

Abstract
Objective:
To precisely delineate clinical risk factors for conversion from idiopathic REM sleep behavior disorder (RBD) to Parkinson disease, dementia with Lewy bodies, and multiple system atrophy, in order to enable practical planning and stratification of neuroprotective trials against neurodegenerative synucleinopathy.
Methods:
In a 10-year prospective cohort, we tested prodromal Parkinson disease markers in 89 patients with idiopathic RBD. With Kaplan-Meier analysis, we calculated risk of neurodegenerative synucleinopathy, and using Cox proportional hazards, tested the ability of prodromal markers to identify patients at higher disease risk. By combining predictive markers, we then designed stratification strategies to optimally select patients for definitive neuroprotective trials.
Results:
The risk of defined neurodegenerative synucleinopathy was high: 30% developed disease at 3 years, rising to 66% at 7.5 years. Advanced age (hazard ratio [HR] = 1.07), olfactory loss (HR = 2.8), abnormal color vision (HR = 3.1), subtle motor dysfunction (HR = 3.9), and nonuse of antidepressants (HR = 3.5) identified higher risk of disease conversion. However, mild cognitive impairment (HR = 1.8), depression (HR = 0.63), Parkinson personality, treatment with clonazepam (HR = 1.3) or melatonin (HR = 0.55), autonomic markers, and sex (HR = 1.37) did not clearly predict clinical neurodegeneration. Stratification with prodromal markers increased risk of neurodegenerative disease conversion by 200%, and combining markers allowed sample size reduction in neuroprotective trials by >40%. With a moderately effective agent (HR = 0.5), trials with fewer than 80 subjects per group can demonstrate definitive reductions in neurodegenerative disease.
Conclusions:
Using stratification with simply assessed markers, it is now not only possible, but practical to include patients with RBD in neuroprotective trials against Parkinson disease, multiple system atrophy, and dementia with Lewy bodies.
REM sleep behavior disorder (RBD) is characterized by loss of REM sleep paralysis, allowing patients to “act out” dreams.1 Idiopathic RBD is an extremely powerful predictor (or, prodromal marker) of neurodegenerative synucleinopathies, including Parkinson disease (PD), dementia with Lewy bodies (DLB), and multiple system atrophy (MSA), and >80% eventually develop neurodegenerative disease.2,3
This has extreme importance for development of neuroprotective therapy against synucleinopathy; no prodromal marker of disease has near RBD's predictive value.4 Patients with idiopathic RBD are ideal candidates for neuroprotective trials against synucleinopathy; one can intervene earlier, before neuronal loss progresses past rescue, and before symptomatic therapies confound assessments.5
However, barriers remain to including patients with idiopathic RBD in neuroprotective trials, namely, (1) disease risk is not yet fully defined; (2) a proportion may be at lower disease risk, therefore their inclusion reduces power; and (3) even at current estimates, conversion rates may still be too low for practical trial design, particularly if duration is limited (e.g., patent-protected agents).
Ten years ago, we commenced a prospective cohort study of patients with idiopathic RBD. We assessed a broad array of potential predictive markers of neurodegenerative synucleinopathy, and repeated assessment annually (several individual markers have been published6–13). We now report the comprehensive 10-year follow-up evaluation, addressing the following questions: (1) What is the true risk of neurodegenerative synucleinopathy? (2) What is the precise predictive value of prodromal neurodegenerative markers in RBD? and (3) Can markers be combined to select high-risk groups for neuroprotective trials?
METHODS
Standard protocol approvals, registrations, and patient consents.
This trial was approved by the local research ethics board, and all patients provided informed written consent to participate.
Patients and evaluations.
Patient selection and evaluation procedures were described in detail elsewhere.14 All patients had idiopathic RBD defined with standard ICSD-II criteria. All had baseline neurologic examination excluding defined neurodegenerative disease. Each annual evaluation included a complete evaluation for parkinsonism (UK Brain Bank criteria) and dementia (Movement Disorder Society dementia criteria15). Follow-up occurred annually until development of neurodegeneration, including home evaluation if patients were unable to come in person; any patients lost to follow-up were analyzed as censored at last visit.
We then evaluated a broad panel of potential prodromal synucleinopathy markers. Annually assessed markers included the following:
Olfactory dysfunction—Cross-Cultural 12-item test16
Color vision testing—Farnsworth-Munsell 100-hue test17
-
Quantitative motor testing6:
a. Alternate tap—hand motor speed
b. Purdue Pegboard—hand dexterity/speed, finger-eye coordination
c. Timed Up and Go—gait/transfer speed
d. Unified Parkinson's Disease Rating Scale, Part III
Autonomic—orthostatic, urinary, erectile, bowel symptoms from the MSA rating scale18 orthostatic hypotension tested using manual measurement, supine and after 1 minute standing
Cognitive testing—annual neuropsychological examination (outlined elsewhere19) evaluating dementia/mild cognitive impairment (MCI),19 Montreal Cognitive Assessment20 since 2006
Baseline markers (not repeated annually) included the following:
Analysis.
Disease risk was estimated with Kaplan-Meier analysis. For testing prodromal markers, Cox proportional hazards was performed, adjusting for baseline age and sex. For stratification, all predictive markers were classified binarily (normal/abnormal). Olfaction was classified abnormal if <80% of age/sex-adjusted normal, and color vision if errors >125% normal.10 Individual motor tests were defined abnormal using cutoffs for ideal sensitivity/specificity defined in our study of parkinsonism in RBD (alternate tap <175 taps, Purdue <11 pegs, Timed Up and Go >7.5 seconds, Unified Parkinson's Disease Rating Scale score >3 excluding action tremor).11 Autonomic symptoms were classified present/abnormal if >0 on the MSA rating scale, and systolic orthostatic blood pressure decrease ≥10 mm Hg was classified abnormal (also based on sensitivity/specificity cutoffs).
For combining markers, we performed Kaplan-Meier analysis, stratified to presence of marker combinations. We calculated percentage disease conversion at defined intervals (3/5 years), and estimated the proportion eligible in each stratification strategy. Finally, we estimated sample size requirements for a theoretical neuroprotective trial. This assumed a categorical definitive endpoint (disease conversion), 2 groups (placebo single-dose treatment), α < 0.05, and 80% power. We used a time-to-event analysis (http://hedwig.mgh.harvard.edu/sample_size/time_to_event/para_time.html, trial accrual = 2 years, follow-up = 2 years [i.e., median follow-up = 3 years], median time to failure). We tested 3 effectiveness assumptions for the neuroprotective agent: moderately effective (hazard ratio [HR] = 0.5), modestly effective (HR = 0.7), and dramatically effective (HR = 0.2). On secondary analysis, we also estimated sample size using binomial probability (2 proportions, http://www.stat.ubc.ca/∼rollin/stats/ssize/b2.html), delineating a 2-year follow-up (total calendar time = 4 years, assuming 2-year accrual), and 50% reduction in total proportion developing disease at study end.
RESULTS
Neurodegenerative outcomes.
Ninety-five patients were seen at baseline between June 2004 and 2012. Eighty-nine (94%) had ≥1 follow-up examination and were eligible for analysis. Of the 6 without follow-up, 2 died before follow-up, and 4 were lost to follow-up. After completing at least one follow-up, an additional 2 patients who were still disease-free died, and 3 were lost to follow-up. Average follow-up to December 2013 was 5.4 ± 2.9 years (disease-free follow-up duration, disease as terminal event = 3.8 ± 2.4 years). Among the 89 patients, a total of 375 annual examinations were performed (86% of perfect attendance). Baseline age was 66.9 ± 9.3 years, and 65 (73%) were male. RBD symptom duration before baseline was 9.2 ± 9.2 years.
Over follow-up to December 2013, 41 patients developed neurodegenerative disease. Twenty-one were diagnosed with primary dementia and 20 with parkinsonism (PD = 17, MSA = 3). Seven were diagnosed at the second annual visit, 6 at year 3, 12 at year 4, 4 at year 5, 5 at year 6, 2 at year 7, 2 at year 8, 1 at year 9, and 2 at year 10. Of the 21 with dementia, 18 had ≥1 cardinal parkinsonism manifestation (11 met full UK Brain Bank criteria for parkinsonism23), and the remaining 3 had abnormal quantitative motor testing; therefore, most, if not all, had DLB.11,16,24 Of 17 patients with PD, 8 had MCI at diagnosis. Therefore, we observed considerable overlap between parkinsonian and cognitive signs.
On Kaplan-Meier analysis (figure 1), we found very high rates of defined neurodegenerative disease: 30% developed neurodegeneration at 3 years, 47% at 5 years, and 66% at 7.5 years.
Figure 1. Development of defined neurodegeneration in idiopathic RBD.
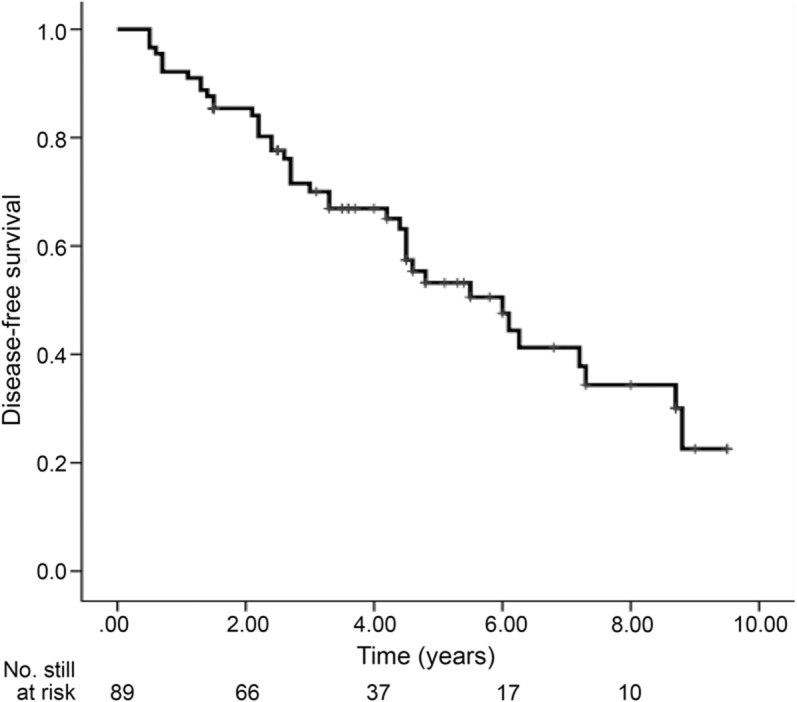
Shown is the Kaplan-Meier plot of disease-free survival of patients with idiopathic REM sleep behavior disorder (RBD). Ticks indicate censoring events.
Outcome according to baseline markers.
Patients who eventually developed disease were older than those who remained disease-free (69.6 vs 64.6). Males and females had similar disease risk. All subsequent analyses were adjusted for age and sex (table 1, figure 2).
Table 1.
Potential predictors of disease at baseline in idiopathic RBD
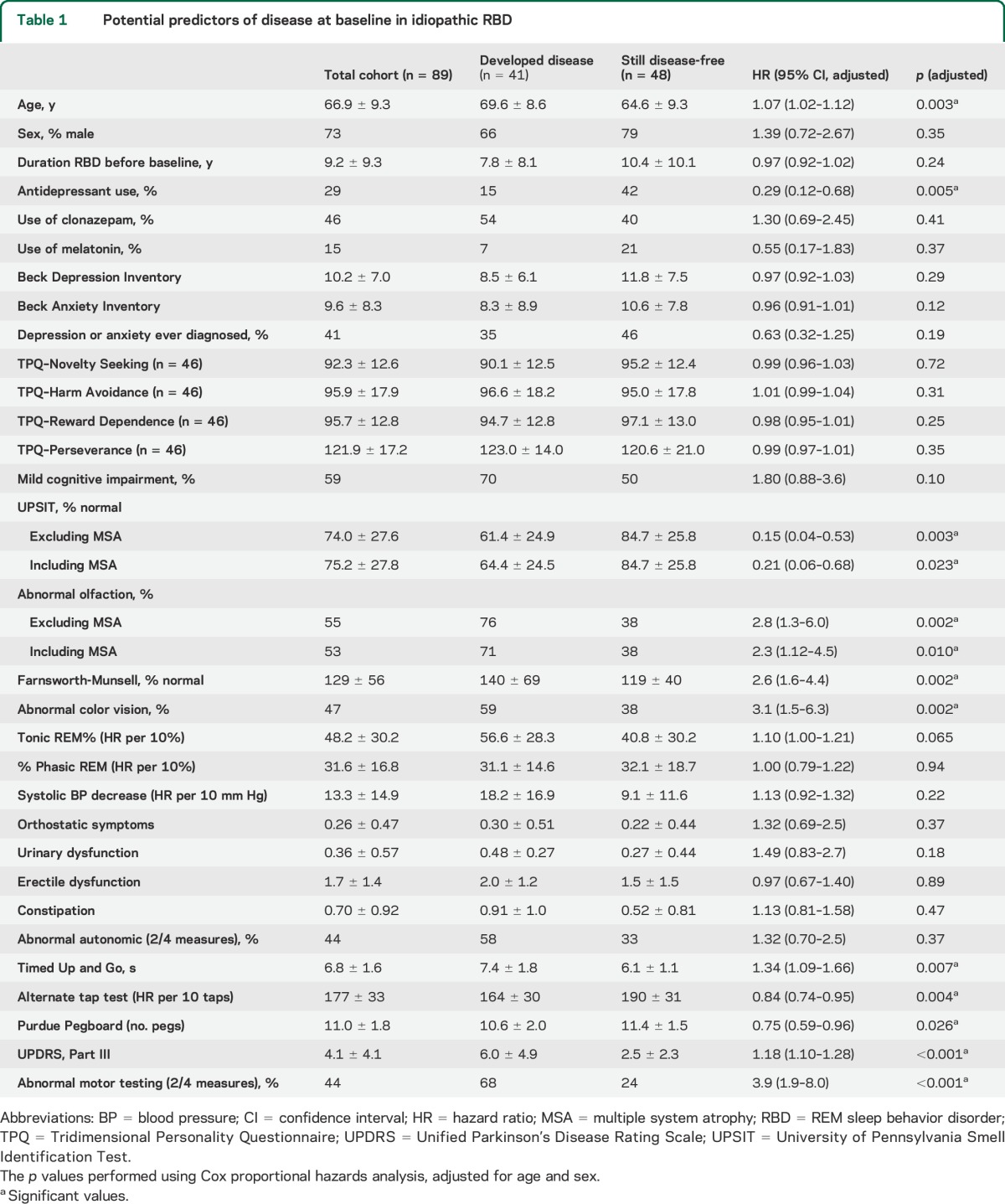
Figure 2. Predictive markers of neurodegeneration in idiopathic RBD.

Shown is the Kaplan-Meier plot of disease-free survival of patients with idiopathic REM sleep behavior disorder (RBD), stratified according to presence of baseline markers: (A) olfaction, (B) color vision, (C) mild motor dysfunction, (D) autonomic dysfunction, (E) depression/anxiety, and (F) REM atonia loss. To mimic a clinical trial recruitment strategy, results are presented according to baseline assessment only (i.e., patients who develop a de novo marker abnormality over the course of the follow-up remain in the “marker-free” group). Solid line indicates patients with normal values, dashed line abnormal values. The 50% REM atonia (F) is for percentage tonic REM. Hazard ratios (HRs) are with Cox proportional hazards, adjusting for age and sex.
Tonic REM% was associated with higher risk on unadjusted, but not adjusted, analysis. Thirty-five patients were taking clonazepam alone, 7 melatonin alone, 6 both, and 41 neither. Symptomatic treatment with either melatonin (HR = 0.55) or clonazepam (HR = 1.30) did not affect neurodegenerative outcome.
As anticipated, motor abnormalities were very strong predictors of neurodegenerative disease. Defining a motor examination as abnormal (2/4 quantitative motor test abnormalities11) increased HR to 3.9 (95% confidence interval = 1.9–8.0). Of note, this was even stronger for dementia diagnosis (HR = 9.8 [2.6–37.4]) than for primary parkinsonism (HR = 2.5 [0.9–7.4]), perhaps consistent with the slower pace of parkinsonism evolution in DLB.11
Abnormal olfaction was associated with an HR for neurodegenerative disease of 2.3 (1.12–4.5). Excluding patients with MSA (olfaction is normal in MSA) produced an HR of 2.8 (1.3–6.0).
Similarly, those with abnormal color vision were at higher conversion risk (HR = 3.1 [1.5–6.3]). This effect was much stronger for primary dementia than parkinsonism (HR = 10.3 [2.8–38.6] vs 1.5 [0.49–4.6]).
We found no clear evidence that baseline MCI could predict clinical PD (HR = 1.1 [0.39–3.0]). However, baseline MCI predicted conversion to primary dementia (HR = 8.0 [1.4–44.2]).
Depressive or anxiety symptoms did not predict who developed disease. A possible antidepressant trigger, however, was associated with lower risk, as recently described.13 Because of numerous reports of a “Parkinson personality,” we assessed personality variables in a subset of patients (n = 46). Although these variables differed compared with controls,14 no personality variable predicted neurodegenerative conversion. No autonomic variables predicted disease risk.
When patients developing Lewy body disease were divided into presentation with “parkinsonism first” vs “dementia first,” there were relatively few differences between groups (table e-1 on the Neurology® Web site at Neurology.org). However, patients presenting dementia first were more likely to have baseline MCI, color vision impairment, and olfactory impairment.
Stratifying risk for clinical trials.
We then tested potential selection strategies to increase likelihood of conversion to clinical neurodegenerative disease (table e-2). Using single markers, stratification could increase likelihood of conversion to disease, sometimes with only minimal loss in screening. Simple measures such as removing possible antidepressant-caused RBD or patients younger than 55 years increased conversion rates modestly. Other clinical signs were more powerful stratifiers, albeit with more recruitment cost. Selecting patients with olfactory loss would allow 52% cohort eligibility and increased 3-year conversion from 30% to 40% and 5-year conversion from 47% to 63%. Selecting those with motor impairment (2/4 quantitative motor tests) increased conversion more dramatically, to 56% at 3 years and 74% at 5 years (45% eligible).
We then explored whether combining markers could further stratify disease risk (table e-3, figure 3). We found numerous potential strategies for increasing conversion rates. For example, removing age younger than 55 years and possible antidepressant-caused RBD, combined with any one of abnormal olfaction, color vision, or motor impairment, increased disease risk to 44% at 3 years and 61% at 5 years, while retaining 66% eligibility. Stricter eligibility requirements could dramatically increase conversion; motor impairment and age/antidepressant exclusion obtained a 65% 3-year rate (36% eligible). Requiring 2/3 abnormalities increased 3-year conversion to 60% and 5-year conversion to 80% (31% eligible), and 3/3 abnormalities identified near-certain conversion (92% at 5 years).
Figure 3. Combining predictive markers of neurodegeneration in idiopathic RBD.

Shown is the Kaplan-Meier plot of disease-free survival of patients with idiopathic REM sleep behavior disorder (RBD), stratified according to combinations of baseline markers: (A) removing antidepressant-triggered RBD and age 55 years and older; (B) addition of olfaction and motor combined; (C) addition of olfaction and color vision combined; and (D) addition of motor and color vision combined. Solid line indicates patients with normal values, dashed lines mean that 1 of 2 measures is abnormal, dotted lines mean both measures are abnormal.
Sample size requirements for clinical trials.
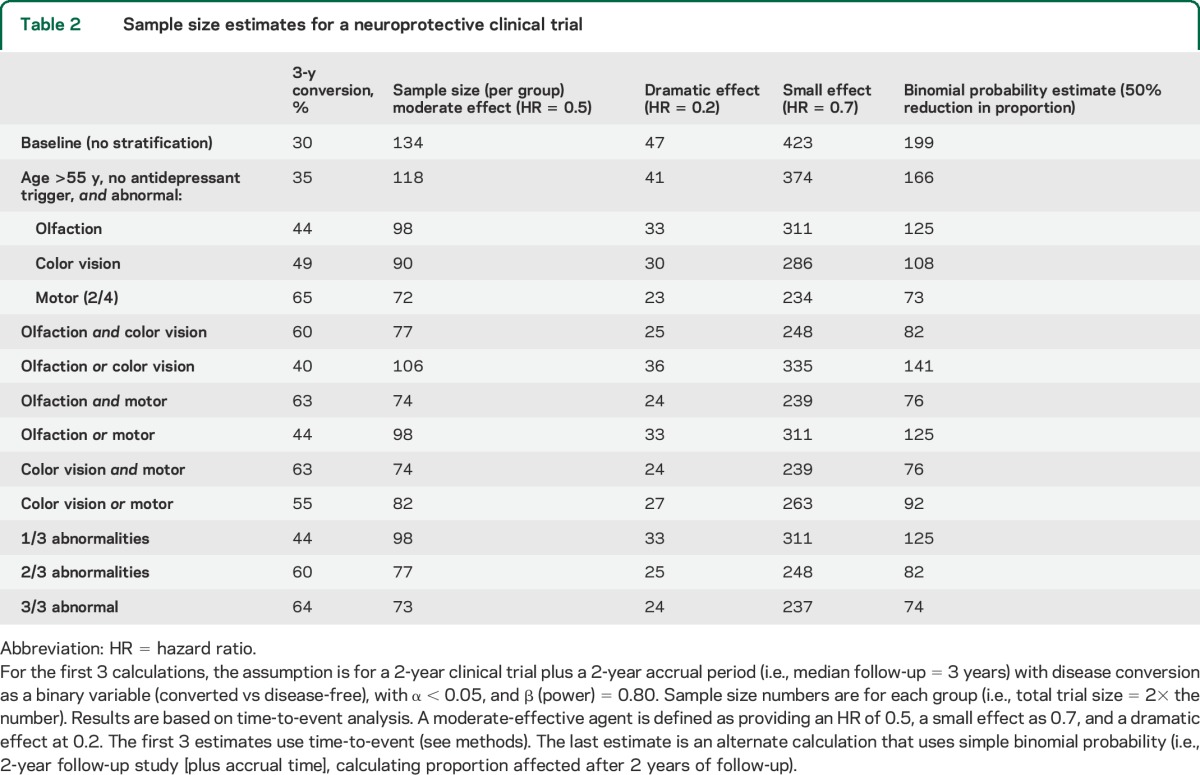
Using unselected patients with RBD, the sample size required for a moderately effective agent was 134 participants per group. This reduced considerably for dramatically effective agents (29 participants) and increased for modestly effective agents (423 participants). Stratification could further reduce sample size requirements (table 2). For example, age/antidepressant exclusion with olfactory, motor, or color vision loss reduced sample size estimates by >25% (98, 33, 311 patients). Age/antidepressant exclusion with motor impairment reduced requirements by >40% (72, 23, 234 patients).
Table 2.
Sample size estimates for a neuroprotective clinical trial
DISCUSSION
The current study has 3 major findings:
When comprehensively examined with annual in-person evaluation, risk of PD and related synucleinopathy is very high in RBD, higher than many earlier reports.7,25,26
This risk is predictable by assessing other markers. We expand on our previous findings that advanced age, olfaction, color vision, quantitative motor function, and nonantidepressant use mark a higher risk of defined neurodegenerative synucleinopathy. Moreover, we assessed new variables: borderline increased risk is associated with MCI, but depression, anxiety, personality, and symptomatic RBD medications do not predict conversion.
By stratifying RBD cohorts with predictive markers, subpopulations can be identified with up to 65% risk in 3 years. Therefore, clinical trials for neuroprotective therapy in synucleinopathy are feasible, even over short time frames.
Our first major finding was the very high conversion rate to clinical disease. This is consistent with 2 key recent follow-up studies indicating 76% and 81% conversion.2,3 Of note, our current rate estimate was higher than our earlier reports.7 This difference may be attributable to:
Follow-up frequency: Our previous reports relied on a combination of annual in-person examination (after 2004) and ad hoc in-person examination/self-report (before 2004). Here, all diagnoses were made on systematic annual in-person examination, often before patients reported substantial symptoms, suggesting that self-reports were insensitive.
Diagnostic sensitivity: Since 2004, all patients were examined for parkinsonism by a movement disorders specialist (R.P.), and neuropsychological examination became standard; this likely allowed earlier disease diagnosis. Also, the current Movement Disorder Society criteria15 allow earlier dementia diagnosis.
Lower loss to follow-up: Our intensive follow-up resulted in very low attrition. We noted that patients who were difficult to locate or refused to attend (requiring home visits) often had dementia or parkinsonism; therefore, loss to follow-up might bias toward lower disease estimates.
RBD diagnosis: Recently, quantification of REM atonia has become more standardized; we now applied a specific REM atonia cutoff.27 Presumably, misdiagnosed patients would have lower disease risk.
Therefore, neurodegenerative disease risk in idiopathic RBD is extremely high; in fact, it appears that only a small minority of patients do not have an underlying neurodegenerative process.
Our second major focus was an examination of potential predictive clinical markers of PD/DLB, using patients with RBD as a test model. We confirmed and expanded on our previous findings for many variables. Olfactory testing predicts disease, but it is not perfect; 2/39 patients with PD/DLB had normal olfaction at diagnosis, and patients with MSA had normal olfaction. Color vision was clearly associated with increased risk, although it may better predict dementia (all patients with dementia had abnormal test at diagnosis, but 8 without dementia tested normally). As we recently published,11 motor dysfunction strongly predicted disease, regardless of parkinsonism/dementia primary diagnosis. Finally, we were again unable to establish autonomic dysfunction as disease predictor in idiopathic RBD.12 An obvious potential explanation is unreliability of these simple autonomic estimates, but we observed similar findings with cardiac autonomic dysfunction on ECG.28 Another explanation could be that autonomic dysfunction is the earliest prodromal marker, and therefore nearly all have loss at RBD onset.
We also assessed novel predictive markers. There has been considerable interest in a putative “Parkinson personality,”29 and we previously found personality differences in patients with RBD compared with controls.14 However, on prospective follow-up, baseline personality variables did not predict conversion risk. Although this could be construed as evidence against the Parkinson personality, it could also suggest that PD personality is lifelong, present equally in idiopathic RBD and established PD.
Although the presence of MCI at baseline predicted dementia, it notably did not predict primary diagnosis of PD, considering the considerable overlap between DLB and PD.16 Note that unlike some cohort studies,3,30 MCI was not delineated a disease outcome in this study (we consider it a marker of cognitive loss that indicates likely transition to dementia,31 analogous to mild parkinsonian signs as predictors of full parkinsonism11). We are currently preparing a full analysis of neuropsychological variables predicting evolution from normal cognition to MCI/dementia in this cohort. Also, although we delineated primary dementia vs parkinsonism, we consider PD and DLB emerging from idiopathic RBD to be pathophysiologically similar, consistent with a recent PD definition statement proposing that they should not be considered mutually exclusive diagnoses.32
We found no evidence that symptomatic treatment for RBD affected neurodegenerative outcome. One might have speculated that RBD itself could accelerate neurodegeneration, perhaps via failure of normal REM sleep maintenance; if so, risk might be lower in those receiving therapy. We found no clear evidence for this possibility.
Whereas depression and anxiety predict PD in population-based studies,4 we found no effect on conversion risk in RBD. This was not simply lack of power; both indices were slightly lower in those who developed disease. One explanation could be confounding by antidepressants, which are associated with lower risk (perhaps because they trigger earlier presentation of RBD13); note, however, that HRs did not change when antidepressants were added to the model (Beck anxiety HR = 0.96/0.96, Beck depression HR = 0.97/0.99).
Our third major finding is that disease risk can be stratified effectively in idiopathic RBD. Moreover, these stratification procedures need not be complex—every marker in this study can be tested in <15 minutes. With some marker combinations identifying >80% 5-year conversion rates, the additive value of more invasive/expensive tests for identifying neuroprotective trial candidates is unclear.
The high conversion rate attainable with stratification has very important implications for the future of neuroprotective therapy. A major reason for the failure to develop neuroprotective synucleinopathy therapy may be that the disease has been present many years before clinical parkinsonism or dementia; it is simply too late to intervene. With idiopathic RBD, one can treat disease in prodromal stages, potentially before irreversible damage, and before symptomatic PD therapy confounds assessment. Two major issues have limited using patients with RBD for clinical trials, which may now no longer apply. First, based on previously published conversion rates, sample size estimates would exceed 300 to 500 participants, even in long-duration trials. Now, stratification using predictive markers enables trials with fewer than 150 participants. Second, patients with RBD were thought to be few and confined to subspecialty sleep clinics. However, as awareness has expanded, diagnosis is more frequent. To illustrate, the RBD study group quickly recruited 347 patients for an epidemiologic study,33 well within sample size calculations (multiple centers would still be required for most trials).
There may be a tradeoff between conversion risk and lead time; selecting for advanced prodromal signs limits the lead time gained by using RBD. Therefore, if a neuroprotective agent might work specifically in very early synucleinopathy, stratification strategies may differ (e.g., exclude subtle motor impairment, at the cost of increased duration/size). Of course, we do not suggest that once neuroprotective therapy is available, only those RBD patients with specific risk factors are candidates; this remains an individual clinical decision (if >80% ultimately develop synucleinopathy,2 one might suggest that all are neuroprotective treatment candidates). Rather, stratification enables selection for trials in settings when financial resources and/or maximal study duration are limited (most settings). There is little doubt that disease conversions will increase further over time, also among those with initially normal prodromal markers. It should also be recognized that patients with PD/DLB who have RBD represent a subgroup of PD34; therefore, results from RBD trials may not completely generalize to other populations. Our primary analysis was for all neurodegenerative disease outcomes; since patients with RBD develop both dementia and parkinsonism, they are perhaps better trial candidates for agents that target synucleinopathy in general.
Our study has some limitations. To maximize generalizability and eventual scalability, we focused on markers assessable in all patients. Many other variables could be assessed (and have been assessed in subcohorts) including MRI,35 SPECT/PET,36,37 transcranial ultrasound,37 MIBG scintigraphy,38 ECG,39 EEG,40 etc. Diagnosis of neurodegenerative disease remains clinical, and the time of “crossed threshold” is difficult to define; other clinicians may have diagnosed disease earlier or later than we did. Although the sample size was large, with a 10-year study duration, there is nonetheless imprecision in all estimates; future neuroprotective trials should choose sample sizes slightly beyond our projections. However, our study has some notable strengths, particularly the annual standardized evaluation up to 10 years, systematic diagnostic protocols, comprehensive evaluation of broadly applicable markers, and limited loss to follow-up.
Therefore, our findings suggest that it is now not only possible, but also practical to include patients with RBD in neuroprotective trials against PD and DLB.
Supplementary Material
GLOSSARY
- DLB
dementia with Lewy bodies
- HR
hazard ratio
- ICSD-II
International Classification of Sleep Disorders–Second Edition
- MCI
mild cognitive impairment
- MSA
multiple system atrophy
- PD
Parkinson disease
- RBD
REM sleep behavior disorder
Footnotes
Supplemental data at Neurology.org
AUTHOR CONTRIBUTIONS
R.B.P. was responsible for study concept, acquisition of data, analysis and interpretation of data, and drafting of the manuscript. J.-F.G. was responsible for study concept, acquisition of data, interpretation of data, and revision of the manuscript. J.-A.B. and D.G.M. were responsible for acquisition of data and revision of the manuscript. J.Y.M. was responsible for study concept, obtaining funding, interpretation of data, and revision of the manuscript.
STUDY FUNDING
This study was supported by the Fonds de la Recherche du Quebec–Santé, the Canadian Institutes of Health Research, and the W. Garfield Weston Foundation.
DISCLOSURE
R. Postuma received personal compensation for travel and speaker fees from Novartis Canada, Allergan Canada, and Teva Neurosciences, and is funded by grants from the Fonds de la Recherche du Québec–Santé, the Parkinson Society of Canada, the Webster Foundation, the W. Garfield Weston Foundation, and by the Canadian Institutes of Health Research. J. Gagnon is funded by grants from the Fonds de la Recherche du Québec–Santé, the W. Garfield Weston Foundation, and the Canadian Institutes of Health Research. D. Génier Marchand and J. Bertrand report no disclosures relevant to the manuscript. J. Montplaisir received personal compensation as consultant (Impax Pharma, Servier, Jazz Pharma, Merck, Valeant), speaker (Valeant), and received financial support for research activities from Merck, GlaxoSmithKline, is funded by grants from the Fonds de la Recherche du Québec–Santé, the W. Garfield Weston Foundation, and by the Canadian Institutes of Health Research. Go to Neurology.org for full disclosures.
REFERENCES
- 1.Schenck CH, Montplaisir JY, Frauscher B, et al. Rapid eye movement sleep behavior disorder: devising controlled active treatment studies for symptomatic and neuroprotective therapy—a consensus statement from the International Rapid Eye Movement Sleep Behavior Disorder Study Group. Sleep Med 2013;14:795–806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Schenck CH, Boeve BF, Mahowald MW. Delayed emergence of a parkinsonian disorder or dementia in 81% of older males initially diagnosed with idiopathic REM sleep behavior disorder (RBD): a 16-year update on a previously reported series. Sleep Med 2013;14:744–748. [DOI] [PubMed] [Google Scholar]
- 3.Iranzo A, Tolosa E, Gelpi E, et al. Neurodegenerative disease status and post-mortem pathology in idiopathic rapid-eye-movement sleep behaviour disorder: an observational cohort study. Lancet Neurol 2013;12:443–453. [DOI] [PubMed] [Google Scholar]
- 4.Postuma RB, Aarsland D, Barone P, et al. Identifying prodromal Parkinson's disease: pre-motor disorders in Parkinson's disease. Mov Disord 2012;27:617–626. [DOI] [PubMed] [Google Scholar]
- 5.Postuma RB, Gagnon JF, Montplaisir JY. REM sleep behavior disorder: from dreams to neurodegeneration. Neurobiol Dis 2012;46:553–558. [DOI] [PubMed] [Google Scholar]
- 6.Postuma RB, Lang AE, Massicotte-Marquez J, Montplaisir J. Potential early markers of Parkinson disease in idiopathic REM sleep behavior disorder. Neurology 2006;66:845–851. [DOI] [PubMed] [Google Scholar]
- 7.Postuma RB, Gagnon JF, Vendette M, Fantini ML, Massicotte-Marquez J, Montplaisir J. Quantifying the risk of neurodegenerative disease in idiopathic REM sleep behavior disorder. Neurology 2009;72:1296–1300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Postuma RB, Lanfranchi PA, Blais H, Gagnon JF, Montplaisir J. Cardiac autonomic dysfunction in idiopathic REM sleep behavior disorder. Mov Disord 2010;25:2304–2310. [DOI] [PubMed] [Google Scholar]
- 9.Postuma RB, Gagnon JF, Rompre S, Montplaisir J. Severity of REM atonia loss in idiopathic REM sleep behavior disorder predicts Parkinson disease. Neurology 2010;74:239–244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Postuma RB, Gagnon JF, Vendette M, Desjardins C, Montplaisir J. Olfaction and color vision identify impending neurodegeneration in REM behavior disorder. Ann Neurol 2011;69:811–818. [DOI] [PubMed] [Google Scholar]
- 11.Postuma RB, Lang AE, Gagnon JF, Pelletier A, Montplaisir JY. How does parkinsonism start? Prodromal parkinsonism motor changes in idiopathic REM sleep behaviour disorder. Brain 2012;135:1860–1870. [DOI] [PubMed] [Google Scholar]
- 12.Postuma RB, Gagnon JF, Pelletier A, Montplaisir J. Prodromal autonomic symptoms and signs in Parkinson's disease and dementia with Lewy bodies. Mov Disord 2013;28:597–604. [DOI] [PubMed] [Google Scholar]
- 13.Postuma RB, Gagnon JF, Tuineaig M, et al. Antidepressants and REM sleep behavior disorder: isolated side effect or neurodegenerative signal? Sleep 2013;36:1579–1585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Postuma RB, Gagnon JF, Vendette M, Montplaisir J. Markers of neurodegeneration in idiopathic REM sleep behavior disorder and Parkinson disease. Brain 2009;132:2298–2307. [DOI] [PubMed] [Google Scholar]
- 15.Dubois B, Burn D, Goetz C, et al. Diagnostic procedures for Parkinson's disease dementia: recommendations from the Movement Disorder Society Task Force. Mov Disord 2007;22:2314–2324. [DOI] [PubMed] [Google Scholar]
- 16.Postuma RB, Gagnon JF, Vendette M, Montplaisir JY. Idiopathic REM sleep behavior disorder in the transition to degenerative disease. Mov Disord 2009;24:2225–2232. [DOI] [PubMed] [Google Scholar]
- 17.Farnsworth D. The Farnsworth 100-hue test and dichotomous tests for color vision. J Optom Soc Am 1943;33:568–578. [Google Scholar]
- 18.Tison F, Seppi K, Sampaio C, et al. Development and validation of the Unified Multiple System Atrophy Rating Scale (UMSARS). Mov Disord 2004;19:1391–1402. [DOI] [PubMed] [Google Scholar]
- 19.Gagnon JF, Vendette M, Postuma RB, et al. Mild cognitive impairment in rapid eye movement sleep behavior disorder and Parkinson's disease. Ann Neurol 2009;66:39–47. [DOI] [PubMed] [Google Scholar]
- 20.Nasreddine ZS, Phillips NA, Bedirian V, et al. The Montreal Cognitive Assessment, MoCA: a brief screening tool for mild cognitive impairment. J Am Geriatr Soc 2005;53:695–699. [DOI] [PubMed] [Google Scholar]
- 21.Beck AT, Ward CH, Mendelson M, Mock J, Erbaugh J. An inventory for measuring depression. Arch Gen Psychiatry 1961;4:561–571. [DOI] [PubMed] [Google Scholar]
- 22.Cloninger CR. A systematic method for clinical description and classification of personality variants: a proposal. Arch Gen Psychiatry 1987;44:573–588. [DOI] [PubMed] [Google Scholar]
- 23.Hughes AJ, Ben-Shlomo Y, Daniel SE, Lees AJ. What features improve the accuracy of clinical diagnosis in Parkinson's disease: a clinicopathologic study. Neurology 1992;42:1142–1146. [DOI] [PubMed] [Google Scholar]
- 24.Boeve BF, Silber MH, Ferman TJ, et al. Clinicopathologic correlations in 172 cases of rapid eye movement sleep behavior disorder with or without a coexisting neurologic disorder. Sleep Med 2013;14:754–762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Schenck CH, Bundlie SR, Mahowald MW. Delayed emergence of a parkinsonian disorder in 38% of 29 older men initially diagnosed with idiopathic rapid eye movement sleep behaviour disorder. Neurology 1996;46:388–393. [DOI] [PubMed] [Google Scholar]
- 26.Wing YK, Li SX, Mok V, et al. Prospective outcome of rapid eye movement sleep behaviour disorder: psychiatric disorders as a potential early marker of Parkinson's disease. J Neurol Neurosurg Psychiatry 2012;83:470–472. [DOI] [PubMed] [Google Scholar]
- 27.Montplaisir J, Gagnon JF, Fantini ML, et al. Polysomnographic diagnosis of idiopathic REM sleep behavior disorder. Mov Disord 2010;25:2044–2051. [DOI] [PubMed] [Google Scholar]
- 28.Postuma RB, Montplaisir J, Lanfranchi P, et al. Cardiac autonomic denervation in Parkinson's disease is linked to REM sleep behavior disorder. Mov Disord 2011;26:1529–1533. [DOI] [PubMed] [Google Scholar]
- 29.Ishihara L, Brayne C. What is the evidence for a premorbid parkinsonian personality: a systematic review. Mov Disord 2006;21:1066–1072. [DOI] [PubMed] [Google Scholar]
- 30.Boot BP, Boeve BF, Roberts RO, et al. Probable rapid eye movement sleep behavior disorder increases risk for mild cognitive impairment and Parkinson disease: a population-based study. Ann Neurol 2012;71:49–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Petersen RC. Clinical practice: mild cognitive impairment. N Engl J Med 2011;364:2227–2234. [DOI] [PubMed] [Google Scholar]
- 32.Berg D, Postuma RB, Bloem B, et al. Time to redefine PD? Introductory statement of the MDS task force on the definition of Parkinson's disease. Mov Disord 2014;29:454–462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Postuma RB, Montplaisir JY, Pelletier A, et al. Environmental risk factors for REM sleep behavior disorder: a multicenter case-control study. Neurology 2012;79:428–434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Postuma RB, Gagnon JF, Vendette M, Charland K, Montplaisir J. Manifestations of Parkinson disease differ in association with REM sleep behavior disorder. Mov Disord 2008;23:1665–1672. [DOI] [PubMed] [Google Scholar]
- 35.Scherfler C, Frauscher B, Schocke M, et al. White and gray matter abnormalities in idiopathic rapid eye movement sleep behavior disorder: a diffusion-tensor imaging and voxel-based morphometry study. Ann Neurol 2011;69:400–407. [DOI] [PubMed] [Google Scholar]
- 36.Dang-Vu TT, Gagnon JF, Vendette M, Soucy JP, Postuma RB, Montplaisir J. Hippocampal perfusion predicts impending neurodegeneration in REM sleep behavior disorder. Neurology 2012;79:2302–2306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Iranzo A, Lomena F, Stockner H, et al. Decreased striatal dopamine transporters uptake and substantia nigra hyperechogenicity as risk markers of synucleinopathy in patients with idiopathic rapid-eye-movement sleep behaviour disorder: a prospective study. Lancet Neurol 2010;9:1070–1077. [DOI] [PubMed] [Google Scholar]
- 38.Miyamoto T, Miyamoto M, Inoue Y, Usui Y, Suzuki K, Hirata K. Reduced cardiac 123I-MIBG scintigraphy in idiopathic REM sleep behavior disorder. Neurology 2006;67:2236–2238. [DOI] [PubMed] [Google Scholar]
- 39.Postuma RB, Lanfranchi PA, Blais H, Gagnon JF, Montplaisir JY. Cardiac autonomic dysfunction in idiopathic REM sleep behavior disorder. Mov Disord 2010;25:2304–2310. [DOI] [PubMed] [Google Scholar]
- 40.Massicotte-Marquez J, Decary A, Vendette M, et al. Cognitive impairment and slowing of waking EEG in RBD patients. J Sleep Res 2006;15(suppl 1):422. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.