins at the yeast rDNA locus. Genes Dev. 2002;16:2479–84. doi: 10.1101/gad.232902. [DOI] [PMC free article] [PubMed] [Google Scholar]; http://f1000.com/prime/1009494
- 38.Patel PK, Arcangioli B, Baker SP, Bensimon A, Rhind N. DNA replication origins fire stochastically in fission yeast. Mol Biol Cell. 2006;17:308–16. doi: 10.1091/mbc.E05-07-0657. [DOI] [PMC free article] [PubMed] [Google Scholar]; http://f1000.com/prime/1032463
- 39.Anglana M, Apiou F, Bensimon A, Debatisse M. Dynamics of DNA replication in mammalian somatic cells: nucleotide pool modulates origin choice and interorigin spacing. Cell. 2003;114:385–94. doi: 10.1016/S0092-8674(03)00569-5. [DOI] [PubMed] [Google Scholar]; http://f1000.com/prime/718689018
- 40.Lebofsky R, Heilig R, Sonnleitner M, Weissenbach J, Bensimon A. DNA replication origin interference increases the spacing between initiation events in human cells. Mol Biol Cell. 2006;17:5337–45. doi: 10.1091/mbc.E06-04-0298. [DOI] [PMC free article] [PubMed] [Google Scholar]; http://f1000.com/prime/1047360
- 41.MacAlpine DM, Bell SP. A genomic view of eukaryotic DNA replication. Chromosome Res. 2005;13:309–26. doi: 10.1007/s10577-005-1508-1. [DOI] [PubMed] [Google Scholar]
- 42.Gilbert DM. Evaluating genome-scale approaches to eukaryotic DNA replication. Nat Rev Genet. 2010;11:673–84. doi: 10.1038/nrg2830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hamlin JL, Mesner LD, Dijkwel PA. A winding road to origin discovery. Chromosome Res. 2010;18:45–61. doi: 10.1007/s10577-009-9089-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lubelsky Y, MacAlpine HK, MacAlpine DM. Genome-wide localization of replication factors. Methods. 2012;57:187–95. doi: 10.1016/j.ymeth.2012.03.022. [DOI] [PubMed] [Google Scholar]; http://f1000.com/prime/1002804
- 45.Wyrick JJ, Aparicio JG, Chen T, Barnett JD, Jennings EG, Young RA, Bell SP, Aparicio OM. Genome-wide distribution of ORC and MCM proteins in S. cerevisiae: high-resolution mapping of replication origins. Science. 2001;294:2357–60. doi: 10.1126/science.1066101. [DOI] [PubMed] [Google Scholar]
- 46.Xu W, Aparicio JG, Aparicio OM, Tavaré S. Genome-wide mapping of ORC and Mcm2p binding sites on tiling arrays and identification of essential ARS consensus sequences in S. cerevisiae. BMC Genomics. 2006;7:276. doi: 10.1186/1471-2164-7-276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.MacAlpine HK, Gordân R, Powell SK, Hartemink AJ, MacAlpine DM. Drosophila ORC localizes to open chromatin and marks sites of cohesin complex loading. Genome Res. 2010;20:201–11. doi: 10.1101/gr.097873.109. [DOI] [PMC free article] [PubMed] [Google Scholar]; http://f1000.com/prime/722875468
- 48.Dellino GI, Cittaro D, Piccioni R, Luzi L, Banfi S, Segalla S, Cesaroni M, Mendoza-Maldonado R, Giacca M, Pelicci PG. Genome-wide mapping of human DNA-replication origins: levels of transcription at ORC1 sites regulate origin selection and replication timing. Genome Res. 2013;23:1–11. doi: 10.1101/gr.142331.112. [DOI] [PMC free article] [PubMed] [Google Scholar]; http://f1000.com/prime/717987460
- 49.Schepers A, Papior P. Why are we where we are? Understanding replication origins and initiation sites in eukaryotes using ChIP-approaches. Chromosome Res. 2010;18:63–77. doi: 10.1007/s10577-009-9087-1. [DOI] [PubMed] [Google Scholar]
- 50.Chesnokov IN. Multiple functions of the origin recognition complex. Int Rev Cytol. 2007;256:69–109. doi: 10.1016/S0074-7696(07)56003-1. [DOI] [PubMed] [Google Scholar]
- 51.Duncker BP, Chesnokov IN, McConkey BJ. The origin recognition complex protein family. Genome Biol. 2009;10:214. doi: 10.1186/gb-2009-10-3-214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hemerly AS, Prasanth SG, Siddiqui K, Stillman B. Orc1 controls centriole and centrosome copy number in human cells. Science. 2009;323:789–93. doi: 10.1126/science.1166745. [DOI] [PMC free article] [PubMed] [Google Scholar]; http://f1000.com/prime/1157119
- 53.Prasanth SG, Shen Z, Prasanth KV, Stillman B. Human origin recognition complex is essential for HP1 binding to chromatin and heterochromatin organization. Proc Natl Acad Sci USA. 2010;107:15093–8. doi: 10.1073/pnas.1009945107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Chakraborty A, Shen Z, Prasanth SG. “ORCanization” on heterochromatin: linking DNA replication initiation to chromatin organization. Epigenetics. 2011;6:665–70. doi: 10.4161/epi.6.6.16179. [DOI] [PubMed] [Google Scholar]
- 55.Hyrien O, Marheineke K, Goldar A. Paradoxes of eukaryotic DNA replication: MCM proteins and the random completion problem. Bioessays. 2003;25:116–25. doi: 10.1002/bies.10208. [DOI] [PubMed] [Google Scholar]
- 56.Blow JJ, Ge XQ, Jackson DA. How dormant origins promote complete genome replication. Trends Biochem Sci. 2011;36:405–14. doi: 10.1016/j.tibs.2011.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Santocanale C, Diffley JF. ORC- and Cdc6-dependent complexes at active and inactive chromosomal replication origins in Saccharomyces cerevisiae. EMBO J. 1996;15:6671–9. [PMC free article] [PubMed] [Google Scholar]
- 58.Lucas I, Palakodeti A, Jiang Y, Young DJ, Jiang N, Fernald AA, Le Beau Michelle M. High-throughput mapping of origins of replication in human cells. EMBO Rep. 2007;8:770–7. doi: 10.1038/sj.embor.7401026. [DOI] [PMC free article] [PubMed] [Google Scholar]; http://f1000.com/prime/1089151
- 59.Karnani N, Taylor CM, Malhotra A, Dutta A. Genomic study of replication initiation in human chromosomes reveals the influence of transcription regulation and chromatin structure on origin selection. Mol Biol Cell. 2010;21:393–404. doi: 10.1091/mbc.E09-08-0707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Mukhopadhyay R, Lajugie J, Fourel N, Selzer A, Schizas M, Bartholdy B, Mar J, Lin CM, Martin MM, Ryan M, Aladjem MI, Bouhassira EE. Allele-specific genome-wide profiling in human primary erythroblasts reveal replication program organization. PLoS Genet. 2014;10:e1004319. doi: 10.1371/journal.pgen.1004319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Hansen RS, Thomas S, Sandstrom R, Canfield TK, Thurman RE, Weaver M, Dorschner MO, Gartler SM, Stamatoyannopoulos JA. Sequencing newly replicated DNA reveals widespread plasticity in human replication timing. Proc Natl Acad Sci USA. 2010;107:139–44. doi: 10.1073/pnas.0912402107. [DOI] [PMC free article] [PubMed] [Google Scholar]; http://f1000.com/prime/1382981
- 62.Chen C, Rappailles A, Duquenne L, Huvet M, Guilbaud G, Farinelli L, Audit B, d’Aubenton-Carafa Y, Arneodo A, Hyrien O, Thermes C. Impact of replication timing on non-CpG and CpG substitution rates in mammalian genomes. Genome Res. 2010;20:447–57. doi: 10.1101/gr.098947.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Baker A, Audit B, Chen C, Moindrot B, Leleu A, Guilbaud G, Rappailles A, Vaillant C, Goldar A, Mongelard F, d’Aubenton-Carafa Y, Hyrien O, Thermes C, Arneodo A. Replication fork polarity gradients revealed by megabase-sized U-shaped replication timing domains in human cell lines. PLoS Comput Biol. 2012;8:e1002443. doi: 10.1371/journal.pcbi.1002443. [DOI] [PMC free article] [PubMed] [Google Scholar]; http://f1000.com/prime/720906639
- 64.Hyrien O, Rappailles A, Guilbaud G, Baker A, Chen C, Goldar A, Petryk N, Kahli M, Ma E, d’Aubenton-Carafa Y, Audit B, Thermes C, Arneodo A. From simple bacterial and archaeal replicons to replication N/U-domains. J Mol Biol. 2013;425:4673–89. doi: 10.1016/j.jmb.2013.09.021. [DOI] [PubMed] [Google Scholar]
- 65.Mesner LD, Crawford EL, Hamlin JL. Isolating apparently pure libraries of replication origins from complex genomes. Mol Cell. 2006;21:719–26. doi: 10.1016/j.molcel.2006.01.015. [DOI] [PubMed] [Google Scholar]; http://f1000.com/prime/1014459
- 66.Mesner LD, Dijkwel PA, Hamlin JL. Purification of restriction fragments containing replication intermediates from complex genomes for 2-D gel analysis. Methods Mol Biol. 2009;521:121–37. doi: 10.1007/978-1-60327-815-7_7. [DOI] [PMC free article] [PubMed] [Google Scholar]; http://f1000.com/prime/718798503
- 67.Mesner LD, Valsakumar V, Karnani N, Dutta A, Hamlin JL, Bekiranov S. Bubble-chip analysis of human origin distributions demonstrates on a genomic scale significant clustering into zones and significant association with transcription. Genome Res. 2011;21:377–89. doi: 10.1101/gr.111328.110. [DOI] [PMC free article] [PubMed] [Google Scholar]; http://f1000.com/prime/8311969
- 68.Mesner LD, Valsakumar V, Cieslik M, Pickin R, Hamlin JL, Bekiranov S. Bubble-seq analysis of the human genome reveals distinct chromatin-mediated mechanisms for regulating early- and late-firing origins. Genome Res. 2013;23:1774–88. doi: 10.1101/gr.155218.113. [DOI] [PMC free article] [PubMed] [Google Scholar]; http://f1000.com/prime/718040523
- 69.Dlaska M, Anderl C, Eisterer W, Bechter OE. Detection of circular telomeric DNA without 2D gel electrophoresis. DNA Cell Biol. 2008;27:489–96. doi: 10.1089/dna.2008.0741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Cohen S, Segal D. Extrachromosomal circular DNA in eukaryotes: possible involvement in the plasticity of tandem repeats. Cytogenet Genome Res. 2009;124:327–38. doi: 10.1159/000218136. [DOI] [PubMed] [Google Scholar]
- 71.Cohen S, Agmon N, Sobol O, Segal D. Extrachromosomal circles of satellite repeats and 5S ribosomal DNA in human cells. Mobile DNA. 2010;1:11. doi: 10.1186/1759-8753-1-11. [DOI] [PMC free article] [PubMed] [Google Scholar]; http://f1000.com/prime/715147800
- 72.Shibata Y, Kumar P, Layer R, Willcox S, Gagan JR, Griffith JD, Dutta A. Extrachromosomal microDNAs and chromosomal microdeletions in normal tissues. Science. 2012;336:82–6. doi: 10.1126/science.1213307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Handeli S, Klar A, Meuth M, Cedar H. Mapping replication units in animal cells. Cell. 1989;57:909–20. doi: 10.1016/0092-8674(89)90329-2. [DOI] [PubMed] [Google Scholar]; http://f1000.com/prime/718700137
- 74.Burhans WC, Vassilev LT, Wu J, Sogo JM, Nallaseth FS, DePamphilis ML. Emetine allows identification of origins of mammalian DNA replication by imbalanced DNA synthesis, not through conservative nucleosome segregation. EMBO J. 1991;10:4351–60. doi: 10.1002/j.1460-2075.1991.tb05013.x. [DOI] [PMC free article] [PubMed] [Google Scholar]; http://f1000.com/prime/720111363
- 75.Burhans WC, Vassilev LT, Caddle MS, Heintz NH, DePamphilis ML. Identification of an origin of bidirectional DNA replication in mammalian chromosomes. Cell. 1990;62:955–65. doi: 10.1016/0092-8674(90)90270-O. [DOI] [PubMed] [Google Scholar]; http://f1000.com/prime/718698921
- 76.Hay RT, DePamphilis ML. Initiation of SV40 DNA replication in vivo: location and structure of 5′ ends of DNA synthesized in the ori region. Cell. 1982;28:767–79. doi: 10.1016/0092-8674(82)90056-3. [DOI] [PubMed] [Google Scholar]; http://f1000.com/prime/718706233
- 77.Hendrickson EA, Fritze CE, Folk WR, DePamphilis ML. The origin of bidirectional DNA replication in polyoma virus. EMBO J. 1987;6:2011–8. doi: 10.1002/j.1460-2075.1987.tb02465.x. [DOI] [PMC free article] [PubMed] [Google Scholar]; http://f1000.com/prime/720113733
- 78.Smith DJ, Whitehouse I. Intrinsic coupling of lagging-strand synthesis to chromatin assembly. Nature. 2012;483:434–8. doi: 10.1038/nature10895. [DOI] [PMC free article] [PubMed] [Google Scholar]; http://f1000.com/prime/14264044
- 79.McGuffee SR, Smith DJ, Whitehouse I. Quantitative, genome-wide analysis of eukaryotic replication initiation and termination. Mol Cell. 2013;50:123–35. doi: 10.1016/j.molcel.2013.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]; http://f1000.com/prime/717998086
- 80.Gerbi SA, Bielinsky AK. Replication initiation point mapping. Methods. 1997;13:271–80. doi: 10.1006/meth.1997.0526. [DOI] [PubMed] [Google Scholar]
- 81.Bielinsky AK, Gerbi SA. Discrete start sites for DNA synthesis in the yeast ARS1 origin. Science. 1998;279:95–8. doi: 10.1126/science.279.5347.95. [DOI] [PubMed] [Google Scholar]
- 82.Gerbi SA, Bielinsky AK, Liang C, Lunyak VV, Urnov FD. Methods to map origins of replication in eukaryotes. In: Cotterill S, editor. Eukaryotic DNA Replication: a Practical Approach. Oxford: Oxford University Press; 1999:. pp. 1–42. [Google Scholar]
- 83.Gerbi SA. Mapping origins of DNA replication in eukaryotes. Methods Mol Biol. 2005;296:167–80. doi: 10.1385/1-59259-857-9:167. [DOI] [PubMed] [Google Scholar]
- 84.Das-Bradoo S, Bielinsky A. Replication initiation point mapping: approach and implications. Methods Mol Biol. 2009;521:105–20. doi: 10.1007/978-1-60327-815-7_6. [DOI] [PubMed] [Google Scholar]
- 85.Bielinsky AK, Gerbi SA. Chromosomal ARS1 has a single leading strand start site. Mol Cell. 1999;3:477–86. doi: 10.1016/S1097-2765(00)80475-X. [DOI] [PubMed] [Google Scholar]
- 86.Gómez M, Antequera F. Organization of DNA replication origins in the fission yeast genome. EMBO J. 1999;18:5683–90. doi: 10.1093/emboj/18.20.5683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Bielinsky AK, Blitzblau H, Beall EL, Ezrokhi M, Smith HS, Botchan MR, Gerbi SA. Origin recognition complex binding to a metazoan replication origin. Curr Biol. 2001;11:1427–31. doi: 10.1016/S0960-9822(01)00444-4. [DOI] [PubMed] [Google Scholar]
- 88.Abdurashidova G, Deganuto M, Klima R, Riva S, Biamonti G, Giacca M, Falaschi A. Start sites of bidirectional DNA synthesis at the human lamin B2 origin. Science. 2000;287:2023–6. doi: 10.1126/science.287.5460.2023. [DOI] [PubMed] [Google Scholar]
- 89.Radding CM. Regulation of lambda exonuclease. I. Properties of lambda exonuclease purified from lysogens of lambda T11 and wild type. J Mol Biol. 1966;18:235–50. doi: 10.1016/S0022-2836(66)80243-7. [DOI] [PubMed] [Google Scholar]
- 90.Little JW. An exonuclease induced by bacteriophage lambda. II. Nature of the enzymatic reaction. J Biol Chem. 1967;242:679–86. [PubMed] [Google Scholar]
- 91.Romero J, Lee H. One-way PCR-based mapping of a replication initiation point (RIP) Nat Protoc. 2008;3:1729–35. doi: 10.1038/nprot.2008.173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Cayrou C, Grégoire D, Coulombe P, Danis E, Méchali M. Genome-scale identification of active DNA replication origins. Methods. 2012;57:158–64. doi: 10.1016/j.ymeth.2012.06.015. [DOI] [PubMed] [Google Scholar]
- 93.Cayrou C, Coulombe P, Vigneron A, Stanojcic S, Ganier O, Peiffer I, Rivals E, Puy A, Laurent-Chabalier S, Desprat R, Méchali M. Genome-scale analysis of metazoan replication origins reveals their organization in specific but flexible sites defined by conserved features. Genome Res. 2011;21:1438–49. doi: 10.1101/gr.121830.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Cayrou C, Coulombe P, Puy A, Rialle S, Kaplan N, Segal E, Méchali M. New insights into replication origin characteristics in metazoans. Cell Cycle. 2012;11:658–67. doi: 10.4161/cc.11.4.19097. [DOI] [PMC free article] [PubMed] [Google Scholar]; http://f1000.com/prime/14227957
- 95.Sequeira-Mendes J, Díaz-Uriarte R, Apedaile A, Huntley D, Brockdorff N, Gómez M. Transcription initiation activity sets replication origin efficiency in mammalian cells. PLoS Genet. 2009;5:e1000446. doi: 10.1371/journal.pgen.1000446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Cadoret J, Meisch F, Hassan-Zadeh V, Luyten I, Guillet C, Duret L, Quesneville H, Prioleau M. Genome-wide studies highlight indirect links between human replication origins and gene regulation. Proc Natl Acad Sci USA. 2008;105:15837–42. doi: 10.1073/pnas.0805208105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Valenzuela MS, Chen Y, Davis S, Yang F, Walker RL, Bilke S, Lueders J, Martin MM, Aladjem MI, Massion PP, Meltzer PS. Preferential localization of human origins of DNA replication at the 5′-ends of expressed genes and at evolutionarily conserved DNA sequences. PLoS ONE. 2011;6:e17308. doi: 10.1371/journal.pone.0017308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Martin MM, Ryan M, Kim R, Zakas AL, Fu H, Lin CM, Reinhold WC, Davis SR, Bilke S, Liu H, Doroshow JH, Reimers MA, Valenzuela MS, Pommier Y, Meltzer PS, Aladjem MI. Genome-wide depletion of replication initiation events in highly transcribed regions. Genome Res. 2011;21:1822–32. doi: 10.1101/gr.124644.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Besnard E, Babled A, Lapasset L, Milhavet O, Parrinello H, Dantec C, Marin J, Lemaitre J. Unraveling cell type-specific and reprogrammable human replication origin signatures associated with G-quadruplex consensus motifs. Nat Struct Mol Biol. 2012;19:837–44. doi: 10.1038/nsmb.2339. [DOI] [PubMed] [Google Scholar]; http://f1000.com/prime/717955792
- 100.Foulk MS, Urban JM, Casella C, Gerbi SA. Characterizing and controlling intrinsic biases of Lambda exonuclease in nascent strand sequencing reveals phasing between nucleosomes and G-quadruplex motifs around a subset of human replication origins. Genome Res. 2015 doi: 10.1101/gr.183848.114. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Picard F, Cadoret J, Audit B, Arneodo A, Alberti A, Battail C, Duret L, Prioleau M. The spatiotemporal program of DNA replication is associated with specific combinations of chromatin marks in human cells. PLoS Genet. 2014;10:e1004282. doi: 10.1371/journal.pgen.1004282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Perkins TT, Dalal RV, Mitsis PG, Block SM. Sequence-dependent pausing of single lambda exonuclease molecules. Science. 2003;301:1914–8. doi: 10.1126/science.1088047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.van Oijen Antoine M, Blainey PC, Crampton DJ, Richardson CC, Ellenberger T, Xie XS. Single-molecule kinetics of lambda exonuclease reveal base dependence and dynamic disorder. Science. 2003;301:1235–8. doi: 10.1126/science.1084387. [DOI] [PubMed] [Google Scholar]; http://f1000.com/prime/1015203
- 104.Conroy RS, Koretsky AP, Moreland J. Lambda exonuclease digestion of CGG trinucleotide repeats. Eur Biophys J. 2010;39:337–43. doi: 10.1007/s00249-009-0502-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Yao Y, Wang Q, Hao Y, Tan Z. An exonuclease I hydrolysis assay for evaluating G-quadruplex stabilization by small molecules. Nucleic Acids Res. 2007;35:e68. doi: 10.1093/nar/gkm194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Han H, Hurley LH, Salazar M. A DNA polymerase stop assay for G-quadruplex-interactive compounds. Nucleic Acids Res. 1999;27:537–42. doi: 10.1093/nar/27.2.537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Lemarteleur T, Gomez D, Paterski R, Mandine E, Mailliet P, Riou J. Stabilization of the c-myc gene promoter quadruplex by specific ligands’ inhibitors of telomerase. Biochem Biophys Res Commun. 2004;323:802–8. doi: 10.1016/j.bbrc.2004.08.150. [DOI] [PubMed] [Google Scholar]
- 108.Potenski CJ, Klein HL. How the misincorporation of ribonucleotides into genomic DNA can be both harmful and helpful to cells. Nucleic Acids Res. 2014;42:10226–34. doi: 10.1093/nar/gku773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Yang W, Li X. Next-generation sequencing of Okazaki fragments extracted from Saccharomyces cerevisiae. FEBS Lett. 2013;587:2441–7. doi: 10.1016/j.febslet.2013.06.014. [DOI] [PubMed] [Google Scholar]
- 110.Aladjem MI, Fanning E. The replicon revisited: an old model learns new tricks in metazoan chromosomes. EMBO Rep. 2004;5:686–91. doi: 10.1038/sj.embor.7400185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Gilbert DM. Origins go plastic. Mol Cell. 2005;20:657–8. doi: 10.1016/j.molcel.2005.11.013. [DOI] [PubMed] [Google Scholar]
- 112.Borowiec JA, Schildkraut CL. Open sesame: activating dormant replication origins in the mouse immunoglobulin heavy chain (Igh) locus. Curr Opin Cell Biol. 2011;23:284–92. doi: 10.1016/j.ceb.2011.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.DePamphilis ML. Eukaryotic DNA replication: anatomy of an origin. Annu Rev Biochem. 1993;62:29–63. doi: 10.1146/annurev.bi.62.070193.000333. [DOI] [PubMed] [Google Scholar]
- 114.Brewer BJ, Fangman WL. Initiation at closely spaced replication origins in a yeast chromosome. Science. 1993;262:1728–31. doi: 10.1126/science.8259517. [DOI] [PubMed] [Google Scholar]
- 115.Brewer BJ, Fangman WL. Initiation preference at a yeast origin of replication. Proc Natl Acad Sci USA. 1994;91:3418–22. doi: 10.1073/pnas.91.8.3418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Marahrens Y, Stillman B. Replicator dominance in a eukaryotic chromosome. EMBO J. 1994;13:3395–400. doi: 10.1002/j.1460-2075.1994.tb06642.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Santocanale C, Sharma K, Diffley JF. Activation of dormant origins of DNA replication in budding yeast. Genes Dev. 1999;13:2360–4. doi: 10.1101/gad.13.18.2360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Vujcic M, Miller CA, Kowalski D. Activation of silent replication origins at autonomously replicating sequence elements near the HML locus in budding yeast. Mol Cell Biol. 1999;19:6098–109. doi: 10.1128/mcb.19.9.6098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Vaughn JP, Dijkwel PA, Hamlin JL. Replication initiates in a broad zone in the amplified CHO dihydrofolate reductase domain. Cell. 1990;61:1075–87. doi: 10.1016/0092-8674(90)90071-L. [DOI] [PubMed] [Google Scholar]
- 120.Dijkwel PA, Hamlin JL. The Chinese hamster dihydrofolate reductase origin consists of multiple potential nascent strand start sites. Mol Cell Biol. 1995;15:3023–31. doi: 10.1128/mcb.15.6.3023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Dijkwel PA, Wang S, Hamlin JL. Initiation sites are distributed at frequent intervals in the Chinese hamster dihydrofolate reductase origin of replication but are used with very different efficiencies. Mol Cell Biol. 2002;22:3053–65. doi: 10.1128/MCB.22.9.3053-3065.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Kalejta RF, Li X, Mesner LD, Dijkwel PA, Lin HB, Hamlin JL. Distal sequences, but not ori-beta/OBR-1, are essential for initiation of DNA replication in the Chinese hamster DHFR origin. Mol Cell. 1998;2:797–806. doi: 10.1016/S1097-2765(00)80294-4. [DOI] [PubMed] [Google Scholar]
- 123.Mesner LD, Li X, Dijkwel PA, Hamlin JL. The dihydrofolate reductase origin of replication does not contain any nonredundant genetic elements required for origin activity. Mol Cell Biol. 2003;23:804–14. doi: 10.1128/MCB.23.3.804-814.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Giacca M, Zentilin L, Norio P, Diviacco S, Dimitrova D, Contreas G, Biamonti G, Perini G, Weighardt F, Riva S. Fine mapping of a replication origin of human DNA. Proc Natl Acad Sci USA. 1994;91:7119–23. doi: 10.1073/pnas.91.15.7119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Paixão S, Colaluca IN, Cubells M, Peverali FA, Destro A, Giadrossi S, Giacca M, Falaschi A, Riva S, Biamonti G. Modular structure of the human lamin B2 replicator. Mol Cell Biol. 2004;24:2958–67. doi: 10.1128/MCB.24.7.2958-2967.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Wang L, Lin C, Brooks S, Cimbora D, Groudine M, Aladjem MI. The human beta-globin replication initiation region consists of two modular independent replicators. Mol Cell Biol. 2004;24:3373–86. doi: 10.1128/MCB.24.8.3373-3386.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Lunyak VV, Ezrokhi M, Smith HS, Gerbi SA. Developmental changes in the Sciara II/9A initiation zone for DNA replication. Mol Cell Biol. 2002;22:8426–37. doi: 10.1128/MCB.22.24.8426-8437.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Sasaki T, Sawado T, Yamaguchi M, Shinomiya T. Specification of regions of DNA replication initiation during embryogenesis in the 65-kilobase DNApolalpha-dE2F locus of Drosophila melanogaster. Mol Cell Biol. 1999;19:547–55. doi: 10.1128/mcb.19.1.547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Saha S, Shan Y, Mesner LD, Hamlin JL. The promoter of the Chinese hamster ovary dihydrofolate reductase gene regulates the activity of the local origin and helps define its boundaries. Genes Dev. 2004;18:397–410. doi: 10.1101/gad.1171404. [DOI] [PMC free article] [PubMed] [Google Scholar]; http://f1000.com/prime/1017892
- 130.Maric C, Prioleau M. Interplay between DNA replication and gene expression: a harmonious coexistence. Curr Opin Cell Biol. 2010;22:277–83. doi: 10.1016/j.ceb.2010.03.007. [DOI] [PubMed] [Google Scholar]
- 131.MacAlpine DM, Rodríguez HK, Bell SP. Coordination of replication and transcription along a Drosophila chromosome. Genes Dev. 2004;18:3094–105. doi: 10.1101/gad.1246404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Rowntree RK, Lee JT. Mapping of DNA replication origins to noncoding genes of the X-inactivation center. Mol Cell Biol. 2006;26:3707–17. doi: 10.1128/MCB.26.10.3707-3717.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Gómez M, Brockdorff N. Heterochromatin on the inactive X chromosome delays replication timing without affecting origin usage. Proc Natl Acad Sci USA. 2004;101:6923–8. doi: 10.1073/pnas.0401854101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Koren A, McCarroll SA. Random replication of the inactive X chromosome. Genome Res. 2014;24:64–9. doi: 10.1101/gr.161828.113. [DOI] [PMC free article] [PubMed] [Google Scholar]; http://f1000.com/prime/718118571
- 135.Danis E, Brodolin K, Menut S, Maiorano D, Girard-Reydet C, Méchali M. Specification of a DNA replication origin by a transcription complex. Nat Cell Biol. 2004;6:721–30. doi: 10.1038/ncb1149. [DOI] [PubMed] [Google Scholar]; http://f1000.com/prime/1001435
- 136.Lubelsky Y, Prinz JA, DeNapoli L, Li Y, Belsky JA, MacAlpine DM. DNA replication and transcription programs respond to the same chromatin cues. Genome Res. 2014;24:1102–14. doi: 10.1101/gr.160010.113. [DOI] [PMC free article] [PubMed] [Google Scholar]; http://f1000.com/prime/718478964
- 137.Lubelsky Y, Sasaki T, Kuipers MA, Lucas I, Le Beau Michelle M, Carignon S, Debatisse M, Prinz JA, Dennis JH, Gilbert DM. Pre-replication complex proteins assemble at regions of low nucleosome occupancy within the Chinese hamster dihydrofolate reductase initiation zone. Nucleic Acids Res. 2011;39:3141–55. doi: 10.1093/nar/gkq1276. [DOI] [PMC free article] [PubMed] [Google Scholar]; http://f1000.com/prime/719377262
- 138.Aggarwal BD, Calvi BR. Chromatin regulates origin activity in Drosophila follicle cells. Nature. 2004;430:372–6. doi: 10.1038/nature02694. [DOI] [PubMed] [Google Scholar]; http://f1000.com/prime/1001436
- 139.Liu J, McConnell K, Dixon M, Calvi BR. Analysis of model replication origins in Drosophila reveals new aspects of the chromatin landscape and its relationship to origin activity and the prereplicative complex. Mol Biol Cell. 2012;23:200–12. doi: 10.1091/mbc.E11-05-0409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Iizuka M, Stillman B. Histone acetyltransferase HBO1 interacts with the ORC1 subunit of the human initiator protein. J Biol Chem. 1999;274:23027–34. doi: 10.1074/jbc.274.33.23027. [DOI] [PubMed] [Google Scholar]
- 141.Burke TW, Cook JG, Asano M, Nevins JR. Replication factors MCM2 and ORC1 interact with the histone acetyltransferase HBO1. J Biol Chem. 2001;276:15397–408. doi: 10.1074/jbc.M011556200. [DOI] [PubMed] [Google Scholar]
- 142.Hartl T, Boswell C, Orr-Weaver TL, Bosco G. Developmentally regulated histone modifications in Drosophila follicle cells: initiation of gene amplification is associated with histone H3 and H4 hyperacetylation and H1 phosphorylation. Chromosoma. 2007;116:197–214. doi: 10.1007/s00412-006-0092-2. [DOI] [PubMed] [Google Scholar]
- 143.Kim JC, Nordman J, Xie F, Kashevsky H, Eng T, Li S, MacAlpine DM, Orr-Weaver TL. Integrative analysis of gene amplification in Drosophila follicle cells: parameters of origin activation and repression. Genes Dev. 2011;25:1384–98. doi: 10.1101/gad.2043111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Dorn ES, Cook JG. Nucleosomes in the neighborhood: new roles for chromatin modifications in replication origin control. Epigenetics. 2011;6:552–9. doi: 10.4161/epi.6.5.15082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Sherstyuk VV, Shevchenko AI, Zakian SM. Epigenetic landscape for initiation of DNA replication. Chromosoma. 2014;123:183–99. doi: 10.1007/s00412-013-0448-3. [DOI] [PubMed] [Google Scholar]
- 146.Zentner GE, Saiakhova A, Manaenkov P, Adams MD, Scacheri PC. Integrative genomic analysis of human ribosomal DNA. Nucleic Acids Res. 2011;39:4949–60. doi: 10.1093/nar/gkq1326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Coffman FD, He M, Diaz M, Cohen S. DNA replication initiates at different sites in early and late S phase within human ribosomal RNA genes. Cell Cycle. 2005;4:1223–6. doi: 10.4161/cc.4.9.1961. [DOI] [PubMed] [Google Scholar]
- 148.Prioleau M, Gendron M, Hyrien O. Replication of the chicken beta-globin locus: early-firing origins at the 5′ HS4 insulator and the rho- and betaA-globin genes show opposite epigenetic modifications. Mol Cell Biol. 2003;23:3536–49. doi: 10.1128/MCB.23.10.3536-3549.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Newlon CS, Theis JF. The structure and function of yeast ARS elements. Curr Opin Genet Dev. 1993;3:752–8. doi: 10.1016/S0959-437X(05)80094-2. [DOI] [PubMed] [Google Scholar]
- 150.Theis JF, Newlon CS. The ARS309 chromosomal replicator of Saccharomyces cerevisiae depends on an exceptional ARS consensus sequence. Proc Natl Acad Sci USA. 1997;94:10786–91. doi: 10.1073/pnas.94.20.10786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Breier AM, Chatterji S, Cozzarelli NR. Prediction of Saccharomyces cerevisiae replication origins. Genome Biol. 2004;5:R22. doi: 10.1186/gb-2004-5-4-r22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Dhar MK, Sehgal S, Kaul S. Structure, replication efficiency and fragility of yeast ARS elements. Res Microbiol. 2012;163:243–53. doi: 10.1016/j.resmic.2012.03.003. [DOI] [PubMed] [Google Scholar]
- 153.Vashee S, Cvetic C, Lu W, Simancek P, Kelly TJ, Walter JC. Sequence-independent DNA binding and replication initiation by the human origin recognition complex. Genes Dev. 2003;17:1894–908. doi: 10.1101/gad.1084203. [DOI] [PMC free article] [PubMed] [Google Scholar]; http://f1000.com/prime/1006207
- 154.Remus D, Beall EL, Botchan MR. DNA topology, not DNA sequence, is a critical determinant for Drosophila ORC-DNA binding. EMBO J. 2004;23:897–907. doi: 10.1038/sj.emboj.7600077. [DOI] [PMC free article] [PubMed] [Google Scholar]; http://f1000.com/prime/720103769
- 155.Schaarschmidt D, Baltin J, Stehle IM, Lipps HJ, Knippers R. An episomal mammalian replicon: sequence-independent binding of the origin recognition complex. EMBO J. 2004;23:191–201. doi: 10.1038/sj.emboj.7600029. [DOI] [PMC free article] [PubMed] [Google Scholar]; http://f1000.com/prime/1017582
- 156.Méchali M. Eukaryotic DNA replication origins: many choices for appropriate answers. Nat Rev Mol Cell Biol. 2010;11:728–38. doi: 10.1038/nrm2976. [DOI] [PubMed] [Google Scholar]
- 157.Leonard AC, Méchali M. DNA replication origins. Cold Spring Harb Perspect Biol. 2013;5:a010116. doi: 10.1101/cshperspect.a010116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Hoshina S, Yura K, Teranishi H, Kiyasu N, Tominaga A, Kadoma H, Nakatsuka A, Kunichika T, Obuse C, Waga S. Human origin recognition complex binds preferentially to G-quadruplex-preferable RNA and single-stranded DNA. J Biol Chem. 2013;288:30161–71. doi: 10.1074/jbc.M113.492504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Liachko I, Youngblood RA, Tsui K, Bubb KL, Queitsch C, Raghuraman MK, Nislow C, Brewer BJ, Dunham MJ. GC-rich DNA elements enable replication origin activity in the methylotrophic yeast Pichia pastoris. PLoS Genet. 2014;10:e1004169. doi: 10.1371/journal.pgen.1004169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Valton A, Hassan-Zadeh V, Lema I, Boggetto N, Alberti P, Saintomé C, Riou J, Prioleau M. G4 motifs affect origin positioning and efficiency in two vertebrate replicators. EMBO J. 2014;33:732–46. doi: 10.1002/embj.201387506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Delgado S, Gómez M, Bird A, Antequera F. Initiation of DNA replication at CpG islands in mammalian chromosomes. EMBO J. 1998;17:2426–35. doi: 10.1093/emboj/17.8.2426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Frum RA, Chastain PD, Qu P, Cohen SM, Kaufman DG. DNA replication in early S phase pauses near newly activated origins. Cell Cycle. 2008;7:1440–8. doi: 10.4161/cc.7.10.5879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Gómez M, Antequera F. Overreplication of short DNA regions during S phase in human cells. Genes Dev. 2008;22:375–85. doi: 10.1101/gad.445608. [DOI] [PMC free article] [PubMed] [Google Scholar]; http://f1000.com/prime/1099354
- 164.Aladjem MI, Rodewald LW, Kolman JL, Wahl GM. Genetic dissection of a mammalian replicator in the human beta-globin locus. Science. 1998;281:1005–9. doi: 10.1126/science.281.5379.1005. [DOI] [PubMed] [Google Scholar]
- 165.Liu G, Malott M, Leffak M. Multiple functional elements comprise a Mammalian chromosomal replicator. Mol Cell Biol. 2003;23:1832–42. doi: 10.1128/MCB.23.5.1832-1842.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Altman AL, Fanning E. Defined sequence modules and an architectural element cooperate to promote initiation at an ectopic mammalian chromosomal replication origin. Mol Cell Biol. 2004;24:4138–50. doi: 10.1128/MCB.24.10.4138-4150.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Guan Z, Hughes CM, Kosiyatrakul S, Norio P, Sen R, Fiering S, Allis CD, Bouhassira EE, Schildkraut CL. Decreased replication origin activity in temporal transition regions. J Cell Biol. 2009;187:623–35. doi: 10.1083/jcb.200905144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Altman AL, Fanning E. The Chinese hamster dihydrofolate reductase replication origin beta is active at multiple ectopic chromosomal locations and requires specific DNA sequence elements for activity. Mol Cell Biol. 2001;21:1098–110. doi: 10.1128/MCB.21.4.1098-1110.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Gray SJ, Liu G, Altman AL, Small LE, Fanning E. Discrete functional elements required for initiation activity of the Chinese hamster dihydrofolate reductase origin beta at ectopic chromosomal sites. Exp Cell Res. 2007;313:109–20. doi: 10.1016/j.yexcr.2006.09.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Lu L, Zhang H, Tower J. Functionally distinct, sequence-specific replicator and origin elements are required for Drosophila chorion gene amplification. Genes Dev. 2001;15:134–46. doi: 10.1101/gad.822101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Zhang H, Tower J. Sequence requirements for function of the Drosophila chorion gene locus ACE3 replicator and ori-beta origin elements. Development. 2004;131:2089–99. doi: 10.1242/dev.01064. [DOI] [PubMed] [Google Scholar]
- 172.Landt SG, Marinov GK, Kundaje A, Kheradpour P, Pauli F, Batzoglou S, Bernstein BE, Bickel P, Brown JB, Cayting P, Chen Y, DeSalvo G, Epstein C, Fisher-Aylor KI, Euskirchen G, Gerstein M, Gertz J, Hartemink AJ, Hoffman MM, Iyer VR, Jung YL, Karmakar S, Kellis M, Kharchenko PV, Li Q, Liu T, Liu XS, Ma L, Milosavljevic A, Myers RM, et al. ChIP-seq guidelines and practices of the ENCODE and modENCODE consortia. Genome Res. 2012;22:1813–31. doi: 10.1101/gr.136184.111. [DOI] [PMC free article] [PubMed] [Google Scholar]; http://f1000.com/prime/717960363