

Abstract
3-Phosphoinositide-dependent protein kinase-1 (PDK1) has been recognized as a promising anticancer target. Thus, it is interesting to identify new inhibitors of PDK1 for anticancer drug discovery. Through a combined use of virtual screening and wet experimental activity assays, we have identified a new PDK1 inhibitor with IC50 = ~200 nM. The anticancer activities of this compound have been confirmed by the anticancer activity assays using 60 cancer cell lines. The obtained new PDK1 inhibitor and its PDK1-inhibitor binding mode should be valuable in future de novo design of novel, more potent and selective PDK1 inhibitors for future development of anticancer therapeutics.
Keywords: Enzyme, enzyme inhibitor, inhibitor identification, anticancer activity
As a pivotal kinase in the phosphotidylinositol-3-kinase (PI3K) pathway, 3-phosphoinositide-dependent protein kinase-1 (PDK1) is a well validated and promising anticancer target [1,2]. PDK1 is over-expressed in pre-malignant and in low to high grade ovarian carcinomas [3]. Elevated levels of PDK1 were also reported in metastasized breast tumors [4]. It has been reported that over-expression of PDK1 in mammary cells resulted in their transformation in vitro and tumor formation in vivo [ 5 ]. Targeting PDK1 with anti-sense oligonucleotides revealed a marked reduction of cell proliferation and survival and also an increased rate of apoptosis than that observed in PI3K or protein kinase B (PKB) inhibition [1]. PDK1-hypomorphic mice which express only 10% of normal levels of PDK1 were reported to be viable and fertile [6]. This finding revealed that inhibition of PDK1 could be achieved without severe toxicity. A more recent PDK1 hypomorphic mice study showed that low levels of PDK1 protected the mice against various tumors [2]. Thus, PDK1 has become a well validated anticancer target. Identifying new PDK1 inhibitors could eventually lead to development of better treatment options for cancers.
In the present work, we have carried out combined virtual screening and experimental studies in order to identity a new PDK1 inhibitor. The virtual screening was based on our previously modeled structure of PDK1 binding with celecoxib [7]. It has been known that celecoxib can inhibit PDK1 with IC50 value of 48 μM [8]. Our modeling of the PDK1-celecoxib complex structure was based on an X-ray crystal structure available in the protein data bank [9] (PDB code: 2BIY with a resolution of 1.95 Å) [10] of PDK1.
Our virtual screening was performed on a subset of ZINC database [11] containing 688,086 compounds from some major commercial compound suppliers including IBScreen and Sigma-Aldrich. First of all, the entire subset of 688,086 compounds was filtered with a pre-screening filter, FILTER v.1.1.1 (OpenEye scientific software, www.eyesopen.com), to eliminate inappropriate or undesirable compounds [12]. The pre-screening Filter is a molecular screening tool that uses a combination of physical property calculations and functional group knowledge to assess libraries and ultimately remove non-lead-like compounds [13]. The default lead-like filter available in this program was used with some minor variations. The main parameters that we used involve: molecular weight (minimal value = 150 Da, Maximum value = 440 Da, and rings (min=0 and max=3), rotatable bonds (min=0 and max=10), allowed elements (H, C, N, O, F, S, Cl, and Br), hydrogen bond donor (max=6), hydrogen bond acceptor (max=10). We filtered out molecules with XlogP greater than 4.0, which violates more than one Lipinski rule of five or if they are known aggregators. The pre-screening filtering led to a final dataset of 157,623 compounds.
Starting from the final dataset of 157,623 compounds, the virtual screening approach used in the present work is essentially the same as that [14] we recently used to identify new inhibitors of microsomal prostaglandine synthase-1 (mPGES-1). Briefly, the 157,623 compounds were first screened by using ligand-based method ROCS [15,16] and rigid docking FRED [15], followed by flexible molecular docking using FlexX [17], molecular dynamics (MD) simulation using the Sander module of AMBER program [18], and molecular mechanics/Poisson-Boltzmann surface area (MM-PBSA) binding free energy calculations [19] as depicted in Figure 1.
Figure 1.

Flowchart of the combined virtual screening and experimental activity assays used.
As shown in Figure 1, of 10,453 compounds selected from the ligand-based screening using the ROCS program, only 3,500 compounds passed the rigid docking screening using the FRED program. Within the 3,500 compounds, the top-1,200 compounds were chosen for flexible docking using the FlexX program. Then, top-120 compounds were chosen for the MD simulations (1 ns for each compound binding with PDK1) and MM-PBSA binding free energy calculations (on 100 snapshots of the simulated structure within the stable MD trajectory for each PDK1-ligand binding system). The final binding free energy calculated for each PDK1-ligand binding structure was taken as the average of the binding free energy values calculated for the 100 snapshots. Based on the calculated binding free energies, one compound (compound 1 depicted in Figure 2A; see Table 1 for the energetic results) was ordered from Analogix, Inc. (Burlington, WI) for wet experimental assays.
Figure 2.
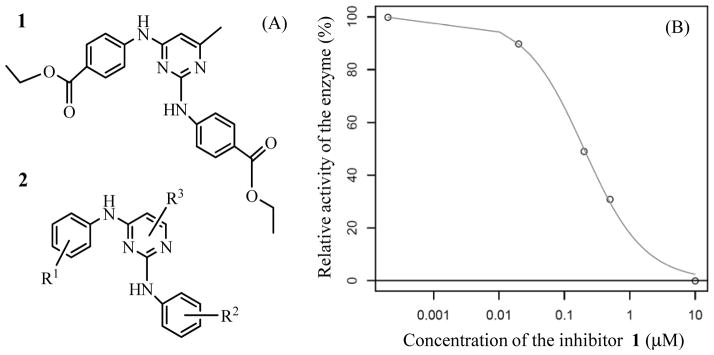
(A) Molecular structures of the new PDK1 inhibitor (compound 1) identified through virtual screening and the corresponding scaffold (2); (B) in vitro inhibitory activity of compound 1 against PDK1.
Table 1.
Binding free energies (kcal/mol) calculated at T = 298.15 K and P = 1 atm for PDK1 binding with celecoxib and compound 1 in comparison with the corresponding experimental data.
| Inhibitor | Calc.a | Expt. | |||
|---|---|---|---|---|---|
| ΔEMM | ΔGsol | -TΔS | ΔGbind | ΔGbind | |
| Celecoxibb | −56.5 | 40.1 | 11.3 | −5.1 | −5.9 |
| Compound 1 | −59.6 | 35.5 | 12.5 | −11.5 | −9.1 |
The MM-PBSA calculations were performed on 100 snapshots along a stable MD trajectory for each PDK1-inhibitor binding complex. The results given in the table are the average values calculated for the 100 snapshots.
The data for celecoxib were reported in reference [7].
The in vitro kinase assay on compound 1 was performed using a fluorescence polarization assay with the Invitrogen PDK1 assay kit (P2884) according to vendor’s instructions. This assay was based on the ability of recombinant PDK1, in the presence of DMSO or the inhibitor, to phosphorylate its substrate peptide (P2925). These phosphopeptides generated during the kinase reaction of PDK1 competes with the fluorescein-labeled phosphopeptides (called as tracer) for binding to anti-phosphothreonine peptide-specific antibodies. This binding is then quantified using fluorescence polarization (FP) technique. FP value was measured using TECAN GENIOS PRO microplate reader in our own lab. We carried out the PDK1 assay along with a standard reference in order to make sure that the inhibitory activity data obtained for the test compound are comparable to the previously reported inhibitory activity of known compound. Our experimental tests revealed that compound 1 can significantly inhibit PDK1. According to the activity data (Figure 2B), we obtained IC50 = ~200 nM for compound 1. Thus, starting with a total of 690,986 molecules, we were able to identify a new inhibitor [20] of PDK1 through a combined use of the computational screening and PDK1 kinase activity tests.
Based on the encouraging result obtained from the PDK1 kinase activity assay, compound 1 was submitted to the developmental therapeutics program (DTP) at national cancer institute (NCI) [21]. The compound was screened against 60 human cancer cell lines, including the multiple myeloma cell lines RPMI8226 etc. In the NCI screen, 60 human cancer cell lines were treated 48 h with 10-fold dilutions of compounds at a minimum of five concentrations (0.01 to 100 μM). The screening was done using a sulforhodamine B protein assay to estimate the cell viability or growth. Using seven absorbance measurements [time zero, (Tz), Control growth, (C), and test growth in the presence of drug at five different concentration levels (Ti)], the percentage growth was calculated at each of the drug concentration levels. The percentage growth was calculated as: [(Ti-Tz)/(C-Tz)]*100 [21]. Compound 1 was found to significantly inhibit many cancer cell lines. The inhibition data of the new compound against four selected cell lines were shown in Figure 3. Compound 1 can also effectively inhibit many other cancer cell lines, such as multiple myeloma cell line RPMI8226, non non-small cell lung cancer cell line HOP92, colon cancer cell line KM12, CNS cancer cells SF268, Melanoma cell LOX IMVI, Ovarian cancer cells OVCAR-3, Renal cancer cells RXF 393, Prostate cancer cell line PC-3, and Breast cancer cell line HS 578T. The main growth inhibitory activity data of compound 1 against various cell lines are summarized in Table 2.
Figure 3.

Growth inhibitory effect of compound 1 against representative cancer cell lines
Table 2.
Growth inhibitory activity of compound 1 against various cell lines in NCI human cancer cell line panel.
| No. | Cell Line (Panel name) | GI50 (μM) a |
|---|---|---|
| 1 | RPMI-8226 (Leukemia) | 3.54 |
| 2 | HOP-92 (Non-small cell lung cancer) | 3.25 |
| 3 | KM12 (Colon cancer) | 3.62 |
| 4 | SF-268 (CNS cancer) | 3.80 |
| 5 | SF-295 (CNS cancer) | 8.64 |
| 6 | U251 (CNS cancer) | 9.88 |
| 7 | LOX IMVI (Melanoma) | 7.24 |
| 8 | OVCAR-3 (Ovarian cancer) | 2.02 |
| 9 | RXF 393 (Renal Cancer) | 5.05 |
| 10 | PC-3 (Prostate cancer) | 6.02 |
| 11 | DU-145 (Prostate cancer) | 5.39 |
| 12 | HS 578T (Breast cancer) | <10.0b |
Data obtained from the NCI-DTP’s in vitro human cancer cell line screen [23]
With compound 1 at 10 μM, the growth of the HS 578T cancel cells was 33% ± 2% (or the 67% ± 2% growth inhibition) according to the NCI-DTP’s assays at 10 μM for two times.
Concerning the absorption, distribution, metabolism, and excretion (ADME) and toxicology of the new PDK1 inhibitor, the i-labs web interface from ACD labs was used in the property prediction [22]. The volume of distribution (Vd) of compound 1 is predicted to be 2.67 L/Kg. Most of the drugs of this category were reported to have moderate Vd with 90% of values in the range of 0.5 to 10 L/Kg. This molecule is also predicted to be devoid of genotoxic potential. It is also predicted to have maximum passive absorption (100%).
Finally, it is interesting to analyze the binding mode of the new PDK1 inhibitor (compound 1) for further lead optimization studies in the future. Figures 4 shows the predicted binding mode of the new compound with PDK1 based on the 1.2 ns MD simulation (Figure 5). Compound 1 has a hydrogen bond interaction with the backbone carbonyl group of Ala162 in the hinge region. The pyrimidine ring is present in the hydrophobic adenine pocket of PDK1. It is surrounded by residues Leu212 and Leu88. There is another hydrogen bond interaction between the backbone carbonyl group of Leu88 and the –NH group of the inhibitor. The ester group is present in the sugar region in the pocket does not interact with any specific residues. Based on the PDK1-inhibitor binding mode, the identified new PDK1 inhibitor may be optimized starting from the scaffold (2) depicted in Figure 2A in future de novo drug design and discovery efforts.
Figure 4.
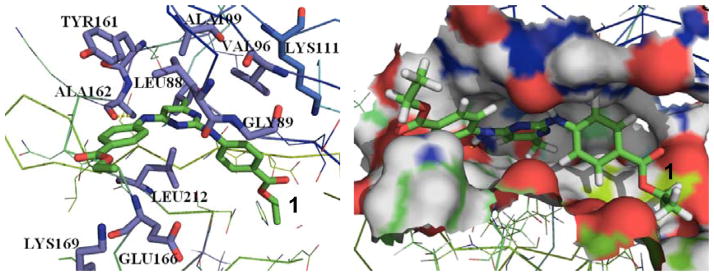
Binding of compound 1 in the PDK1 active site. Compound 1 is shown in sticks with carbon atoms in green color.
Figure 5.
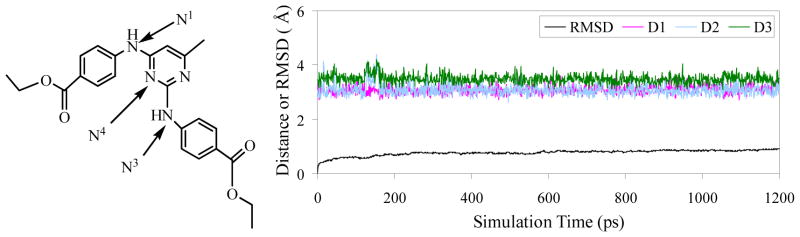
Plots of MD-simulated internuclear distances versus simulation time for PDK1 binding with compound 1. D1 refers to the distance between N1 atom of compound 1 and the carbonyl oxygen of Ala162 backbone, D2 the distance between N3 atom of compound 1 and the carbonyl oxygen of Leu88 backbone, and D3 the distance between N4 atom of compound 1 and the carbonyl oxygen of Leu88 backbone.
Supplementary Material
Acknowledgments
The research was supported in part by the NIH (grant RC1MH088480 to Zhan), Kentucky Science & Engineering Foundation (grant KSEF-925-RDE-008 to Zhan) and the Center for Computational Sciences (CCS) at University of Kentucky. The entire work was carried out in Zhan’s lab at University of Kentucky. Wenchao Yang worked in Zhan’s lab at University of Kentucky as an exchange graduate student (2005–2009) or a postdoctoral fellow (since January 2010) from Central China Normal University. The authors also acknowledge the Center for Computational Sciences (CCS) at University of Kentucky for supercomputing time on IBM X-series Cluster with 340 nodes or 1,360 processors and a Dell Supercomputer Cluster consisting of 388 nodes or 4,816 processors.
References and Notes
- 1.Flynn P, Wongdagger M, Zavar M, Dean NM, Stokoe D. Curr Biol. 2000;10:1439. doi: 10.1016/s0960-9822(00)00801-0. [DOI] [PubMed] [Google Scholar]
- 2.Bayascas JR, Leslie NR, Parsons R, Fleming S, Alessi DR. Curr Biol. 2005;15:1839. doi: 10.1016/j.cub.2005.08.066. [DOI] [PubMed] [Google Scholar]
- 3.Ahmed N, Riley C, Quinn MA. Britis J Cancer. 2008;98:1415. doi: 10.1038/sj.bjc.6604306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lin HJ, Hsieh FC, Song H, Lin J. British J cancer. 2005;93:1372. doi: 10.1038/sj.bjc.6602862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zeng X, Xu H, Glazer RI. Cancer Res. 2002;62:3538. [PubMed] [Google Scholar]
- 6.Lawlor MA, Mora A, Ashby PR, Williams MR, Murray-trait V, Malone L, Prescott AR, Lucocq JM, Alessi DR. EMBO J. 2002;21:3728. doi: 10.1093/emboj/cdf387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.AbdulHameed MDM, Hamza A, Zhan C-GJ. Phys Chem B. 2006;110:26365. doi: 10.1021/jp065207e. [DOI] [PubMed] [Google Scholar]
- 8.Zhu J, Huang JW, Tseng PH, Yang YT, Fowble J, Shiau CW, Shaw YJ, Kulp SK, Chen CS. Cancer Res. 2004;64:4309. doi: 10.1158/0008-5472.CAN-03-4063. [DOI] [PubMed] [Google Scholar]
- 9.Berman HM, Westbrook J, Feng Z, Gilliland G, Bhat TN, Weissig H, Shindyalov IN, Bourne PE. Nucleic Acids Res. 2000;28:235. doi: 10.1093/nar/28.1.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Komander D, Kular G, Deak M, Alessi DR, VanAalten DMF. J Biol Chem. 2005;280:18797. doi: 10.1074/jbc.M500977200. [DOI] [PubMed] [Google Scholar]
- 11.Irwin JJ, Shoichet BK. J Chem Inf Model. 2005;45:177. doi: 10.1021/ci049714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.FILTER v.2.0.1. OpenEye scientific software; www.eyesopen.com. [Google Scholar]
- 13.Miteva MA, Lee WH, Montes MO, Villoutreix BO. J Med Chem. 2005;48:6012. doi: 10.1021/jm050262h. [DOI] [PubMed] [Google Scholar]
- 14.Hamza A, Zhao X, Tong M, Tai HH, Zhan CG. Bioorg Med Chem. 2011;19:6077. doi: 10.1016/j.bmc.2011.08.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.ROCS, version 2.3.1. OpenEye Scientific Software, Inc; Santa Fe, NM, USA: 2007. www.eyesopen.com. [Google Scholar]
- 16.Rush TS, Grant JA, Mosyak L, Nicholls A. J Med Chem. 2005;48:1489. doi: 10.1021/jm040163o. [DOI] [PubMed] [Google Scholar]
- 17.Rarey M, Kramer B, Lengauer T, Kleb GJ. Mol Biol. 1996;261:470. doi: 10.1006/jmbi.1996.0477. [DOI] [PubMed] [Google Scholar]
- 18.Case DA, Darden TA, Cheatham TE, III, Simmerling CL, Wang J, Duke RE, Luo R, Merz KM, Wang B, Pearlman DA, Crowley M, Brozell S, Tsui V, Gohlke H, Mongan J, Hornak V, Cui G, Beroza P, Schafmeister C, Caldwell JW, Ross WS, Kollman PA. AMBER 8. University of California; San Francisco: 2004. [Google Scholar]
- 19.All docking simulations were based on the default parameters. For all of the MD simulations, AMBER ff03 force field was used. See Supporting Information for the details of the MD simulations.
- 20.Compared to the known PDK1 inhibitors reported in literature, compound 1 was found to have a low 2D-similarity, with Tanimoto score 0.18; no one was associated with a Tanimoto score better than 0.18. The 2D-similarity was calculated using extended connectivity fingerprints (ECFP4) in pipeline pilot.
- 21.http://dtp.nci.nih.gov/screening.html
- 22.https://ilab.acdlabs.com/iLab2/index.php (12-21-2011).
- 23.Monks A, Scudiero D, Skehan P, Shoemaker R, Paull K, Vistica D, Hose C, Langley J, Cronise P, Vaigro-Wolff A. J Natl Cancer Inst. 1991;86:1853. doi: 10.1093/jnci/83.11.757. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.