

Abstract
In this project, we aimed to bring large-scale gene identification findings into a developmental psychopathology framework. Using a family-based sample, we tested whether polygenic scores for externalizing disorders—based on single nucleotide polymorphism weights derived from genome-wide association study results in adults (n = 1,249)—predicted externalizing disorders, subclinical externalizing behavior, and impulsivity-related traits adolescents (n = 248) and young adults (n = 207), and whether parenting and peer factors in adolescence moderated polygenic risk to predict externalizing disorders. Polygenic scores predicted externalizing disorders in adolescents and young adults, even after controlling for parental externalizing disorder history. Polygenic scores also predicted subclinical externalizing behavior and impulsivity traits in the adolescents and young adults. Adolescent parental monitoring and peer substance use moderated polygenic scores to predict externalizing disorders. This illustrates how state of the science genetics can be integrated with psychological science to identify how genetic risk contributes to the development of psychopathology.
Keywords: externalizing disorders, impulsivity, gene-by-environment, gene-by-development, polygenic, developmental, Collaborative Study on the Genetics of Alcoholism (COGA)
With widespread recognition of the importance of genetic factors on psychopathology, a growing number of psychological studies are incorporating genetic components. However, there are two critical disconnects between the fields of genetics and psychological science. First, the majority of psychological research on measured gene-by-development effects (i.e., how genetic predispositions unfold across time) as well as gene-by-environment effects (i.e., the role of environmental factors in moderating genetic predispositions) has not kept pace with our understanding of the polygenic basis of complex behavioral outcomes. Despite widespread adoption of candidate gene approaches—where a researcher studies genetic variants in a pre-specified gene of interest based on its putative biological function (such as the serotonin-transporter-linked polymorphic region in SLC6A4)—very few well-replicated associations have emerged from these hypothesized genes of interest (Bosker et al., 2011; Collins, Kim, Sklar, O'Donovan, & Sullivan, 2012). Exceptions to this include variants in the alcohol dehydrogenase (ADH) gene cluster and alcohol outcomes (Gelernter et al., 2013; Thomasson et al., 1991) and nicotinic receptor genes for smoking outcomes (Broms et al., 2012; Tobacco and Genetics Consortium, 2010). Candidate gene approaches are also at odds with our understanding that behavioral outcomes are likely to have a polygenic architecture, meaning that they include the effects of many common variants of small magnitude across the genome (Plomin, Haworth, & Davis, 2009). Now that there are relatively inexpensive methods for genotyping hundreds of thousands of genetic variants across the genome, candidate-gene approaches no longer represent the state of the science for examining measured gene-by-development and gene-by-environment effects.
Second, the nature of large-scale gene identification efforts for many common disorders has created a gap in our understanding of how the emerging findings fit into a developmental psychopathology framework. In one common gene identification study design, known as a genome-wide association study (GWAS), researchers systematically examine whether genotype frequencies for variants across the genome differ between individuals who are affected and unaffected with a disorder. In the area of externalizing disorder research, adults are often the focus of these investigations because the average age at onset for these disorders is in late adolescence or early adulthood (Hingson, Heeren, & Winter, 2006; Warner, Kessler, Hughes, Anthony, & Nelson, 1995). This makes it important to use adult samples that have passed through the period of greatest risk for developing the disorder in these gene identification efforts in order to increase the probability that individuals who are classified as “unaffected” are truly unaffected. Still, it is widely recognized that externalizing disorders do not typically appear de novo in adulthood; rather, they are often foreshadowed by precursor disorders, personality traits, or patterns of behavior (Cicchetti, 1984), some of which are genetically correlated with the eventual adult disorder (i.e., gene-by-development effects). For example, twin studies have demonstrated that adolescent conduct disorder and adult alcohol dependence partly share genetic influences (Slutske et al., 1998), and analyses of a specific gene, GABRA2, provided further evidence that was the case, yielding association with conduct symptoms in adolescents and alcohol problems in adults (Dick et al., 2006). Likewise, polygenic scores that include the effects of many genes illustrate that genetic influences on smoking behaviors differ as a function of developmental stage (Belsky et al., 2013; Vrieze, McGue, & Iacono, 2012). These latter two examples notwithstanding, there have been limited attempts to bring the results from large-scale gene-identification efforts into a developmental psychopathology framework to examine how these genetic predispositions unfold across time, making this an important gap in the literature.
Bridging these disconnects and bringing together the best theories and practices in genetics and clinical psychological science is central for understanding how genetic predispositions for behavioral outcomes manifest across development and interface with environmental factors. In the present study we address these critical gaps in the area of externalizing disorder research by taking a polygenic approach. This approach considers the contributions of many common genetic variants of small magnitude across the genome to examine gene-by-development and gene-by-environment effects and has demonstrated predictive power in cases where individual genes do not (The International Schizophrenia Consortium, 2009). It typically uses results from a GWAS. Selecting a liberal p-value threshold, a polygenic score for each individual is calculated by summing over the number of alleles for each SNP—the most common type of genetic variation that involves a single nucleotide substitution in a DNA sequence, such as an adenine substitution for a guanine base—weighted by the effect size drawn from a GWAS. The score then represents the composite additive effect of these multiple variants. As an example, consider two SNPs with the following alleles: SNP 1 (C/T) and SNP 2 (A/G). If a GWAS indicates that increasing copies of the T and G alleles are associated with a higher level of externalizing disorders (betas = 0.06 and 0.08), then an individual who carries two copies for the T and G alleles will have a polygenic score equivalent to 2 * 0.06 + 2 * 0.08 = 0.28; and an individual who carries one copy for the T and G alleles will have a polygenic score equivalent to 1 * 0.06 + 1* 0.08 = 0.14. A higher polygenic score indicates a greater genetic predisposition toward that trait.
We first tested whether polygenic risk for adulthood externalizing disorders predicted externalizing disorders in adolescents and young adults, and whether any observed polygenic effects were robust after controlling for parental externalizing disorder history. These analyses addressed the question of whether polygenic risk for adulthood disorders manifest at a clinical level in adolescents and young adults. We then ran a series of analyses to examine whether polygenic risk for adulthood externalizing disorders predicted subclinical levels of externalizing behavior as well as impulsivity-related traits in adolescents and young adults. These analyses addressed the possibility that polygenic risk for adulthood externalizing disorders may not be related to clinical-level externalizing disorders in adolescents and young adults, but rather subclinical levels of externalizing behaviors (measured using the Achenbach Externalizing Scale) and broadband indicators of impulse control. We focused on impulsivity-related traits since the inability to control impulses is the defining feature of the highly heritable genetic factor shared across externalizing disorders (Kendler, Prescott, Myers, & Neale, 2003; Krueger et al., 2002; Young, Stallings, Corley, Krauter, & Hewitt, 2000). We hypothesized that adolescents and young adults who are genetically predisposed toward adulthood externalizing disorders would have higher levels of subclinical externalizing behaviors and impulsivity. We used a range of impulsivity measures, including impulsiveness, conscientiousness (i.e., constraint) and sensation seeking, in order to capture impulsivity's multi-faceted nature (Depue & Collins, 1999; Evenden, 1999; Miller, Joseph, & Tudway, 2004; Whiteside & Lynam, 2001).
Finally, we examined whether adolescents' reports of parental monitoring and perceived peer substance use moderated polygenic scores to predict externalizing disorders. Our gene-by-environment hypotheses were informed by twin studies, which have demonstrated that permissive environments may allow for the expression of latent genetic predispositions for externalizing psychopathology that more restrictive environments inhibit (Shanahan & Hofer, 2005). Two of the more robust moderation effects in this area are observed for adolescent parental monitoring (i.e., parents' knowledge of one's plans, whereabouts, and friends) and peer deviance (i.e., friends' engagement in substance use or criminal activity). Previous studies indicate that genetic influences on externalizing increase under conditions of low parental monitoring or high peer deviance (Button et al., 2007; Harden, Hill, Turkheimer, & Emery, 2008; Hicks, South, DiRago, Iacono, & McGue, 2009). However, this has only been demonstrated in twin data, where genetic influences are inferred by comparisons of relative types. It has not been explored with respect to measured genome-wide indices of risk. We hypothesized that polygenic predispositions toward externalizing disorders would be more pronounced under conditions of lower parental monitoring and higher peer substance use.
Method
Sample
Participants came from the Collaborative Study on the Genetics of Alcoholism (COGA) (Begleiter et al., 1995), whose objective is to identify genes involved in alcohol dependence and related disorders. Probands were identified through alcohol treatment programs at six U.S. sites and were invited to participate if they had a sufficiently large family (usually sibships > 3 with parents available) with two or more members in the COGA catchment area. The Institutional Review Boards at all sites approved this study and written consent was obtained from all participants. As shown in Figure 1, the present analyses included a subset of 118 European-American COGA families densely affected with alcohol dependence (at least 3+ affected members) and for whom genome-wide association data were available (Kang et al., 2013; Wang et al., 2013). By design, this sample was limited to European-American individuals in order to avoid false positives in the GWAS driven by population stratification (i.e., differences in allele frequencies between populations) (for a review see Cardon & Palmer, 2003). We focused on the adolescent (Adolescent subsample; ages 12-17) and young adult (Young Adult subsample; ages 18-24) participants in the family-based sample who are also part of the COGA Prospective Study of the developmental antecedents of alcohol dependence (Dick et al., 2013). The adolescent and young adult prospective participants were recruited into the Prospective Study beginning in 2004, and have been followed-up biennially ever since.
Figure 1.

Schematic of study sample. The Adult genome-wide association study (GWAS) subsample included adult participants who were 29 years of age and older at the time of their psychiatric interviews. The GWAS weights from the adult subsample were used to calculate polygenic scores in the Adolescent and Young Adult subsamples.
Subclinical externalizing behavior, impulsivity-related traits, parental monitoring, and perceived peer substance use data came from prospective participants' first assessment of these constructs [adolescents: age = 14.44 years (SD = 1.78; range 12-17 years), n = 248 (54% male); young adults: age= 19.86 years (SD = 1.41; range 18-24 years), n = 207, (47% male)]. We note that parental monitoring and peer substance use data were available for adolescent prospective participants only. Owing to the longitudinal design of the Prospective Study, prospective participants have completed the externalizing disorders psychiatric interview multiple times. We used data from the interview where they endorsed the greatest number of alcohol dependence criteria to create the externalizing disorder composite, and we note that the mean ages at which these groups completed their psychiatric interviews were 16.74 (SD = 1.87) and 21.63 (SD = 2.95), respectively. Our scientific rationale for using externalizing disorder data from a single interview (versus calculating average scores across multiple assessments) was that in a high-risk sample such as COGA we wanted to measure individuals' greatest expression of their predisposition to the externalizing disorder (i.e., alcohol dependence) on which the sample was originally ascertained.
For the GWAS of adult externalizing disorders, we used the participants who were 29 years of age and older at the time of their psychiatric interviews (Adult genome-wide association subsample; n = 1,249; 45% male; Mage = 44.97; SD = 12.56, range = 29-88). The age cutoff was selected because the oldest prospective participant's externalizing disorder data came from an age 28 psychiatric interview. As described in Wang et al. (2013), the sample was genotyped on the Illumina Human OmniExpress array 12.VI. In total, 587,378 SNPs with minor allele frequency > 5% and genotyping success rate per individual (> 75%) were analyzed.
Measures
Externalizing disorder composite
We created an externalizing disorder composite based on DSM-IV alcohol dependence and abuse criteria, DSM-IV illicit drug (cocaine, marijuana, sedatives, stimulants, opiates, and other drugs) dependence and abuse criteria, and antisocial behavior criteria (measured with DSM-IV or DSM-III-R antisocial personality disorder or conduct disorder criteria, depending on age). Criterion counts were obtained from the reliable (kappas for individual diagnoses range from 0.70 - 0.90) and valid Semi-Structured Assessment for the Genetics of Alcoholism (SSAGA) (for those 18 years of age or older) or the adolescent version of the SSAGA (for those between 12-17) (Bucholz et al., 1994; Hesselbrock, Easton, Bucholz, Schuckit, & Hesselbrock, 1999). We did not include ADHD given the mixed literature on the genetic epidemiology of ADHD vis a vis other externalizing disorders (Young et al., 2000). Antisocial behavior was measured using criterion counts for DSM-IV antisocial personality disorder (ASPD) or ASPD DSM-III-R criteria modified to match ASPD DSM-IV criteria. Because ASPD is only assessed in those 18 years and older, DSM-III-R or DSM-IV conduct disorder (CD) criteria were substituted for those under 18. Because ASPD and CD have different numbers of criteria, CD criterion counts were proportionally scored in order to create a comparable range (0-7) with ASPD scores (e.g., a participant endorsing 7/15 CD criteria would receive a proportional score of 3.23/7). Illicit drug dependence and abuse were measured with separate sum scores of DSM-IV cocaine, marijuana, sedatives, stimulants, opiates, and other drugs dependence and abuse criterion counts. For participants with data on the alcohol and illicit drug dependence and abuse and antisocial behavior variables [2148 individuals (93%) in the family-based sample, 47% male], we extracted component scores from a principal components analysis. A single component accounted for 68% of the common variance among the externalizing disorder variables with the following loadings: alcohol dependence, 0.86; alcohol abuse, 0.86; antisocial behavior, 0.80; illicit drug dependence, 0.82; illicit drug abuse, 0.79.
Parental externalizing disorder history
Parental externalizing disorder data were available for 435 (96%) of the mothers and 326 (72%) of the fathers of prospective participants. We used a 3-category parental externalizing disorder history measure. Those for whom either parent had externalizing disorder diagnosis [352 (77%); of these, 151 (33%) had two affected parents, 87 (19%) only the father was affected/known to be affected, and 114 (25%) only the mother was affected/known to be affected)] were set as the reference group and were compared to those for whom neither parent had an externalizing disorder diagnosis [47 (10%)] and those for whom there were incomplete parental externalizing disorder history information (i.e., one parent was unaffected and the other parent was missing externalizing disorder data) [55 (12%)].
Achenbach Externalizing Adult Self-Report and Youth Self-Report
Participants were asked how well a series of items described their externalizing behavior in the past six months on a three-point scale anchored 0 (not true) to 2 (very true or often true). Young adult prospective participants were administered the Adult Self-Report (ASR) form (Achenbach & Rescorla, 2001) and their adolescent counterparts were administered the Youth Self-Report (YSR) form (Achenbach, 2003). The YSR and ASR included 32 and 35 items, respectively, tapping rule-breaking, aggressive, and intrusive (ASR only) behavior. Sum scores were used.
Barratt Impulsiveness Scale
The Barratt Impulsiveness Scale (Patton, Stanford, & Barratt, 1995) asked participants how well a series of 30 statements related to attentional, motor, and nonplanning impulsiveness described their behavior on a 4-point scale ranging from 1 (rarely/never) to 4 (usually). The adolescents completed a modified version so that the questions were more appropriate to adolescents. A sum score was used.
Conscientiousness
Participants responded to the 60-item NEO Five Factor Inventory (Costa & McCrae, 1986) and indicated how well a series of statements described their behavior on a 5-point scale ranging from Strongly Disagree to Strongly Agree. We used sum scores from the conscientiousness subscale.
Sensation Seeking Scale (SSS-V)
The Sensation Seeking Scale (Zuckerman, 1984) presented participants with 40 pairs of items related to differences in stimulation and arousal. A 26-item version was used for adolescents that included modified substance use and sexual activity items (Russo et al., 1993). Sum scores were used.
Parental monitoring and perceived peer substance use (adolescents only)
The parental monitoring measure asked participants three questions (how much their parent figures know about their plans, interests, and where/with whom they spend time when not at home) from Chassin et al. (1993). Responses were made on a 4-point scale ranging from 1 (Always) to 4 (Rarely), and the inter-item correlations ranged from 0.45-0.56 (all p-values < 0.0001). The four perceived peer substance use questions came from FinnTwin12 (Kaprio, Pulkkinen, & Rose, 2002), and asked participants about how many of their friends smoke, use alcohol, use marijuana, or use other drugs using a 4-point scale ranging from 1 (None of them) to 4 (All of them). The inter-item correlations for the peer substance use questions ranged from 0.45-0.65 (all p-values < 0.0001). Items were summed to create separate parental monitoring and peer substance use composites. Prior to summing, the parental monitoring items were reverse scored so that higher scores indicated more parental monitoring.
Statistical Analyses
GWAS and computation of polygenic scores
We ran a family-based GWAS using the GWAF package (Chen & Yang, 2010) on the adult subsample (n = 1,249), which accounts for familial nesting and genetic distance using a kinship matrix. Covariates included gender, age at interview, and cohort. We then used GWAS estimates to calculate polygenic scores for the adolescent and young adult prospective participants using the --score procedure in PLINK (Purcell et al., 2007), which computes a linear function of the number of score alleles an individual possesses weighted by the associated GWAS t-statistic.
Because there are no set criteria for establishing a threshold to create maximally informative scores (Evans, Visscher, & Wray, 2009), we conducted preliminary analyses using a series of p-value thresholds ranging from 0.05 to 0.50 to evaluate what threshold maximized the variance accounted for (R2) in the externalizing disorder composite in the adolescent subsample. We maximized R2 in this subsample because it was the one in which we subsequently evaluated gene-by-environment effects, and previous work suggests that SNPs with a nominal main effect may be enriched for gene-by-environment interaction (Thomas, 2010). There was little variability in the percent variance accounted for (range 5.5% to 5.9%); p < 0.30 (176,562 SNPs) maximized R2 and was used to create polygenic scores for adolescents and young adults.
Prospective sample polygenic association and environmental moderation analyses
Association and moderation analyses for the prospective sample were conducted using the SURVEYREG procedure in SAS, which accounts for the nesting of individuals within families (90 and 84 families were represented in the adolescent and young adult subsamples, respectively). Covariates included gender and age at assessment. For the association analyses, we examined the variance (R2) in the externalizing disorder composite and impulsivity-related traits explained by polygenic scores in the adolescent and young adult prospective subsamples. We examined these associations in adolescents and young adults separately because of measurement differences and in view of the numerous social and legal changes during the transition to adulthood that may change gene-behavior associations. We then tested for gene-environment interaction with parental monitoring and peer substance use in the adolescent subsample (i.e., the subsample for which these environmental measures were available and likely to be developmentally relevant). It has been suggested that variants having a nominal association with an outcome are likely to be enriched for gene-by-environment interaction (Thomas, 2010). Accordingly, we focused our gene-by-environment analyses in adolescents on an externalizing disorder composite because we constructed our polygenic scores using estimates from a GWAS of an externalizing disorder composite in adults. We used p < 0.05 as suggestive evidence for association and moderation because our tests were not independent (owing to correlated phenotypes) and a Bonferroni correction would be too stringent.
Results
Descriptive Statistics, GWAS Results, and Polygenic Association Analyses
The descriptive statistics and correlations across all phenotypes for adolescents and young adults are available in Tables S1 and S2 in the Supplemental Material available online. The phenotypes were moderately to highly correlated across both age groups, although conscientiousness did not relate to the substance use and antisocial behavior variables in young adults. Low parental monitoring and high peer substance use were associated with higher externalizing disorder composite scores (rs = -0.38 and 0.56, ps < 0.01).
The GWAS results are summarized in Figure 2 and the quantile-quantile plot is shown in Figure S1 in the Supplemental Material available online. The genomic inflation factor, which is mathematically defined as the ratio of the median of the observed distribution of the GWAS test statistic to the expected median (Devlin & Roeder, 1999) was acceptable, λ = 1.01. Extreme deviations in λ indicate an excess false positive rate, (i.e., inflation) in a GWAS, which may be attributable to technical problems in the genotyping or uncontrolled population stratification. Thus, in our study, technical issues and population stratification did not appear to inflate the results. The most associated SNP (p = 2.37 × 10-07) was located in zinc finger protein 407 (ZNF407) on chromosome 18.
Figure 2.
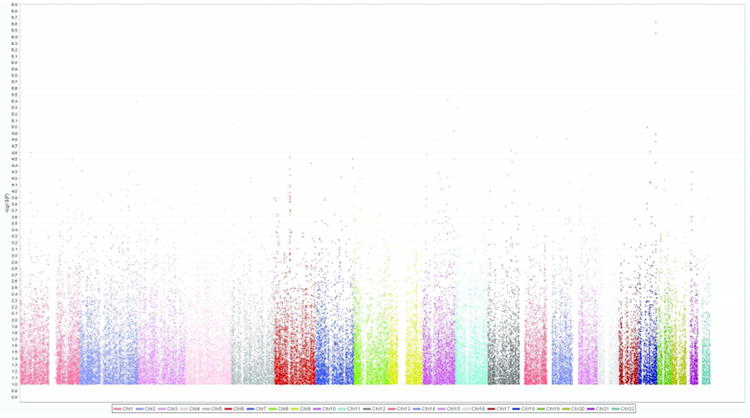
Association results from the GWAS of the externalizing disorder composite in the adult sample. On the x-axis are chromosomes 1-22. On the y-axis are inverse logarithms of the p-values for each SNP.
Polygenic scores predicted the externalizing disorder composite in adolescents and young adults and accounted for 6% of the variance after controlling for gender and age (Table 1). Polygenic scores continued to predict the externalizing disorder composite after controlling for parental externalizing disorder history (Table 1). Table 2 summarizes the variance (R2) in subclinical externalizing behavior and impulsivity-related traits explained by polygenic scores after controlling for gender and age. Higher polygenic scores predicted higher Achenbach externalizing and impulsiveness in adolescents and young adults. Higher polygenic scores predicted lower conscientiousness in adolescents, but not young adults. Higher polygenic scores predicted higher sensation seeking in young adults but not adolescents.
Table 1.
Regressions of the externalizing disorder composite onto polygenic scores and covariates in adolescents and young adults. Model I includes gender and age as covariates. Model II includes gender, age, and parental externalizing disorder (ED) history as covariates.
| ADOLESCENTS | ||||||
|---|---|---|---|---|---|---|
|
| ||||||
| Model I (N = 246) | Model II (N = 246) | |||||
|
|
||||||
| B | SE B | t | B | SE B | t | |
|
|
||||||
| Intercept | -2.48 | 0.24 | -10.45 | -2.57 | 0.24 | -10.75 |
| Polygenic score | 6.33 | 1.33 | 4.74 | 5.85 | 1.51 | 3.87 |
| Gender | -0.10 | 0.04 | -2.34 | -0.09 | 0.04 | -2.06 |
| Age | 0.12 | 0.01 | 8.34 | 0.12 | 0.01 | 8.61 |
| Parental history | ||||||
| Either parent ED | -- | -- | -- | set as reference | ||
| Neither parent ED | -- | -- | -- | -0.08 | 0.05 | -1.68 |
| Incomplete parent ED history | -- | -- | -- | 0.12 | 0.06 | 2.09 |
|
| ||||||
| YOUNG ADULTS | ||||||
|
| ||||||
| Model I (N = 190) | Model II (N = 190) | |||||
|
| ||||||
| B | SE B | t | B | SE B | t | |
|
| ||||||
| Intercept | 0.36 | 0.49 | 0.46 | 0.54 | 0.50 | 1.08 |
| Polygenic score | 12.62 | 3.00 | 4.21 | 10.49 | 3.34 | 3.14 |
| Gender | 0.00 | 0.10 | 0.04 | 0.01 | 0.10 | 0.06 |
| Age | -0.02 | 0.02 | -1.06 | -0.03 | 0.22 | -1.25 |
| Parental history | ||||||
| Either parent ED | -- | -- | -- | set as reference | ||
| Neither parent ED | -- | -- | -- | -0.18 | 0.11 | -1.53 |
| Incomplete parent ED history | -- | -- | -- | -0.30 | 0.11 | -2.61 |
Note. Bolded statistics significant at p < .05. Bolded and italicized statistics significant at p < .01. The ‘neither parent ED’ contrast was not significant in the multivariate model for adolescents or young adults; however, it did have univariate main effects in the respective models (B = -0.25, t = -6.68, p < 0.001 and B = -0.44, t = -3.21, p < 0.001, respectively).
Table 2. Variance explained (R2) in subclinical externalizing behavior and impulsivity-related traits as a function of polygenic scores.
| Measure | Adolescents | Young Adults |
|---|---|---|
| Achenbach externalizing | 0.05 | 0.01 |
| Barratt Impulsiveness | 0.07 | 0.03 |
| Conscientiousness | 0.02 | 0.00 |
| Sensation seeking | 0.01 | 0.02 |
Note. Bolded values significant at p < .05. Bolded and italicized values significant at p ≤ .01.
Polygenic Score × Environmental Moderators Analyses
Separate moderated multiple regression analyses using the adolescent subsample indicated that parental monitoring and peer substance use moderated polygenic risk to predict the externalizing disorder composite after controlling for gender, age, and the main effects of the polygenic score and parental monitoring and peer substance use. However, we note that the moderation effect was only marginally significant for parental monitoring [Bparents × polygene = -1.30, t(154) = -1.88, p = 0.06, R2 = 1%; Bpeers × polygene = 1.81, t(155) = 2.68, p < .01, R2 = 2%]. Genetic effects were more pronounced at low levels of parental monitoring and high levels of peer substance use compared to high levels of parental monitoring and low levels of peer substance use (Figure 3).
Figure 3.

Adolescent parental monitoring (Panel A) and perceived peer substance use (Panel B) moderate polygenic risk to predict the externalizing disorder composite.
To address concerns that these gene-by-environment effects were due to gene-environment correlation (polygenic scores predicted 1% and 2% of the variance in parental monitoring and peer substance use), we re-ran our gene-by-environment analyses using residualized polygenic score, parental monitoring, and peer substance use variables (e.g., by saving the residuals from a regression of polygenic scores onto parental monitoring and vice versa). Using residualized variables statistically eliminates gene-environment correlation in the model because the genetic and environmental effects have been partialled from one another. The gene-by-environment effects continued to be significant [Bparents × polygene = -1.60, t(154) = -1.88, p = 0.02; Bpeers × polygene = 1.97, t(155) = 2.84, p < .01], and again indicated that genetic variance increased at low levels of parental monitoring and high levels of peer substance use.
Discussion
We used genome-wide SNP data to examine how polygenic predispositions for adulthood externalizing disorders manifest in earlier developmental stages and whether key environmental factors moderate genetic influences to predict externalizing disorders. We found a modestly sized effect whereby genetic predispositions toward externalizing disorders in adulthood also manifest as clinical-level problems at younger ages. Identifying whether the small effect size reflects a modest genetic correlation between adolescent and adult externalizing disorders or is attributable to the fact that GWAS-derived polygenic scores only account for common (versus rare; Gibson, 2012) genetic variation is an important direction for future research. It is especially impressive that polygenic scores predicted adolescents' and young adults' externalizing disorder composite even after accounting for parental externalizing disorder history. We also found that genetic predispositions for adult externalizing disorders predicted subclinical externalizing behavior and multiple facets of impulsivity in adolescents and young adults. This nicely maps onto evidence from twin studies that a highly heritable behavioral disinhibition factor broadly predisposes individuals to a range of externalizing disorders (Kendler et al., 2003; Krueger et al., 2002). The differential age associations for conscientiousness and sensation seeking (whereby polygenic scores predicted lower levels of adolescent conscientiousness (i.e., constraint) and higher levels of young adult sensation seeking, but not the reverse) suggest that the genetic influences on distinct impulsivity dimensions may change across development.
Finally, polygenic scores had differential associations with the externalizing disorder composite in the context of different environments, with stronger effects for peer substance use. Genetic differences were more pronounced under conditions of low parental monitoring or high peer substance versus conditions of high parental monitoring or low peer substance use. This parallels findings from twin studies showing that genetic influences for externalizing behavior increase under conditions of low parental monitoring and high peer deviance (Button et al., 2007; Harden et al., 2008; Hicks et al., 2009). Such moderation effects likely reflect conditions or processes that may limit the expression of an individual's genetic predispositions toward externalizing (Shanahan & Hofer, 2005). We also note that there was evidence for gene-environment correlation, which is common for psychiatric disorders (Kendler & Baker, 2007). Although gene-environment correlations can produce spurious gene-environment interactions, that concern is mitigated by previous twin analyses in this area that yield evidence for gene-environment interaction after accounting for gene-environment correlation (Button et al., 2007; Hicks et al., 2009) and by our supplementary analyses where we partialed for gene-environment correlation.
As a whole, the present study brings large-scale gene identification efforts into a developmental psychopathology framework to better understand gene-behavior associations. Applying GWAS results for adult externalizing disorders to younger samples and conducting more fine-grained gene-by-development and gene-by-environment analyses is an important integration of the current best practices in genetics research with the types of research questions that are of key interest to psychologists. It is encouraging to see that measured genotypic results are consistent with gene-by-development and gene-by-environment findings from twin samples, particularly in view of the criticisms of twin methods (for a review of these critiques see Tenesa & Haley, 2013). Twin models characterize “genetic influence” latently, by inferring genetic influence based on differences between relatives with varying degrees of genetic sharing. Polygenic risk scores provide a specific measure of genetic risk based on measured genotypes. The fact that divergent methods produce convergent results provides compelling evidence for the developmental and gene-by-environment effects reported here. Further, polygenic approaches have the advantage of providing a more global index of genetic risk goes beyond widely-criticized and only nominally informative candidate gene studies (Duncan & Keller, 2011). Although polygenic approaches do not—by design—identify specific genes associated with an outcome, complementary methods such as gene set analyses may identify the potential biological mechanisms underlying their effects (e.g., through examination of whether SNPs included in polygenic scores are located in functionally related genes (Wang, Jia, Wolfinger, Chen, & Zhao, 2011)). We return to this point shortly.
Despite enthusiasm for using genome-wide information for personalized medicine (Hamburg & Collins, 2010), our results echo the conclusions from previous work (Yan et al., 2013) that it would be premature to use empirically-derived polygenic scores to predict an individual's risk for developing externalizing disorders. Rather, our results highlight the potential clinical utility of environmentally focused preventions and interventions for moderating genetic predispositions toward externalizing disorders. Those who are genetically predisposed toward externalizing disorders may particularly benefit from such intervention efforts (see Brody, Beach, Philibert, Chen, & Murry, 2009 for an example).
Our study has several limitations. First, our participants included only individuals of European-American descent. We selected a homogenous sample in order reduce GWAS false positives due to population stratification; however, the GWAS weights we used to calculate the externalizing disorder polygenic scores in the present study may have limited generalizability as a result. This is because GWAS findings across a number of complex traits from individuals of European ancestry do not replicate in individuals of African ancestry (Marigorta & Navarro, 2013); relatedly, recent findings from alcohol dependence GWAS implicate the same alcohol-metabolizing pathways and genes in African- and European-Americans; however the specific genetic variants differ across populations (Gelernter et al., 2013). Thus, the GWAS weights used to create the polygenic scores in the current study may not be appropriate for constructing externalizing disorder polygenic scores in samples of diverse ancestry. Second, although we find evidence for our hypothesized main and interactive effects, they only account for a small amount of the variance. Third, we relied on a modest set of self-report measures of the parenting and peer environments, and incorporating information from additional reporters or using additional methods is important. Finally, our adolescent and young adult subsamples included relatively broad age ranges, and we were unable to test whether our associations changed as a function of age due to limited sample size.
In summary, we found that polygenic risk for externalizing disorders in adulthood is associated with externalizing disorders, subclinical externalizing behavior, and several impulsivity-related traits in adolescents and young adults, and is moderated by adolescent parenting and peer factors. Examining how polygenic predispositions for externalizing disorders manifest across development and interact with salient environmental moderators is a potentially useful way to characterize pathways toward disorder. These results raise several high-priority directions for future research. We highlight here two directions that we believe have the greatest promise for advancing our understanding of how genetic predispositions toward adult externalizing disorders manifest earlier in development and interact with environmental factors. First, developing theory-driven, holistic measures of the environment is central for moving this area of research forward. In the present study, as in many gene-by-environment interaction studies, we considered parental monitoring and peer substance use in isolation. However, environmental risk factors are often related to one another, and there is no unified framework for measuring or modeling cumulative risk in gene-by-environment interaction studies. A corollary to this point is that to understand gene-by-environment interactions for clinical outcomes across development—an approach that is important for creating effective early preventive-intervention efforts—holistic models of the environment will need to incorporate a developmental perspective. For example, exposure to risk factors later in development may be offset by experiencing protective factors earlier in development (and vice versa) (Rönkä, Oravala, & Pulkkinen, 2002; Salvatore, Haydon, Simpson, & Collins, 2013; Sroufe, Egeland, & Kreutzer, 1990). Bronfenbrenner's (1979) classic ecological model, which conceptualizes the environment as a series of transactional, nested environments that both affect and are affected by one another, provides a useful heuristic for what we are suggesting. Translating this conceptual model into an empirical one is critical for moving beyond “candidate environments” toward an integrative, developmentally sensitive understanding of how multiple, nested environments are likely to interface with genetic predispositions to predict the onset and course of clinical disorders. Psychologists are in a strong position to contribute to these efforts.
Second, as we begin to identify the sets of genetic variants contributing to the polygenic architecture of complex behavioral outcomes—efforts that are currently being led through large GWAS collaborations such as the Psychiatric Genomics Consortium— the next step will be to characterize their underlying biology. Gene-network analyses, which permit examination of whether variants included in polygenic scores are located in functionally related genes (Wang et al., 2011) will be critical to these efforts. Relatedly, there are exciting cross-disciplinary opportunities to identify whether the genetic variants that contribute to polygenic scores are located in regulatory regions of the genome (i.e., regions that include enhancers, promoters, insulators, and silencers) using data from the ENCODE project (ENCODE Project Consortium, 2004). Initial findings from the ENCODE project indicate that many of the top variants that have emerged from GWAS of complex diseases (e.g., multiple sclerosis and ulcerative colitis) are located in potential regulatory DNA regions (ENCODE Project Consortium, 2012). Whether this is the case for common psychiatric disorders, such as the externalizing disorders examined here, remains to be seen. Moving beyond gene identification to identify how sets of genetic variants are implicated in gene regulation will provide critical insights into the biological processes underlying pathways to complex behavioral outcomes.
In closing, continuing to bridge the gaps between the fields of genetics and psychology is imperative so that genetic-psychological science truly represents the state of the science in each field. The nature of large-scale gene identification studies conducted in adult samples has historically precluded a more fine-grained, developmentally informed understanding of how genetic risk for externalizing disorders contributes to psychopathology. Our approach represents a substantial departure from candidate gene studies and illustrates how recent advances in the world of genetics can be successfully integrated into a developmentally-sensitive psychological study to enhance our understanding of how genetic risk unfolds across time and interfaces with environmental factors.
Supplementary Material
Figure S1 (see Supplemental Material available online). Quantile-quantile plot for the externalizing disorder composite genome-wide association analysis.
{kind=link}
Acknowledgments
The Collaborative Study on the Genetics of Alcoholism (COGA), Principal Investigators B. Porjesz, V. Hesselbrock, H. Edenberg, L. Bierut, includes ten different centers: University of Connecticut (V. Hesselbrock); Indiana University (H.J. Edenberg, J. Nurnberger Jr., T. Foroud); University of Iowa (S. Kuperman, J. Kramer); SUNY Downstate (B. Porjesz); Washington University in St. Louis (L. Bierut, A. Goate, J. Rice, K. Bucholz); University of California at San Diego (M. Schuckit); Rutgers University (J. Tischfield); Texas Biomedical Research Institute (L. Almasy), Howard University (R. Taylor) and Virginia Commonwealth University (D. Dick). Other COGA collaborators include: L. Bauer (University of Connecticut); D. Koller, S. O'Connor, L. Wetherill, X. Xuei (Indiana University); Grace Chan (University of Iowa); S.Kang, N. Manz, M. Rangaswamy (SUNY Downstate); J. Rohrbaugh, J-C Wang (Washington University in St. Louis); A. Brooks (Rutgers University); and F. Aliev (Virginia Commonwealth University). A. Parsian and M. Reilly are the NIAAA Staff Collaborators. We continue to be inspired by our memories of Henri Begleiter and Theodore Reich, founding PI and Co-PI of COGA, and also owe a debt of gratitude to other past organizers of COGA, including Ting-Kai Li, currently a consultant with COGA, P. Michael Conneally, Raymond Crowe, and Wendy Reich, for their critical contributions. This national collaborative study is supported by NIH Grant U10AA008401 from the National Institute on Alcohol Abuse and Alcoholism (NIAAA) and the National Institute on Drug Abuse (NIDA). We thank the Genome Technology Access Center in the Department of Genetics at Washington University School of Medicine for help with genomic analysis. The Center is partially supported by NCI Cancer Center Support Grant #P30 CA91842 to the Siteman Cancer Center and by ICTS/CTSA Grant# UL1RR024992 from the National Center for Research Resources (NCRR), a component of the National Institutes of Health (NIH), and NIH Roadmap for Medical Research. This work was also supported by K02AA018755 (DMD), K02DA032573 (AA), and T32MH20030-14 and F32AA022269 (JES). This publication is solely the responsibility of the authors and does not necessarily represent the official view of NCRR or NIH.
Footnotes
Author Contributions: The authors JES and DMD were responsible for the study concept and design, and drafting the manuscript. JES and FA conducted statistical analyses. KB, AA, VH, MH, LB, SK, MAS, JK, HJE, and TMF provided critical revision of the manuscript for important intellectual content. All authors critically reviewed content and approved final version for publication.
Contributor Information
Jessica E. Salvatore, Department of Psychiatry, Virginia Commonwealth University
Fazil Aliev, Department of Psychiatry, Virginia Commonwealth University.
Kathleen Bucholz, Department of Psychiatry, Washington University.
Arpana Agrawal, Department of Psychiatry, Washington University.
Victor Hesselbrock, Department of Psychiatry, University of Connecticut.
Michie Hesselbrock, School of Social Work, University of Connecticut.
Lance Bauer, Department of Psychiatry, University of Connecticut.
Samuel Kuperman, Department of Psychiatry, University of Iowa.
Marc A. Schuckit, Department of Psychiatry, University of California-San Diego
John Kramer, Department of Psychiatry, University of Iowa.
Howard J. Edenberg, Department of Biochemistry and Molecular Biology, Indiana University
Tatiana M. Foroud, Department of Medical and Molecular Genetics, Indiana University
Danielle M. Dick, Department of Psychiatry, Virginia Commonwealth University
References
- Achenbach TM. Manual for the Adult Self-Report. Burlington, VT: University of Vermont Department of Psychiatry; 2003. [Google Scholar]
- Achenbach TM, Rescorla LA. Manual for ASEBA School-Age Forms & Profiles. University of Vermont, Research Center for Children, Youth, & Families; Burlington, VT: 2001. [Google Scholar]
- Begleiter H, Reich T, Hesselbrock V, Porjesz B, Li TK, Schuckit MA, et al. Rice JP. The Collaborative Study on the Genetics of Alcoholism. Alcohol Health & Research World. 1995;19:228–236. [PMC free article] [PubMed] [Google Scholar]
- Belsky DW, Moffitt TE, Baker TB, Biddle AK, Evans JP, Harrington H, et al. Caspi A. Polygenic risk and the developmental progression to heavy, persistent smoking and nicotine dependence: evidence from a 4-decade longitudinal study. JAMA Psychiatry. 2013;70:534–542. doi: 10.1001/jamapsychiatry.2013.736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bosker FJ, Hartman CA, Nolte IM, Prins BP, Terpstra P, Posthuma D, et al. Nolen WA. Poor replication of candidate genes for major depressive disorder using genome-wide association data. Mol Psychiatry. 2011;16:516–532. doi: 10.1038/mp.2010.38. [DOI] [PubMed] [Google Scholar]
- Brody GH, Beach SRH, Philibert RA, Chen YF, Murry VM. Prevention effects moderate the association of 5-HTTLPR and youth risk behavior initiation: Gene × environment hypotheses tested via a randomized prevention design. Child Development. 2009;80:645–661. doi: 10.1111/j.1467-8624.2009.01288.x. [DOI] [PubMed] [Google Scholar]
- Broms U, Wedenoja J, Largeau MR, Korhonen T, Pitkaniemi J, Keskitalo-Vuokko K, et al. Loukola A. Analysis of detailed phenotype profiles reveals CHRNA5-CHRNA3-CHRNB4 gene cluster association with several nicotine dependence traits. Nicotine Tob Res. 2012;14:720–733. doi: 10.1093/ntr/ntr283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bronfenbrenner U. The ecology of human development: Experiments by nature and design. Cambridge, Massachusetts: Harvard University Press; 1979. [Google Scholar]
- Bucholz KK, Cadoret R, Cloninger CR, Dinwiddie SH, Hesselbrock VM, Nurnberger JL, et al. Schuckit MA. A new, semi-structured psychiatric interview for use in genetic linkage studies: A report on the reliability of the SSAGA. Journal of Studies on Alcohol. 1994;55:149–158. doi: 10.15288/jsa.1994.55.149. [DOI] [PubMed] [Google Scholar]
- Button TMM, Corley RP, Rhee SH, Hewitt JK, Young SE, Stallings MC. Delinquent peer affiliation and conduct problems: A twin study. Journal of Abnormal Psychology. 2007;116:554–564. doi: 10.1037/0021-843x.116.3.554. [DOI] [PubMed] [Google Scholar]
- Cardon LR, Palmer LJ. Population stratification and spurious allelic association. Lancet. 2003;361:598–604. doi: 10.1016/S0140-6736(03)12520-2. [DOI] [PubMed] [Google Scholar]
- Chassin L, Pillow DR, Curran PJ, Molina BSG, Barrera M. Relation of parental alcoholism to early adolescent substance use: A test of 3 mediating mechanisms. Journal of Abnormal Psychology. 1993;102:3–19. doi: 10.1037//0021-843x.102.1.3. [DOI] [PubMed] [Google Scholar]
- Chen MH, Yang Q. GWAF: an R package for genome-wide association analyses with family data. Bioinformatics. 2010;26:580–581. doi: 10.1093/bioinformatics/btp710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cicchetti D. The emergence of developmental psychopathology. Child Development. 1984;55:1–7. [PubMed] [Google Scholar]
- Collins AL, Kim Y, Sklar P, O'Donovan MC, Sullivan PF. Hypothesis-driven candidate genes for schizophrenia compared to genome-wide association results. Psychological Medicine. 2012;42:607–616. doi: 10.1017/s0033291711001607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costa PT, McCrae RR. Cross-sectional studies of personality in a national sample: Development and validation of survey measures. Psychology and Aging. 1986;1:140–143. doi: 10.1037//0882-7974.1.2.140. [DOI] [PubMed] [Google Scholar]
- Depue RA, Collins PF. Neurobiology of the structure of personality: dopamine, facilitation of incentive motivation, and extraversion. Behavioral and Brain Sciences. 1999;22:491–517. doi: 10.1017/S0140525X99002046. [DOI] [PubMed] [Google Scholar]
- Devlin B, Roeder K. Genomic control for association studies. Biometrics. 1999;55:997–1004. doi: 10.1111/j.0006-341x.1999.00997.x. [DOI] [PubMed] [Google Scholar]
- Dick DM, Aliev F, Latendresse S, Porjesz B, Schuckit M, Rangaswamy M, et al. Kramer J. How phenotype and developmental stage affect the genes we find: GABRA2 and impulsivity. Twin Research and Human Genetics. 2013;16:661–669. doi: 10.1017/thg.2013.20.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dick DM, Bierut L, Hinrichs A, Fox L, Bucholz KK, Kramer J, et al. Foroud T. The role of GABRA2 in risk for conduct disorder and alcohol and drug dependence across developmental stages. Behavior Genetics. 2006;36:577–590. doi: 10.1007/s10519-005-9041-8. [DOI] [PubMed] [Google Scholar]
- Duncan L, Keller MC. A critical review of the first ten years of measured gene-by-environment interaction research in psychiatry. American Journal of Psychiatry. 2011;168:1041–1049. doi: 10.1176/appi.ajp.2011.11020191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ENCODE Project Consortium. The ENCODE(ENCyclopedia Of DNA Elements) Project. Science. 2004;306:636–640. doi: 10.1126/science.1105136. [DOI] [PubMed] [Google Scholar]
- ENCODE Project Consortium. An integrated encyclopedia of DNA elements in the human genome. Nature. 2012;489:57–74. doi: 10.1038/nature11247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans D, Visscher PM, Wray N. Harnessing the information contained within genome-wide association studies to improve individual prediction of complex disease risk. Human Molecular Genetics. 2009;18:3525–3531. doi: 10.1093/hmg/ddp295. [DOI] [PubMed] [Google Scholar]
- Evenden JL. Varieties of impulsivity. Psychopharmacology. 1999;146:348–361. doi: 10.1007/pl00005481. [DOI] [PubMed] [Google Scholar]
- Gelernter J, Kranzler HR, Sherva R, Almasy L, Koesterer R, Smith AH, et al. Farrer LA. Genome-wide association study of alcohol dependence:significant findings in African- and European-Americans including novel risk loci. Molecular Psychiatry. 2013 doi: 10.1038/mp.2013.145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibson G. Rare and common variants: Twenty arguments. Nature Reviews Genetics. 2012;13:135–145. doi: 10.1038/nrg3118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamburg MA, Collins FS. The path to personalized medicine. New England Journal of Medicine. 2010;363:301–304. doi: 10.1056/NEJMp1006304. [DOI] [PubMed] [Google Scholar]
- Harden KP, Hill JE, Turkheimer E, Emery RE. Gene-environment correlation and interaction in peer effects on adolescent alcohol and tobacco use. Behavior Genetics. 2008;38:339–347. doi: 10.1007/s10519-008-9202-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hesselbrock M, Easton C, Bucholz KK, Schuckit M, Hesselbrock V. A validity study of the SSAGA--a comparison with the SCAN. Addiction. 1999;94:1361–1370. doi: 10.1046/j.1360-0443.1999.94913618.x. [DOI] [PubMed] [Google Scholar]
- Hicks BM, South SC, DiRago AC, Iacono WG, McGue M. Environmental adversity and increasing genetic risk for externalizing disorders. Archives of General Psychiatry. 2009;66:640–648. doi: 10.1001/archgenpsychiatry.2008.554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hingson RW, Heeren T, Winter MR. Age of alcohol-dependence onset: Associations with severity of dependence and seeking treatment. Pediatrics. 2006;118:E755–E763. doi: 10.1542/peds.2006-0223. [DOI] [PubMed] [Google Scholar]
- Kang S, Rangaswamy M, Chorlian D, Manz N, Porjesz B COGA Collaborators. Family-based genome-wide association study of resting EEG identifies UROC1 and neurotransmitter biosynthetic process; Paper presented at the XXIst World Congress of Psychiatric Genetics; Boston. 2013. [Google Scholar]
- Kaprio J, Pulkkinen L, Rose RJ. Genetic and environmental factors in health-related behaviors: Studies on Finnish twins and twin families. Twin Research. 2002;5:366–371. doi: 10.1375/twin.5.5.366. [DOI] [PubMed] [Google Scholar]
- Kendler KS, Baker JH. Genetic influences on measures of the environment: a systematic review. Psychological Medicine. 2007;37:615–626. doi: 10.1017/s0033291706009524. [DOI] [PubMed] [Google Scholar]
- Kendler KS, Prescott CA, Myers J, Neale MC. The structure of genetic and environmental risk factors for common psychiatric and substance use disorders in men and women. Archives of General Psychiatry. 2003;60:929–937. doi: 10.1001/archpsyc.60.9.929. [DOI] [PubMed] [Google Scholar]
- Krueger RF, Hicks BM, Patrick CJ, Carlson SR, Iacono WG, McGue M. Etiological connections among substance dependence, antisocial behavior, and personality: Modeling the externalizing spectrum. Journal of Abormal Psychology. 2002;111:411–424. doi: 10.1037//0021-843X.111.3.411. [DOI] [PubMed] [Google Scholar]
- Marigorta UM, Navarro A. High trans-ethnic replicability of GWAS results implies common causal variants. PLoS Genetics. 2013;9:1–13. doi: 10.1371/journal.pgen.1003566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller E, Joseph S, Tudway J. Assessing the component structure of four self-report measures of impulsivity. Personality and Individual Differences. 2004;37:349–358. doi: 10.1016/j.paid.2003.09.008. [DOI] [Google Scholar]
- Patton JH, Stanford MS, Barratt ES. Factor structure of the Barratt Impulsiveness Scale. Journal of Clinical Psychology. 1995;51:768–774. doi: 10.1002/1097-4679(199511)51:6<768∷AID-JCLP2270510607>3.0.CO;2-1. [DOI] [PubMed] [Google Scholar]
- Plomin R, Haworth CMA, Davis OSP. Common disorders as quantitative traits. Nature Reviews Genetics. 2009;10:872–878. doi: 10.1038/nrg2670. [DOI] [PubMed] [Google Scholar]
- Purcell S, Neale B, Todd-Brown K, Thomas L, Ferreira M, Bender D, et al. Sham P. PLINK: a tool set for whole-genome association and population-based linkage analyses. American Journal of Human Genetics. 2007;81:559–575. doi: 10.1086/519795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rönkä A, Oravala S, Pulkkinen L. “I met this wife of mine and things got onto a better track” Turning points in risk development. Journal of Adolescence. 2002;25:47–63. doi: 10.1006/jado.2001.0448. [DOI] [PubMed] [Google Scholar]
- Russo MF, Stokes GS, Lahey BB, Christ MAG, McBurnett K, Loeber R, et al. Green SM. A Sensation Seeking Scale for children: further refinement and psychometric development. 1993;15:69–86. doi: 10.1007/BF00960609. [DOI] [Google Scholar]
- Salvatore JE, Haydon KC, Simpson JA, Collins WA. The distinctive role of romantic relationships in moderating the effects of early caregiving on adult anxious-depressed symptoms over nine years. Development and Psychopathology. 2013;25:843–856. doi: 10.1017/S0954579413000205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shanahan MJ, Hofer SM. Social context in gene-environment interactions: retrospect and prospect. Journals of Gerontology Series B-Psychological Sciences and Social Sciences. 2005;60B:65–76. doi: 10.1093/geronb/60.Special_Issue_1.65. [DOI] [PubMed] [Google Scholar]
- Slutske WS, Heath AC, Dinwiddie SH, Madden PAF, Bucholz KK, Dunne MP, et al. Martin NG. Common genetic risk factors for conduct disorder and alcohol dependence. Journal of Abnormal Psychology. 1998;107:363–374. doi: 10.1037/0021-843X.107.3.363. [DOI] [PubMed] [Google Scholar]
- Sroufe LA, Egeland B, Kreutzer T. The fate of early experience following developmental change: Longitudinal approaches to individual adaptation in childhood. Child Development. 1990;61:1363–1373. doi: 10.2307/1130748. [DOI] [PubMed] [Google Scholar]
- Tenesa A, Haley CS. The heritability of human disease: Estimation, uses, and abuses. Nature Reviews Genetics. 2013;14:139–149. doi: 10.1038/nrg3377. [DOI] [PubMed] [Google Scholar]
- The International Schizophrenia Consortium. Common polygenic variation contributes to risk of schizophrenia and bipolar disorder. Nature. 2009;460:748–752. doi: 10.1038/nature08185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas D. Gene-environment-wide association studies: Emerging approaches. Nature Reviews Genetics. 2010;11:259–272. doi: 10.1038/nrg2764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomasson HR, Edenberg HJ, Crabb DW, Mai XL, Jerome RE, Li TK, et al. Yin SJ. Alcohol and aldehyde dehydrogenase genotypes and alcoholism in Chinese men. American Journal of Human Genetics. 1991;48:667–681. [PMC free article] [PubMed] [Google Scholar]
- Tobacco and Genetics Consortium. Genome-wide meta-analyses identify multiple loci associated with smoking behavior. Nature Genetics. 2010;42 doi: 10.1038/ng.571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vrieze SI, McGue M, Iacono WG. The interplay of genes and adolescent development in substance use disorders: Leveraging findings from GWAS meta-analyses to test developmental hypotheses about nicotine consumption. Human Genetics. 2012;131:791–801. doi: 10.1007/s00439-012-1167-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang JC, Foroud T, Hinrichs AL, Le NX, Bertelsen S, Budde JP, et al. Goate AM. A genome-wide association study of alcohol-dependence symptom counts in extended pedigrees identifies C15orf53. Molecular Psychiatry. 2013;18:1218–1224. doi: 10.1038/mp.2012.143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L, Jia P, Wolfinger RD, Chen X, Zhao Z. Gene set analysis of genome-wide association studies: Methodological issues and perspectives. Genomics. 2011;98:1–8. doi: 10.1016/j.ygeno.2011.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warner LA, Kessler RC, Hughes M, Anthony JC, Nelson CB. Prevalence and correlates of drug-use and dependence in the United States: Results from the National Comborbidity Survey. Archives of General Psychiatry. 1995;52:219–229. doi: 10.1001/archpsyc.1995.03950150051010. [DOI] [PubMed] [Google Scholar]
- Whiteside SP, Lynam DR. The Five Factor Model and impulsivity: Using a structural model of personality to understand impulsivity. Personality and Individual Differences. 2001;30:669–689. doi: 10.1016/S0191-8869(00)00064-7. [DOI] [Google Scholar]
- Yan J, Aliev F, Webb BT, Kendler KS, Edenberg HJ, Agrawal A, et al. Dick DM. Using genetic information from candidate gene and genome wide association studies in risk prediction for alcohol dependence. Addiction Biology. 2013 doi: 10.1111/adb.12035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young SE, Stallings MC, Corley RP, Krauter KS, Hewitt JK. Genetic and environmental influences on behavioral disinhibition. American Journal of Medical Genetics. 2000;96:684–695. doi: 10.1002/1096-8628(20001009)96:5<684∷aid-ajmg16>3.0.co;2-g. [DOI] [PubMed] [Google Scholar]
- Zuckerman M. Sensation seeking: A comparative approach to a human trait. Behavioral and Brain Sciences. 1984;7:413–471. doi: 10.1017/S0140525X00018938. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1 (see Supplemental Material available online). Quantile-quantile plot for the externalizing disorder composite genome-wide association analysis.