

Abstract
Since its initial reports in the 19th century, neuromyelitis optica (NMO) had been thought to involve only the optic nerves and spinal cord. However, the discovery of highly specific anti–aquaporin-4 antibody diagnostic biomarker for NMO enabled recognition of more diverse clinical spectrum of manifestations. Brain MRI abnormalities in patients seropositive for anti–aquaporin-4 antibody are common and some may be relatively unique by virtue of localization and configuration. Some seropositive patients present with brain involvement during their first attack and/or continue to relapse in the same location without optic nerve and spinal cord involvement. Thus, characteristics of brain abnormalities in such patients have become of increased interest. In this regard, MRI has an increasingly important role in the differential diagnosis of NMO and its spectrum disorder (NMOSD), particularly from multiple sclerosis. Differentiating these conditions is of prime importance because early initiation of effective immunosuppressive therapy is the key to preventing attack-related disability in NMOSD, whereas some disease-modifying drugs for multiple sclerosis may exacerbate the disease. Therefore, identifying the MRI features suggestive of NMOSD has diagnostic and prognostic implications. We herein review the brain, optic nerve, and spinal cord MRI findings of NMOSD.
Neuromyelitis optica (NMO) is an inflammatory disease of the CNS that is characterized by severe attacks of optic neuritis (ON) and longitudinally extensive transverse myelitis (LETM).1 The past decade has witnessed dramatic advances in our understanding of NMO. Such advances were initiated by the discovery of the disease-specific autoantibody, NMO–immunoglobulin G (NMO-IgG), and subsequent identification of the main target autoantigen, aquaporin-4 (AQP4), which has distinguished NMO as a distinct disease from multiple sclerosis (MS).2
Current diagnostic criteria, however, still require both ON and myelitis for an NMO diagnosis.3 Nevertheless, the identification of anti-AQP4 antibodies beyond the current diagnostic criteria of NMO indicates a broader clinical phenotype of this disorder, so-called “NMO spectrum disorder” (NMOSD).4,5 The NMOSD encompasses anti-AQP4 antibody seropositive patients with limited or inaugural forms of NMO and with specific brain abnormalities. It also includes anti-AQP4 antibody seropositive patients with other autoimmune disorders such as systemic lupus erythematosus and Sjögren syndrome.4 In this regard, MRI has an increasingly important role in differentiating NMOSD from other inflammatory disorders of the CNS, particularly from MS.6,7 Differentiating these conditions is critical because treatments are distinct. Furthermore, recent advanced MRI techniques are detecting additional specific markers and help elucidate the underlying mechanisms of tissue damage in NMOSD.
We herein summarize the MRI findings of NMOSD and discuss their diagnostic and prognostic implications.
BRAIN MRI FINDINGS IN NMOSD
Since the early studies using brain MRI in NMO,8,9 unexplained clinically silent and nonspecific white matter abnormalities were found in some patients. With the advent of AQP4-IgG assays, it became clear that a high proportion of patients with NMOSD harbored brain MRI abnormalities, frequently located in areas associated with high AQP4 expression.10,11 However, brain abnormalities also occurred in areas where AQP4 expression is not particularly high.12 Although nonspecific small dots and patches of hyperintensity in subcortical and deep white matter on T2-weighted or fluid-attenuated inversion recovery sequences are the most common findings in NMOSD, certain lesions have a location or appearance characteristic for NMOSD.6,7,11–15
Before the discovery of anti-AQP4 antibody, brain MRI abnormalities were reported in only 13% to 46% of patients with NMO.1,8,16 However, when excluding the brain MRI criteria, the incidence of brain MRI abnormalities increased to 50% to 85% using the revised 2006 NMO diagnostic criteria3,11,13,17,e1–e3 and to 51% to 89% in seropositive patients with NMOSD.5,12,18,19,e4,e5 Furthermore, brain MRI abnormalities at onset have been reported in 43% to 70% of patients with NMOSD.5,7,11 One of the explanations for discrepancies in frequency between studies may be that brain MRI abnormalities become more frequent with duration of disease. In a published series of 88 seropositive children, brain abnormalities were observed in 68% of the children with available MRI studies, and were predominantly located within periventricular regions of the third (diencephalic) and fourth ventricles (brainstem), supratentorial and infratentorial white matter, midbrain, and cerebellum.20 This is consistent with the observation that 45% to 55% of children with NMOSD show episodic cerebral symptoms, including ophthalmoparesis, intractable vomiting and hiccups, altered consciousness, severe behavioral changes, narcolepsy, ataxia, and seizures.20
Classification of brain MRI findings seen in NMOSD.
Periependymal lesions surrounding the ventricular system.
Diencephalic lesions surrounding the third ventricles and cerebral aqueduct.
Diencephalic lesions surrounding the third ventricles and cerebral aqueduct, which include the thalamus, hypothalamus, and anterior border of the midbrain have been reported in NMOSD (figure 1A).10,12 These lesions frequently are asymptomatic, but some patients may present with a syndrome of inappropriate antidiuretic hormone secretion,e6 narcolepsy,e7 hypothermia, hypotension, hypersomnia, obesity,e8 hypothyroidism, hyperprolactinemia, secondary amenorrhea, galactorrhea, and behavioral changes.e9
Figure 1. MRI lesions characteristic of neuromyelitis optica spectrum disorder.
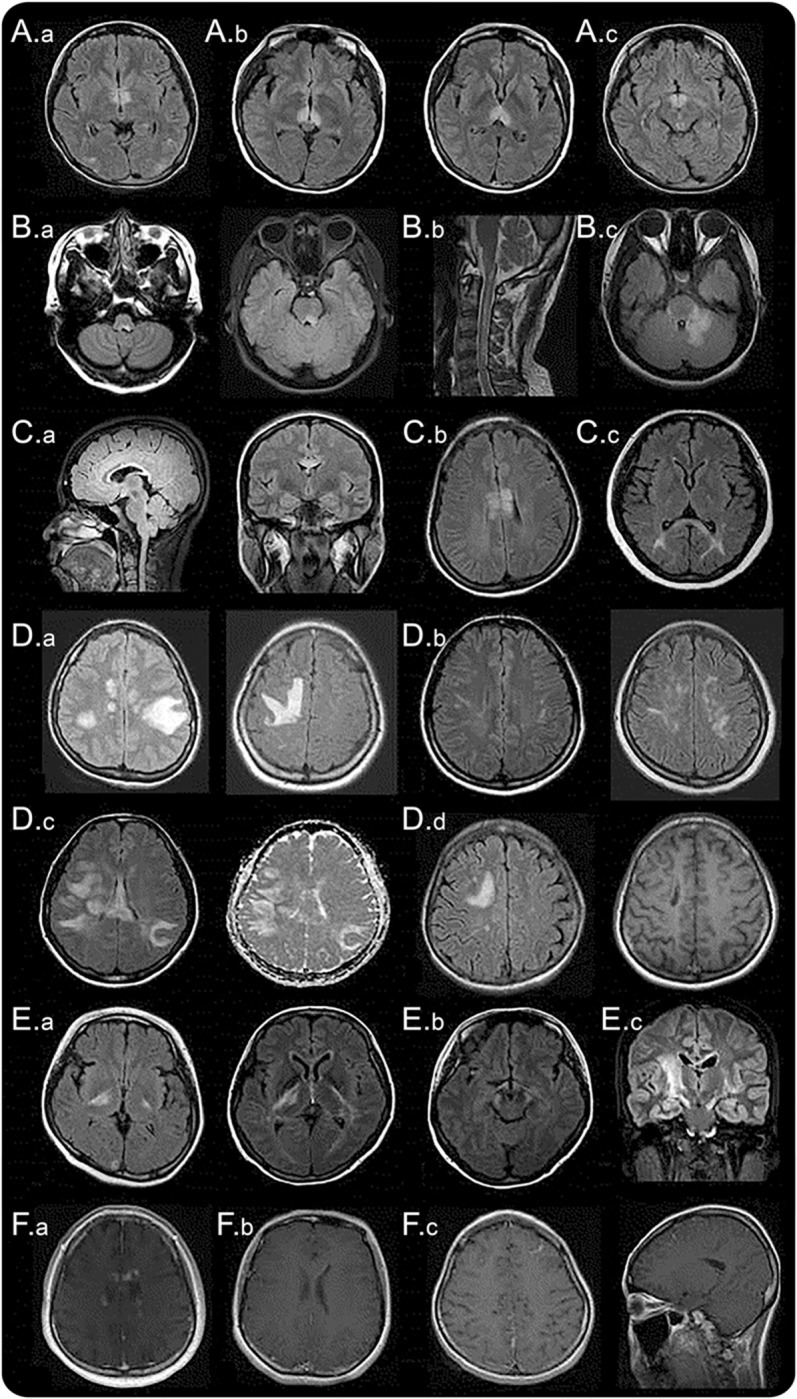
Diencephalic lesions surrounding (A.a) the third ventricles and cerebral aqueduct, (A.b) which include thalamus, hypothalamus, and (A.c) anterior border of the midbrain. (B.a) Dorsal brainstem lesion adjacent to the fourth ventricle, (B.b) linear medullary lesion that is contiguous with cervical cord lesion, (B.c) edematous and extensive dorsal brainstem lesion involving the cerebellar peduncle. (C.a) Callosal lesion immediately next to the lateral ventricle, following the ependymal lining, (C.b) “marbled pattern” callosal lesion, (C.c) “arch bridge pattern” callosal lesion. (D.a) Tumefactive hemispheric white matter lesions, (D.b) a long spindle-like or radial-shape lesion following white matter tracts, (D.c) extensive and confluent hemispheric lesions show increased diffusivity on apparent diffusion coefficient maps suggesting vasogenic edema, (D.d) hemispheric lesions in the chronic phase showing cystic-like cavitary changes. (E.a) Corticospinal tract lesions involving the posterior limb of the internal capsule and (E.b) cerebral peduncle of the midbrain, (E.c) longitudinally extensive lesion following the pyramidal tract. (F.a) Cloud-like enhancement, (F.b) linear enhancement of the ependymal surface of the lateral ventricles, (F.c) meningeal enhancement.
Dorsal brainstem lesions adjacent to the fourth ventricle.
One of the most specific brain MRI abnormalities in patients with NMOSD is a lesion in the dorsal brainstem adjacent to the fourth ventricle including the area postrema and the nucleus tracts solitarius. Such lesions are highly associated with intractable hiccups, nausea, and vomiting,10,12,21 and have been reported in 7% to 46% of patients with NMOSD.12,15,e1,e10 This area, the emetic reflex center, has a less restrictive blood-brain barrier, making it more accessible to AQP4-IgG attack. The MRI as well as clinical evidence support the notion that area postrema is an important point of attack in patients with NMOSD and further suggests that this area is a portal for entry of circulating IgG into the CNS.22,23 Pathologic abnormalities were noted in this region in 40% of patients with NMO, but there was no obvious neuronal, axonal, or myelin loss.21 Medullary lesions are often contiguous with cervical cord lesion, usually taking a linear shape (figure 1B.b). These lesions may be associated with the first symptoms of the disease22,24 or herald acute exacerbation.25 Various symptoms corresponding to a brainstem lesion may develop, such as nystagmus, dysarthria, dysphagia, ataxia, or ophthalmoplegia.15,20,e11,e12
Periependymal lesions surrounding the lateral ventricles.
Lesions in the corpus callosum have been described in 12% to 40% of patients with NMOSD.12,15,26 Because both NMO and MS frequently have callosal lesions, location by itself is not a unique finding that differentiates NMOSD from MS. However, while the callosal lesions in MS are discrete, ovoid, and perpendicular to the ventricles and involve inferior aspects of the corpus callosum (figure 2A),e13,e14 NMOSD lesions are located immediately next to the lateral ventricles, following the ependymal lining (figure 1C.a).12 The acute callosal lesions in NMOSD are often edematous and heterogeneous, creating a “marbled pattern”26 and sometimes involving the complete thickness of splenium in a unique “arch bridge pattern” (figure 1, C.b and C.c).12 Sometimes, the callosal lesions extend into the cerebral hemisphere, forming an extensive and confluent white matter lesion.12 In the chronic phase of NMOSD, the callosal lesions tend to reduce in size and intensity and may even disappear26; however, cystic changes and atrophy of the corpus callosum have been described.e15 Certain clinical symptoms, such as dysfunctions of cognition and motor coordination, may be attributed to callosal lesions, but they have not been well evaluated yet.
Figure 2. MRI lesions characteristic of MS.

(A) Contrasting MS periventricular and callosal lesions, which are discrete, ovoid, and perpendicular to the ventricles. (B) Contrasting MS enhancing lesions, which are ovoid or open-ring gadolinium-enhanced lesions with well-defined borders. MS = multiple sclerosis.
Hemispheric white matter lesions.
Extensive and confluent hemispheric white matter lesions are often tumefactive (>3 cm in longest diameter) or have long spindle-like or radial-shape following white matter tracts (figure 1D).12 Mass effect is usually absent.e16 Increased lesion diffusivity on apparent diffusion coefficient maps suggests vasogenic edema in association with acute inflammation (figure 1D.c),12,27 occasionally mimicking posterior reversible encephalopathy syndrome28 or Baló lesions.e17,e18 These extensive lesions have been found more frequently in anti-AQP4 antibody seropositive than seronegative patients.29 In the chronic phase, these large lesions tend to shrink and even disappear, but in some cases, cystic-like or cavitary changes are revealed (figure 1D.d).e19,e20 These lesions may cause various symptoms such as hemiparesis, encephalopathy, and visual field defects depending on the area they involve. Large confluent hemispheric white matter lesions are not uncommon in children with NMOSD. Tumefactive lesions with a surrounding zone of edema and variable mass effect may resemble acute disseminated encephalomyelitis20,30 or CNS malignancies.31
Lesions involving corticospinal tracts.
Lesions involving the corticospinal tracts can be unilateral or bilateral, and may extend from the deep white matter in the cerebral hemisphere through the posterior limb of the internal capsule to reach the cerebral peduncles of the midbrain or the pons (figure 1E).12 These lesions are contiguous and often longitudinally extensive, following the pyramidal tracts (figure 1E.c). Corticospinal tract lesions have been found in 23% to 44% in some cohorts of patients with NMOSD12,e2 and have occasionally been reported in other cohorts.11,13 It is of interest that, unlike circumventricular areas, corticospinal tracts are not the areas where the AQP4 is highly expressed; it is unknown why these regions are also frequently involved in NMOSD.
Nonspecific lesions: Not unique, but most common.
Nonspecific punctate or small (<3 mm) dots or patches of hyperintensities on T2-weighted or fluid-attenuated inversion recovery sequences in the subcortical or deep white matter have been described most frequently on brain imaging studies of NMOSD (35%–84%)11,12,17 and are usually asymptomatic.
Enhancing lesions.
Although the exact frequency is unclear, previous studies have described a variable percentage of gadolinium-enhancing brain lesions (9%–36%) in patients with NMOSD.12,15,e2,e3 Most of the enhancement was displayed in a poorly marginated, subtle, and multiple patchy pattern, a so-called “cloud-like” enhancement (figure 1F.a).18 These cloud-like enhanced lesions differ from the ovoid or ring/open-ring gadolinium-enhancing lesions with well-defined borders that are more typical of MS (figure 2). A linear enhancement of the ependymal surface of the lateral ventricles (pencil-thin lesion) has also been described in NMOSD (figure 1F.b).e21 Rarely, well-marginated nodular enhancement or meningeal enhancement has been reported in NMOSD (figure 1F.c).12,e16
OPTIC NERVE MRI FINDINGS IN NMOSD
MRI studies have reported nonspecific optic nerve sheath thickening, optic nerve hyperintensities on T2-weighted sequences, and gadolinium enhancement on T1-weighted sequences in acute ON of NMOSD.14,17 However, as similar findings also have been described in ON of MS,e22 these findings are not considered diagnostic of NMOSD. Recent studies have looked at the differential MRI features of the optic nerve lesion between MS and NMOSD.32,33 A trend to more posterior involvement of the optic nerve including chiasm, and simultaneous bilateral disease, has been observed in NMOSD (figure 3).32,33 Thus, long-segment inflammation of the optic nerve, particularly when simultaneous bilateral and extending posteriorly into the optic chiasm, should lead us to suspect the diagnosis of NMOSD in the appropriate clinical context.
Figure 3. Optic nerve MRI lesions characteristic of neuromyelitis optica spectrum disorder.
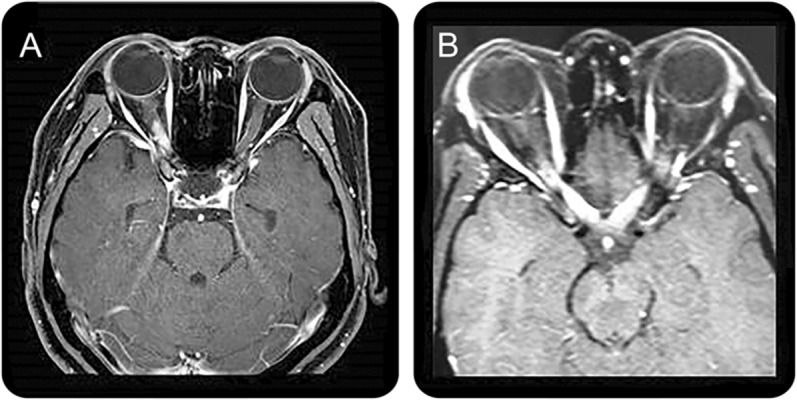
(A) Dense gadolinium-enhancing lesion at the posterior part of the right optic nerve. (B) Extensive gadolinium-enhancing lesion at the bilateral posterior part of the optic nerve/chiasm.
SPINAL CORD MRI FINDINGS IN NMOSD
The inflammatory process of NMOSD in spinal cord MRI is characterized by hyperintensity on T2-weighted sequences and by hypointensity on T1-weighted sequences. These abnormalities in the spinal cord MRI have been reported to be, in general, more frequently present in the cervical and the upper thoracic spinal cord segments than the lower thoracic and lumbar regions23,34,e23 with a preferential involvement in the central gray matter.34,35 In the spinal cord, AQP4 is abundant in the gray matter and in glial cell processes adjacent to the ependymal cells of the central canal and to a lesser degree in the white matter of the spinal cord.e24
The most distinct manifestation of NMO is LETM, defined as a lesion that spans over 3 or more contiguous vertebral segments and predominantly involves central gray matter on the spinal cord MRI (figure 4).4 However, not all LETM is NMOSD and several studies of patients with LETM have observed significant differences in demographic and clinical features between anti-AQP4 antibody positive compared with negative patients with LETM.19,36–38 LETM seems to be less specific for NMO in children than in adults. LETM is frequently observed in children with acute disseminated encephalomyelitis,39,40 but also in 17% of those with MS,e25 and in 67% to 88% of children with monophasic transverse myelitis.e26,e27 Therefore, it is important to bear in mind that numerous other differential diagnoses than NMOSD need to be considered when a patient presents with LETM.
Figure 4. Spinal cord MRI lesions characteristic of neuromyelitis optica spectrum disorder.
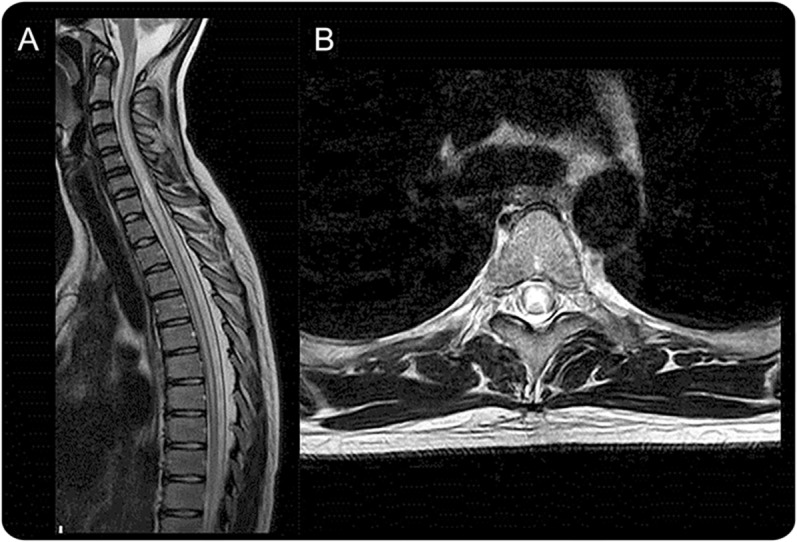
(A) Longitudinally extensive cord lesion involving thoracic cord. (B) Exclusive involvement of gray matter (H-shaped cord lesion).
Spinal cord lesions during follow-up of NMOSD.
MRI changes of LETM have been observed over the course of NMOSD and MRI data indicate that LETM lesions may evolve into multiple shorter lesions during remission or after treatment with high-dose steroids.23,41 In addition, spinal cord atrophy as a consequence of recurrent myelitis has been reported and may correlate with neurologic disability.23 Consequently, the timing of MRI may be important for the demonstration of LETM.42
COMPARING THE IMAGING OF NMOSD WITH MS
In clinical practice, the main differential diagnosis of NMO is MS, particularly disease limited to the optic nerves and spinal cord. Differentiating these conditions is of prime importance because of differences in prognosis and therapy, as some MS therapies can exacerbate NMO.43–45 Thus, it is important to improve the methods and analysis by which to distinguish these conditions to facilitate early and accurate diagnosis. Contrasting features between the 2 conditions may further improve our understanding of the different pathogenic processes.
Whereas it is possible to select patients with NMOSD using the specific marker (serum anti-AQP4 antibodies), there is no corresponding specific biomarker for MS. Studies contrasting NMO and MS have often used different selection criteria, particularly whether they have restricted the NMO inclusion criteria to patients positive for anti-AQP4 antibody or not, and this may influence the results. Conflicting data may also be partly explained by the use of various assays for anti-AQP4 antibodies, which differ in sensitivity and are confounded by differences in the duration of follow-up.
As previously described, the most important imaging hallmark of NMO is the LETM, but a few patients may have centrally located short myelitis.46 Other MRI features of the spinal cord lesion that appear to differ between NMOSD and MS are summarized in the table.
Table.
Comparison of characteristic MRI findings between NMOSD and MS

The 2006 NMO diagnostic criteria include a brain MRI that is nondiagnostic for MS (using the Paty criteria) at onset as support for NMO. However, it is now known that MS-like lesions may appear in 10% to 12.5% of cases,11,e3 and 5% to 42% of patients with NMO fulfill the Barkhof criteria.6,14,15,47 A recent report showed that 13% and 9% of patients with NMOSD, respectively, met Barkhof and the European Magnetic Imaging in MS diagnostic criteria for MS on brain MRI at onset.7 Lesion probability maps have not found statistically significant lesion locations in patients positive for anti-AQP4 antibody over those with MS.6 However, distinguishing features were identified on MS brain MRI that were sensitive and specific, such as the presence of a lateral ventricle and inferior temporal lobe lesion, Dawson fingers, or an S-shaped U-fiber lesion, to classify the patient as MS. Imaging sensitive to cortical lesions has revealed their absence in NMO (excluding one Japanese study of NMO pathology48), whereas they are seen in the majority of patients with MS.49,50 Characteristic MS brain lesions surround central venule in >80% on high-strength MRI.50,51 In NMO lesions, this is less frequent, reported in 9% to 35% of cases50,52 and likely indicates the different pathogenic mechanisms of the disease.
The frequency of silent lesion formation appears to differ between the 2 diseases. Patients with NMOSD are less likely to develop clinically silent MRI lesions than patients with MS. However, new silent MRI lesions do occur in a small proportion of patients with NMOSD. In addition, most studies show that nonlesional tissue damage as measured on nonconventional imaging such as diffusion tensor imaging is well recognized in MS and may not occur in NMO except in the connecting tracts up and downstream of lesions.53,54 Collectively, these findings support the clinical observation that NMO, in contrast to MS, may be a lesion-dependent disease that produces relapses without more generalized neurodegenerative pathology, and hence the lack of a progressive phase.
The differences noted between NMO and MS may relate to the CNS-specific antibody-mediated pathology against astrocytes rather than a T-cell–predominant inflammatory condition targeting myelin. In support of this possibility, a marker of astrocytic function, myo-inositol was reduced in cervical cord lesions of patients positive for anti-AQP4 antibody, but not in patients with MS. In contrast, N-acetyl-aspartate, a marker of myelin- and neurofilament-specific injury, was significantly reduced in patients with MS compared with controls and nonsignificantly reduced in the patients positive for anti-AQP4 antibody.55
Important comparisons between NMOSD and MS scans are summarized in the table. Because long-term systematic imaging studies in NMO have not yet been performed, the reported cross-sectional differences compared with MS require further confirmation. Developing algorithms using the brain criteria described by Matthews et al.6 in combination with spinal cord and optic nerve imaging features and possibly nonconventional imaging may further improve the sensitivity and specificity.
PROGNOSTIC IMPLICATION OF MRI ABNORMALITIES
Anti-AQP4 antibody positivity is established as a prognostic marker, and its positivity indicates a high risk of further relapses of ON and myelitis.56,57 Because of the presence of many imaging features suggesting severe damage of spinal cord, such as T1 hypointensity with edema or cavitation and atrophy, patients with NMOSD are more likely to have a poor recovery, refractory pain,e28 and a high risk of permanent disability. In addition, patients with NMOSD who have lesions in the upper cervical region extending to the brainstem may be at risk of respiratory failure.
High levels of glial fibrillary acidic protein in the CSF of patients with NMOSD during acute attacks correlated with length of MRI spinal cord lesion and Expanded Disability Status Scale score 6 months after those attacks. This correlation suggests that imaging findings may be proportional to the amount of astrocyte damage and have potential prognostic implications.e29 The presence of extensive brain lesions might predict a higher rate of relapse and increased disability at follow-up.e30 Longitudinal follow-up studies are required to confirm whether patients with brain lesions have a worse prognosis than those without brain lesions. At this point, there are no individual MRI parameters that can predict the prognosis of NMO.
More recently, antibodies against myelin-oligodendrocyte glycoprotein (MOG) have been found in some patients with clinical features of NMOSD, but who lack anti-AQP4 antibodies.58 Patients exhibiting the anti-MOG–positive and anti-AQP4–negative serotype have been suggested to have fewer attacks, bilateral ON, more caudal myelitis, and recover better than patients with anti-AQP4 antibodies and those who are seronegative for both antibodies.59,60 Therefore, patients presenting with an NMOSD phenotype with anti-MOG antibodies may have a distinct underlying disease mechanism presumably with a better prognosis than those with anti-AQP4 antibodies, although this needs to be confirmed by further studies.
OUTLOOK: MRI FINDINGS IN THE CONTEXT OF NMO DIAGNOSTIC CRITERIA
The notion that brain MRI abnormalities are frequent in patients with NMOSD refutes the older doctrine that a normal brain MRI is a prerequisite for a diagnosis of NMO. Herein, we have reviewed the advances in our knowledge on the spectrum of imaging findings in NMOSD. However, sensitivity and specificity of these imaging features for NMOSD have not been systematically investigated in a prospective manner, and none of the findings can be considered pathognomonic or evidentiary for NMOSD. Therefore, as with other inflammatory CNS conditions, imaging findings should prompt broad differential diagnostic consideration—a topic that is beyond the scope of this review. The actual utility of lesion probability maps to distinguish NMO and MS is limited by an unclear definition of some traditional criteria for MS-suggestive findings, such as “Dawson fingers.”e31 The picture is further complicated by recent observations of patients with anti-MOG antibodies that some, but not all, have considered part of the NMO spectrum.58,59 Some commonalities but also differences in clinical presentation, epidemiology, and imaging have been reported between these 2 conditions, suggesting that NMOSD may not be a homogeneous nosologic entity. In addition, because NMOSD can coexist with other autoimmune diseases and antibodies to other CNS antigens such as anti-NMDA receptor antibodies may be present in patients seropositive for anti-AQP4 antibody, it is possible that autoimmunity against multiple CNS autoantigens may participate in the formation of inflammatory lesions of NMOSD.e32,e33 The emerging heterogeneity of NMOSD is mirrored by the broad range of neuroimaging findings summarized in this article. Areas for improved imaging that may facilitate more specific diagnostic, prognostic, therapeutic efficacy or other patient care benefit include higher-resolution imaging methods, 3-dimensional imaging of site-specific lesions, and potential computationally guided analysis of images for quantitative comparisons. International collaborative efforts are now under way that will permit accrual of sufficiently large, carefully characterized NMO/NMOSD patients to better understand the frequency of brain involvement and to more thoroughly appreciate the implications of MRI abnormalities in clinical diagnosis and prognosis.
Supplementary Material
ACKNOWLEDGMENT
The authors thank The Guthy-Jackson Charitable Foundation for its support in organizing the NMO International Clinical Consortium & Biorepository. The authors thank Drs. Brian Weinshenker and Jack Simon for their comments and Dr. Valerie Pasquetto for her assistance.
GLOSSARY
- AQP4
aquaporin-4
- IgG
immunoglobulin G
- LETM
longitudinally extensive transverse myelitis
- MOG
myelin-oligodendrocyte glycoprotein
- MS
multiple sclerosis
- NMO
neuromyelitis optica
- NMOSD
neuromyelitis optica spectrum disorder
- ON
optic neuritis
Footnotes
Supplemental data at Neurology.org
Contributor Information
Collaborators: Friedemann Paul, Jens Wuerfel, Philippe Cabre, Romain Marignier, Jérôme de Seze, Thomas Tedder, Juan P. Garrahan, Silvia Tenembaum, Danielle van Pelt, Simon Broadley, Albert Saiz, Pablo Villoslada, Michael Levy, Tanuja Chitnis, Eric C. Klawiter, Dean Wingerchuk, Ho Jin Kim, Lekha Pandit, Ilya Kister, Maria Isabel Leite, Jacqueline Palace, Metha Apiwattanakul, Ingo Kleiter, Naraporn Prayoonwiwat, May Han, Kerstin Hellwig, Brenda Banwell, Katja van Herle, Jacinta Behne, Gareth John, D. Craig Hooper, Kazuo Fujihara, Ichiro Nakashima, Douglas Sato, Anthony Traboulsee, Michael R. Yeaman, Emmanuelle Waubant, Scott Zamvil, Jeffrey Bennett, Marco Lana-Peixoto, Benjamin Greenberg, Olaf Stuve, Orhan Aktas, Jens Wuerfel, Terry J. Smith, Nasrin Asgari, Anu Jacob, and Kevin O'Connor
AUTHOR CONTRIBUTIONS
Dr. H.J. Kim conceived and designed the work, analyzed the literature, wrote the manuscript, critically reviewed and revised the manuscript, and approved the final manuscript. Dr. F. Paul conceived the work, analyzed the literature, wrote the manuscript, critically reviewed and revised the manuscript, and approved the final manuscript. Dr. M.A. Lana-Peixoto analyzed the literature, wrote the manuscript, critically reviewed and revised the manuscript, and approved the final manuscript. Dr. N. Asgari analyzed the literature, wrote the manuscript, critically reviewed and revised the manuscript, and approved the final manuscript. Dr. S. Tenembaum analyzed the literature, wrote the manuscript, critically reviewed and revised the manuscript, and approved the final manuscript. Dr. J. Palace analyzed the literature, wrote the manuscript, critically reviewed and revised the manuscript, and approved the final manuscript. Dr. E.C. Klawiter analyzed the literature, wrote the manuscript, critically reviewed and revised the manuscript, and approved the final manuscript. Dr. D.K. Sato analyzed the literature, wrote the manuscript, critically reviewed and revised the manuscript, and approved the final manuscript. Dr. J. de Seze critically reviewed and revised the manuscript, and approved the final manuscript. Dr. J. Wuerfel critically reviewed and revised the manuscript, and approved the final manuscript. Dr. B.L. Banwell critically reviewed and revised the manuscript, and approved the final manuscript. Dr. P. Villoslada critically reviewed and revised the manuscript, and approved the final manuscript. Dr. A. Saiz critically reviewed and revised the manuscript, and approved the final manuscript. Dr. K. Fujihara critically reviewed and revised the manuscript, and approved the final manuscript. Dr. S.-H. Kim analyzed the literature, wrote the manuscript, critically reviewed and revised the manuscript, and approved the final manuscript.
STUDY FUNDING
No targeted funding reported.
DISCLOSURE
H. Kim has given talks, consulted, and received honoraria and/or research support from Bayer Schering Pharma, Biogen Idec, Genzyme, Kael-GemVax, Merck Serono, Novartis, Teva-Handok, and UCB. He serves on a steering committee for MedImmune. F. Paul has received funding from German Research Council, German Ministry of Education and Research (Competence Network Multiple Sclerosis “KKNMS”), and The Guthy-Jackson Charitable Foundation. He has received travel compensation, speaker honoraria, and research support from Biogen, Bayer, Teva, Merck, Novartis, and Sanofi, and has served as steering committee member of the OCTIMS Study sponsored by Novartis. M. Lana-Peixoto reports no disclosures relevant to the manuscript. S. Tenembaum has provided consulting services to Genzyme Corporation and Biogen Idec and received lecture fees from Merck Serono. N. Asgari reports no disclosures relevant to the manuscript. J. Palace is partly funded by highly specialized services to run a national congenital myasthenia service and a neuromyelitis service. She has received support for scientific meetings and honorariums for advisory work from Merck Serono, Biogen Idec, Novartis, Teva, Chugai Pharma, and Bayer Schering, and unrestricted grants from Merck Serono, Novartis, Biogen Idec, and Bayer Schering. Her hospital trust receives funds for her role as clinical lead for the RSS, and she has received grants from the MS Society and Guthy-Jackson Foundation for unrelated research studies. E. Klawiter has received research funding from Roche. He has received consulting fees and/or speaking honoraria from Biogen Idec, Bayer Healthcare, Genzyme Corporation, and Teva Neuroscience. D. Sato receives scholarship funds from the Ministry of Education, Culture, Sports, Science and Technology (MEXT) of Japan and has received research support from Ichiro Kanehara Foundation. J. de Seze has received honoraria from Bayer Schering, Biogen Idec, LFB, Merck Serono, Novartis, Sanofi-Aventis, and Teva. He serves as a consultant for Alexion and Chugai. J. Wuerfel serves on advisory boards for Novartis and Biogen Idec. He received a research grant from Novartis, and speaker honoraria from Bayer, Novartis, and Biogen Idec. He is supported by the German Ministry of Science (BMBF/KKNMS). B. Banwell serves as a senior editor for Multiple Sclerosis and Related Disorders and on the editorial board of Neurology®. She serves as a consultant for Biogen Idec, Novartis, Teva Neuroscience, and Merck Serono. She has been funded by the Canadian MS Research Foundation, the Canadian MS Society, and CIHR. P. Villoslada serves as a board member for Roche, Novartis, Neurotec Pharma, Bionure Farma, and as a consultant for Novartis, Roche, TFS, Heidelberg Engineering, MedImmune, Digna Biotech, and Neurotec Pharma. He has received research support from European Commission, Instituto Salud Carlos III, Marato TV3, Novartis, and Roche and travel expenses from Novartis. He holds patents with Digna Biotech, Bionure Farma, and stock/stock options of Bionure Farma. A. Saiz has received compensation for consulting services and speaking from Bayer Schering, Merck Serono, Biogen Idec, Sanofi-Aventis, Teva Pharmaceutical Industries, and Novartis. K. Fujihara serves on scientific advisory boards for Bayer Schering Pharma, Biogen Idec, Mitsubishi Tanabe Pharma Corporation, Novartis Pharma, Chugai Pharmaceutical, Ono Pharmaceutical, Nihon Pharmaceutical, Merck Serono, Alexion Pharmaceuticals, MedImmune, and Medical Review; has received funding for travel and speaker honoraria from Bayer Schering Pharma, Biogen Idec, Eisai Inc., Mitsubishi Tanabe Pharma Corporation, Novartis Pharma, Astellas Pharma Inc., Takeda Pharmaceutical Company Limited, Asahi Kasei Medical Co., Daiichi Sankyo, and Nihon Pharmaceutical; serves as an editorial board member of Clinical and Experimental Neuroimmunology (2009–present) and an advisory board member of Sri Lanka Journal of Neurology; has received research support from Bayer Schering Pharma, Biogen Idec Japan, Asahi Kasei Medical, The Chemo-Sero-Therapeutic Research Institute, Teva Pharmaceutical, Mitsubishi Tanabe Pharma, Teijin Pharma, Chugai Pharmaceutical, Ono Pharmaceutical, Nihon Pharmaceutical, and Genzyme Japan; and is funded as the secondary investigator (22229008, 2010–2015) by the Grants-in-Aid for Scientific Research from the Ministry of Education, Science and Technology of Japan and as the secondary investigator by the Grants-in-Aid for Scientific Research from the Ministry of Health, Welfare and Labor of Japan (2010–present). S. Kim reports no disclosures relevant to the manuscript. Go to Neurology.org for full disclosures.
REFERENCES
- 1.Wingerchuk DM, Hogancamp WF, O'Brien PC, Weinshenker BG. The clinical course of neuromyelitis optica (Devic's syndrome). Neurology 1999;53:1107–1114. [DOI] [PubMed] [Google Scholar]
- 2.Lennon VA, Wingerchuk DM, Kryzer TJ, et al. A serum autoantibody marker of neuromyelitis optica: distinction from multiple sclerosis. Lancet 2004;364:2106–2112. [DOI] [PubMed] [Google Scholar]
- 3.Wingerchuk DM, Lennon VA, Pittock SJ, Lucchinetti CF, Weinshenker BG. Revised diagnostic criteria for neuromyelitis optica. Neurology 2006;66:1485–1489. [DOI] [PubMed] [Google Scholar]
- 4.Wingerchuk DM, Lennon VA, Lucchinetti CF, Pittock SJ, Weinshenker BG. The spectrum of neuromyelitis optica. Lancet Neurol 2007;6:805–815. [DOI] [PubMed] [Google Scholar]
- 5.Kim SH, Kim W, Li XF, Jung IJ, Kim HJ. Clinical spectrum of CNS aquaporin-4 autoimmunity. Neurology 2012;78:1179–1185. [DOI] [PubMed] [Google Scholar]
- 6.Matthews L, Marasco R, Jenkinson M, et al. Distinction of seropositive NMO spectrum disorder and MS brain lesion distribution. Neurology 2013;80:1330–1337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Huh SY, Min JH, Kim W, et al. The usefulness of brain MRI at onset in the differentiation of multiple sclerosis and seropositive neuromyelitis optica spectrum disorders. Mult Scler 2014;20:695–704. [DOI] [PubMed] [Google Scholar]
- 8.O'Riordan JI, Gallagher HL, Thompson AJ, et al. Clinical, CSF, and MRI findings in Devic's neuromyelitis optica. J Neurol Neurosurg Psychiatry 1996;60:382–387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Filippi M, Rocca MA, Moiola L, et al. MRI and magnetization transfer imaging changes in the brain and cervical cord of patients with Devic's neuromyelitis optica. Neurology 1999;53:1705–1710. [DOI] [PubMed] [Google Scholar]
- 10.Pittock SJ, Weinshenker BG, Lucchinetti CF, Wingerchuk DM, Corboy JR, Lennon VA. Neuromyelitis optica brain lesions localized at sites of high aquaporin 4 expression. Arch Neurol 2006;63:964–968. [DOI] [PubMed] [Google Scholar]
- 11.Pittock SJ, Lennon VA, Krecke K, Wingerchuk DM, Lucchinetti CF, Weinshenker BG. Brain abnormalities in neuromyelitis optica. Arch Neurol 2006;63:390–396. [DOI] [PubMed] [Google Scholar]
- 12.Kim W, Park MS, Lee SH, et al. Characteristic brain magnetic resonance imaging abnormalities in central nervous system aquaporin-4 autoimmunity. Mult Scler 2010;16:1229–1236. [DOI] [PubMed] [Google Scholar]
- 13.Bichuetti DB, Rivero RL, Oliveira DM, et al. Neuromyelitis optica: brain abnormalities in a Brazilian cohort. Arq Neuropsiquiatr 2008;66:1–4. [DOI] [PubMed] [Google Scholar]
- 14.Wang F, Liu Y, Duan Y, Li K. Brain MRI abnormalities in neuromyelitis optica. Eur J Radiol 2011;80:445–449. [DOI] [PubMed] [Google Scholar]
- 15.Chan KH, Tse CT, Chung CP, et al. Brain involvement in neuromyelitis optica spectrum disorders. Arch Neurol 2011;68:1432–1439. [DOI] [PubMed] [Google Scholar]
- 16.Ghezzi A, Bergamaschi R, Martinelli V, et al. Clinical characteristics, course and prognosis of relapsing Devic's neuromyelitis optica. J Neurol 2004;251:47–52. [DOI] [PubMed] [Google Scholar]
- 17.Cabrera-Gomez JA, Quevedo-Sotolongo L, Gonzalez-Quevedo A, et al. Brain magnetic resonance imaging findings in relapsing neuromyelitis optica. Mult Scler 2007;13:186–192. [DOI] [PubMed] [Google Scholar]
- 18.Ito S, Mori M, Makino T, Hayakawa S, Kuwabara S. “Cloud-like enhancement” is a magnetic resonance imaging abnormality specific to neuromyelitis optica. Ann Neurol 2009;66:425–428. [DOI] [PubMed] [Google Scholar]
- 19.Jarius S, Ruprecht K, Wildemann B, et al. Contrasting disease patterns in seropositive and seronegative neuromyelitis optica: a multicentre study of 175 patients. J Neuroinflammation 2012;9:14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.McKeon A, Lennon VA, Lotze T, et al. CNS aquaporin-4 autoimmunity in children. Neurology 2008;71:93–100. [DOI] [PubMed] [Google Scholar]
- 21.Popescu BF, Lennon VA, Parisi JE, et al. Neuromyelitis optica unique area postrema lesions: nausea, vomiting, and pathogenic implications. Neurology 2011;76:1229–1237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Apiwattanakul M, Popescu BF, Matiello M, et al. Intractable vomiting as the initial presentation of neuromyelitis optica. Ann Neurol 2010;68:757–761. [DOI] [PubMed] [Google Scholar]
- 23.Asgari N, Skejoe HP, Lillevang ST, Steenstrup T, Stenager E, Kyvik KO. Modifications of longitudinally extensive transverse myelitis and brainstem lesions in the course of neuromyelitis optica (NMO): a population-based, descriptive study. BMC Neurol 2013;13:33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kim W, Kim SH, Lee SH, Li XF, Kim HJ. Brain abnormalities as an initial manifestation of neuromyelitis optica spectrum disorder. Mult Scler 2011;17:1107–1112. [DOI] [PubMed] [Google Scholar]
- 25.Takahashi T, Miyazawa I, Misu T, et al. Intractable hiccup and nausea in neuromyelitis optica with anti-aquaporin-4 antibody: a herald of acute exacerbations. J Neurol Neurosurg Psychiatry 2008;79:1075–1078. [DOI] [PubMed] [Google Scholar]
- 26.Nakamura M, Misu T, Fujihara K, et al. Occurrence of acute large and edematous callosal lesions in neuromyelitis optica. Mult Scler 2009;15:695–700. [DOI] [PubMed] [Google Scholar]
- 27.Matsushita T, Isobe N, Matsuoka T, et al. Extensive vasogenic edema of anti-aquaporin-4 antibody-related brain lesions. Mult Scler 2009;15:1113–1117. [DOI] [PubMed] [Google Scholar]
- 28.Magana SM, Matiello M, Pittock SJ, et al. Posterior reversible encephalopathy syndrome in neuromyelitis optica spectrum disorders. Neurology 2009;72:712–717. [DOI] [PubMed] [Google Scholar]
- 29.Matsushita T, Isobe N, Piao H, et al. Reappraisal of brain MRI features in patients with multiple sclerosis and neuromyelitis optica according to anti-aquaporin-4 antibody status. J Neurol Sci 2010;291:37–43. [DOI] [PubMed] [Google Scholar]
- 30.Eichel R, Meiner Z, Abramsky O, Gotkine M. Acute disseminating encephalomyelitis in neuromyelitis optica: closing the floodgates. Arch Neurol 2008;65:267–271. [DOI] [PubMed] [Google Scholar]
- 31.O'Mahony J, Bar-Or A, Arnold DL, et al. Masquerades of acquired demyelination in children: experiences of a national demyelinating disease program. J Child Neurol 2013;28:184–197. [DOI] [PubMed] [Google Scholar]
- 32.Khanna S, Sharma A, Huecker J, Gordon M, Naismith RT, Van Stavern GP. Magnetic resonance imaging of optic neuritis in patients with neuromyelitis optica versus multiple sclerosis. J Neuroophthalmol 2012;32:216–220. [DOI] [PubMed] [Google Scholar]
- 33.Storoni M, Davagnanam I, Radon M, Siddiqui A, Plant GT. Distinguishing optic neuritis in neuromyelitis optica spectrum disease from multiple sclerosis: a novel magnetic resonance imaging scoring system. J Neuroophthalmol 2013;33:123–127. [DOI] [PubMed] [Google Scholar]
- 34.Cassinotto C, Deramond H, Olindo S, Aveillan M, Smadja D, Cabre P. MRI of the spinal cord in neuromyelitis optica and recurrent longitudinal extensive myelitis. J Neuroradiol 2009;36:199–205. [DOI] [PubMed] [Google Scholar]
- 35.Nakamura M, Miyazawa I, Fujihara K, et al. Preferential spinal central gray matter involvement in neuromyelitis optica: an MRI study. J Neurol 2008;255:163–170. [DOI] [PubMed] [Google Scholar]
- 36.Kitley J, Leite MI, Kuker W, et al. Longitudinally extensive transverse myelitis with and without aquaporin 4 antibodies. JAMA Neurol 2013;70:1375–1381. [DOI] [PubMed] [Google Scholar]
- 37.Chang KH, Lyu RK, Chen CM, et al. Distinct features between longitudinally extensive transverse myelitis presenting with and without anti-aquaporin 4 antibodies. Mult Scler 2013;19:299–307. [DOI] [PubMed] [Google Scholar]
- 38.Sepulveda M, Blanco Y, Rovira A, et al. Analysis of prognostic factors associated with longitudinally extensive transverse myelitis. Mult Scler 2013;19:742–748. [DOI] [PubMed] [Google Scholar]
- 39.Tenembaum S, Chitnis T, Ness J, Hahn JS; International Pediatric MSSG. Acute disseminated encephalomyelitis. Neurology 2007;68:S23–S36. [DOI] [PubMed] [Google Scholar]
- 40.Banwell B, Tenembaum S, Lennon VA, et al. Neuromyelitis optica-IgG in childhood inflammatory demyelinating CNS disorders. Neurology 2008;70:344–352. [DOI] [PubMed] [Google Scholar]
- 41.Krampla W, Aboul-Enein F, Jecel J, et al. Spinal cord lesions in patients with neuromyelitis optica: a retrospective long-term MRI follow-up study. Eur Radiol 2009;19:2535–2543. [DOI] [PubMed] [Google Scholar]
- 42.Iorio R, Damato V, Mirabella M, et al. Distinctive clinical and neuroimaging characteristics of longitudinally extensive transverse myelitis associated with aquaporin-4 autoantibodies. J Neurol 2013;260:2396–2402. [DOI] [PubMed] [Google Scholar]
- 43.Kim SH, Kim W, Li XF, Jung IJ, Kim HJ. Does interferon beta treatment exacerbate neuromyelitis optica spectrum disorder? Mult Scler 2012;18:1480–1483. [DOI] [PubMed] [Google Scholar]
- 44.Min JH, Kim BJ, Lee KH. Development of extensive brain lesions following fingolimod (FTY720) treatment in a patient with neuromyelitis optica spectrum disorder. Mult Scler 2012;18:113–115. [DOI] [PubMed] [Google Scholar]
- 45.Jacob A, Hutchinson M, Elsone L, et al. Does natalizumab therapy worsen neuromyelitis optica? Neurology 2012;79:1065–1066. [DOI] [PubMed] [Google Scholar]
- 46.Sato DK, Nakashima I, Takahashi T, et al. Aquaporin-4 antibody-positive cases beyond current diagnostic criteria for NMO spectrum disorders. Neurology 2013;80:2210–2216. [DOI] [PubMed] [Google Scholar]
- 47.Asgari N, Lillevang ST, Skejoe HP, Falah M, Stenager E, Kyvik KO. A population-based study of neuromyelitis optica in Caucasians. Neurology 2011;76:1589–1595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Saji E, Arakawa M, Yanagawa K, et al. Cognitive impairment and cortical degeneration in neuromyelitis optica. Ann Neurol 2013;73:65–76. [DOI] [PubMed] [Google Scholar]
- 49.Calabrese M, Oh MS, Favaretto A, et al. No MRI evidence of cortical lesions in neuromyelitis optica. Neurology 2012;79:1671–1676. [DOI] [PubMed] [Google Scholar]
- 50.Sinnecker T, Dorr J, Pfueller CF, et al. Distinct lesion morphology at 7-T MRI differentiates neuromyelitis optica from multiple sclerosis. Neurology 2012;79:708–714. [DOI] [PubMed] [Google Scholar]
- 51.Kilsdonk ID, de Graaf WL, Soriano AL, et al. Multicontrast MR imaging at 7T in multiple sclerosis: highest lesion detection in cortical gray matter with 3D-FLAIR. AJNR Am J Neuroradiol 2013;34:791–796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kister I, Herbert J, Zhou Y, Ge Y. Ultrahigh-field MR (7 T) imaging of brain lesions in neuromyelitis optica. Mult Scler Int 2013;2013:398259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Pichiecchio A, Tavazzi E, Poloni G, et al. Advanced magnetic resonance imaging of neuromyelitis optica: a multiparametric approach. Mult Scler 2012;18:817–824. [DOI] [PubMed] [Google Scholar]
- 54.Klawiter EC, Xu J, Naismith RT, et al. Increased radial diffusivity in spinal cord lesions in neuromyelitis optica compared with multiple sclerosis. Mult Scler 2012;18:1259–1268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Ciccarelli O, Thomas DL, De Vita E, et al. Low myo-inositol indicating astrocytic damage in a case series of neuromyelitis optica. Ann Neurol 2013;74:301–305. [DOI] [PubMed] [Google Scholar]
- 56.Matiello M, Lennon VA, Jacob A, et al. NMO-IgG predicts the outcome of recurrent optic neuritis. Neurology 2008;70:2197–2200. [DOI] [PubMed] [Google Scholar]
- 57.Weinshenker BG, Wingerchuk DM, Vukusic S, et al. Neuromyelitis optica IgG predicts relapse after longitudinally extensive transverse myelitis. Ann Neurol 2006;59:566–569. [DOI] [PubMed] [Google Scholar]
- 58.Kitley J, Woodhall M, Waters P, et al. Myelin-oligodendrocyte glycoprotein antibodies in adults with a neuromyelitis optica phenotype. Neurology 2012;79:1273–1277. [DOI] [PubMed] [Google Scholar]
- 59.Sato DK, Callegaro D, Lana-Peixoto MA, et al. Distinction between MOG antibody-positive and AQP4 antibody-positive NMO spectrum disorders. Neurology 2014;82:474–481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Kitley J, Waters P, Woodhall M, et al. Neuromyelitis optica spectrum disorders with aquaporin-4 and myelin-oligodendrocyte glycoprotein antibodies: a comparative study. JAMA Neurol 2014;71:276–283. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.