

Abstract
Traditional descriptions of the basal forebrain cholinergic projection system to the cortex have focused on neuromodulatory influences, that is, mechanisms that modulate cortical information processing but are not necessary for mediating discrete behavioral responses and cognitive operations. This review summarises and conceptualises the evidence in support of more deterministic contributions of cholinergic projections to cortical information processing. Through presynaptic receptors expressed on cholinergic terminals, thalamocortical and corticocortical projections can evoke brief cholinergic release events. These acetylcholine (ACh) release events occur on a fast, sub-second to seconds-long time scale (‘transients’). In rats performing a task requiring the detection of cues as well as the report of non-cue events cholinergic transients mediate the detection of cues specifically in trials that involve a shift from a state of monitoring for cues to cue-directed responding. Accordingly, ill-timed cholinergic transients, generated using optogenetic methods, force false detections in trials without cues. We propose that the evidence is consistent with the hypothesis that cholinergic transients reduce detection uncertainty in such trials. Furthermore, the evidence on the functions of the neuromodulatory component of cholinergic neurotransmission suggests that higher levels of neuromodulation favor staying-on-task over alternative action. In other terms, higher cholinergic neuromodulation reduces opportunity costs. Evidence indicating a similar integration of other ascending projection systems, including noradrenergic and serotonergic systems, into cortical circuitry remains sparse, largely because of the limited information about local presynaptic regulation and the limitations of current techniques in measuring fast and transient neurotransmitter release events in these systems.
Keywords: acetylcholine, attention, cortex, neuromodulation, noradrenaline, serotonin
Introduction
The ascending neuromodulator systems include the brainstem noradrenergic, serotonergic and cholinergic nuclei and their widespread ascending projections, as well as the cholinergic and non-cholinergic projections from the basal forebrain to telencephalic regions. Descriptions of the anatomical properties of brainstem ascending systems often emphasised that these projections originate from relatively small numbers of neurons and that they innervate large regions in the forebrain via their high degree of axonal collateralisation (Fallon & Loughlin, 1982; Espana & Berridge, 2006; Waselus ~ et al., 2011). The presence and degree of collateralised cholinergic projections arising from the basal forebrain has remained in dispute (e.g., Chandler et al., 2013) but generally these neurons exhibit less axonal branching than those arising from the brainstem, and the terminals of individual neurons tend to cluster in the cortical innervation space (Zaborszky, 2002; Briand et al., 2007; Hasselmo & Sarter, 2011; Zaborszky et al., 2012). Moreover, volume neurotransmission has been viewed as dominating the synaptic communication of neuromodulatory systems (for discussion see Sarter et al., 2009). When taken together, these considerations have supported the conceptualisation of ascending systems as exerting powerful modulatory, but primarily nonspecific, functions such as ‘arousal’, ‘activation’, ‘information gating’, or ‘increasing the signal-to-noise ratio’. The intuitive allure of these traditional views persists in the contemporary literature (e.g. Hornung, 2003; Eggermann & Feldmeyer, 2009; Lee & Dan, 2012; Sara & Bouret, 2012; Moran et al., 2013; Varela, 2013).
The usefulness of such poorly-defined functional concepts for guiding research on the functions of ascending systems has been questioned (Robbins & Everitt, 1995). Moreover, newer evidence concerning the basal forebrain system indicates a highly structured and topographic organisation of efferent projections and the presence of clusters of cholinergic terminals in the cortical innervation space (e.g., Zaborszky, 2002; Zaborszky et al., 2008, 2013). The presence of phasic actions of ascending neurotransmitter systems (Dayan & Yu, 2006; Parikh et al., 2007; Howells et al., 2012) further challenges the classification of the neurotransmitters of ascending projection systems as strictly neuromodulators (Parikh & Sarter, 2008; Dayan, 2012; Marder, 2012; Picciotto et al., 2012; Sun et al., 2013).
Below we review the available evidence in support of the hypothesis that basal forebrain cholinergic projections to the cortex form an integral part of cortical circuitry, capable of mediating, as opposed to modulating, discrete cognitive and behavioral functions. In other words, cortical and subcortical projections employ cholinergic inputs to contribute to cortical information processing (Fig. 1). Furthermore, these cholinergic inputs themselves are subject to neuromodulation by cortical and subcortical input (Fig. 1; below).
Fig. 1.
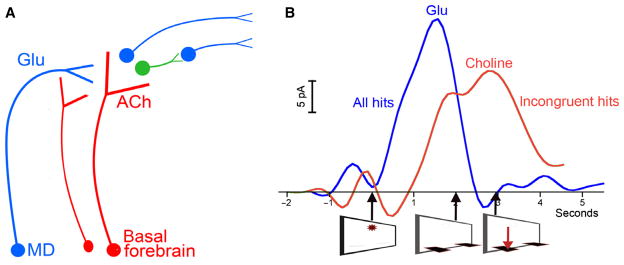
Illustration of (A) the integration of cortical cholinergic inputs into cortical circuitry and (B) cue-evoked glutamatergic and cholinergic transients. The illustrations are based on evidence from our electrochemical studies (Parikh et al., 2007, 2010; Parikh & Sarter, 2008; Howe et al., 2013). In attentional contexts, all cues that are detected (see text for definition of ‘detection’) elicit a glutamatergic (Glu) transient from mediodorsal thalamic afferents (MD in A). Such glutamatergic transients are necessary, but not sufficient, to generate cholinergic transients (ACh), perhaps via ionotropic glutamate receptors that may be expressed at cholinergic terminals (see text). Glutamate may elicit cholinergic transients regardless of cholinergic depolarisation (Kunz et al., 2013), thereby integrating cholinergic inputs into cortical circuitry and employing these terminals for cortical information processing. (B) Glutamatergic and cholinergic transients (spline-interpolated traces) recorded in the medial prefrontal thalamic input layers during incongruent hits. As detailed in the text, all cues yielding hits, regardless of trial sequence, evoke glutamatergic transients. These transients peak at around the time the levers are extended and prior to the response. The absence of a cue-evoked glutamatergic transient invariably predicts a miss. Cholinergic transients are observed only during cued trials yielding hits that are preceded by trials ending with correct rejections or misses (‘incongruent hits’), but not in trials preceded by a hit (‘consecutive hits’). Thus, it is hypothesised that cholinergic transients mediate cue detection during trials involving a shift from perceptual attention to cue-oriented behavior (Howe et al., 2013). As indicated by gamma oscillations seen during such trials, the interactions between glutamatergic and cholinergic transients during incongruent hits synchronises cortical output assemblies to forward the processing of the cue to further telencephalic regions, thereby mediating an attentional mode shift and the detection of the cue in trials requiring such a shift. In addition to this deterministic function of ACh, basal forebrain cholinergic projections also modulate the glutamatergic–cholinergic transient interactions via stimulating alpha4beta2* nAChRs; it is hypothesised that these cholinergic neurons form a separate population of basal forebrain cholinergic neurons (A). Cholinergic neuromodulatory upregulation of glutamatergic–cholinergic interactions is a functions of attentional effort (e.g., St Peters et al., 2011; see also main text). Thus, such upregulation restores or increases detection rates.
This review does not cover the basic organisation of the cholinergic system and evidence indicating neuromodulatory functions (Wenk, 1997; Deco & Thiele, 2008; Schliebs & Arendt, 2011; Picciotto et al., 2012). Rather, we will focus specifically on the evidence in support of the idea that cortical circuitry integrates a component of the ascending systems to support cortical information processing and therefore has deterministic functions. By reviewing the evidence in support of this hypothesis we are not rejecting the importance or presence of a neuromodulatory component of ascending systems, including a component of the cortically-projecting basal forebrain cholinergic system (St Peters et al., 2011; see also further below for a conceptualisation of cholinergic neuromodulation). Rather, we propose that separate from and in addition to their role as a neuromodulator, these ascending projections take part in highly specialised cortical information processing (Aston-Jones & Cohen, 2005; Zaborszky et al., 2005; Unal et al., 2012).
Heteroreceptors – local control of cortical cholinergic activity
Cholinergic inputs to cortical regions are capable of generating complex neurophysiological effects via multiple muscarinic and nicotinergic acetylcholine (ACh) receptor subtypes (mAChR and nAChR). In turn, the release of ACh is itself under the control of heteroreceptors. Such heteroreceptor-mediated control of neurotransmitter release involves ionotropic as well as metabotropic receptors situated near the active presynaptic zone, activating either ion channels or second-messenger mechanisms to influence or even determine neurotransmitter release (for reviews see MacDermott et al., 1999; Schicker et al., 2008). Presynaptic control of neurotransmitter release can occur via depolarisation-dependent modulation of release levels as well as the induction of release in the absence of action potentials (Kunz et al., 2013). However, the intracellular mechanisms mediating depolarisation-independent release remain poorly understood.
Early experiments measuring ACh release from cerebral synaptosomal preparations and slices demonstrated that it is subject to GABAergic modulation; however, these studies did not indicate a consistent set of effects (e.g., Bonanno et al., 1991). Evidence from in vivo microdialysis studies suggested that local GABAergic activity directly inhibits basal ACh release from cortical terminals (Giorgetti et al., 2000). However, ascending cholinergic projections also target GABAergic interneurons which in turn inhibit release from cholinergic terminals (Disney & Aoki, 2008; Kruglikov & Rudy, 2008; Disney et al., 2012). Furthermore, local GABAergic activity also modulates changes in cholinergic activity that are evoked by local noradrenergic and serotonergic mechanisms (Moroni et al., 1983; Beani et al., 1986; Ramírez et al., 1996). Clearly, the mechanisms involved in cerebral GABAergic modulation of ACh release remain very poorly understood.
Our own recent research has focused on local mechanisms contributing to the generation of brief cholinergic release events in pre-frontal cortex. We demonstrated that glutamate released from thalamic afferents is necessary to evoke brief, seconds-based or ‘transient’ cholinergic release events (Parikh et al., 2008). Furthermore, glutamate release from these thalamic inputs is itself modulated by cholinergic activity and stimulation of nAChRs (Gioanni et al., 1999; Lambe et al., 2003; Howe et al., 2010; Parikh et al., 2010). We exploited this mechanism to study the relationships between cholinergic neuromodulation and cholinergic transients by determining the effects of nAChR stimulation on glutamatergic and cholinergic transients in prefrontal cortex. As expected based on the presence of nAChRs on glutamatergic terminals and our hypothesis about cortical glutamatergic–cholinergic interactions (Fig. 1), stimulation of alpha4beta2* nAChRs evokes both transient glutamate release and ACh transients. Furthermore, cholinergic transients evoked by such stimulation are abolished by removing thalamic inputs or by blocking ionotropic glutamate receptors in the prefrontal recording region (Parikh et al., 2008, 2010). Although the assumption that ionotropic glutamate receptors are expressed by cholinergic terminals would provide a straightforward mechanism underlying these glutamatergic–cholinergic transient interactions, to our knowledge the presence of ionotropic glutamate receptors on cholinergic terminals has not been investigated. Thus, a more complex, multi-synaptic mechanism underlying the relationship between prefrontal cholinergic and glutamatergic signaling cannot be excluded. We will return to the discussion of potential synaptic mechanisms further below following the discussion of the cognitive functions of cholinergic transients.
Functions of cholinergic transients
Cholinergic inputs to the cortex are necessary for attentional performance and specifically for the detection and use of instructive cues to guide decisions about ongoing behavior (Muir et al., 1992; McGaughy et al., 1996; Turchi & Sarter, 1997; Dalley et al., 2004; Botly & De Rosa, 2009). The use of cues to guide behavior henceforth is termed ‘detection’, as defined in Posner et al. (1980). Importantly, this definition integrates the perceptual with the cognitive processes involved in the decision to report a signal – ‘By detection, we will mean the entry of information concerning the presence of a signal into a system that allows the subject to report the existence of the signal by an arbitrary response indicated by the experimenter’ (Posner et al., 1980, p. 162).
Cholinergic activity in the cortex serves both neuromodulatory and deterministic functions, albeit via separate mechanisms. Our current model assumes that that the cholinergic neurons that modulate cortical circuitry form a separate population from those that generate the transient release events that are integrated into cortical information processing and exert deterministic functions (Hasselmo & Sarter, 2011; see also Hasselmo & Bower, 1992). This assumption awaits further testing, but separate cholinergic cell populations may be revealed based on, for example, their topographic organisation in the basal forebrain, differential histological markers, and/or their differential cortical vs. subcortical afferent organisation (Unal et al., 2012; Zaborszky, 2002; Zaborszky et al., 2005, 2013; Fig. 1).
In the present context, the neuromodulatory component of cholinergic activity is hypothesised to influence the probability and amplitude of cortical glutamatergic–cholinergic transients, primarily via stimulation of nAChRs (as described above). The level of this neuromodulatory influence has been shown to co-vary with demands on attentional control, not level of performance. That is, performance-associated increases are highest when performance is low as a result of distractors, extended time on task, or pharmacological challenges (Kozak et al., 2006; Sarter et al., 2006; St Peters et al., 2011).
While the neuromodulatory component appears to be more closely linked to demand (and attempts to maintain performance in the face of such demand), we propose that transient ACh release events play a more direct and immediate role in controlling detection performance. This hypothesis initially arose from our studies using fixed-potential amperometry to record medial prefrontal cholinergic transients in rats performing a cued appetitive response task. Cue presentations were separated by ~ 90-s intervals during which animals were free to engage in task-irrelevant behavior. Cues that were detected and thus evoked a shift from ongoing behavior (e.g., grooming) to cue-directed behavior produced transient increases in ACh release (Parikh et al., 2007). In contrast, cues that were not detected (‘misses’) failed to evoke cholinergic transients. Several control experiments demonstrated that reward delivery and reward retrieval do not contribute to the generation of cholinergic transients. Furthermore, we showed that cue-evoked cholinergic transients emerged during the learning of this task, as cues began to control behavior.
Subsequent experiments recorded both glutamatergic and cholinergic activity in rats performing an operant sustained-attention task (SAT). This task consists of separate trials during which visual cues (or signals) are presented, or not, followed by the extension of the levers into the operant chamber which triggers a response. Rats press one lever to report the presence of the cue and another to report the cue’s absence (nonsignal trial). Correct responses are ‘hits’ on signal trials and ‘correct rejections’ on nonsignal or blank trials. In the thalamic input layer of the prelimbic cortex, all cues that resulted in hits evoked glutamatergic transients (W.M. Howe, H. Gritton & M. Sarter, unpublished observations; Fig. 1B).
Although glutamatergic transients were found for all hit trials, cholinergic transients occurred for only a proportion (~ 60%) of cues yielding hits. Thus, glutamatergic transients, while required for cholinergic transients, were not sufficient for their generation. Instead, the presence or absence of cholinergic events during cue-hit trials depended on the previous trial type (Howe et al., 2013). Specifically, cholinergic transients were only evoked by cues in hit trials when those trials were preceded by a missed cue or correct rejection trial. In other words, transients only occurred when hits (correct indication of a signal trial) were preceded by an actual (correct rejection) or perceived (miss) nonsignal trial. We therefore refer to these particular hit trials as ‘incongruent hits’ or ‘shift-hits’, i.e., the signal response on these trials is incongruent with nonsignal response on the prior trial, and requires a shift in task representation and response. Cholinergic transients were not evoked by cues that were presented consecutively and reliably detected (‘consecutive hits’; Howe et al., 2013).
We have interpreted this cholinergic signal as forcing a shift, away from a state of monitoring that dominates attentional performance during the absence of cues, to cue-directed attention. This same state would also have been engaged during the long inter-trial intervals in the task described in Parikh et al. (2007). Importantly, parallel experiments employing functional MRI in human subjects revealed coincident basal forebrain and prefrontal activation during incongruent hits, as well as in prefrontal oxygen levels in rats (for details see Howe et al., 2013). Combined, these data support the presence a prefrontal cholinergic mechanism that is preserved across species and supports attentional performance by forcing shifts from monitoring to cue-directed attention.
Evidence for the deterministic role of cholinergic transients in attentional performance was obtained from a subsequent set of studies that demonstrated that the generation or suppression of such transients, using optogenetic methods, enhances or reduces, respectively, hit rates in SAT-performing mice (H. Gritton, W.M. Howe & M. Sarter, unpublished observations). Specifically, if transients are evoked to coincide with cues, hit rates increase; this is most robustly demonstrated for trials in which cue illumination is briefest in duration. Correspondingly, if endogenously generated cholinergic transients are suppressed using opsins that inhibit depolarisation, animals detect fewer cues. These data suggest that cholinergic transients promote a shift to cue-associated response representations.
In what is perhaps an even more direct demonstration of the causal relationship between phasic cholinergic signaling and cue ‘detection’, artificially generating a cholinergic transient on non-signal trials increases the likelihood of a false alarm. These induced, ill-timed transients produce false alarms in as many as 50% of such trials (as opposed to < 10% at baseline). This finding supports the hypothesis that cholinergic transients increase the probability for a discrete behavioral response, the reporting of a signal. Generating transients in the absence of signals ‘inserts’ the cholinergic activity normally generated by a detected, incongruent cue. Thus, we hypothesise that cholinergic transients are a sufficient cause for incongruent hits. Clearly, this hypothesis requires more testing, including more stringent manipulations of cholinergic transient activity during controlled sequences of signal and nonsignal trials.
The timecourse of cholinergic transients (Fig. 1B) leads to additional speculation about their function. Specifically, cholinergic activity extends beyond the completion of incongruent hits and persists into the subsequent inter-trial interval, peaking at ~ 6 s following the cue (see fig. 2 in Howe et al., 2013). This ongoing activity is not likely to be related to the mediation of the actual hit in that particular trial. Rather, such prolonged cholinergic activity may serve as a reporter that binds action selection with outcome. Thus, the extended portion of the transient could act as a ‘teaching signal’, confirming the accuracy of the response choice in such trials and thereby increasing the likelihood of future shifts from monitoring for cues to cue-directed behavior. Again, however, further experiments are necessary to test this theory.
Absence of cholinergic transients during consecutive hits
The synaptic mechanisms responsible for the generation of cue-evoked cholinergic transients during incongruent hits remain largely speculative. The evidence supports the general idea that a cue that will be detected is ‘inserted’ into cortical circuitry via cue-evoked glutamatergic transients from mediodorsal thalamic projections (Fig. 1). As discussed above, cue-evoked glutamatergic transients, evoked by all cues yielding hits irrespective of trial sequence, are necessary, but not sufficient, for generating the cholinergic transients; the latter being evoked only by cues yielding incongruent hits. Thus, it needs to be determined whether cholinergic transients are actively suppressed during consecutive hits or whether such transients are generated specifically during incongruent hits and based on additional, currently unknown, circuitry.
One possibility is that cholinergic transients are not generated on consecutive hits because the signal-associated task response condition is already activated, and thus there is no need for a ‘shift’. On the other hand, there is evidence consistent with the alternative possibility that cholinergic transients are actively suppressed during consecutive hits. Cholinergic transients may depolarise GABAergic interneurons and thereby contribute to their own subsequent suppression (see above; Xiang et al., 1998). Furthermore, muscarinic mechanisms have been demonstrated to maintain persistent firing of neurons (Klink & Alonso, 1997; Egorov et al., 2002). Some of these neurons may be inhibitory interneurons, and thus this mechanism could contribute to the persistent suppression of cholinergic transients during strings of consecutive hits. Our own preliminary evidence supports the hypothesis that local GAB-Aergic activity can suppress cholinergic transients (Berry et al., 2011). In this scheme, a nonsignal event would be speculated to terminate such suppression of cholinergic transients, ‘releasing’ glutamatergic–cholinergic transient interactions from inhibition and therefore allowing a subsequent cue, if detected, to again evoke a cholinergic transient. The mechanisms that would terminate this proposed persistent suppression of cholinergic transients remain entirely unknown.
Postsynaptic mechanisms – high-frequency oscillations
To this point, our discussion has focused largely on presynaptic mechanisms and cognitive contexts associated with the generation of cholinergic transients. An additional, and equally important consideration focuses on the postsynaptic effects of these release events. What effect do transient increases in glutamatergic and cholinergic activity have on the state of local prefrontal networks, and how might these changes relate to task performance? One possible answer originates from experiments in which we recorded local field potentials in prefrontal networks in animals performing the SAT. These recordings indicated enhanced synchronous activity at gamma frequencies during cue detection. However, we also found that cue detection on incongruent hits coincided with more synchronised gamma activity that was sustained through the reward period consistent with the timeline noted for long-lasting cholinergic transients (Howe et al., 2011). Thus, we hypothesise that the increased gamma power during incongruent hits, reflecting the postsynaptic impact of combined glutamatergic–cholinergic activity, relays the local processing of the cue across a distributed network that in turn recruits the circuitry required to execute the motor response. In the absence of a cholinergic transient, gamma synchrony is attenuated, the likelihood for a successful attentional mode shift is reduced, and cues in such trials are more likely to be missed.
The hypotheses described above align with the idea that cortical circuitry integrates the ascending cholinergic system into local circuitry to support cognitive operations (Fig. 1). Stimulation of intra-cortical and efferent neurons by cholinergic transients, in conjunction with glutamatergic activity, increases synchronous high-frequency oscillatory activity (as described above). Such enhanced coordination of local activity fosters the formation of cell assemblies to relay output across a distributed network in support of cue detection (see also Fan et al., 2007; Gulledge et al., 2009) and, more generally, the ability of such a cue to control behavior (Engel & Singer, 2001; Rodriguez et al., 2004, 2010; Fries, 2005; Briggs et al., 2013). In the absence of cholinergic transients and synchronous high-frequency activity, hit rates are predicted to be reduced, specifically in cued trials requiring an attentional mode shift.
Our hypothesis has been deduced from recordings in rats performing the SAT and thus suggests a cortical cholinergic function required for a specific cognitive operation that underlies SAT trial-sequence-based performance. However, this hypothesis may be readily generalised to other cognitive operations involving cue detection and cue-directed behavior. For example, in rats performing a cross-modal divided-attention task (McGaughy et al., 1994), cholinergic activity is necessary for shifting between cues of different modalities but not for shifting between cues within modalities (see also Turchi & Sarter, 1997). Although cholinergic transients in animals performing this task have not been recorded, the present data would predict that cues involving cross-modal shifts likewise generate cholinergic transients to orchestrate cue-related processing (see also Senkowski et al., 2008; Schneider et al., 2011).
Cholinergic transients require wired neurotransmission, or does it matter?
The hypothesis that cholinergic transients mediate a specific component of the cortical processing of cues entails the characteristics of wired neurotransmission. That is, this hypothesis predicts that the transient cholinergic signal stimulates a defined set of postsynaptic receptors as opposed to a more persistent stimulation of cholinergic receptors across a larger cortical region and involving receptors located away from the presynaptic release sites (volume neurotransmission; for an illustration of the two transmission modes see fig. 3 in Sarter et al., 2009).
Our electrochemical evidence suggests that all newly released ACh is hydrolyzed by endogenous ACh esterase (AChE; Giuliano et al., 2008). In other words, this evidence suggests that because of the abundance and extraordinary potency of AChE (ACh esterase), little or no ACh remains available for volume neurotransmission, certainly not the high nanomolar to low micromolar ACh concentrations that were proposed to support volume neurotransmission (Descarries, 1998). However, the presence vs. absence of volume neurotransmission is extremely difficult to resolve experimentally. We suggested that this issue may be of secondary importance when compared to the significance of transient release events (see the discussion in Sarter et al., 2009). It appears more important to understand how the time course of these transients maps onto behavior and information processing, rather than deciphering the degree to which extra-synaptic neurotransmission underlies the ability of a cue to be detected and shift attentional modes.
Cortical cholinergic signaling – computational conceptualisation
This section provides a reductionist description of the information-processing steps that require cholinergic transients in prefrontal cortex. Furthermore, the impact of the neuromodulatory component of cholinergic neurotransmission on the generation of cholinergic transients will be described in computational terms of attentional effort.
Cholinergic transients reduce detection uncertainty
As detailed above, our evidence from electrochemical recordings and optogenetic experiments indicate that for cues to yield hits after an extended period of nonsignal processing, these cues need to produce a cholinergic transient. The perceptual component of the detection process may depend on the glutamatergic transient and does not require a prefrontal cholinergic transient; consecutive cues, if reliably detected, do not evoke cholinergic transients. Instead, the specific association of cholinergic transients with hits that follow extended nonsignal processing, as well as the increase in false alarms on non-cued trials during which such transients were optogenetically generated (described above), suggests that these transients instigate, or at least increase the probability of, a shift away from monitoring for cues and towards the processes needed to generate the cue-directed response. As also described above, we hypothesise that the increase in gamma power triggered by cholinergic transients represents a postsynaptic efferent mechanism for executing hits in these trials.
The function of cholinergic transients could also be phrased in terms consistent with the theory of Yu & Dayan (2005) that cholinergic activity in the cortex indicates expected uncertainty (see also Dayan, 2012), though with some modifications. First, rather than the global probability with which a cue is predicted by a practiced performer, the current conceptualisation emphasises the uncertainty of the detection process as a function of trial sequence, a concept perhaps more akin to the response bias in signal detection theory. Second, it is not the neuromodulatory component that signals the level of predicted uncertainty (see below for a discussion of neuromodulatory effects); rather, it is solely the cholinergic transient that affects the certainty of detection. Third, the cholinergic transient does not merely signal the degree of predicted uncertainty in incongruently cued trials; instead it reduces such uncertainty. In other words, the presence of a cholinergic transient shifts the performer toward adopting a riskier detection criterion, thereby enhancing the probability that detection occurs in cued trials that follow non-cued trials.
Reducing uncertainty of detection does not tap purely perceptual or purely behavioral operations; rather, it concerns the integration of the two, as captured by the definition of detection (detailed above) in Posner et al. (1980). Therefore, a neuronal mechanism that is designed to reduce detection uncertainty must be closely connected to, and to a degree depend on, the actual perceptual mechanisms. The finding that the generation of a cholinergic transient depends upon thalamic glutamatergic input, that is relayed to the prefrontal cortex by all cues that yield hits, reflects this close connection between perceptual and decisional mechanisms. Moreover, as illustrated rather drastically by the ability of artificially generated cholinergic transients to force hits on nonsignal trials (above), a cholinergic transient appears to be capable of overriding perception and triggering a decision to report a cue even in its absence.
What then would be the costs of cholinergic transients if evoked on consecutively-cued trials? What would be the costs of further reducing detection uncertainty when the perceptual process already established that a cue was present, as indicated by the finding that glutamatergic transients reliably predict hits (Fig. 1B)? We speculate that the presence of cholinergic transients during consecutively cued hits would nearly completely abolish any residual detection uncertainty and thereby strongly bias performance to the reporting of signals. As a consequence, the ability to respond accurately to subsequent nonsignal trials could be impaired. In other words, cholinergic transients during consecutively-cued trials would reduce the flexibility to accurately perform a task that presents cued and non-cued trials at equal probability. Certainly, manipulating such probability will be an important experimental means of further testing our hypothesis. The speculation that the absence of cholinergic transients during consecutive hits maintains the flexibility to shift from executing hits to reporting the absence of cues would also be consistent with the proposition that transients are actively suppressed during consecutively cued trials.
Our evidence from animals and humans (Howe et al., 2013) indicates that cholinergic transients serve to shift the performance from a state of monitoring for signals to responding to cues. Here we suggest that cholinergic transients increase the likelihood for accurate responding during such shifts by reducing the uncertainty with which a cue is detected. The hypothesis that cholinergic transients reduce detection uncertainty in trials in which such uncertainty is high allows for interesting predictions of the consequences of dys-regulated cholinergic transients (Sarter et al., 2012). A robust attenuation or absence of such transients predicts failures in detecting cues specifically in situations involving dynamic cue probabilities (Perry & Hodges, 1999). Conversely, ill-timed cholinergic transients enhance the ability of random and behaviorally irrelevant cues to control behavior and cognitive activity (Nuechterlein et al., 2009; Luck et al., 2012).
Cholinergic modulation reduces opportunity costs
Our collective evidence indicates that attentional-performance associated levels of cholinergic neuromodulation are highest in the presence of distractors and when performance is relatively low (e.g., St Peters et al., 2011; see also Kozak et al., 2006). On the other hand, such levels are attenuated in animals exhibiting relatively poor and highly fluctuating performance as a trait (Paolone et al., 2013). We have previously conceptualised this cholinergic neuromodulatory function as a top-down modulation of cortical detection circuitry as a function of attentional effort (Sarter et al., 2006). As an important technical corollary, the evidence supports the view that cholinergic transients and the more tonically active neuromodulatory component that is measured by microdialysis and varies on a scale of tens of seconds to minutes, are separate phenomena. ACh levels in dialy-sates do not reflect the sum of transients over one or several minutes (Paolone et al., 2010; Sarter et al., 2010).
We have previously conceptualised attentional effort as a set of mechanisms designed to cope with, or combat the consequences of, limited attentional resources (Sarter et al., 2006). An arguably more informative conceptualisation of the attentional effort construct considers such effort as the experience of mentally calculating the utility of continuing performance of the present task relative to the costs and benefits of discontinuing performance of or reallocating resources to alternative tasks (Kurzban et al., 2013). This view begins to explain important observations from our research. For example, rodents performing versions of the basic SAT do not exhibit significant within-session performance decline. The absence of performance decrements is not well explained by speculations about limited demands on attentional resources or limited cognitive load imposed by such a task. Such hypotheses are also quite difficult to reject. Rather, the absence of behavioral-cognitive alternatives, combined with high levels of motivation to stay on task and not engage in task-unrelated behavior keeps ‘opportunity costs’ relatively low (Kurzban et al., 2013). As attentional effort and the associated sensation of fatigue and boredom result from monitoring and accruing opportunity costs, a motivated subject routinely performing a single task, with no alternative action in sight, accrues little to no such costs and thus performance will not degrade.
We repeatedly observed relatively stable levels of cholinergic neuromodulatory activity over 40–60 min of SAT performance (Arnold et al., 2002; St Peters et al., 2011). As an alternative to hypothesising that these levels indicate the stable and limited demands on top-down control of attention in subjects performing the standard SAT, these stable levels of cholinergic neuromodulation may index the output of estimating the utility of the current over alternative actions, in short, the low opportunity costs that are accrued by subjects having access only to the regular SAT. Because opportunity costs are already low in the absence of alternative tasks, we now understand why lowering the demands on performance (animals had access to only one response lever) failed to alter levels of cholinergic neuromodulation (Himmelheber et al., 2001).
In contrast, staying on task in the presence of a distractor and regaining high performance levels thereafter requires activation of diverse neuronal mechanisms to enhance the processing of cues and filter distractors and to monitor prediction errors (see Sarter et al., 2006). Even in the absence of an alternative task, distractors therefore increase the costs for staying on task and the relatively utility of discontinuing performance. The presentation of distractors may also trigger the actual monitoring of these relative utilities. It is in such situations that we observed highest levels of cholinergic neuromodulation. Moreover, and importantly, higher cholinergic levels were correlated with better (residual) performance (St Peters et al., 2011). Thus, we hypothesise that higher levels of cholinergic neuromodulation shift the cost/benefit calculation for staying on task, relative to the utility for switching to an alternative task or, in our experimental settings, over discontinuation of performance. Higher levels of cholinergic neuromodulation reduce opportunity costs and perhaps also the subjective and aversive experience of computing these costs (mental effort), thereby decreasing the likelihood for discontinuing performance or, if available, switching to alternative action. As elevated levels of cholinergic neuromodulation are recruited in part via mesolimbic–basal forebrain interactions (St Peters et al., 2011; see also Neigh et al., 2004; Zmarowksi et al., 2005), it is conceivable that the cholinergic modulation of opportunity costs primarily increases the computed value of earned rewards, as opposed to decreasing the costs of discontinuing performance and omitting rewards.
Consistent with the hypothesis that higher levels of cholinergic neuromodulatory activity reduce opportunity costs in rats performing an attention task, such levels were found to correlate with the degree of task compliance under taxing conditions (Passetti et al., 2000). Furthermore, this hypothesis also predicts the relatively poor and fluctuating levels of attentional performance in rats exhibiting relatively low levels of cholinergic neuromodulation during such performance (see Paolone et al., 2013). Likewise, this hypothesis predicts that humans who carry a minor allele of the choline transporter gene, which may limit the dynamic range of neuromodulatory cholinergic activation, self-report greater levels of distractibility in situations that readily allow for discontinuation of performance and engagement on alternative behavioral of cognitive activities (e.g. are easily distracted by a TV or radio playing in the next room). In contrast, such vulnerability to distraction may be more difficult to demonstrate in situations that demand high levels of attention but are relatively devoid of competitive alternatives (Berry et al., 2013). In other words, compared with humans expressing the wild-type gene for this transporter, the variant-expressing subjects, assuming that expression of this allele limits the capacity for cholinergic neurotransmission, may experience higher opportunity costs and assign relative greater utility to engaging in alternative mental or behavioral action.
Reducing opportunity costs reduces detection uncertainty
As discussed above, the results of our research cumulatively support the hypothesis that increases in cholinergic neuromodulation enhance prefrontal glutamatergic–cholinergic transient interactions (Fig. 1) and that stimulation of nAChRs ‘import’ the neuromodulatory impact on transients. Our studies on the beneficial effects of alpha4beta2* nAChR agonists on cholinergic transients and SAT performance demonstrated that such benefits are restricted to SAT performance that is burdened by the presence of a distractor. Furthermore, nAChR agonist-induced increase in hits was due primarily to an increase in hits on trials where a signal followed extended periods of nonsignal processing, that is, hits for which cholinergic transients are required (Howe et al., 2010). Thus, higher levels of cholinergic neuromodulation increase the probability for cholinergic transients and thus for incongruent hits. These considerations are consistent with the hypothesis that higher levels of cholinergic neuromodulatory activity lower opportunity costs in part by reducing detection uncertainty, thereby stabilising and restoring hit rates and thus performance outcome. In simpler words, this means that an operator that computes relatively lower opportunity costs, perhaps by assigning relatively greater value to continuing performance and obtaining rewards, will then also obtain more rewards.
Cortical integration of other ascending systems
Evidence from the cholinergic system reminds us that the local, cortical control of release events via presynaptic heteroreceptors allows for specificity even if these afferents originate from a relatively small number of neurons (see also Zaborszky et al., 2013). The neuromodulatory impact of brainstem ascending systems on cortical functions has been extensively demonstrated in recent decades (e.g., Berridge & Arnsten, 2013) and it would not be surprising if future studies reveal other discrete cognitive operations that are mediated via presynaptic mechanisms that control local transient neurotransmitter release events. The presence of discrete, cortically-generated and cognitive-operation-associated activity in branches of noradrenergic and serotonergic systems would be consistent with the increasingly refined hypotheses about their functions (Aston-Jones & Cohen, 2005; Aznar & Klein, 2013).
Acknowledgments
The authors’ research was supported by PHS Grants R01MH086530 and PO1 DA031656. W.M.H. is now at Pfizer (Cambridge, MA, USA) and H.G. is now at Boston University (Boston, MA, USA). A.S.B. was supported by an NSF Graduate Research Fellowship.
Abbreviations
- ACh
acetylcholine
- AChE
ACh esterase
- mAChR
muscarinic ACh receptor subtype
- nAChR
nicotinergic ACh receptor subtype
- SAT
sustained attention task
References
- Arnold HM, Burk JA, Hodgson E, Sarter M, Bruno JP. Differential increases in cortical acetylcholine release in rats performing a sustained attention task versus behavioral control tasks that do not explicitly tax attention. Neuroscience. 2002;114:451–460. doi: 10.1016/s0306-4522(02)00292-0. [DOI] [PubMed] [Google Scholar]
- Aston-Jones G, Cohen JD. An integrative theory of locus coeruleus-norepinephrine function: adaptive gain and optimal performance. Annu Rev Neurosci. 2005;28:403–450. doi: 10.1146/annurev.neuro.28.061604.135709. [DOI] [PubMed] [Google Scholar]
- Aznar S, Klein AB. Regulating prefrontal cortex activation: an emerging role for the 5-HT2A serotonin receptor in the modulation of emotion-based actions? Mol Neurobiol. 2013;48:841–853. doi: 10.1007/s12035-013-8472-0. [DOI] [PubMed] [Google Scholar]
- Beani L, Tanganelli S, Antonelli T, Bianchi C. Noradrenergic modulation of cortical acetylcholine release is both direct and gamma-aminobutyric acid-mediated. J Pharmacol Exp Ther. 1986;236:230–236. [PubMed] [Google Scholar]
- Berridge CW, Arnsten AF. Psychostimulants and motivated behavior: arousal and cognition. Neurosci Biobehav R. 2013;37:1976–1984. doi: 10.1016/j.neubiorev.2012.11.005. [DOI] [PubMed] [Google Scholar]
- Berry AS, St Peters MM, Uremek N, Gritton H, Grupe M, Mirza NR, Sarter M. Selective GABA Inverse Agonist RO4938581 Modulates Nicotine-Evoked Transients in Prefrontal Cortex. Paper presented at the Society for Neuroscience Annual Meeting. Soc. Neurosci. Ann. Meeting; Washington, DC. 2011. [Google Scholar]
- Berry AS, Isaacs Y, Demeter E, Blakely RD, Sarter M, Lustig C. Selective Vulnerability to Distraction Associated with Choline Transporter Gene. Cogn. Neurosci. Soc. Ann. Meeting; San Francisco, CA. 2013. [Google Scholar]
- Bonanno G, Ruelle A, Andrioli GC, Raiteri M. Cholinergic nerve terminals of human cerebral cortex possess a GABA transporter whose activation induces release of acetylcholine. Brain Res. 1991;539:191–195. doi: 10.1016/0006-8993(91)91620-g. [DOI] [PubMed] [Google Scholar]
- Botly LC, De Rosa E. Cholinergic deafferentation of the neocortex using 192 IgG-saporin impairs feature binding in rats. J Neurosci. 2009;29:4120–4130. doi: 10.1523/JNEUROSCI.0654-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Briand LA, Gritton H, Howe WM, Young DA, Sarter M. Modulators in concert for cognition: modulator interactions in the prefrontal cortex. Prog Neurobiol. 2007;83:69–91. doi: 10.1016/j.pneurobio.2007.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Briggs F, Mangun GR, Usrey WM. Attention enhances synaptic efficacy and the signal-to-noise ratio in neural circuits. Nature. 2013;499:476–480. doi: 10.1038/nature12276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chandler DJ, Lamperski CS, Waterhouse BD. Identification and distribution of projections from monoaminergic and cholinergic nuclei to functionally differentiated subregions of prefrontal cortex. Brain Res. 2013;1522:38–58. doi: 10.1016/j.brainres.2013.04.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dalley JW, Theobald DE, Bouger P, Chudasama Y, Cardinal RN, Robbins TW. Cortical cholinergic function and deficits in visual attentional performance in rats following 192 IgG-saporin-induced lesions of the medial prefrontal cortex. Cereb Cortex. 2004;14:922–932. doi: 10.1093/cercor/bhh052. [DOI] [PubMed] [Google Scholar]
- Dayan P. Twenty-five lessons from computational neuromodulation. Neuron. 2012;76:240–256. doi: 10.1016/j.neuron.2012.09.027. [DOI] [PubMed] [Google Scholar]
- Dayan P, Yu AJ. Phasic norepinephrine: a neural interrupt signal for unexpected events. Network. 2006;17:335–350. doi: 10.1080/09548980601004024. [DOI] [PubMed] [Google Scholar]
- Deco G, Thiele A. Cholinergic control of cortical network interactions enables feedback-mediated attentional modulation. Eur J Neurosci. 2011;34:146–157. doi: 10.1111/j.1460-9568.2011.07749.x. [DOI] [PubMed] [Google Scholar]
- Descarries L. The hypothesis of an ambient level of acetylcholine in the central nervous system. J Physiol Paris. 1998;92:215–220. doi: 10.1016/s0928-4257(98)80013-2. [DOI] [PubMed] [Google Scholar]
- Disney AA, Aoki C. Muscarinic acetylcholine receptors in macaque V1 are most frequently expressed by parvalbumin-immunoreactive neurons. J Comp Neurol. 2008;507:1748–1762. doi: 10.1002/cne.21616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Disney AA, Aoki C, Hawken MJ. Cholinergic suppression of visual responses in primate V1 is mediated by GABAergic inhibition. J Neurophysiol. 2012;108:1907–1923. doi: 10.1152/jn.00188.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eggermann E, Feldmeyer D. Cholinergic filtering in the recurrent excitatory microcircuit of cortical layer 4. Proc Natl Acad Sci USA. 2009;106:11753–11758. doi: 10.1073/pnas.0810062106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Egorov AV, Hamam BN, Fransen E, Hasselmo ME, Alonso AA. Graded persistent activity in entorhinal cortex neurons. Nature. 2002;420:173–178. doi: 10.1038/nature01171. [DOI] [PubMed] [Google Scholar]
- Engel AK, Singer W. Temporal binding and the neural correlates of sensory awareness. Trends Cogn Sci. 2001;5:16–25. doi: 10.1016/s1364-6613(00)01568-0. [DOI] [PubMed] [Google Scholar]
- Espana RA, Berridge CW. Organization of noradrenergic effer- ~ ents to arousal-related basal forebrain structures. J Comp Neurol. 2006;496:668–683. doi: 10.1002/cne.20946. [DOI] [PubMed] [Google Scholar]
- Fallon JH, Loughlin SE. Monoamine innervation of the fore-brain: collateralization. Brain Res Bull. 1982;9:295–307. doi: 10.1016/0361-9230(82)90143-5. [DOI] [PubMed] [Google Scholar]
- Fan J, Byrne J, Worden MS, Guise KG, McCandliss BD, Fossella J, Posner MI. The relation of brain oscillations to attentional networks. J Neurosci. 2007;27:6197–6206. doi: 10.1523/JNEUROSCI.1833-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fries P. A mechanism for cognitive dynamics: neuronal communication through neuronal coherence. Trends Cogn Sci. 2005;9:474–480. doi: 10.1016/j.tics.2005.08.011. [DOI] [PubMed] [Google Scholar]
- Gioanni Y, Rougeot C, Clarke PB, Lepousé C, Thierry AM, Vidal C. Nicotinic receptors in the rat prefrontal cortex: increase in glutamate release and facilitation of mediodorsal thalamo-cortical transmission. Eur J Neurosci. 1999;11:18–30. doi: 10.1046/j.1460-9568.1999.00403.x. [DOI] [PubMed] [Google Scholar]
- Giorgetti M, Bacciottini L, Giovannini MG, Colivicchi MA, Goldfarb J, Blandina P. Local GABAergic modulation of acetylcholine release from the cortex of freely moving rats. Eur J Neurosci. 2000;12:1941–1948. doi: 10.1046/j.1460-9568.2000.00079.x. [DOI] [PubMed] [Google Scholar]
- Giuliano C, Parikh V, Ward JR, Chiamulera C, Sarter M. Increases in cholinergic neurotransmission measured by using choline-sensitive microelectrodes: enhanced detection by hydrolysis of acetylcholine on recording sites? Neurochem Int. 2008;52:1343–1350. doi: 10.1016/j.neuint.2008.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gulledge AT, Bucci DJ, Zhang SS, Matsui M, Yeh HH. M1 receptors mediate cholinergic modulation of excitability in neocortical pyramidal neurons. J Neurosci. 2009;29:9888–9902. doi: 10.1523/JNEUROSCI.1366-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hasselmo ME, Bower JM. Cholinergic suppression specific to intrinsic not afferent fiber synapses in rat piriform (olfactory) cortex. J Neurophysiol. 1992;67:1222–1229. doi: 10.1152/jn.1992.67.5.1222. [DOI] [PubMed] [Google Scholar]
- Hasselmo ME, Sarter M. Modes and models of forebrain cholinergic neuromodulation of cognition. Neuropsychopharmacology. 2011;36:52–73. doi: 10.1038/npp.2010.104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Himmelheber AM, Sarter M, Bruno JP. The effects of manipulations of attentional demand on cortical acetylcholine release. Brain Res Cogn Brain Res. 2001;12:353–370. doi: 10.1016/s0926-6410(01)00064-7. [DOI] [PubMed] [Google Scholar]
- Hornung JP. The human raphe nuclei and the serotonergic system. J Chem Neuroanat. 2003;26:331–343. doi: 10.1016/j.jchemneu.2003.10.002. [DOI] [PubMed] [Google Scholar]
- Howe WM, Ji J, Parikh V, Williams S, Mocaer E, Trocmé-Thibierge C, Sarter M. Enhancement of attentional performance by selective stimulation of alpha4beta2* nAChRs: underlying cholinergic mechanisms. Neuropsychopharmacology. 2010;35:1391–1401. doi: 10.1038/npp.2010.9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howe WM, Gritton H, Berke J, Sarter M. Soc f Neurosci Ann Meet. 2011. Attention-demanding cues evoke prefrontal gamma oscillations and are differentially modulated by prefrontal muscarinic and nicotinic receptors. (abstract # 197.09) [Google Scholar]
- Howe WM, Berry AS, Francois J, Gilmour G, Carp JM, Tricklebank M, Lustig C, Sarter M. Prefrontal cholinergic mechanisms instigating shifts from monitoring for cues to cue-guided performance: converging electrochemical and fMRI evidence from rats and humans. J Neurosci. 2013;33:8742–8752. doi: 10.1523/JNEUROSCI.5809-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howells FM, Stein DJ, Russell VA. Synergistic tonic and phasic activity of the locus coeruleus norepinephrine (LC-NE) arousal system is required for optimal attentional performance. Metab Brain Dis. 2012;27:267–274. doi: 10.1007/s11011-012-9287-9. [DOI] [PubMed] [Google Scholar]
- Klink R, Alonso A. Muscarinic modulation of the oscillatory and repetitive firing properties of entorhinal cortex layer II neurons. J Neurophysiol. 1997;77:1813–1828. doi: 10.1152/jn.1997.77.4.1813. [DOI] [PubMed] [Google Scholar]
- Kozak R, Bruno JP, Sarter M. Augmented prefrontal acetylcholine release during challenged attentional performance. Cereb Cortex. 2006;16:9–17. doi: 10.1093/cercor/bhi079. [DOI] [PubMed] [Google Scholar]
- Kruglikov I, Rudy B. Perisomatic GABA release and thalamocortical integration onto neocortical excitatory cells are regulated by neuromodulators. Neuron. 2008;58:911–924. doi: 10.1016/j.neuron.2008.04.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kunz PA, Roberts AC, Philpot BD. Presynaptic NMDA receptor mechanisms for enhancing spontaneous neurotransmitter release. J Neurosci. 2013;33:7762–7769. doi: 10.1523/JNEUROSCI.2482-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurzban R, Duckworth A, Kable JW, Myers J. An opportunity cost model of subjective effort and task performance. Behav Brain Sci. 2013;36:661–679. doi: 10.1017/S0140525X12003196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lambe EK, Picciotto MR, Aghajanian GK. Nicotine induces glutamate release from thalamocortical terminals in prefrontal cortex. Neuropsychopharmacology. 2003;28:216–225. doi: 10.1038/sj.npp.1300032. [DOI] [PubMed] [Google Scholar]
- Lee SH, Dan Y. Neuromodulation of brain states. Neuron. 2012;76:209–222. doi: 10.1016/j.neuron.2012.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luck SJ, Ford JM, Sarter M, Lustig C. CNTRICS final biomarker selection: control of attention. Schizophrenia Bull. 2012;38:53–61. doi: 10.1093/schbul/sbr065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacDermott AB, Role LW, Siegelbaum SA. Presynaptic ionotropic receptors and control of transmitter release. Nat Rev Neurosci. 1999;22:443–485. doi: 10.1146/annurev.neuro.22.1.443. [DOI] [PubMed] [Google Scholar]
- Marder E. Neuromodulation of neuronal circuits: back to the future. Neuron. 2012;76:1–11. doi: 10.1016/j.neuron.2012.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGaughy J, Turchi J, Sarter M. Crossmodal divided attention in rats: effects of chlordiazepoxide and scopolamine. Psychopharmacology. 1994;115:213–220. doi: 10.1007/BF02244774. [DOI] [PubMed] [Google Scholar]
- McGaughy J, Kaiser T, Sarter M. Behavioral vigilance following infusions of 192 IgG-saporin into the basal forebrain: selectivity of the behavioral impairment and relation to cortical ache-positive fiber density. Behav Neurosci. 1996;110:247–265. doi: 10.1037//0735-7044.110.2.247. [DOI] [PubMed] [Google Scholar]
- Moran RJ, Campo P, Symmonds M, Stephan KE, Dolan RJ, Friston KJ. Free energy, precision and learning: the role of cholinergic neuromodulation. J Neurosci. 2013;33:8227–8236. doi: 10.1523/JNEUROSCI.4255-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moroni F, Tanganelli S, Antonelli T, Carla V, Bianchi C, Beani L. Modulation of cortical acetylcholine and gamma-aminobutyric acid release in freely moving guinea pigs: effects of clonidine and other adrenergic drugs. J Pharmacol Exp Ther. 1983;227:435–440. [PubMed] [Google Scholar]
- Muir JL, Dunnett SB, Robbins TW, Everitt BJ. Attentional functions of the forebrain cholinergic systems: effects of intraventricular hemicholinium, physostigmine, basal forebrain lesions and intracortical grafts on a multiple-choice serial reaction time task. Exp Brain Res. 1992;89:611–622. doi: 10.1007/BF00229886. [DOI] [PubMed] [Google Scholar]
- Neigh G, Arnold MH, Rabenstein R, Sarter M, Bruno JP. Neuronal activity in the nucleus accumbens is necessary for performance-related increases in cortical acetylcholine release. Neuroscience. 2004;123:635–645. doi: 10.1016/j.neuroscience.2003.10.006. [DOI] [PubMed] [Google Scholar]
- Nuechterlein KH, Luck SJ, Lustig C, Sarter M. CNTRICS final task selection: control of attention. Schizophrenia Bull. 2009;35:182–196. doi: 10.1093/schbul/sbn158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paolone G, Howe MW, Gopalakrishnan M, Decker MW, Sarter M. Regulation and function of the tonic component of cortical acetylcholine release. In: Westerink B, Clinckers R, Smolders I, Sarre S, Michotte Y, editors. Monitoring Molecules in Neuroscience. Vrije Universiteit Brussels; Brussels, Belgium: 2010. pp. 363–365. [Google Scholar]
- Paolone G, Angelakos CC, Meyer P, Robinson TE, Sarter M. Cholinergic control over attention in rats prone to attribute incentive salience to reward cues. J Neurosci. 2013;33:8321–8335. doi: 10.1523/JNEUROSCI.0709-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parikh V, Sarter M. Cholinergic mediation of attention: contributions of phasic and tonic increases in prefrontal cholinergic activity. Ann NY Acad Sci. 2008;1129:225–235. doi: 10.1196/annals.1417.021. [DOI] [PubMed] [Google Scholar]
- Parikh V, Kozak R, Martinez V, Sarter M. Prefrontal acetylcholine release controls cue detection on multiple timescales. Neuron. 2007;56:141–154. doi: 10.1016/j.neuron.2007.08.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parikh V, Man K, Decker MW, Sarter M. Glutamatergic contributions to nicotinic acetylcholine receptor agonist-evoked cholinergic transients in the prefrontal cortex. J Neurosci. 2008;28:3769–3780. doi: 10.1523/JNEUROSCI.5251-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parikh V, Ji J, Decker MW, Sarter M. Prefrontal beta2 subunit-containing and alpha7 nicotinic acetylcholine receptors differentially control glutamatergic and cholinergic signaling. J Neurosci. 2010;30:3518–3530. doi: 10.1523/JNEUROSCI.5712-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Passetti F, Dalley JW, O’Connell MT, Everitt BJ, Robbins TW. Increased acetylcholine release in the rat medial prefrontal cortex during performance of a visual attentional task. Eur J Neurosci. 2000;12:3051–3058. doi: 10.1046/j.1460-9568.2000.00183.x. [DOI] [PubMed] [Google Scholar]
- Perry RJ, Hodges JR. Attention and executive deficits in Alzheimer’s disease. A critical review Brain. 1999;122:383–404. doi: 10.1093/brain/122.3.383. [DOI] [PubMed] [Google Scholar]
- Picciotto M, Higley M, Mineur Y. Acetylcholine as a neuromodulator: cholinergic signaling shapes nervous system function and behavior. Neuron. 2012;76:116–129. doi: 10.1016/j.neuron.2012.08.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Posner MI, Snyder CR, Davidson BJ. Attention and the detection of signals. J Exp Psychol. 1980;109:160–174. [PubMed] [Google Scholar]
- Ramírez MJ, Cenarruzabeitia E, Lasheras B, Del Río J. Involvement of GABA systems in acetylcholine release induced by 5-HT3 receptor blockade in slices from rat entorhinal cortex. Brain Res. 1996;712:274–280. doi: 10.1016/0006-8993(95)01471-3. [DOI] [PubMed] [Google Scholar]
- Robbins TW, Everitt BJ. Arousal systems and attention. In: Gazzaniga MS, editor. The Cognitive Neurosciences. MIT Press; Cambridge, MA: 1995. pp. 703–720. [Google Scholar]
- Rodriguez R, Kallenbach U, Singer W, Munk MH. Short- and long-term effects of cholinergic modulation on gamma oscillations and response synchronization in the visual cortex. J Neurosci. 2004;24:10369–10378. doi: 10.1523/JNEUROSCI.1839-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez R, Kallenbach U, Singer W, Munk MH. Stabilization of visual responses through cholinergic activation. Neuroscience. 2010;165:944–954. doi: 10.1016/j.neuroscience.2009.10.059. [DOI] [PubMed] [Google Scholar]
- Sara S, Bouret S. Orienting and reorienting: the locus coeruleus mediates cognition through arousal. Neuron. 2012;76:130–141. doi: 10.1016/j.neuron.2012.09.011. [DOI] [PubMed] [Google Scholar]
- Sarter M, Gehring WJ, Kozak R. More attention must be paid: the neurobiology of attentional effort. Brain Res Rev. 2006;51:145–160. doi: 10.1016/j.brainresrev.2005.11.002. [DOI] [PubMed] [Google Scholar]
- Sarter M, Parikh V, Howe WM. Phasic acetylcholine release and the volume transmission hypothesis: time to move on. Nat Rev Neurosci. 2009;10:383–390. doi: 10.1038/nm2635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarter M, Parikh V, Howe MW, Gritton H, Paolone G, Lee TM. Multiple time scales and variable spaces: synaptic neurotransmission in vivo. In: Westerink B, Clinckers R, Smolders I, Sarre S, Michotte Y, editors. Monitoring Molecules in Neuroscience. Vrije Universiteit Brussels; Brussels, Belgium: 2010. pp. 7–9. [Google Scholar]
- Sarter M, Lustig C, Taylor SF. Cholinergic contributions to the cognitive symptoms of schizophrenia and the viability of cholinergic treatments. Neuropharmacology. 2012;62:1544–1553. doi: 10.1016/j.neuropharm.2010.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schicker KW, Dorostkar MM, Boehm S. Modulation of transmitter release via presynaptic ligand-gated ion channels. Curr Mol Pharmacol. 2008;1:106–129. doi: 10.2174/1874467210801020106. [DOI] [PubMed] [Google Scholar]
- Schliebs R, Arendt T. The cholinergic system in aging and neuronal degeneration. Behav Brain Res. 2011;221:555–563. doi: 10.1016/j.bbr.2010.11.058. [DOI] [PubMed] [Google Scholar]
- Schneider TR, Lorenz S, Senkowski D, Engel AK. Gamma-Band activity as a signature for cross-modal priming of auditory object recognition by active haptic exploration. J Neurosci. 2011;31:2502–2510. doi: 10.1523/JNEUROSCI.6447-09.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Senkowski D, Schneider TR, Foxe JJ, Engel AK. Crossmodal binding through neural coherence: implications for multisensory processing. Trends Neurosci. 2008;31:401–409. doi: 10.1016/j.tins.2008.05.002. [DOI] [PubMed] [Google Scholar]
- St Peters M, Demeter E, Lustig C, Bruno JP, Sarter M. Enhanced control of attention by stimulating mesolimbic-corticopetal cholinergic circuitry. J Neurosci. 2011;31:9760–9771. doi: 10.1523/JNEUROSCI.1902-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun YG, Pita-Almenar JD, Wu CS, Renger JJ, Uebele VN, Lu HC, Beierlein M. Biphasic cholinergic synaptic transmission controls action potential activity in thalamic reticular nucleus neurons. J Neurosci. 2013;33:2048–2059. doi: 10.1523/JNEUROSCI.3177-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turchi J, Sarter M. Cortical acetylcholine and processing capacity: effects of cortical cholinergic deafferentation on crossmodal divided attention in rats. Brain Res Cogn Brain Res. 1997;6:147–158. doi: 10.1016/s0926-6410(97)00027-x. [DOI] [PubMed] [Google Scholar]
- Unal CT, Golowasch JP, Zaborszky L. Adult mouse basal fore-brain harbors two distinct cholinergic populations defined by their electro-physiology. Front Behav Neurosci. 2012;6:21. doi: 10.3389/fnbeh.2012.00021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varela C. The gating of neocortical information by modulators. J Neurophysiol. 2013;109:1229–1232. doi: 10.1152/jn.00701.2012. [DOI] [PubMed] [Google Scholar]
- Waselus M, Valentino RJ, Van Bockstaele EJ. Collateralized dorsal raphe nucleus projections: a mechanism for the integration of diverse functions during stress. J Chem Neuroanat. 2011;41:266–280. doi: 10.1016/j.jchemneu.2011.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wenk GL. The nucleus basalis magnocellularis cholinergic system: one hundred years of progress. Neurobiol Learn Mem. 1997;67:85–95. doi: 10.1006/nlme.1996.3757. [DOI] [PubMed] [Google Scholar]
- Xiang Z, Huguenard JR, Prince DA. Cholinergic switching within neocortical inhibitory networks. Science. 1998;281:985–988. doi: 10.1126/science.281.5379.985. [DOI] [PubMed] [Google Scholar]
- Yu AJ, Dayan P. Uncertainty, neuromodulation and attention. Neuron. 2005;46:681–691. doi: 10.1016/j.neuron.2005.04.026. [DOI] [PubMed] [Google Scholar]
- Zaborszky L. The modular organization of brain systems. Basal fore-brain: the last frontier. Prog Brain Res. 2002;136:359–372. doi: 10.1016/s0079-6123(02)36030-8. [DOI] [PubMed] [Google Scholar]
- Zaborszky L, Buhl DL, Pobalashingham S, Bjaalie JG, Nadasdy Z. Three-dimensional chemoarchitecture of the basal forebrain: spatially specific association of cholinergic and calcium binding protein-containing neurons. Neuroscience. 2005;136:697–713. doi: 10.1016/j.neuroscience.2005.05.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaborszky L, Hoemke L, Mohlberg H, Schleicher A, Amunts K, Zilles K. Stereotaxic probabilistic maps of the magnocellular cell groups in human basal forebrain. NeuroImage. 2008;42:1127–1141. doi: 10.1016/j.neuroimage.2008.05.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaborszky L, van den Pol A, Gyengesi E. The basal forebrain cholinergic projection system in mice. In: Watson C, Paxinos G, Puelles L, editors. The Mouse Nervous System. Academic Press; San Diego, CA: 2012. pp. 684–718. [Google Scholar]
- Zaborszky L, Csordas C, Mosca K, Kim J, Gielow MR, Vasdasz C, Nadasdy Z. Neurons in the basal forebrain project to the cortex in a complex topographic organization that reflects cortico-cortical connectivity patterns: an experimental study based on retrograde tracing and 3D reconstruction. Cereb Cortex. 2013 doi: 10.1093/cercor/bht210. Epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zmarowksi A, Sarter M, Bruno JP. NMDA and dopamine interactions in the nucleus accumbens modulate cortical acetylcholine release. Eur J Neurosci. 2005;22:1731–1740. doi: 10.1111/j.1460-9568.2005.04333.x. [DOI] [PubMed] [Google Scholar]