

Abstract
Aim
Conducting PK studies in pregnant women is challenging. Therefore, we asked if a physiologically-based pharmacokinetic (PBPK) model could be used to predict the disposition in pregnant women of drugs cleared by multiple CYP enzymes.
Methods
We expanded and verified our previously published pregnancy PBPK model by incorporating hepatic CYP2B6 induction (based on in vitro data), CYP2C9 induction (based on phenytoin PK) and CYP2C19 suppression (based on proguanil PK), into the model. This model accounted for gestational age-dependent changes in maternal physiology and hepatic CYP3A, CYP1A2 and CYP2D6 activity. For verification, the pregnancy-related changes in the disposition of methadone (cleared by CYP2B6, 3A and 2C19) and glyburide (cleared by CYP3A, 2C9 and 2C19) were predicted.
Results
Predicted mean post-partum to second trimester (PP : T2) ratios of methadone AUC, Cmax and Cmin were 1.9, 1.7 and 2.0, vs. observed values 2.0, 2.0 and 2.6, respectively. Predicted mean post-partum to third trimester (PP : T3) ratios of methadone AUC, Cmax and Cmin were 2.1, 2.0 and 2.4, vs. observed values 1.7, 1.7 and 1.8, respectively. Predicted PP : T3 ratios of glyburide AUC, Cmax and Cmin were 2.6, 2.2 and 7.0 vs. observed values 2.1, 2.2 and 3.2, respectively.
Conclusions
Our PBPK model integrating prior physiological knowledge, in vitro and in vivo data, allowed successful prediction of methadone and glyburide disposition during pregnancy. We propose this expanded PBPK model can be used to evaluate different dosing scenarios, during pregnancy, of drugs cleared by single or multiple CYP enzymes.
Keywords: CYP2B6, CYP2C19, CYP2C9, PBPK, pharmacokinetics, pregnancy
What is Already Known about this Subject
During pregnancy, changes in physiology and absorption, distribution, metabolism and excretion (ADME) processes can significantly affect the disposition of drugs. Such changes may require adjustments in the dosing regimen of the drug.
Since conducting pharmacokinetic (PK) studies in pregnant women is challenging, alternative approaches that can predict adjustment of drug dosing regimens (if any) are highly desirable.
What this Study Adds
We expanded and verified a physiologically based PK model, which incorporated gestational age-dependent changes in maternal physiology and major hepatic CYP enzyme activity, to predict disposition during pregnancy of drugs cleared by multiple CYP enzymes (e.g. glyburide and methadone).
Introduction
Considerable literature data demonstrate that the pharmacokinetics (PK) of drugs can be significantly affected by pregnancy. Such changes may lead to either underdosing or overdosing of medication with the standard adult dose, with varying consequences for safety and efficacy 1. For example, in pregnant opioid-dependent women, the oral clearance (CLoral) of methadone (METH) is induced by ∼105% during the second trimester (T2) vs. post-partum (PP) and ∼65% during third trimester (T3) vs. PP 2. Given the established link between plasma methadone concentration and the pharmacological effects 3, these changes during pregnancy may require careful evaluation of patient complaints of a dose ‘not holding’. Similarly, in gestational diabetic subjects, the steady-state AUC(0,12 h) and Cmax of glyburide (GLB), a drug with an established plasma concentration–effect relationship at therapeutic doses 4, are 53% and 55% lower during T3 vs. non-pregnant type 2 diabetic subjects 5. These results suggest that patients with inadequate glucose control might benefit from increased GLB dosage. Obviously, for drugs operating at the plateau phase of the plasma concentration–effect curve, the pregnancy-induced change in their PK would be of less concern.
Altered drug disposition can be attributed to pregnancy-induced changes in drug absorption (e.g. gastric pH, transporters), distribution (e.g. plasma protein binding and transporters), metabolism (e.g. cytochrome P450 (CYP) metabolism) and excretion (e.g. renal secretion via transporters) (ADME). Further, data in the literature suggest that the magnitude of change in maternal hepatic enzyme activity is CYP isoform specific and gestational age dependent 6. The underlying mechanism for the hepatic isoform-specific and gestational state specific induction during pregnancy is not fully understood. It has been postulated that the rising concentrations of various hormones in maternal blood, including placental growth hormone, progesterone, corticosteroids and estrogens, are responsible for induction of hepatic enzyme activity 7.
Since it is logistically impossible to delineate the changes in PK of all drugs administered to pregnant women, alternative approaches such as mechanistic modelling, to predict drug disposition and adjustment of drug dosing regimens (if any) are highly desirable. These approaches, in conjunction with opportunistic PK studies in pregnant women of drugs administered for therapeutic purposes, should improve our ability to adjust drug dosing regimens for pregnant women. To this end, we have focused on a systems pharmacology approach, i.e. physiologically-based pharmacokinetic (PBPK) modelling. PBPK modelling has the advantage of incorporating both physiological parameters that are important for ADME processes and drug-specific parameters (e.g. physico-chemical and drug disposition characteristics) into a quantitative predictive model 8,9. A maternal, whole-body PBPK model, incorporating prior physiological parameters as well as maternal hepatic CYP activity in each trimester was recently developed 10,11. We refined this PBPK model and showed that the PBPK model populated with CYP3A, 1A2 and 2D6 activity change, based on CLoral of midazolam, caffeine and metoprolol, respectively, could accurately predict the disposition during pregnancy of other drugs cleared primarily by the same CYP enzyme, namely CYP3A-metabolized drugs nifedipine and indinavir (CYP3A) 12, CYP1A2-metabolized drug theophylline and CYP2D6-metabolized drugs paroxetine, dextromethorphan and clonidine 13. In the current study, we expanded and verified the established PBPK model, by incorporating CYP2B6 induction (based on in vitro data), CYP2C9 induction (based on phenytoin PK), and CYP2C19 suppression (based on proguanil PK). For model verification, the PBPK model was used to predict the disposition during pregnancy of drugs cleared via these multiple CYP pathways including methadone (METH) and glyburide (GLB).
Methods
General workflow of PBPK model development and verification criterion
A general workflow (Figure 1) of PBPK modelling and simulation of test compounds in non-pregnant subjects consisted of the following steps: (i) comparison of mean plasma concentration–time (C–T) profiles simulated using a 13-compartment full PBPK model (Simcyp® Population-based Simulator Version 12.1, Simcyp Limited, Sheffield, UK) with those obtained from in vivo studies including i.v. dosing, single and multiple oral dosing (to qualify the drug-specific parameters); (ii) refinement (hence referred to as modified model) of the drug-specific parameters (e.g. fm) if the prediction in (i) above deviates significantly (<0.8-fold or >1.25-fold) from that observed. Such refinements were often based on changes in mean AUC and mean concentration–time profiles in the presence of inhibitors or genetic polymorphism of the enzymes clearing the drug; (iii) populating the time-varying full PBPK model constructed in Matlab v. 7.10® (2010, Mathworks®, Natick, MA) with these qualified drug-specific parameters and pregnancy-induced CYP activity changes (see below). Once the drug model was qualified in the non-pregnant population, the drug-specific parameters were fixed for simulating PK of drugs during pregnancy and post-partum. The post-partum systemic exposure to drugs was considered to represent drug exposure in the non-pregnant female population.
Figure 1.
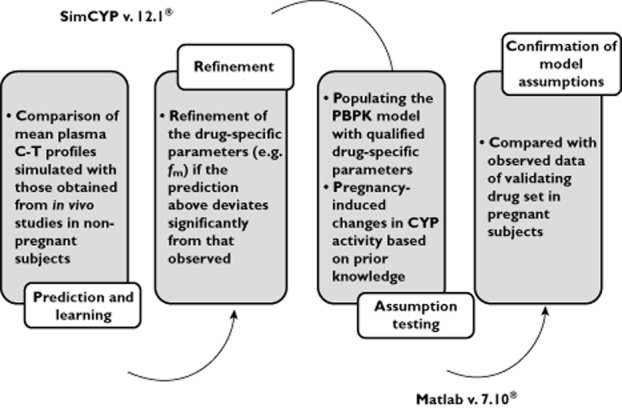
A diagram depicting the general workflow of PBPK modeling and simulation. Our modelling approach entails a two step model validation procedure, first to demonstrate adequacy of the model for the non-pregnant population and second for the pregnant population. Simcyp was used to for the first step, i.e. to qualify the drug-specific parameters in the non-pregnant population. For the second step of model validation, these fixed drug-specific parameters were fed into the MATLAB model to predict C–T profiles in the pregnant population. The MATLAB model was populated with known pregnancy-dependent system changes, including presumed pregnancy effect on CYP enzyme activity
Verification of the established PBPK model was primarily based on AUC because achieving equivalent drug exposure in pregnant and non-pregnant women was our primary focus. The term ‘verification’ is used in place of ‘validation’ to acknowledge the complexity of the PBPK model that requires more than plasma data to accomplish proper validation. In addition, prediction of Cmax and Cmin was considered because achieving similar drug Cmax and Cmin may be important for some drugs where these measures are related to drug efficacy and/or toxicity. For model verification, mean AUC, Cmax and Cmin of METH and GLB during pregnancy were predicted and compared with published studies in pregnant subjects. We chose the criterion of PK bioequivalence as the criterion for successful verification of the model, namely, the predicted mean population PK parameters of the drug (as described above), should fall within 80% to 125% of the observed value, i.e. 0.80 ≤ predicted/observed ≤1.25.
General pregnancy PBPK model structure and key assumptions
The general pregnancy PBPK model structure and key assumptions were described in detail previously 10,12. Briefly, the placental-fetal unit was represented as a single lumped compartment because the focus of the current study was to predict maternal PK of drugs during pregnancy. The gestational age dependent changes in physiological parameters (e.g. cardiac output, glomerular filtrate rate, etc) were incorporated into an existing PBPK model (Jamei et al. 9). Maternal glomerular filtration rate (GFR) was assumed to increase during pregnancy by 19%, 40% and 37% during T1, T2 and T3, respectively 11. The change in drug unbound fraction in plasma (fu,p) during pregnancy, as a function of serum albumin or α1-AGP concentrations, was accounted for in the model as described previously 12. The established PBPK model also assumed that hepatic CYP3A activity increased by 2.0-fold (measured by midazolam CLoral) during T3 12. The magnitude of CYP3A during T2 was assumed to be the same as that of T3, as suggested by similar T2 vs. T3 effect on indinavir CLoral 14. The PBPK model was further expanded to incorporate pregnancy-induced induction of CYP2B6 and CYP2C9, as well as suppression of CYP2C19 as described below. Maternal hepatic CYP2B6 was assumed to increase during pregnancy by 1.1-fold, 1.4-fold and 1.9-fold (based on in vitro to in vivo extrapolation, see results) during T1, T2 and T3, respectively. Maternal CYP2C9 activity was assumed to increase by 1.4-fold, 1.5-fold and 1.6-fold (reported by phenytoin PK) during T1, T2 and T3 15,16 (see results). Maternal CYP2C19 activity, as reported by CLoral of the anti-malarial agent proguanil 17,18, was assumed to decrease by 62% and 68% during T2 and T3, respectively (see below). These changes in CYP activity were considered static for each trimester. Hepatic organic anion transporter OATP1B1 or 2B1 activity was assumed to remain constant throughout pregnancy. These changes were accomplished in Matlab v7.10®.
The mass balance differential equations (except for the permeability-limited liver model) used in the model have been described previously 10,12. Briefly, all tissues except maternal liver were considered to be well-stirred compartments, i.e. that the unbound tissue concentration is at equilibrium with the unbound concentration in the emergent blood. The maternal liver was modelled as a permeability-limited tissue, incorporating scaled active uptake (PSint,OATP) and scaled passive diffusion clearances (CLpd) of unbound drug at the sinusoidal membrane, and scaled metabolic clearance (CLint,u,h) of unbound drug (Equations 1 and 2). The liver compartment was subdivided into extracellular (EC) and intracellular compartments (IC). The EC combined tissue blood (11.5% of liver volume) and interstitial fluid (16.3% of liver volume). Biliary excretion of unchanged drug is negligible for the tested drugs and was therefore not considered in the current model.
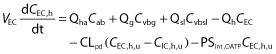 |
1 |
| 2 |
For drugs that are not actively transported into the hepatocytes (such as METH and PHT), equation 3 was used. Equation 4 was used to describe the apparent intrinsic clearance. Equation 5 was used to describe the dynamic change of the enzyme amount as a result of auto-induction by drugs such as PHT.
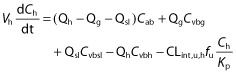 |
3 |
| 4 |
| 5 |
where Cab, Ch, Cvbsl, Cvbg and Cvbh represent the concentrations in the arterial blood, liver, spleen-blood, gut-blood and hepatic outlet (or liver-blood), respectively; Qh, Qg and Qsl represent the blood flows of the liver, gut and spleen respectively; Vh, VEC and VIC represent the volume of the liver, extracellular compartment and intracellular compartment of the liver, respectively; fu and fu,h represents the fractions unbound in plasma and liver (fu,h = fu/Kp for drugs with no active uptake or efflux). CLint,u,h, Vmax and Km represent the unbound hepatic intrinsic clearance, maximum velocity and the Michaelis-Menten constant for metabolism, respectively, and ACYP and ACYP,0 represent the total hepatic amount of CYP isoform at a given time and at baseline. Kdeg represents the apparent first order degradation rate constant of the CYP enzyme, EC50,u and Emax represent the unbound concentration at which 50% of maximum induction is reached, and the maximum induction potential, respectively. Pregnancy-induced change, either induction or suppression of CYP enzyme activity was reflected in unbound hepatic intrinsic clearance CLint,u,h for drugs with linear kinetics or ACYP,0 for drugs associated with saturable hepatic metabolism.
METH PBPK model construction
Methadone (METH), a drug used to treat opiate addiction, is mainly eliminated via hepatic metabolism by CYP2B6, CYP3A and CYP2C19, with a significant contribution from renal elimination (16–27%) 19,20. A number of studies have shown that methadone distribution and disposition are influenced by its chirality. R-METH has lower plasma protein binding level (binding to α1-AGP), a larger volume of distribution, and higher total body clearance 21,22. Therefore, PBPK models for R-METH and S-METH were constructed, respectively, to account for the differential disposition of each enantiomer. Physiochemical and protein binding parameters (log Po : w, pKa, B : P ratio, fu,p), absorption (Fa, ka, tlag), distribution (Vss), elimination (CLIV) and excretion (CLr) were obtained from the literature (see Table 1). Initial scaling of in vitro metabolic data 19,23 did not fully recover the observed R- and S-METH metabolic clearance, CLH. Therefore hepatic unbound intrinsic clearance CLint,u was back-calculated from observed CLH (= CLIV − CLr) using the well-stirred liver model. Because the in vitro Km for CYP2B6, CYP3A and CYP2C19 23 is significantly greater than the therapeutic METH concentration of 1–2 μm, saturation of hepatic CYP enzymes in the clinically used dose range is unlikely. This speculation is also supported by comparable mean CLoral following chronic dosing vs. single dosing of the racemate 20,21,24–26. For these reasons saturable metabolism was not incorporated into the model. The contribution from individual CYP to total metabolic clearance obtained in vitro is 43.4%, 45.3% and 9.1% (R-METH) and 69.2%, 37.1% and 10.4% (S-METH), for CYP2B6, 3A and 2C19, respectively 19. In vivo fm for individual CYP was calculated by taking the product of fm (= 1 − fe) and the fractional contribution of individual CYP. Urine pH has a significant effect on renal clearance. When urine pH is below 6, the contribution of renal clearance becomes significant, accounting for around 16–27% of total body clearance 20,27. The average literature values for CLr and fe of R-METH and S-METH were used in the final model 20,21,28. The constructed drug model was verified by comparing predicted mean AUC, Cmax and Cmin of R-METH and S-METH following single and multiple oral dosing of the racemate in non-pregnant subjects to reported literature values. The drug-dependent parameters of METH are listed in Table 1.
Table 1.
Summary of methadone (MET) drug-dependent parameters
| Parameter | Value | Methods/reference |
|---|---|---|
| Molecular weight | 309.4 | a |
| Log Po:w | 3.93 | b |
| pKa | 9.2 | a |
| B : P ratio | 0.75 | c |
| fu,p | R = 0.16; S = 0.12 | d |
| Fa | 0.88 | e |
| ka (h−1) | 0.59 | f |
| Fg | R = 0.98; S = 0.99 | Predicted by Qgut modelg |
| tlag (h) | 0.295 | f |
| Vss (l kg−1) | R = 6.2; S = 4.7 | Predictedh |
| CLi.v. (l h−1) | R = 8.6; S = 6.8 | i |
| CLr (l h−1) | R = 1.8; S = 1.1 | j |
| CLint,u (μl min−1/pmol) | R: CLint,u,2B6 = 0.306 CLint,u,3A = 0.039 | k |
| CLint,u,2C19 = 0.078 | ||
| S: CLint,u,2B6 = 0.427 CLint,u,3A = 0.028 | ||
| CLint,u,2C19 = 0.078 | ||
| fm and fe | R: fm,2B6 = 35.1% fm,3A = 36.6%fm,2C19 = 7.3% fe = 21% S: fm,2D6 = 49.8% fm,3A = 26.7% fm,2C19 = 7.5% fe = 16% | l,j |
aReported 62. bhttp://www.chemspider.com/Chemical-Structure.3953.html. cReported value 27. dAverage value for R-methadone (R) and S-methadone (S) in the literature 62–65. eBack-calculated from mean reported Fa × Fg (= 0.78 − 0.79) 26 and predicted Fg (= 0.98 − 0.99). Reported mean F is 0.82 (range 0.41–0.99) 20,26,64. fMean reported values 21,65. gQgut model is provided in Simcyp simulator. It retains the form of the ‘well-stirred’ liver model, but the flow term (Qgut) is a hybrid of both permeability through the enterocyte membrane and villous blood flow 66. hPredicted according to Rodgers & Rowland 67. Reported Vss following i.v. dosing is 5.9 ± 1.4 (R-METH) and 3.4 ± 0.9 (R-METH) 20 or 4.74 ± 1.94 l kg−1 (racemic METH) 26. iReported 20. jUrine pH has a significant effect on renal clearance. When urine pH is below 6, the contribution of renal clearance becomes significant, accounting for around 16–30% of total body clearance 27. Average of the literature values was used 20,21,28. kIn vitro HLM or recombinant CYP data 19,23 did not fully recover observed metabolic clearance. Therefore, CLint,h,u for CYP2B6, 3A and 2C19 were back calculated from hepatic CL (= CL − CLr), fm for individual CYP (see below), and ‘average’ population values for liver weight and hepatic CYP enzyme abundance of 17, 137 and 14 pmol mg−1 protein for CYP2B6, 3A and 2C19, respectively. lThe contribution from individual CYP to total metabolic clearance obtained in vitro was 43.4%, 45.3% and 9.1% (R-METH) and 69.2%, 37.1% and 10.4% (S-METH), for CYP2B6, 3A and 2C19, respectively 19. In vivo fm for individual CYP was calculated by taking the product of fm (= 1 − fe) and the percent contribution from individual CYP.
In vitro to in vivo extrapolation (IVIVE) of CYP2B6 induction during pregnancy
Hepatic CYP2B6 mRNA expression and enzyme activity was shown to be induced by estradiol in a concentration-dependent manner in human hepatocytes, with EC50,u of 1.9 ± 0.5 μm and Emax (fold-increase) of 34 ± 22 (based on CYP2B6 mRNA expression), respectively 29. The corresponding values based on CYP2B6 activity data are 4.1 ± 4.1 μm and 20 ± 21, respectively 29. Because estradiol depletion in human hepatocytes is significant with a reported half-life of 0.57 ± 0.12 h 30, the reported EC50,u was corrected for estradiol depletion by calculating the average concentration (AUC(0,τ)/τ) during an incubation period of 24 h. In the same experiment, rifampicin and carbamazepine were included as positive controls. Since the in vivo Emax of CYP2B6 induction by rifampicin or carbamazepine is not known, the in vivo Emax for estradiol-mediated CYP2B6 induction was not calibrated. Based on meta-analysis of literature data, the average estradiol concentration in plasma is 9.3, 33.3 and 59.6 nm during T1, T2 and T3 11. The reported mean unbound fraction in plasma of estradiol is 2% (range 1–3%) in the non-pregnant population 31. Estradiol mainly binds to albumin 31 and its unbound fraction in plasma is predicted to increase from the average pre-pregnancy level of 2% to 2.1%, 2.3% and 2.9% during T1, T2 and T3. These correspond to unbound estradiol concentrations of 0.20, 0.76 and 1.72 nm, respectively. The magnitude of CYP2B6 induction based on CYP2B6 mRNA data was predicted using equation 6.
| 6 |
GLB PBPK model construction
GLB has dose-proportional kinetics following single and multiple oral dosing in the clinical dose range 32. GLB is almost exclusively cleared via hepatic metabolism (fe < 0.1%) 33. Data in the literature regarding the contribution from individual CYP, particularly CYP2C9, to the metabolic clearance of GLB are inconsistent. In one study, selective inhibition studies in human liver microsomes showed that CYP3A plays a major role in GLB metabolism (∼53%), followed by CYP2C9 (∼28%) and CYP2C19 (<20%) 34. In another study, using a similar experimental approach, the individual CYP contribution to GLB metabolism was as follows: CYP3A (∼50%)> CYP2C8 (∼33%)> CYP2C19 (∼17%) 35. In vivo studies, on the other hand, suggested a significant role for CYP2C9 in the clearance of GLB. Single and multiple coadministration of fluvastatin (40 mg), a CYP2C9 inhibitor, increased the plasma AUC of GLB by 23% 36. CYP2C9 is a highly polymorphic enzyme. Kirchheiner et al. showed that the total oral clearance of GLB in the CYP2C9 *3/*3 subjects (n = 3) was approximately 40% of that in the CYP2C9*1/*1 subjects (n = 4) 4. Niemi et al. reported that the oral clearance of GLB in individuals heterozygous for the CYP2C9*3 allele (n = 2) was 35.7% of the respective value in the CYP2C9*1/*1 subjects 37. These clinical studies suggest the contribution of CYP2C9 to GLB metabolism in vivo (fm,2C9) falls in the range of 18.6% to 63%. Considering all the in vitro and in vivo evidence, in the final GLB model, the assigned contribution from individual CYP to total GLB metabolic clearance was 50%, 30% and 20% for CYP3A, 2C9 and 2C19, respectively. GLB has also been identified in vitro as a substrate of OTAP2B1 38 and an inhibitor of OATP1B1 39. It is likely that GLB is also a substrate of OATP1B1, although this has not been shown in literature to date. In vivo, 600 mg i.v. rifampicin over 30 min immediately followed by 1.25 mg oral GLB increased GLB plasma AUC by 118% and Cmax by 81% 33. Because rifampicin is a potent pan OATP inhibitor, this significant drug–drug interaction could be attributed to the inhibition of OATP2B1 and/or OATP1B1-mediated hepatic uptake clearance of GLB. These results also indicate the fraction of transport-mediated clearance towards total hepatic uptake clearance is at least 54.5%, assuming complete inhibition of OATPs by rifampicin.
The PBPK modelling strategy for GLB was as follows: First, the metabolic clearance of GLB (= 3.6 l h−1) was predicted via IVIVE from two independent in vitro studies 34,40 and similar clearance values were obtained. Definitive in vitro transport studies of GLB are not available, therefore the following parameters were estimated simultaneously using mean glyburide plasma PK data reported in subjects taking a single oral dose of 1.75–5 mg glyburide: hepatic bidirectional permeability clearance (CLpd), and hepatic intrinsic uptake clearance by OATP (or the permeability surface area product, PSint,OATP). As discussed above, both OATP2B1 and 1B1 could be important in mediating the uptake of the drug into hepatocytes and therefore a lumped clearance PSint,OATP, was used. Two automated sensitivity analyses were conducted to identify the initial estimates for fitting PSint,OATP and CLpd (Figure S1). There are many combinations of PSint,OATP and CLpd values that can result in an AUC within the range of 0.46–0.73 ng ml−1 h following 1.75 mg orally, which is 80–125% of the mean AUC observed in vivo (0.58 ng ml−1 h). Consequently the combination with PSint,OATP > CLpd (PSint,OATP 50 μl min−1/106 cells; CLpd 30 μl min−1/106 cells) were selected and used as initial estimates for data fitting, as the fraction of transport-mediated clearance towards total hepatic uptake clearance is at least 54.5% as discussed earlier. Other drug-specific parameters, including physiochemical and protein binding parameters (log Po:w, pKa, fu,p), absorption (Fa, ka, tlag), distribution (Vss) and excretion (CLr) were obtained from literature. The drug-dependent parameters of GLB are listed in Table 2. To predict the drug–drug interaction between GLB and rifampicin, a rifampicin drug model provided in Simcyp simulator was updated with an in vitro Ki of rifampicin against hepatic OATP2B1 or 1B1. The in vitro Ki sourced from the online database UCSF-FDATransPortal (http://bts.ucsf.edu/fdatransportal) ranged from 0.41–17 μm. Varma et al. showed using PBPK modelling, the in vivo Ki of rifampin to be 0.41–0.6 μM, essentially the most potent in vitro Ki values reported. These Ki values best recovered the DDI between RIF and pravastatin, a well-established substrate drug of hepatic OATP1B1 41. Hence we used in vivo Ki of 0.41 μm in our prediction.
Table 2.
Summary of glyburide (GLB) drug-dependent parameters
| Parameter | Value | Methods/reference |
|---|---|---|
| Molecular weight | 494.0 | a |
| Log Po:w | 4.79 | b |
| pKa | 5.3 | c |
| B : P ratio | 0.55 | Assumed d |
| fu,p | 0.015 | e |
| Fa | 0.84 | a |
| ka (h−1) | 0.756 | f |
| tlag (h) | 0.39–0.46 | f |
| Fg | 0.91 | Predicted by Qgut model |
| Vss (l kg)−1 | 0.1 | Predictedg |
| CLIV (l h−1) | 4.4 | h |
| CLr (l h−1) | 0.001 | i |
| CLint,CYP (μl min−1/pmol) | CLint,CYP3A4 = 0.238 | j |
| CLint,2C9 = 0.268 | ||
| CLint,CYP2C19 = 0.931 | ||
| CLpd (μl min−1/106 cells) | 24 | Parameter estimationk |
| PSint,OATP2B1 (μl min−1/106 cells) | 43.5 | Parameter estimationk |
| fm | fm,3A = 50% | l |
| fm,2C9 = 30% | ||
| fm,2C19 = 20% |
aReported 32. bhttp://www.drugbank.ca/drugs/DB01016. cLiterature value 68. dThere is no evidence in the literature that glyburide partitions into erythrocytes. Therefore B : P was calculated from the equation: E : P = [B : P − (1 − Hct)]/Hct, where E : P refers to erythrocyte partition coefficient and Hct refers to haematocrit value (mean value of 0.45 used). eAverage of reported value in the literature 5,69. fOptimized in the range of 0.39–0.46 h. The reported value is 0.46 h 70. gPredicted Vss according to Rodgers & Rowland is 0.61 l kg−1 67. This value was further optimized by applying a global Kp scalar of 0.1, in order to match the reported Vss of 0.077 ± 0.013 l kg−1 following i.v. infusion 59. hReported value is 4.4 ± 0.56 l h−1 (n = 8) 59. iReported value 33. jBack calculation from hepatic clearance, fm for individual CYP (see below), and ‘average’ population values for liver weight and hepatic CYP enzyme abundance of 137, 73 and 14 pmol mg−1 protein for CYP3A, 2C9 and 2C19, respectively. Hepatic intrinsic metabolic clearance (= 252.9 l h−1) was predicted using in vitro to in vivo extrapolation. Briefly, in vitro CLint,u determined in HLM 40 was scaled by ‘average’ population values for liver weight and microsomal protein of 1618 g and 38.9 (mg g−1 liver), respectively. Alternatively, in vitro Vmax and Km determined in recombinant CYP enzyme system 34 were scaled by ‘average’ population values for liver weight and respective hepatic CYP enzyme abundance as described above. Both approaches yielded similar values and the mean hepatic intrinsic clearance was used. kHepatic bidirectional permeability clearance (CLpd), and hepatic intrinsic uptake clearance by OATP (or the permeability surface area product, PSint,OATP), were estimated simultaneously using mean glyburide plasma PK data reported in subjects taking a single oral dose of 1.75–5 mg glyburide. lThe contribution from individual CYP obtained using HLM with selective chemical inhibitors is 53%, 28% and 19% for 3A, 2C9 and 2C19, respectively 34. In another study, the contribution from CYP3A was approximately 50%, whereas 2C8 and 2C19 combined contribute 50% 35. However, in vivo drug–drug interaction studies using fluvastatin as the perpetrator (a 2C9 inhibitor) reported AUC % change of 23% 36. In addition, a 2.7-fold higher glyburide AUC in subjects with CYP2C9*1/*3 and CYP2C9*2/*3 vs. CYP2C9*1/*1 was observed 37. This evidence suggests that CYP2C9 plays a significant role in glyburide metabolism in vivo. Therefore, the in vitro contribution from individual CYPs was adjusted accordingly and 50%, 30% and 20% for 3A, 2C9 and 2C19, respectively, was assigned in the final model.
Back-calculation of the magnitude of CYP2C19 suppression during pregnancy based on proguanil PK
CYP2C19 mediates the biotransformation of the anti-malarial agent, proguanil, to its active metabolite cycloguanil. Proguanil is also excreted as unchanged drug in urine (fe = 32%) 42. Proguanil is extensively absorbed from the gastrointestinal tract and exhibits linear PK over the dose range 100 mg to 400 mg 43. The drug is 75% bound to plasma proteins and highly partitions into the red blood cells (blood : plasma concentration ratio or B : P = ∼5) 44. The CLoral of proguanil decreases by 60% during T2 and T3 in patients with uncomplicated P. falciparum malaria, as compared with non-pregnant healthy subjects 17. A possible disease effect on CLoral of proguanil cannot be completely ruled out. In another study, where the plasma metabolic ratio of proguanil : cycloguanil was used as a CYP2C19 marker, this metabolic ratio was 62% higher during T3 vs. post-partum in subjects phenotyped as CYP2C19 extensive metabolizers 18. The observed decrease in proguanil CLoral during pregnancy, was then used to back-calculate the magnitude of decrease in 2C19 activity after accounting for other pregnancy related components of proguanil CLoral (CLr, B : P ratio and plasma protein binding). Specifically, Fa × FG was assumed to be equal to unity and remain unchanged during pregnancy. The CLr of proguanil greatly exceeds (>11-fold) the renal filtration clearance (= fu,p × GFR), suggesting proguanil is actively transported in the kidney. Because the renal transport mechanism of proguanil is unknown and because CLr has not been assessed in pregnant women, we assumed that only the filtration clearance was increased during pregnancy. The B : P ratio was predicted to decrease from a baseline value of 5.0 to 4.4 during T3 following a decrease in haematocrit value. Lastly, fu,p was predicted to increase from baseline value of 0.25 to 0.33 during T3 as a function of plasma albumin concentration. Using the well-stirred liver model shown below, the back-calculated CLint,h,u, i.e. CYP2C19 activity, is decreased by 62% in T2 and 68% in T3.
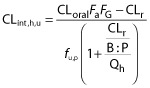 |
7 |
Results
In vitro to in vivo extrapolation (IVIVE) of CYP2B6 induction during pregnancy
Using human hepatocytes, it was recently shown that hepatic CYP2B6 mRNA expression and enzyme activity, can be induced by estradiol following a 48 h incubation period, with an EC50,u of 1.9 ± 0.5 μm and Emax (fold-increase) of 34 ± 22 (based on CYP2B6 mRNA expression), respectively 29. The average EC50,u was 61.4 nm after correcting for estradiol depletion (see Methods). For reasons outlined in Methods, the Emax for estradiol-mediated CYP2B6 induction was not calibrated based on positive control data including rifampicin and carbamazepine. At the unbound estradiol concentration in plasma of 0.20, 0.76 and 1.72 nm, during T1, T2 and T3, respectively, the predicted magnitude of CYP2B6 induction based on CYP2B6 mRNA data was 1.1-, 1.4- and 1.9-fold, respectively. The predicted magnitude based on CYP2B6 activity data was 1.0-, 1.1- and 1.3-fold, respectively. Because the increase in the rate of enzyme translation following administration of an inducer is secondary to its effect on the rate of transcription, there may be a delay, for example, due to turnover rate of nuclear receptors mediating the increased level of protein synthesis, between the manifestation of these two processes 45. Considering these factors, the predicted magnitude of CYP2B6 induction based on CYP2B6 mRNA data, was used in subsequent PBPK modelling of methadone disposition.
Methadone (METH) PK prediction in pregnancy using PBPK model incorporating CYP2B6 induction based on in vitro data
The mean AUC, Cmax and Cmin of R-METH and S-METH following a single oral dose of racemic METH of 9.9 mg in non-pregnant subjects were quantitatively predicted by the constructed R-METH and S-METH models (i.e. predicted/observed in the range of 0.8–1.2, Figure 2A and Table 3). At steady-state, the mean AUC, Cmax and Cmin of R-METH and S-METH following chronic dosing of racemic METH of 70 mg once daily or 100 mg once daily were also quantitatively predicted, with the exception of AUC(0,τss) (70 mg once daily) of S-METH and Cmin,ss (70 mg once daily) of R-METH (Figure 2B and Table 3). Nevertheless, the corresponding predicted/observed of the latter was 1.26 and 0.75, respectively. Then, METH exposure in pregnant women was predicted based on the study design described 2. In this study, the disposition of racemic METH was studied in nine pregnant subjects who had been on METH maintenance therapy. R-METH and S-METH plasma concentrations following an average dose of 30 mg once daily of racemic METH were predicted respectively, and the summed concentrations of each enantiomer were compared with the observed data (Figure 3 and Table 4). The model predicted mean AUCR (PP : T2) of 1.9, mean Cmax ratio (PP : T2) of 1.7 and mean Cmin ratio (PP : T2) of 2.0, compared with observed mean ratios 2.0, 2.0 and 2.6, respectively. The model predicted mean AUCR (PP : T3) of 2.1, mean Cmax ratio (PP : T3) of 2.0 and mean Cmin ratio (PP : T3) of 2.4, compared with observed mean ratios 1.7, 1.7 and 1.8, respectively. The PBPK model successfully predicted racemic methadone PK during T2, i.e. predicted/observed of mean AUC, Cmax and Cmin in the range of 0.80–1.11 (Table 4). T3 AUC and Cmin were slightly under-predicted, with pred./obs. of 0.79 and 0.73, respectively. R-METH and S-METH plasma unbound fraction was predicted to increase by 23% and 25% during T2, and 27% and 29% during T3 vs. post-partum, as a result of reduced plasma concentration of α1-AGP, the major binding protein. The reported pregnancy effect on racemic METH plasma unbound fraction was not significant, although there was a trend towards increased unbound fraction by 19–31% during pregnancy vs. post-partum. The predicted R-METH and S-METH CLr was 1.7-fold and 1.8-fold higher during T3 vs. post-partum, as a result of increased GFR and plasma unbound fraction, comparable with the reported value of two-fold of the racemate.
Figure 2.
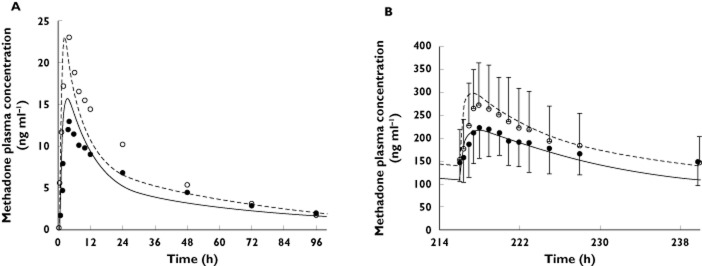
Predicted and observed plasma concentration–time profiles of the methadone (METH) enantiomers R-METH and S-METH following the administration of single oral dose of 9.9 mg (A) and multiple oral doses of 70 mg day−1 (B) to non-pregnant subjects. The solid and dashed lines represent predicted mean R-METH and S-METH profile, respectively. Mean observed data are overlaid (• R-METH profile; ○ S-METH profile) 20,21. Error bars represent SDs
Table 3.
Methadone PK parameters in non-pregnant healthy volunteers
| R-methadone | S-methadone | ||||||
|---|---|---|---|---|---|---|---|
| AUC(0,∞) | Observed | Predicted | Predicted/ | AUC(0,∞) | Observed | Predicted | Predicted/ |
| (ng ml−1 h) | observed | ng ml−1 h | observed | ||||
| 9.9 mg* | 461 ± 72 | 463.0 | 1.00 | 9.9 mg* | 639 ± 165 | 591.7 | 0.93 |
| Cmax (ng ml−1) | Cmax (ng ml−1) | ||||||
| 9.9 mg | 13 ± 3 | 15.8 | 1.22 | 9.9 mg | 23 ± 6 | 19.5 | 0.85 |
| Cmin,96h (ng ml−1) | Cmin,96h (ng ml−1) | ||||||
| 9.9 mg | 1.9 | 1.63 | 0.85 | 9.9 mg | 1.7 | 2.1 | 1.21 |
| AUC(0,τss) | AUC(0,τss) | ||||||
| 70 mg once daily† | 3484 ± 1846 | 3777 | 1.08 | 70 mg once daily† | 3797 ± 2088 | 4767 | 1.26 |
| 100 mg once daily‡ | 5540 ± 2100 | 5305 | 0.96 | 100 mg once daily‡ | 5730 ± 2580 | 6740 | 1.18 |
| Cmax,ss (ng ml−1) | Cmax,ss (ng ml−1) | ||||||
| 70 mg once daily | 251 ± 68 | 217 | 0.86 | 70 mg once daily | 303 ± 97 | 299.1 | 0.99 |
| 100 mgonce daily | 322 ± 109 | 299 | 0.93 | 100 mg once daily | 366 ± 128 | 402.6 | 1.10 |
| Cmin,ss (ng ml−1) | Cmin,ss (ng ml−1) | ||||||
| 70 mg once daily | 148 ± 50 | 110.5 | 0.75 | 70 mg once daily | 146 ± 61 | 140.4 | 0.96 |
| 100 mg once daily | 184.5 | 163.1 | 0.88 | 100 mg once daily | 177.0 | 215.2 | 1.22 |
Figure 3.
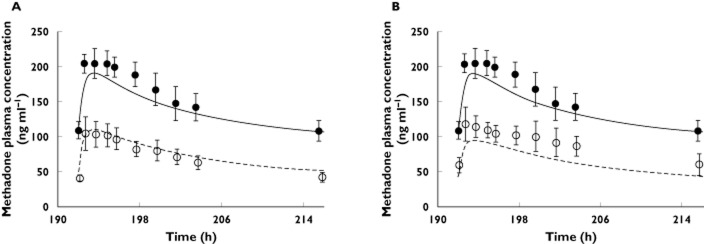
Predicted and observed plasma concentration–time profiles of racemic methadone (METH) in pregnant subjects on METH maintenance therapy of 30 mg day−1. (A) The solid and dashed lines represent predicted mean METH profile during post-partum and during the second trimester (T2), respectively. (B) The solid and dashed lines represent predicted mean METH profile during post-partum and during the third trimester (T3), respectively. Mean observed data are overlaid (• post-partum profile; ○ T2 or T3 profile) 2. Error bars represent SDs
Table 4.
Methadone PK parameters (30 mgonce daily) during T3 and post-partum
| AUC(0,τ) | Observed* | Predicted | Predicted/observed |
|---|---|---|---|
| (ng ml−1 h) | |||
| Pregnancy(T2) | 1607.7 | 1778.7 | 1.11 |
| Pregnancy(T3) | 1953.1 | 1539.0 | 0.79 |
| Postpartum(PP) | 3225.8 | 3298.1 | 1.02 |
| AUCPP/AUCT2 | 2.0 | 1.9 | 0.92 |
| AUCPP/AUCT3 | 1.7 | 2.1 | 1.30 |
| Cmax (ng ml−1) | |||
| Pregnancy(T2) | 104.3 ± 23.2 | 108.9 | 1.04 |
| Pregnancy(T3) | 117.7 ± 24.0 | 94.5 | 0.80 |
| Postpartum(PP) | 204.4 ± 17.6 | 190.2 | 0.93 |
| Cmax,PP/Cmax,T2 | 2.0 | 1.7 | 0.89 |
| Cmax,PP/Cmax,T3 | 1.7 | 2.0 | 1.16 |
| Cmin,24h (ng ml−1) | |||
| Pregnancy(T2) | 42.2 ± 6.7 | 51.6 | 1.04 |
| Pregnancy(T3) | 60.1 ± 16.1 | 43.9 | 0.73 |
| Post-partum (PP) | 108.3 ± 14.7 | 105.6 | 0.98 |
| Cmin,PP/Cmax,T2 | 2.6 | 2.0 | 0.94 |
| Cmin,PP/Cmax,T3 | 1.8 | 2.4 | 1.33 |
*AUC was calculated based on reported mean CLoral and mean dose of 30 mg (n = 9) 2. Cmax and Cmin,24h were extracted from published dose-adjusted mean concentration–time profiles, using ‘digitize’, a MATLAB tool for digitizing images that is freely available on http://www.mathworks.com/matlabcentral/.
PHT prediction to inform CYP2C9 induction during pregnancy
The PHT model provided in the Simcyp simulator was used without modification. The major 4-hydroxy metabolite was attributed to CYP2C9 (fm,2C9 = 72%) and CYP2C19 (fm,2C19 = 8%) mediated clearance, whereas the non 4-hydroxy (fm,other = 15–20%) clearance was unassigned. Furthermore, the model also accounted for saturable metabolism by CYP2C9 and 2C19, as well as auto-induction of CYP2C9 (see Table 5). To confirm in vivo fm,2C9, we predicted the effect of fluconazole inhibition (200 mg day−1 orally for 14 days) on PHT PK (250 mg day−1 orally for 4 days). The fluconazole model provided in the Simcyp simulator was used without modification. The predicted PHT AUCR (AUCinhibited/AUCcontrol) of 2.15 (data not shown) was comparable with the observed AUCR of 1.75 46. Furthermore, the mean AUC, Cmax, Cmin and Cpre-dose of PHT (300 mg once daily) in non-pregnant subjects were quantitatively predicted by the PHT model (i.e., predicted/observed in the range of 0.83–1.02, Figure 4A and Table 6). In studies conducted in the pregnant subjects, PHT plasma trough concentration (i.e. Cpre-dose) is invariably used as a marker of CYP2C9 activity. Hence the change in PHT Cpre-dose was used here to back calculate the magnitude of increase in CYP2C9 activity. In pregnant epileptic patients receiving a fixed PHT dose of 300 mg daily, PHT Cpre-dose is decreased by 43%, 51% and 61% during T1, T2 and T3, respectively 15,16. Through sensitivity analysis, CYP2C9 fold-induction was 1.4-, 1.5- and 1.6-fold, during T1, T2 and T3, respectively, to recover the reported changes in PHT Cpre-dose (Figure 4B). The predicted PHT plasma unbound fraction was 15%, 26% and 30% during T1, T2 and T3, compared with the reported values of 14%, 16–38% and 26–40%, respectively 16,47.
Table 5.
Summary of phenytoin (PHT) drug-dependent parameters
| Parameter | Value |
|---|---|
| Molecular weight | 252.28 |
| Log Po:w | 2.47 |
| pKa | 8.15 |
| B : P ratio | 0.61 |
| fu,p | 0.1 |
| Fa | 0.9 |
| ka (h−1) | 0.53 |
| Fg | 1.0 |
| Vss (l kg−1) | 0.573 |
| CLIV (l h−1) | 2.07 |
| CLr (l h−1) | 0.015 |
| Vmax (μl min−1/pmol) | Vmax,2C9 = 0.24 |
| Vmax,2C19 = 1.53 | |
| Km (μm) | Km,2C9 = 4.1 |
| Km,2C19 = 36.8 | |
| CLint,other (μl min−1 mg−1) | 0.97 |
| Emax (CYP2C9) | 1.9 |
| EC50 (μm) (CYP2C9) | 15.3 |
| fm and fe | fm,2C9 = 72% |
| fm,2C19 = 8% | |
| fm,other = 19% | |
| fe = 1% |
Figure 4.
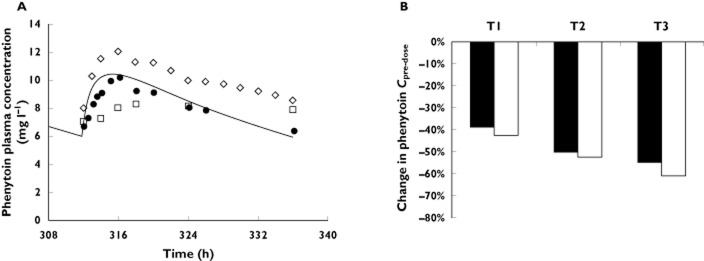
Predicted and observed phenytoin (PHT) plasma concentrations following multiple oral doses of 300 mg day−1. (A) Predicted (solid line) and observed (symbols) 56–58 PHT plasma concentration–time profiles following chronic dosing of 300 mg day−1 (orally) in non-pregnant healthy subjects. (B) The grey bars represent predicted percent decrease in PHT trough concentration (Cpre-dose) and the white bars represent observed mean percent decrease in PHT Cpre-dose in pregnant women during pregnancy (T1, T2 and T3) vs. post-partum 16. □, Lim et al. 56; ◊, Purkins et al. 58; •, Randinitis et al. 57
Table 6.
Phenytoin (300 mg Q.D.) PK parameters in non-pregnant subjects
| Observed* | Predicted | Predicted/observed | |
|---|---|---|---|
| AUC(0,τss) (ng ml−1 h) | 207.6 ± 25.5 | 201.6 | 0.97 |
| Cmax,ss (ng ml−1) | 10.3 ± 1.5 | 10.6 | 1.03 |
| Cmin,ss (ng ml−1) | 7.6 ± 1.5 | 6.3 | 0.83 |
| Cpre-dose (ng ml−1) | 7.3 ± 1.1 | 6.3 | 0.87 |
GLB PBPK model prediction incorporating OATP uptake
The constructed GLB model, accounting for metabolism by CYP3A, CYP2C9 and 2C19, as well as hepatic OATP-mediated uptake, was verified against the disposition kinetics following the administration of a single oral dose of 1.75–5 mg to non-pregnant healthy volunteers. Model-predicted AUC(0,∞), Cmax and Cmin,12 h all met the verification criterion, with predicted/observed in the range of 0.87–1.25 (Figure 5A and Table 7). Cmin,24 h of GLB (5 mgorally) was under-predicted, with a predicted/observed of 0.21. Following rifampicin treatment (600 mg i.v. over 30 min), the predicted GLB (1.25 mg orally) plasma AUCR and Cmax ratios were 1.88 and 1.47, respectively, compared with the observed ratios of 2.2 ± 0.51 and 1.8 ± 0.28, respectively 33 (Figure S2A). The rifampicin plasma concentration–time profile was also in reasonable agreement with the observed data 48 (Figure S2B).
Figure 5.
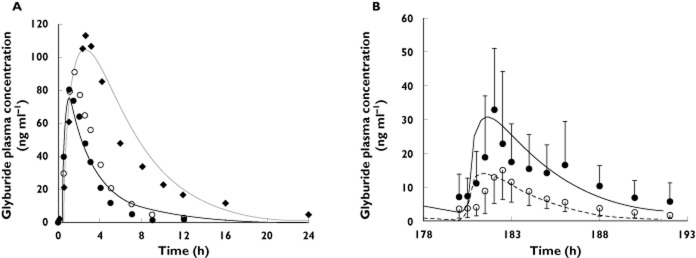
(A) Predicted and observed plasma concentration–time profiles of glyburide (GLB) after administration of a single oral dose of 1.75–5 mg to non-pregnant subjects. The black and grey solid lines represent predicted mean profile in subjects receiving 1.75 mg and 5 mg, respectively. Mean observed data after the administration of 1.75 mg GLB (• and ○) and 5 mg GLB (◆) are overlaid [37],[71],[72]. (B) Predicted and observed plasma concentration–time profiles of glyburide (GLB) following chronic dosing of 1 mg day−1 orally during the third trimester (T3) and post-partum. The solid and dashed lines represent predicted mean GLB profile during post-partum and during T3, respectively. Mean observed data 5 are overlaid (•: non-pregnant type 2 diabetic subjects profile or T2DM; ○: gestational diabetic subjects T3 profile or GDM). Error bars represent SDs
Table 7.
Glyburide PK parameters after oral dosing in non-pregnant subjects
| 1.75 mg* | Observed | Predicted | Predicted/observed |
|---|---|---|---|
| AUC(0,∞) (ng ml−1 h) | 273.9 | 281.0 | 1.03 |
| Cmax (ng ml−1) | 85.8 | 75.0 | 0.87 |
| Cmin,12h (ng ml−1) | 2.6 | 3.2 | 1.25 |
| 5 mg† | |||
| AUC(0,∞) (ng ml−1 h) | 809 ± 388 | 800.8 | 0.99 |
| Cmax (ng ml−1) | 109 ± 44 | 104.8 | 0.96 |
| Cmin,24h (ng ml−1) | 5.0 | 1.0 | 0.21 |
| Glyburide PK parameters (1 mg twice daily) during T3 and post-partum | |||
| AUC(0,τ) | |||
| (ng ml−1 h) | Obs.‡ | Pred. | Pred./Obs. |
| Pregnancy (T3) | 72 ± 27 | 62.4 | 0.87 |
| Post-partum (PP) | 153 ± 69 | 161.1 | 1.05 |
| AUCPP/AUCT3 | 2.1 | 2.6 | 1.2 |
| AUC(0,τ) | Obs.‡ | Pred. | Pred./Obs. |
| Pregnancy (T3) | 15 ± 8 | 14.2 | 0.95 |
| Post-partum (PP) | 33 ± 21 | 30.8 | 0.93 |
| Cmax,PP/Cmax,T3 | 2.2 | 2.2 | 0.99 |
| Cmin,24h (ng ml−1) | |||
| Pregnancy (T3) | 1.82 | 0.41 | 0.23 |
| Post-partum (PP) | 5.85 | 2.87 | 0.49 |
| Cmin,PP/Cmax,T3 | 3.2 | 7.0 | 2.17 |
*Reported mean values in non-pregnant, healthy subjects with CYP2C9*1/*1 or *1/*2 genotype (n = 18 total) 37,71. †Reported mean ± SD values (n = 24). The genotype of the subjects is unknown 72. ‡Reported mean ± SD values normalized to 1 mg twice daily in gestational diabetic subjects during the third trimester (n = 40) and non-pregnant type 2 diabetic subjects (n = 26) 5. CYP2C9 genotype had no impact on GLB disposition in this study.
The GLB plasma concentration–time profile following a fixed dose of 1 mg once daily in pregnant women was predicted based on the study design described 5. In this study, the steady-state GLB disposition was evaluated in gestational diabetic subjects (n = 40) during T3 and compared with non-pregnant type 2 diabetic subjects (n = 26). Predicted mean GLB AUCR (PP : T3), Cmax ratio (PP : T3) and Cmin ratio (PP : T3) were 2.6, 2.2 and 7.0, compared with the observed values of 2.1, 2.2 and 3.2. Model-predicted AUC(0,τ) and Cmax during T3 and post-partum met the verification criterion, with predicted/observed in the range of 0.87–1.05 (Figure 5B and Table 7). Cmin,12h of GLB during T3 and post-partum was under-predicted, with predicted/observed of 0.23 and 0.49, respectively. The GLB plasma unbound fraction was predicted to increase from 1.5% to 2.1% during T3. In comparison, the reported GLB plasma unbound fraction in gestational diabetic subjects during T3 (1.6 ± 0.1%) was not different from that of the non-pregnant type 2 diabetic subjects (1.5 ± 0.1%).
Discussion
Many studies in the literature have utilized model (probe) drugs that report CYP enzyme activities to delineate the magnitude of change in activity of major CYP enzymes, mostly during the third trimester (e.g. caffeine for CYP1A2, metoprolol for CYP2D6, midazolam for CYP3A, PHT for CYP2C9) 49. However, for hepatic CYP2B6 enzymes, such a probe drug study (e.g. bupropion) to delineate in vivo CYP2B6 activity during pregnancy has not been reported. Therefore, we utilized a bottom-up approach based on mechanistic studies (i.e. IVIVE). Based on the gradually rising concentration of estradiol in plasma, we predicted that hepatic CYP2B6 induction during pregnancy increased gradually and peaked during T3 at 1.9-fold of the post-partum value. It is noteworthy that estradiol concentrations vary considerably between individuals during pregnancy 50, and, as such, the magnitude of CYP2B6 induction could vary between women during pregnancy. For example, at the highest reported estradiol plasma concentration of ∼140nm, the predicted fold-induction of CYP2B6 is 3.1-fold. We then confirmed the predicted magnitude of CYP2B6 induction in vivo by predicting METH disposition during pregnancy. To do so, a respective METH PBPK model accounting for stereo-selective disposition of R-METH and S-METH was constructed and verified against observed PK data following single or multiple oral dosing of the racemate to the non-pregnant population. The PBPK model accounting for CYP2B6 induction, as well as known pregnancy effect on other clearance pathways including CYP3A, CYP2C19, and CLr, successfully predicted METH disposition during T2 as compared with the observed data 2. Model prediction of METH disposition during T3 was less robust. We suspect this is due to the fact that the reported mean T2 CLoral is 21% higher than mean T3 CLoral. This observed trend in vivo in METH CLoral during T2 vs. T3, although not statistically significant, is opposite to what we predicted based on the IVIVE approach. Other clinical studies conducted in pregnant subjects on METH maintenance therapy showed a similar or greater magnitude of increment in METH CLORAL during pregnancy vs. post-partum (2.9–3.6 fold) 51, or vs. historical controls (∼1.6 fold) 52. However because plasma concentration–time profiles were not reported in those studies and they had a limited sample size, these data were not used for model verification. One limitation of our study is that because METH CLoral reports the change in multiple clearance pathways, it might not be a sensitive reporter of CYP2B6 activity. As suspected, sensitivity analysis showed CYP2B6 fold-induction in the range of 1.0–3.0-fold and 1.0–2.0-fold during T2 and T3, respectively, could recover the observed plasma AUC of METH during pregnancy (i.e. predicted/observed within 0.80–1.25). These results also indicate that IVIVE based on CYP2B6 activity data, which predicted a smaller induction potential vs. that of mRNA data, could also recover the observed pregnancy effect on METH disposition. Future studies with probe drug bupropion during various stages of pregnancy and post-partum are highly desirable to further confirm the in vivo fold-induction of CYP2B6 predicted from in vitro data. Nevertheless, the above successful prediction of METH disposition during pregnancy shows for the first time that quantitative PK predictions of drugs cleared by multiple CYP enzymes is possible, through a PBPK model that integrates prior physiological knowledge such as CYP activity, in vitro and in vivo data.
CYP2B6 is also an important clearance pathway for the HIV drug efavirenz. In vitro, CYP2B6 has been identified as the main isoform metabolizing efavirenz, with smaller contributions from CYP2A6, CYP3A and UGT2B7. In vivo, CYP2B6 genetic variations are one of the main sources of efavirenz PK variability 53. Interestingly, the AUC and Cmax of efavirenz did not differ during pregnancy and post-partum in a Thai population and only Cmin,24 h was lower during T3 54. A possible explanation for the lack of effect of pregnancy on efavirenz PK, at least in this Thai population, could be that the PK variability introduced by the CYP2B6 genotype reduced the power of the study to detect the pregnancy effect. Auto-induction of CYP2B6 and CYP3A4 is another potential explanation. For CYP3A4, in vitro and in vivo data support an inverse correlation between the baseline enzyme activity and the fold induction of enzyme activity, that is, the magnitude of induction depends on the baseline enzyme activity 45. If the same holds true for CYP2B6, for an individual with high 2B6 activity pre-pregnancy (due to auto-induction), pregnancy-related increase in CYP2B6 activity may be smaller.
Estradiol (∼157 nm) enhanced activities of CYP2C9 (diclofenac 4'-hydroxylation) by 1.3–2.0 fold in human hepatocytes, a magnitude comparable with that of known inducers of CYP2C9 such as phenobarbital 30. Estradiol (100 nm) was shown to down-regulate CYP2C19 mRNA expression by ∼30% in human hepatocytes via activation of estrogen receptor 55. These mechanistic studies provide a potential explanation for the mechanisms by which CYP2C9 is increased and CYP2C19 activity is decreased during pregnancy. However, a concentration-dependent induction or suppression relationship (i.e. EC50 and Emax) has not been established for these CYP isoforms. Therefore the IVIVE of the magnitude of CYP2C9 and 2C19 induction or suppression in each trimester is not possible.
To model the PK changes of the CYP2C9 substrate drug GLB during pregnancy, we used the probe drug PHT as the model training drug. PHT PK are complicated by two balancing factors, saturable metabolism by CYP2C9 and CYP2C19 reducing its metabolic clearance at higher concentrations and auto-induction of CYP2C9 increasing its metabolic clearance at higher concentrations. The established PHT drug model accounting for these mechanisms quantitatively predicted PHT PK, including Cpre-dose, following chronic dosing in the non-pregnant population. Numerous studies have assessed PHT Cpre-dose in epileptic patients during pregnancy 15,16,47. Although the overall trend of the change in PHT Cpre-dose during each trimester was consistent across various studies, the exact magnitude differed. For example, Yerby et al. reported that the drop in PHT total and free concentration was 37%, 49%, 49% and 32%, 41% and 38%, respectively, during T1, T2 and T3 47. PHT dose used during pregnancy was increased (453 mg day−1) compared with post-partum (333 mg day−1). We selected Tomson et al.'s study as the model training set because of its superior study design: a large sample size (n = 29), fixed PHT dose, monotherapy, and trough sampling. Interestingly, following the same fixed dose of 300 mg day−1, PHT mean Cpre-dose during the post-partum period (10.27 ± 5.25 μg l−1) reported in this study was 41% higher than the reported mean Cpre-dose in the non-pregnant, healthy population (∼7.3 μg l−1, n = 31) 56–58. The difference in Cpre-dose between epileptic patients and healthy subjects may be reflective of disease-related factors on PHT disposition. As a result of this difference, the PHT model quantitatively predicted Cpre-dose in the non-pregnant healthy population (Table 6), but under-predicted Cpre-dose in the pregnant epileptic patients (data not shown). However, it is expected that at steady-state, CYP2C9 activity change is proportional to the percent change in PHT Cpre-dose. Therefore, the difference in the absolute value of baseline Cpre-dose (predicted vs. observed) should not affect the capacity of the model to recover the change in CYP2C9 activity. The modest induction of CYP2C9 recovered from in vivo data was also comparable with the magnitude observed in in vitro studies, of 1.3–2.0 fold in human hepatocytes 30.
The commonly used proton pump inhibitors including omeprazole, lansoprazole and pantoprazole, are primarily metabolized by CYP2C19. The effect of pregnancy on the PK of these drugs has not been studied. Pregnancy-induced suppression of CYP2C19 activity was back-calculated from the magnitude of decrease in CLoral of proguanil after accounting for other pregnancy related components of proguanil CLoral (see Methods). One caveat of this approach, however, is that renal clearance is a significant clearance pathway of proguanil, especially during pregnancy when CYP2C19 activity is suppressed. The pregnancy effect on CLr of proguanil is unknown, therefore we assumed that only the filtration clearance (∼10% of renal clearance) was increased during pregnancy. To assess definitively the magnitude of CYP2C19 induction during pregnancy, we propose that a PK study with the 2C19 probe omeprazole be conducted during various stages of pregnancy and post-partum in the future.
GLB has long been considered as primarily metabolized by CYP2C9. Pharmacogenetic studies appear to support this and suggest that in vivo CYP2C9 contributes significantly (up to 63%) to GLB metabolism, based on a limited number of subjects with reduced catalytic activity of CYP2C9 37. However, recent in vitro studies 34,40, including from our laboratory 35, suggest that CYP3A is the major isoform metabolizing GLB, contributing ∼50% to GLB metabolism, followed by ∼30% contribution from CYP2C9 and ∼20% contribution from CYP2C19. The constructed GLB model, accounting for metabolism by these multiple CYPs, also incorporated hepatic OATP-mediated uptake in order to explain a significant drug–drug interaction caused by rifampicin treatment. Hepatic OATP-mediated uptake of the drug into hepatocytes is not likely to be the sole rate-limiting step of drug clearance from the body, since GLB i.v. clearance of 4.4 l h−1 59 can be well-estimated by metabolic clearance. Consistent with this speculation, the estimated mean GLB sinusoidal uptake clearance of 572 l h−1 (Figure S2C) is greater than the hepatic intrinsic metabolic clearance of 252.9 l h−1 (see footnotes to Table 2). Following rifampicin treatment (600 mg i.v. over 30 min), GLB plasma AUCR and Cmax ratio were well predicted by the constructed PBPK model. It is not known whether pregnancy affects hepatic OATP expression or activity. Therefore no change in OATP activity was assumed in the current model. However, we conducted a sensitivity analysis to assess the impact of hepatic OATP activity on GLB plasma AUC during T3. The latter was shown to be sensitive to change in hepatic OATP activity, when no pregnancy-related CYP induction or suppression was assumed (Figure S2D). Therefore, we cannot rule out the possibility that hepatic OATP activity is affected during pregnancy. The GLB plasma concentration–time profile following a fixed dose of 1 mg once daily in pregnant women was quantitatively predicted. Because Cmax prediction met verification criterion, the under-prediction of GLB steady-state Cmin,12 h during T3 and post-partum could possibly be due to inadequate prediction of tissue distribution (hence the t1/2 of drugs) which could result in inadequate prediction of Cmin. The absorption of GLB was assumed to follow first order kinetics with a lag time. Under this assumption, the absorption phase of the GLB plasma profile following a single oral dose in non-pregnant subjects (Figure 4A) was adequately described. However this was not the case in pregnant subjects following multiple doses (Figure 4B). Other absorption models, such as an inverse Gaussian density function, have been used to describe GLB absorption 5. It is not clear as to what factors contribute to the apparent non-linear absorption of GLB. Saturation of intestinal transporters such as P-glycoprotein and the breast cancer resistance protein are possible explanations because GLB has been identified as a substrate for both transporters 60. However, when evaluating GLB Cmax in the dose range from 0.875 mg–10 mg oral dose in the literature (University of Washington DIDB, http://www.druginteractioninfo.org/), we did not observe dose-dependent change in Cmax with increasing dose. Similar to METH CLoral, because GLB CLoral reports the change in multiple clearance pathways, it will not be a sensitive reporter of CYP2C9 activity. CYP2C9 and CYP2C19 together contribute ∼50% of GLB metabolic clearance. We performed a sensitivity analysis by simultaneously varying CYP2C9 activity in the range of 1.0-fold to 3.0-fold induction, and a reduction in CYP2C19 activity of 0% to 80%, respectively. Multiple combinations of CYP2C9 and 2C19 activity change, for example, 2.7-fold induction of CYP2C9 activity coupled with 80% reduction of CYP2C19 activity, could recover the observed plasma AUC of GLB during T3 (i.e. predicted/observed within 0.80–0.93). This highlights the importance of conducting PK studies in pregnant women with a well-established probe drug, to delineate definitively the change in hepatic enzyme activity during pregnancy. Nevertheless, our successful prediction of GLB AUCR (PP : T3) during pregnancy illustrates the utility of using a PBPK model, incorporating pregnancy-dependent changes in CYP activities, to predict the disposition of a drug cleared by multiple pathways.
In summary, we have shown for the first time that our PBPK model 1) can quantitatively predict the disposition of drugs during pregnancy when they are cleared via multiple CYP pathways using prior knowledge of a pregnancy effect on various clearance pathways, 2) extrapolates in vitro data on estradiol-mediated CYP2B6 induction to its in vivo effect on the CYP2B6 substrate drug METH and 3) allows generalization beyond the model drugs studied (e.g. PHT, proguanil) to other drugs with well-characterized ADME characteristics (e.g. GLB). Previously, we have shown that such extrapolation can also be made for CYP3A 12, 1A2 and 2D6-metabolized drugs 61. Nevertheless, the magnitude of in vivo induction of hepatic CYP2B6 and suppression of hepatic CYP2C19 activity in pregnant women needs to be confirmed. Since conducting PK studies in pregnant women is challenging, our expanded PBPK model provides a valuable tool to evaluate different dosing regimens of drugs cleared primarily by single or multiple CYP enzymes during pregnancy. Future expansion of the presented PBPK model could incorporate variability in the predicted drug exposure measures when necessary data on the variability (and covariance) in the system- and drug-dependent parameters are better defined. While achieving equivalent drug (total or unbound) exposure in pregnant and non-pregnant women is often the primary focus of maternal pharmacotherapy, it is equally important to evaluate carefully fetal and neonatal safety, especially for drugs such as GLB that can cross the placenta. A comprehensive fetal model could be incorporated into this PBPK model in the future to predict fetal exposure to drugs. Using such systems a pharmacology approach can potentially allow us to identify drugs whose maternal-fetal PK, and therefore their efficacy and toxicity for the mother and/or the fetus, are likely to be affected by pregnancy. Conducting the trial in silico before its execution in vivo can be helpful in optimizing the design of a ‘first in pregnancy’ PK study including prioritizing study period (first, second or third trimester), sample size and dosage selection. The generated information or hypotheses can be rigorously tested, and the model can be further refined as data become available from such PK studies. Ultimately, the proposed approach may help support the design of rational dosing regimen for pregnant women and their offspring.
Competing Interests
All authors have completed the Unified Competing Interest form at http://www.icmje.org/coi_disclosure.pdf (available on request from the corresponding author) and declare ABK was supported by the Office of Women's Health, US Food and Drug Administration, JDU was a member of the Simcyp (now part of Certara) scientific advisory board and no other relationships or activities that could appear to have influenced the submitted work.
Acknowledgments
The authors acknowledge funding from the FDA Office of Women's Health and a visiting fellowship from SimCYP (ABK). The authors would like to note that the pregnancy structural model has now been incorporated into Simcyp Simulator version 12.2. The original Matlab model was developed by SimCYP Limited, Sheffield, UK and therefore SimCYP holds the IP rights to the model. Since the model is a commercial product, it cannot be made freely available to the public without protecting the IP rights of SimCYP. However, the company has indicated that the developed model (Matlab code for the structural model) will be made available for scientific research on a case by case basis, after reaching an agreement about IP with the requestor. The drug file (drug information in executable format for Matlab) can be obtained from the corresponding author upon written request.
Supporting Information
Additional Supporting Information may be found in the online version of this article at the publisher's web-site:
Figure S1
Changes in glyburide AUC (1.75 mg orally) as a function of hepatic bidirectional permeability clearance (CLpd), and hepatic intrinsic uptake clearance by OATP (PSint,OATP). There are many combinations of PSint,OATP and CLpd values that can result in AUC within the range of 0.46–0.73 ng ml−1 h following 1.75 mg orally, which is 80–125% of the mean AUC observed in vivo (0.58 ng ml−1 h). Consequently, PSint,OATP > CLpd (PSint,OATP 50 μl min−1/106 cells; CLpd 30 μl min−1/106 cells) were selected and used as initial estimates for data fitting
Figure S2
(A) Predicted plasma concentration–time profiles of GLB after administration of a single oral dose of 1.25 mg to non-pregnant subjects, in the presence and absence of rifampin (RIF) treatment (600 mg i.v.) (control: solid line; RIF treatment: dashed line). (B) Predicted (solid line) and observed (●) [50] plasma concentration–time profiles of rifampin (600 mg i.v. over 30 min infusion). (C) Predicted time course of GLB hepatic sinusoidal uptake clearance in the presence and absence of RIF treatment (control: solid line; RIF treatment: dashed line). (D) Change in GLB AUC (1 mg day−1 orally) during T3 as a function of hepatic OATP activity. While hepatic OATP activity was increased up to 10-fold of the value used in the final GLB model, pregnancy-induced changes in CYP activities were ignored. The dashed line represents the mean observed value
References
- 1.van Hasselt JG, Andrew MA, Hebert MF, Tarning J, Vicini P, Mattison DR. The status of pharmacometrics in pregnancy: highlights from the 3(rd) American Conference on Pharmacometrics. Br J Clin Pharmacol. 2012;74:932–939. doi: 10.1111/j.1365-2125.2012.04280.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Pond SM, Kreek MJ, Tong TG, Raghunath J, Benowitz NL. Altered methadone pharmacokinetics in methadone-maintained pregnant women. J Pharmacol Exp Ther. 1985;233:1–6. [PubMed] [Google Scholar]
- 3.Shiran MR, Lennard MS, Iqbal MZ, Lagundoye O, Seivewright N, Tucker GT, Rostami-Hodjegan A. Pharmacokinetic-pharmacodynamic modeling of mood and withdrawal symptoms in relation to plasma concentrations of methadone in patients undergoing methadone maintenance treatment. J Clin Psychopharmacol. 2012;32:666–671. doi: 10.1097/JCP.0b013e3182664ecd. [DOI] [PubMed] [Google Scholar]
- 4.Kirchheiner J, Brockmoller J, Meineke I, Bauer S, Rohde W, Meisel C, Roots I. Impact of CYP2C9 amino acid polymorphisms on glyburide kinetics and on the insulin and glucose response in healthy volunteers. Clin Pharmacol Ther. 2002;71:286–296. doi: 10.1067/mcp.2002.122476. [DOI] [PubMed] [Google Scholar]
- 5.Hebert MF, Ma X, Naraharisetti SB, Krudys KM, Umans JG, Hankins GD, Caritis SN, Miodovnik M, Mattison DR, Unadkat JD, Kelly EJ, Blough D, Cobelli C, Ahmed MS, Snodgrass WR, Carr DB, Easterling TR, Vicini P. Are we optimizing gestational diabetes treatment with glyburide? The pharmacologic basis for better clinical practice. Clin Pharmacol Ther. 2009;85:607–614. doi: 10.1038/clpt.2009.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hodge LS, Tracy TS. Alterations in drug disposition during pregnancy: implications for drug therapy. Expert Opin Drug Metab Toxicol. 2007;3:557–571. doi: 10.1517/17425225.3.4.557. [DOI] [PubMed] [Google Scholar]
- 7.Jeong H. Altered drug metabolism during pregnancy: hormonal regulation of drug-metabolizing enzymes. Expert Opin Drug Metab Toxicol. 2010;6:689–699. doi: 10.1517/17425251003677755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rowland M, Peck C, Tucker G. Physiologically-based pharmacokinetics in drug development and regulatory science. Annu Rev Pharmacol Toxicol. 2011;51:45–73. doi: 10.1146/annurev-pharmtox-010510-100540. [DOI] [PubMed] [Google Scholar]
- 9.Jamei M, Dickinson GL, Rostami-Hodjegan A. A framework for assessing inter-individual variability in pharmacokinetics using virtual human populations and integrating general knowledge of physical chemistry, biology, anatomy, physiology and genetics: a tale of ‘bottom-up’ vs ‘top-down’ recognition of covariates. Drug Metab Pharmacokinet. 2009;24:53–75. doi: 10.2133/dmpk.24.53. [DOI] [PubMed] [Google Scholar]
- 10.Lu G, Abduljalil K, Jamei M, Johnson TN, Rostami-Hodjegan A. A pregnancy physiologically-based pharmacokinetic (p-PBPK) model for disposition of drugs metabolized by CYP1A2, CYP3A4 and CYP2D6. Br J Clin Pharmacol. 2012;74:873–885. doi: 10.1111/j.1365-2125.2012.04363.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Abduljalil K, Furness P, Johnson TN, Rostami-Hodjegan A, Soltani H. Anatomical, physiological and metabolic changes with gestational age during normal pregnancy: a database for parameters required in physiologically based pharmacokinetic modelling. Clin Pharmacokinet. 2012;51:365–396. doi: 10.2165/11597440-000000000-00000. [DOI] [PubMed] [Google Scholar]
- 12.Ke AB, Nallan SC, Zhao P, Rostami-Hodjegan A, Unadkat JD. A PBPK model to predict disposition of CYP3A-metabolized drugs in pregnant women: verification and discerning the site of CYP3A induction. Clin Pharmacol Ther. 2012;1 doi: 10.1038/psp.2012.2. . Available at http://www.nature.com/psp/journal/v1/n9/index.html (last accessed 6 August 2013) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ke AB, Nallani SC, Zhao P, Rostami-Hodjegan A, Isoherranen N, Unadkat JD. A physiologically based pharmacokinetic model to predict disposition of CYP2D6 and CYP1A2 metabolized drugs in pregnant women. Drug Metab Dispos. 2013;41:801–813. doi: 10.1124/dmd.112.050161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cressey TR, Best BM, Achalapong J, Stek A, Wang J, Chotivanich N, Yuthavisuthi P, Suriyachai P, Prommas S, Shapiro DE, Watts DH, Smith E, Capparelli E, Kreitchmann R, Mirochnick M. Reduced indinavir exposure during pregnancy. Br J Clin Pharmacol. 2013;76:475–483. doi: 10.1111/bcp.12078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tomson T, Lindbom U, Ekqvist B, Sundqvist A. Epilepsy and pregnancy: a prospective study of seizure control in relation to free and total plasma concentrations of carbamazepine and phenytoin. Epilepsia. 1994;35:122–130. doi: 10.1111/j.1528-1157.1994.tb02921.x. [DOI] [PubMed] [Google Scholar]
- 16.Tomson T, Lindbom U, Ekqvist B, Sundqvist A. Disposition of carbamazepine and phenytoin in pregnancy. Epilepsia. 1994;35:131–135. doi: 10.1111/j.1528-1157.1994.tb02922.x. [DOI] [PubMed] [Google Scholar]
- 17.McGready R, Stepniewska K, Edstein MD, Cho T, Gilveray G, Looareesuwan S, White NJ, Nosten F. The pharmacokinetics of atovaquone and proguanil in pregnant women with acute falciparum malaria. Eur J Clin Pharmacol. 2003;59:545–552. doi: 10.1007/s00228-003-0652-9. [DOI] [PubMed] [Google Scholar]
- 18.McGready R, Stepniewska K, Seaton E, Cho T, Cho D, Ginsberg A, Edstein MD, Ashley E, Looareesuwan S, White NJ, Nosten F. Pregnancy and use of oral contraceptives reduces the biotransformation of proguanil to cycloguanil. Eur J Clin Pharmacol. 2003;59:553–557. doi: 10.1007/s00228-003-0651-x. [DOI] [PubMed] [Google Scholar]
- 19.Totah RA, Sheffels P, Roberts T, Whittington D, Thummel K, Kharasch ED. Role of CYP2B6 in stereoselective human methadone metabolism. Anesthesiology. 2008;108:363–374. doi: 10.1097/ALN.0b013e3181642938. [DOI] [PubMed] [Google Scholar]
- 20.Kharasch ED, Hoffer C, Whittington D, Walker A, Bedynek PS. Methadone pharmacokinetics are independent of cytochrome P4503A (CYP3A) activity and gastrointestinal drug transport: insights from methadone interactions with ritonavir/indinavir. Anesthesiology. 2009;110:660–672. doi: 10.1097/ALN.0b013e3181986a9a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Foster DJ, Somogyi AA, Dyer KR, White JM, Bochner F. Steady-state pharmacokinetics of (R)- and (S)-methadone in methadone maintenance patients. Br J Clin Pharmacol. 2000;50:427–440. doi: 10.1046/j.1365-2125.2000.00272.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Eap CB, Buclin T, Baumann P. Interindividual variability of the clinical pharmacokinetics of methadone: implications for the treatment of opioid dependence. Clin Pharmacokinet. 2002;41:1153–1193. doi: 10.2165/00003088-200241140-00003. [DOI] [PubMed] [Google Scholar]
- 23.Totah RA, Allen KE, Sheffels P, Whittington D, Kharasch ED. Enantiomeric metabolic interactions and stereoselective human methadone metabolism. J Pharmacol Exp Ther. 2007;321:389–399. doi: 10.1124/jpet.106.117580. [DOI] [PubMed] [Google Scholar]
- 24.Cao YJ, Smith PF, Wire MB, Lou Y, Lancaster CT, Causon RC, Bigelow GE, Martinez E, Fuchs EJ, Radebaugh C, McCabe S, Hendrix CW. Pharmacokinetics and pharmacodynamics of methadone enantiomers after coadministration with fosamprenavir-ritonavir in opioid-dependent subjects. Pharmacotherapy. 2008;28:863–874. doi: 10.1592/phco.28.7.863. [DOI] [PubMed] [Google Scholar]
- 25.Liu P, Foster G, Labadie R, Somoza E, Sharma A. Pharmacokinetic interaction between voriconazole and methadone at steady state in patients on methadone therapy. Antimicrob Agents Chemother. 2007;51:110–118. doi: 10.1128/AAC.00559-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kharasch ED, Hoffer C, Whittington D, Sheffels P. Role of hepatic and intestinal cytochrome P450 3A and 2B6 in the metabolism, disposition, and miotic effects of methadone. Clin Pharmacol Ther. 2004;76:250–269. doi: 10.1016/j.clpt.2004.05.003. [DOI] [PubMed] [Google Scholar]
- 27.Garrido MJ, Troconiz IF. Methadone: a review of its pharmacokinetic/pharmacodynamic properties. J Pharmacol Toxicol Methods. 1999;42:61–66. doi: 10.1016/s1056-8719(00)00043-5. [DOI] [PubMed] [Google Scholar]
- 28.Boulton DW, Arnaud P, DeVane CL. Pharmacokinetics and pharmacodynamics of methadone enantiomers after a single oral dose of racemate. Clin Pharmacol Ther. 2001;70:48–57. doi: 10.1067/mcp.2001.116793. [DOI] [PubMed] [Google Scholar]
- 29.Dickmann LJ, Isoherranen N. Quantitative prediction of CYP2B6 induction by estradiol during pregnancy, potential explanation for increased methadone clearance during pregnancy. Drug Metab Dispos. 2012;41:270–274. doi: 10.1124/dmd.112.047118. [DOI] [PubMed] [Google Scholar]
- 30.Choi SY, Koh KH, Jeong H. Isoform-specific regulation of cytochromes P450 expression by estradiol and progesterone. Drug Metab Dispos. 2012;41:263–269. doi: 10.1124/dmd.112.046276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Product label for Activella® (estradiol/norethindrone acetate) tablets. Available at http://www.accessdata.fda.gov/drugsatfda_docs/label/2006/022001lbl.pdf (last accessed 5 August 2013)
- 32. GLIMEL® (glibenclamide) Tablets Product Label. Available at http://www.pbs.gov.au/meds%2Fpi%2Fafpglime10808.pdf (last accessed 5 August 2013)
- 33.Zheng HX, Huang Y, Frassetto LA, Benet LZ. Elucidating rifampin's inducing and inhibiting effects on glyburide pharmacokinetics and blood glucose in healthy volunteers: unmasking the differential effects of enzyme induction and transporter inhibition for a drug and its primary metabolite. Clin Pharmacol Ther. 2009;85:78–85. doi: 10.1038/clpt.2008.186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zharikova OL, Fokina VM, Nanovskaya TN, Hill RA, Mattison DR, Hankins GD, Ahmed MS. Identification of the major human hepatic and placental enzymes responsible for the biotransformation of glyburide. Biochem Pharmacol. 2009;78:1483–1490. doi: 10.1016/j.bcp.2009.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zhou L, Naraharisetti SB, Liu L, Wang H, Lin YS, Isoherranen N, Unadkat JD, Hebert MF, Mao Q. Contributions of human cytochrome P450 enzymes to glyburide metabolism. Biopharm Drug Dispos. 2010;31:228–242. doi: 10.1002/bdd.706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Appel S, Rufenacht T, Kalafsky G, Tetzloff W, Kallay Z, Hitzenberger G, Kutz K. Lack of interaction between fluvastatin and oral hypoglycemic agents in healthy subjects and in patients with non-insulin-dependent diabetes mellitus. Am J Cardiol. 1995;76:29A–32A. doi: 10.1016/s0002-9149(05)80012-8. [DOI] [PubMed] [Google Scholar]
- 37.Niemi M, Cascorbi I, Timm R, Kroemer HK, Neuvonen PJ, Kivisto KT. Glyburide and glimepiride pharmacokinetics in subjects with different CYP2C9 genotypes. Clin Pharmacol Ther. 2002;72:326–332. doi: 10.1067/mcp.2002.127495. [DOI] [PubMed] [Google Scholar]
- 38.Satoh H, Yamashita F, Tsujimoto M, Murakami H, Koyabu N, Ohtani H, Sawada Y. Citrus juices inhibit the function of human organic anion-transporting polypeptide OATP-B. Drug Metab Dispos. 2005;33:518–523. doi: 10.1124/dmd.104.002337. [DOI] [PubMed] [Google Scholar]
- 39.Hirano M, Maeda K, Shitara Y, Sugiyama Y. Drug-drug interaction between pitavastatin and various drugs via OATP1B1. Drug Metab Dispos. 2006;34:1229–1236. doi: 10.1124/dmd.106.009290. [DOI] [PubMed] [Google Scholar]
- 40.Naritomi Y, Terashita S, Kagayama A. Identification and relative contributions of human cytochrome P450 isoforms involved in the metabolism of glibenclamide and lansoprazole: evaluation of an approach based on the in vitro substrate disappearance rate. Xenobiotica. 2004;34:415–427. doi: 10.1080/00498250410001685728. [DOI] [PubMed] [Google Scholar]
- 41.Varma MV, Lai Y, Feng B, Litchfield J, Goosen TC, Bergman A. Physiologically based modeling of pravastatin transporter-mediated hepatobiliary disposition and drug-drug interactions. Pharm Res. 2012;29:2860–2873. doi: 10.1007/s11095-012-0792-7. [DOI] [PubMed] [Google Scholar]
- 42.Funck-Brentano C, Becquemont L, Lenevu A, Roux A, Jaillon P, Beaune P. Inhibition by omeprazole of proguanil metabolism: mechanism of the interaction in vitro and prediction of in vivo results from the in vitro experiments. J Pharmacol Exp Ther. 1997;280:730–738. [PubMed] [Google Scholar]
- 43. Product label for MALARONE® (250 mg Atovaquone + 100 mg Proguanil Hydrochloride) Tablets. Available at http://www.gsk.ca/english/docs-pdf/Malarone_PM_EN_20071102.pdf (last accessed 5 August 2013)
- 44.Wattanagoon Y, Taylor RB, Moody RR, Ochekpe NA, Looareesuwan S, White NJ. Single dose pharmacokinetics of proguanil and its metabolites in healthy subjects. Br J Clin Pharmacol. 1987;24:775–780. doi: 10.1111/j.1365-2125.1987.tb03245.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Almond LM, Yang J, Jamei M, Tucker GT, Rostami-Hodjegan A. Towards a quantitative framework for the prediction of DDIs arising from cytochrome P450 induction. Curr Drug Metab. 2009;10:420–432. doi: 10.2174/138920009788498978. [DOI] [PubMed] [Google Scholar]
- 46.Blum RA, Wilton JH, Hilligoss DM, Gardner MJ, Henry EB, Harrison NJ, Schentag JJ. Effect of fluconazole on the disposition of phenytoin. Clin Pharmacol Ther. 1991;49:420–425. doi: 10.1038/clpt.1991.49. [DOI] [PubMed] [Google Scholar]
- 47.Yerby MS, Friel PN, McCormick K, Koerner M, Van Allen M, Leavitt AM, Sells CJ, Yerby JA. Pharmacokinetics of anticonvulsants in pregnancy: alterations in plasma protein binding. Epilepsy Res. 1990;5:223–228. doi: 10.1016/0920-1211(90)90042-t. [DOI] [PubMed] [Google Scholar]
- 48.Lau YY, Huang Y, Frassetto L, Benet LZ. effect of OATP1B transporter inhibition on the pharmacokinetics of atorvastatin in healthy volunteers. Clin Pharmacol Ther. 2007;81:194–204. doi: 10.1038/sj.clpt.6100038. [DOI] [PubMed] [Google Scholar]
- 49.Anderson GD. Pregnancy-induced changes in pharmacokinetics: a mechanistic-based approach. Clin Pharmacokinet. 2005;44:989–1008. doi: 10.2165/00003088-200544100-00001. [DOI] [PubMed] [Google Scholar]
- 50.O'Leary P, Boyne P, Flett P, Beilby J, James I. Longitudinal assessment of changes in reproductive hormones during normal pregnancy. Clin Chem. 1991;37:667–672. [PubMed] [Google Scholar]
- 51.Wolff K, Boys A, Rostami-Hodjegan A, Hay A, Raistrick D. Changes to methadone clearance during pregnancy. Eur J Clin Pharmacol. 2005;61:763–768. doi: 10.1007/s00228-005-0035-5. [DOI] [PubMed] [Google Scholar]
- 52.Jarvis MA, Wu-Pong S, Kniseley JS, Schnoll SH. Alterations in methadone metabolism during late pregnancy. J Addict Dis. 1999;18:51–61. doi: 10.1300/J069v18n04_05. [DOI] [PubMed] [Google Scholar]
- 53.Siccardi M, Almond L, Schipani A, Csajka C, Marzolini C, Wyen C, Brockmeyer NH, Boffito M, Owen A, Back D. Pharmacokinetic and pharmacodynamic analysis of efavirenz dose reduction using an in vitroin vivo extrapolation model. Clin Pharmacol Ther. 2012;92:494–502. doi: 10.1038/clpt.2012.61. [DOI] [PubMed] [Google Scholar]
- 54.Cressey TR, Stek A, Capparelli E, Bowonwatanuwong C, Prommas S, Sirivatanapa P, Yuthavisuthi P, Neungton C, Huo Y, Smith E, Best BM, Mirochnick M. Efavirenz pharmacokinetics during the third trimester of pregnancy and postpartum. J Acquir Immune Defic Syndr. 2012;59:245–252. doi: 10.1097/QAI.0b013e31823ff052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Mwinyi J, Cavaco I, Pedersen RS, Persson A, Burkhardt S, Mkrtchian S, Ingelman-Sundberg M. Regulation of CYP2C19 expression by estrogen receptor alpha: implications for estrogen-dependent inhibition of drug metabolism. Mol Pharmacol. 2010;78:886–894. doi: 10.1124/mol.110.065540. [DOI] [PubMed] [Google Scholar]
- 56.Lim ML, Min SS, Eron JJ, Bertz RJ, Robinson M, Gaedigk A, Kashuba AD. Coadministration of lopinavir/ritonavir and phenytoin results in two-way drug interaction through cytochrome P-450 induction. J Acquir Immune Defic Syndr. 2004;36:1034–1040. doi: 10.1097/00126334-200408150-00006. [DOI] [PubMed] [Google Scholar]
- 57.Randinitis EJ, Alvey CW, Koup JR, Rausch G, Abel R, Bron NJ, Hounslow NJ, Vassos AB, Sedman AJ. Drug interactions with clinafloxacin. Antimicrob Agents Chemother. 2001;45:2543–2552. doi: 10.1128/AAC.45.9.2543-2552.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Purkins L, Wood N, Ghahramani P, Love ER, Eve MD, Fielding A. Coadministration of voriconazole and phenytoin: pharmacokinetic interaction, safety, and toleration. Br J Clin Pharmacol. 2003;56(Suppl 1):37–44. doi: 10.1046/j.1365-2125.2003.01997.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Rydberg T, Jonsson A, Melander A. Comparison of the kinetics of glyburide and its active metabolites in humans. J Clin Pharm Ther. 1995;20:283–295. doi: 10.1111/j.1365-2710.1995.tb00664.x. [DOI] [PubMed] [Google Scholar]
- 60.Pollex EK, Anger G, Hutson J, Koren G, Piquette-Miller M. Breast cancer resistance protein (BCRP)-mediated glyburide transport: effect of the C421A/Q141K BCRP single-nucleotide polymorphism. Drug Metab Dispos. 2010;38:740–744. doi: 10.1124/dmd.109.030791. [DOI] [PubMed] [Google Scholar]
- 61.Ke AB, Nallan SC, Zhao P, Rostami-Hodjegan A, Isoherranen N, Unadkat JD. A PBPK model to predict disposition of P450 2D6 and P450 1A2 metabolized drugs in pregnant women. Drug Metab Dispos. 2013;41:801–813. doi: 10.1124/dmd.112.050161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Gerber JG, Rosenkranz S, Segal Y, Aberg J, D'Amico R, Mildvan D, Gulick R, Hughes V, Flexner C, Aweeka F, Hsu A, Gal J. Effect of ritonavir/saquinavir on stereoselective pharmacokinetics of methadone: results of AIDS Clinical Trials Group (ACTG) 401. J Acquir Immune Defic Syndr. 2001;27:153–160. doi: 10.1097/00126334-200106010-00010. [DOI] [PubMed] [Google Scholar]
- 63.Eap CB, Cuendet C, Baumann P. Binding of d-methadone, l-methadone, and dl-methadone to proteins in plasma of healthy volunteers: role of the variants of alpha 1-acid glycoprotein. Clin Pharmacol Ther. 1990;47:338–346. doi: 10.1038/clpt.1990.37. [DOI] [PubMed] [Google Scholar]
- 64.Dale O, Sheffels P, Kharasch ED. Bioavailabilities of rectal and oral methadone in healthy subjects. Br J Clin Pharmacol. 2004;58:156–162. doi: 10.1111/j.1365-2125.2004.02116.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Wolff K, Rostami-Hodjegan A, Hay AW, Raistrick D, Tucker G. Population-based pharmacokinetic approach for methadone monitoring of opiate addicts: potential clinical utility. Addiction. 2000;95:1771–1783. doi: 10.1046/j.1360-0443.2000.951217717.x. [DOI] [PubMed] [Google Scholar]
- 66.Yang J, Jamei M, Yeo KR, Tucker GT, Rostami-Hodjegan A. Prediction of intestinal first-pass drug metabolism. Curr Drug Metab. 2007;8:676–684. doi: 10.2174/138920007782109733. [DOI] [PubMed] [Google Scholar]
- 67.Rodgers T, Rowland M. Mechanistic approaches to volume of distribution predictions: understanding the processes. Pharm Res. 2007;24:918–933. doi: 10.1007/s11095-006-9210-3. [DOI] [PubMed] [Google Scholar]
- 68.Savolainen J, Jarvinen K, Taipale H, Jarho P, Loftsson T, Jarvinen T. Co-administration of a water-soluble polymer increases the usefulness of cyclodextrins in solid oral dosage forms. Pharm Res. 1998;15:1696–1701. doi: 10.1023/a:1011900527021. [DOI] [PubMed] [Google Scholar]
- 69.Obach RS, Lombardo F, Waters NJ. Trend analysis of a database of intravenous pharmacokinetic parameters in humans for 670 drug compounds. Drug Metab Dispos. 2008;36:1385–1405. doi: 10.1124/dmd.108.020479. [DOI] [PubMed] [Google Scholar]
- 70.Rydberg T, Jonsson A, Karlsson MO, Melander A. Concentration–effect relations of glibenclamide and its active metabolites in man: modelling of pharmacokinetics and pharmacodynamics. Br J Clin Pharmacol. 1997;43:373–381. doi: 10.1046/j.1365-2125.1997.00571.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Niemi M, Backman JT, Neuvonen M, Neuvonen PJ, Kivisto KT. Effects of rifampin on the pharmacokinetics and pharmacodynamics of glyburide and glipizide. Clin Pharmacol Ther. 2001;69:400–406. doi: 10.1067/mcp.2001.115822. [DOI] [PubMed] [Google Scholar]
- 72.Karim A, Laurent A, Munsaka M, Wann E, Fleck P, Mekki Q. Coadministration of pioglitazone or glyburide and alogliptin: pharmacokinetic drug interaction assessment in healthy participants. J Clin Pharmacol. 2009;49:1210–1219. doi: 10.1177/0091270009338938. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1
Changes in glyburide AUC (1.75 mg orally) as a function of hepatic bidirectional permeability clearance (CLpd), and hepatic intrinsic uptake clearance by OATP (PSint,OATP). There are many combinations of PSint,OATP and CLpd values that can result in AUC within the range of 0.46–0.73 ng ml−1 h following 1.75 mg orally, which is 80–125% of the mean AUC observed in vivo (0.58 ng ml−1 h). Consequently, PSint,OATP > CLpd (PSint,OATP 50 μl min−1/106 cells; CLpd 30 μl min−1/106 cells) were selected and used as initial estimates for data fitting
Figure S2
(A) Predicted plasma concentration–time profiles of GLB after administration of a single oral dose of 1.25 mg to non-pregnant subjects, in the presence and absence of rifampin (RIF) treatment (600 mg i.v.) (control: solid line; RIF treatment: dashed line). (B) Predicted (solid line) and observed (●) [50] plasma concentration–time profiles of rifampin (600 mg i.v. over 30 min infusion). (C) Predicted time course of GLB hepatic sinusoidal uptake clearance in the presence and absence of RIF treatment (control: solid line; RIF treatment: dashed line). (D) Change in GLB AUC (1 mg day−1 orally) during T3 as a function of hepatic OATP activity. While hepatic OATP activity was increased up to 10-fold of the value used in the final GLB model, pregnancy-induced changes in CYP activities were ignored. The dashed line represents the mean observed value